

Abstract
The production of metallo-β-lactamases is the most important strategy by which pathogenic bacteria become resistant to currently known β-lactam antibiotics. The emergence of these enzymes is particularly concerning for the future treatment of bacterial infections. There are no clinically available drugs capable of inhibiting any of the metallo-β-lactamases, so there is an urgent need to find such inhibitors. In this review, an up-to-date status of the inhibitors investigated for the inhibition of metallo-β-lactamases has been given so that this rich source of structural information of presently known metallo-β-lactamases could be helpful in generating a broad-spectrum potent inhibitor of metallo-β-lactamases.
Keywords: Metallo-beta-lactamases, Inhibition, Thiol, β-Lactam analogues, Peptides, Biphenyl tetrazoles, Triazoles
Introduction
β-Lactam antibiotics are the most widely used antibacterial agents for treating bacterial infections. However, many pathogenic bacteria have developed resistance against these antibiotics through mechanisms such as a decrease in cell wall permeability, efflux pump, and hydrolysis of the β-lactam ring by β-lactamases [1, 2]. β-Lactamases of class B are metallo-proteins, also called metallo-β-lactamases. These enzymes use a zinc-bound hydroxyl group as the nucleophile [3] to promote the hydrolysis of a very broad range of β-lactam antibiotics, including penicillins, cephalosporins, and carbapenems [4].
Serine-β-lactamase inhibitors do not inhibit metallo-β-lactamases and there are no clinically approved inhibitors of metallo-β-lactamases. So there is an urgent need for the development of such an inhibitor that could inhibit all metallo-β-lactamases. Here, we discuss currently available MBL inhibitors from different groups to provide a collective view of the chemical structures of these inhibitors, which could be helpful in designing inhibitors capable of meeting the serious biological threat, resistance of pathogenic bacteria to β-lactam antibiotics.
Phenazines
Gilpin et al., [5] isolated two novel phenazines (SB 212021 and SB212305) from a streptomyces in 1995 and screened them against three metallo-β-lactamases Xanthomonas maltophilia L-l, Bacteroides fragilis 262 CfiA, and Bacillus cereus II. These compounds are non-specific and appear to chelate the zinc, so when zinc levels are increased, enzyme activity recovers. In the absence of added zinc, both compounds had IC50s of 1∼75 μM for the B. fragilis 262 CfiA and X. maltophilia L-1 metallo-β-lactamases. SB212305, which contains a thio group, has the lowest IC50 = 1 μM for X. maltophilia L-1, while in the case of B. cereus II, the phenazine SB212021 has the lowest IC50 = 37 μM (Figure 1).
Fig. 1.
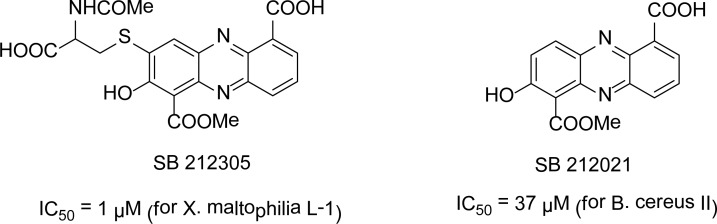
Phenazine inhibitors
Thiol derivatives
Due to the high affinity of sulfur for zinc, it has been found that compounds having a thiol group showed promising inhibition against MBLs. A series of mercaptoacetic acid thiol esters [6] was synthesized and identified as metallo-β-lactamase inhibitors. Mass spectrometric data suggest that mercaptoacetic thiol esters act as mechanism-based inhibitors for BcII by generating mercaptoacetic acid in situ, which forms a disulfide linkage with a cysteine residue in the active site of the enzyme. The inhibitors of this series showed a broad range of potencies (IC50’s varied from mM to μM) against the enzymes.
A series of thioesters and thiols synthesized by a novel solid-phase Mitsunobu reaction were screened [7] against the CcrA and IMP-1 enzymes. These compounds showed better inhibition for IMP-1 than for CcrA MBL. In the thiodepsipeptides series, the most potent inhibitor was compound 1 with an IC50 of 0.25 μM against IMP-1. However, the thiol moieties of the thiodepsipeptides showed better inhibition, with IC50s of 0.086–0.023 μM against IMP-1. On the other hand, the simple acetyl and benzoyl thioesters were found to be significantly more potent inhibitors than the thiols themselves. This comparison of activity of thiols and thioesters is shown in the Figure 2.
Fig. 2.

Comparison of IC50 values of thiols and thiol esters
In 1998, S. Bounaga and his co-workers [8] reported N-(2′-mercaptoethyl)-2-phenylacetamide 6 as a competitive inhibitor of β-lactamase II from B. cereus with a Ki of 70 mM. This compound was also identified as a competitive inhibitor of L1 with a Ki of 50 ± 3 μM [9].
Thiomandelic acid [10] was identified as a broad spectrum inhibitor with a sub-micromolar Ki value for the nine MBLs tested, except that from the Aeromonas hydrophila enzyme. The Ki values of R and S-thiomandelic acids for B. cereus enzyme were found 0.09 and 1.28 μM, respectively (Figure 3). Kurosaki at al., [11] reported the two irreversible inhibitors 7 and 8 with Ki values of 3.452 ± 0.030 μM and 0.423 ± 0.013 μM, respectively for the IMP-1 enzyme (Figure 3).
Fig. 3.

Thiols inhibitors of MBLs
Siemann et al., also studied thiols as classical inhibitors of MBLs and reported mercaptoacetic acid 9 (IC50 = 1.3 μM) and 1,2-benzenedimethanethiol 10 (IC50 = 2.3 μM) as inhibitors of the IMP-1 enzyme [12]. Demonstrating the efficiency of the Dynamic Combinatorial Mass Spectrometry (DCMS) technique, N-Benzoyl-D-cysteine 11[13] was identified as the potent inhibitor for BcII enzyme with a Ki value of 740 nM. D-captopril [14] was reported as a broad spectrum potent inhibitor of subclass B1 and B3, but its potency against subclass B2 is low (Ki = 72 μM).
In search of potent inhibitors of all subclasses of MBLs, Liénard and his co-workers [15] synthesized compounds containing thiol function(s). Compounds 12 and 13 were found to inhibit MBLs from all three subclasses. For the monozinc CphA MBL, compounds 12 and 13 were reported to be the most potent inhibitors with Ki = 90 nM and Ki = 50 nM, respectively. Lassaux et al., [16] synthesized mercaptophosphonate compounds and screened them against all subclasses of MBLs. Most of their synthesized compounds were found to be competitive inhibitors for all three subclasses B1, B2, and B3 MBLs. With some exceptions, all the mercaptophosphonate derivatives showed good inhibitory effects on the CphA, a subclass B2 enzyme with low inhibition constants (Ki < 15 μM). The most potent broad spectrum inhibitor 14 of this series is given in Figure 3.
The mercaptocarboxylate, PhenylC3SH (Figure 3) [17], acts as a potent inhibitor of the VIM-2 enzyme with a Ki value of 220 nM, whereas it is less active for the IMP-1 enzyme (Ki = 1660 nM) [18].
Concha and others [19] determined the crystal structure of the IMP-1- mercaptocarboxylate 15 complex and found that this mercaptocarboxylate inhibits the IMP-1, B. fragilis, and L1 enzymes with IC50 values between 100 and 500 nM. W. Jin [18] and his colleagues studied the inhibitory effect of two series of compounds, 2-ω-phenylalkyl-3-mercaptopropionic acids and N-[(7-chloro-quinolin-4-ylamino)-alkyl]-3-mercapto-propionamides, on IMP-1 and VIM-2 metallo-β-lactamases. Among the first series, PhenylC4SH (Figure 3) was found to be the potent inhibitor of both IMP-1 and VIM-2 with IC50 values of 1.2 and 1.1 μM, respectively, whereas QuinolineC4SH (Figure 3) of the second series showed maximum inhibition for the IMP-1 (IC50 = 2.5 μM) and VIM-2 (IC50 = 2.4 μM) enzymes.
Trifluoromethyl alcohols and ketones
Trifluoromethyl ketones are reported as serine proteases [20, 21] and zinc dependent carboxypeptidase [22] enzymes. Walter et al., [23, 24] reported the trifluoromethyl compounds as the first synthetic inhibitors of different strains of MBLs. They synthesized different N-phenoxyacetyl-substituted trifluoromethyl ketones and alcohols and screened against MBLs from X. maltophilia ULA-511, A. hydrophila AE036, B. cereus 569H, and Pseudomonas aeruginosa 101 as inhibitors. Among these trifluoromethyl ketones and alcohols, the most potent inhibitors 16–19 are given in Figure 4 along with the Ki values.
Fig. 4.

Ki values of trifluoromethyl ketones and alcohol
Biphenyl tetrazoles
Screening of the Merck chemical collection by Toney et al., [25] led to the identification of biphenyl tetrazoles as potent competitive inhibitors of metallo-β-lactamase (B. fragilis). The structure-activity relationships of biphenyl tetrazoles showed that unsubstituted BPT is a poor inhibitor with IC50 value of 860 ± 60 μM. It was found that the ortho position of the tetrazole group, relative to the biphenyl ring system, is important in enzyme inhibition, because movement of the tetrazole group to the meta or para positions led to the loss of inhibitory activity [25]. It was also found that substitution at the 4′-position of the BPT further increased the inhibitory activity of the BPTs. Figure 5 shows the IC50 values comparison of the parent BPT and the substituted BPTs. To further explore the potency of BPTs as MBLs inhibitors, in 1999, Toney et al., [26] screened a series of BPTs containing 3-n-butyl-1-phenylpyrazole-5-carboxylate against B. fragilis and IMP-1 metallo-β-lactamases. The parent BPT 20 was found to be a weak inhibitor with an IC50 of ∼ 200 ± 8 μM of B. fragilis MBL, while against IMP-1, it was found to be inactive (IC50 >200 μM). The substitution upon the phenyl ring and esterification of the carboxylic group of compound 20 lowered the IC50 value up to 60 ± 30 μM 21 for IMP-1 and 18 ± 2 μM 22 for B. fragilis MBL (Figure 5).
Fig. 5.

Comparison of IC50 values of BPT and substituted BPTs
Succinic and phthalic acid derivatives
The IMP-1 enzyme is a plasmid-borne zinc metalloenzyme responsible for the hydrolysis of β-lactam antibiotics, including carbapenems, rendering them ineffective. To protect the broad spectrum antibiotics from hydrolysis by IMP-1, Toney et al., [27] have identified a series of 2,3-(S,S)-disubstituted succinic acids as potent inhibitors of IMP-1. Among this series, the most potent inhibitor is 23 with an IC50 of 0.0027 μM. In 2005, Toney [28] and others reported several novel succinic acid derivatives, as IMP-1 inhibitors showing mixed inhibition. They reported compound 20707 with lowest Ki value of 3.3 ± 1.7 μM.
Using docking methodologies and experimental enzyme kinetics, Olsen [29] and his coworkers identified several succinic acid derivatives as the di-zinc metallo-β-lactamase inhibitors. The potent inhibitors, 24 and 25 (Figure 6), of this series have an IC50 value range of 10-100 μM. Hiraiwa et al., [30] synthesized substituted phthalic acids and screened against the IMP-1 enzyme for inhibitory activity. Phthalic acid has almost no inhibitory activity against IMP-1, but 3-substituted phthalic acid derivatives are potent inhibitors of this enzyme. Some of the most potent inhibitors 26–29 of this series, along with their IC50 values, are given in Figure 6.
Fig. 6.

Succinic and phthalic acid derivatives as MBL inhibitors
Hydroxamates
Walter et al., [31] synthesized amino acid-derived hydroxamates and screened against different MBLs for inhibitory activity and found several compounds as the inhibitors of clinically relevant enzymes from A. hydrophila. In 2006, B. M. R. Liénard and his colleagues [32] synthesized a series of derivatives of benzohydroxamic acid 30 and tested them against FEZ-1, IMP-1, BcII, CphA, and L1 MBLs for inhibitory activity. Their study resulted in the identification of selective inhibitors of FEZ-1 metallo-β-lactamase. The most potent selective inhibitor of FEZ-1 from this series was identified as 2,5-substituted benzophenone hydroxamic acid 31 (Figure 7) with a Ki value of 6.1 ± 0.7 μM.
Fig. 7.
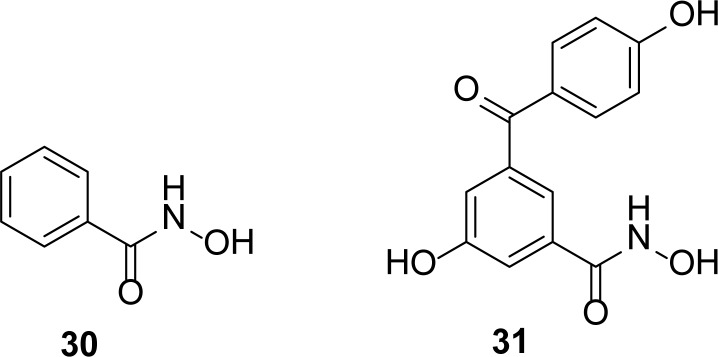
Hydroxamates as FEZ-1 inhibitor
β-Lactam analogues
In search of broad spectrum potent inhibitors of MBLs, a variety of 1β-methylcarbapenem conjugates were tested against different MBLs [33]. The kinetic studies showed that 1β-methylcarbapenems having dithiocarbamate, benzothienythio, or pyrrolidinylthio moieties at the C-2 position showed promising inhibition against MBLs. The most potent inhibitor among these compounds is J-110,441, which simultaneously targets class A, B, and C β-lactamases. It almost acts as a broad spectrum inhibitor of MBLs with Ki values of 0.83, 1.00, 0.23, and 0.0037 μM for II from B. cereus, L1from Stenotrophomonas maltophilia, CcrA from B. fragilis, and IMP-1 MBLs, respectively [33]. A novel 1β-methylcarbapenem with a trans-3,5-disubstituted pyrrolidinylthio moiety at the C-2 position (J-111,225) inhibits the IMP-1 enzyme with a Ki of 0.18 μM [34]. F. V. Hovel et al., [35] also reported penicillin and their rearranged products as potential inhibitors of B. cereus MBL.
Buynak et al., [36] synthesized penicillin-derived inhibitors that simultaneously inhibit both serine and metallo-β-lactamases. The 6-(mercaptomethyl)penicillinates 32–35 (Figure 8) were found as good inhibitors of L1 and BCII MBLs with a Ki range 0.10–32.1 μM. Tsang et al., [37] reported 8-thioxocephalosporins as weak competitive inhibitors (Ki ∼ 700 μM) of B.cereus MBL. Interestingly, the hydrolysis product of thioxocephalosporin, a thioacid, acts as a competitive inhibitor with a Ki = 96 μM, while the cyclic thioxo-piperazinedione, formed by intramolecular aminolysis of thioxo-cephalexin, inhibits the same enzyme with a Ki of 29 μM. Badarau [38] and his coworkers also reported that the hydrolysis products of cephalosporins and thiols inhibit the B.cereus MBL at the micromolar range. Beharry et al.,[39] identified 6-alkylidene-2-substituted penam sulfones as inhibitors of BIa2 with IC50 values less than 10 μM. Compound 36 is the representative of this series with the lowest IC50 of 1.0 μM. A series of cephalosporin-derived reverse hydroxamates and oximes were prepared and tested against MBLs for inhibitory activity. The reverse hydroxamates were found to inhibit the GIM-1 MBL at the submicromolar level [40]. Cyclobutanone analogues of β-lactams [41] (37 and 38) were also reported as the inhibitors of the IMP-1 enzyme.
Fig. 8.

β-Lactam-derived MBL inhibitors
Peptides
Sanschagrin et al., [42] reported a peptide as an inhibitor for metallo-β-lactamases. Cys-Val-His-Ser-Pro-Asn-Arg-Glu-Cys was identified as a promising inhibitor of the L1 enzyme showing mixed inhibition, Ki competitive of 16 ± 4 μM and a Ki uncompetitive of 9 ± 1μM.
Bounaga et al.,[23] synthesized several cysteinyl peptides and identified N-carbobenzoxy-D-cysteinyl-D-phenylalanine 39 as the most potent reversible competitive inhibitor of the B. cereus MBL with a Ki value of 3.0 μM. A library of homo-cysteinyl peptides [44] was synthesized and screened for inhibitory activity against L1 metallo-β-lactamase. It was found that homo-cysteinyl peptides are more active than the cysteinyl peptides. The most active compound of the homo-cysteinyl peptides is 40 with a Ki value of 2.1 nM (Figure 9).
Fig. 9.

Cysteinyl peptides and pyridine dicarboxylic acid inhibitors of MBLs
Pyridine Dicarboxylates
Different pyridine dicarboxylates were tested against different MBLs. 2-picolinic and pyridine-2,4-dicarboxylic acids 41 and 42 (Figure 9) were identified as competitive inhibitors of CphA MBL with Ki values of 5.7 and 4.5 μM, respectively [45]. Roll et al., [46] screened natural products, pyridine monothiocaraboxylic acid analogues, for inhibitory activity against MBLs. Among these naturally isolated compounds, dithioacid 43 (Figure 9) was found to be the strongest inhibitor of CcrA from B. fragilis and L1 from S. maltophilia.
Natural Products
Screening of an extract from a strain of Chaetomium funicola against B. cereus II resulted in the identification of tricyclic natural products (SB238569, SB236050, and SB236049) [47] (Figure 10) as MBL inhibitors. The most active of these natural products was the SB238569 with Ki values of 3.4, 17.0, and 79.0 μM for B. fragilis CfiA, P. aeruginosa IMP-1, and B. cereus II MBL, respectively. The flavonoids galangin and quercetin[48] (Figure 10) were also reported as the inhibitors of MBL from S. maltophilia.
Fig. 10.

Natural product-based inhibitors of MBLs
Triazoles and N-acylated thiosemicarbazides
The VIM-2 enzyme is the most commonly found MBL in clinical isolates worldwide [49–51]. In search of a potent inhibitor of VIM-2, Minond [52] and others screened a library of pharmacologically active compounds and identified two potent and competitive novel sulphonyl-triazoles, 44 (Ki = 0.41 ± 0.03 μM) and 45 (Ki = 1.4 ± 0.10 μM), which are inhibitors of VIM-2 MBL. To improve the potency of compound 44, Weide et al., [53] varied the substitutions on the triazole ring and generated the most potent inhibitor 46 of this series with a Ki of 0.01 ± 0.001 μM. Some other inhibitors 47–49 resulting from this work are given in Figure 11.
Fig. 11.

Triazoles and N-acylated thiosemicarbazide inhibitors of VIM-2 and IMP-1 MBLs
Vella et al., [2] recently screened a 500 compound Maybridge™ library for several new classes of leading inhibitors against the IMP-1 MBL, and considered the 4-methyl-5-(trifluoromethyl)-4H-1,2,4-triazole-3-thiol 50 (Ki = 0.97 ± 0.60 mM) the most promising for further study. We elaborated this ring system to increase the potency of this compound and identified the mercapto triazole 51 as showing mixed inhibition (Ki competitive = 75 ± 30 μM and Ki uncompetitive = 56 ± 10 μM) for the IMP-1 enzyme. We found that the N-acylated thiosemicarbazide intermediates in the synthesis of mercapto triazoles are also potent inhibitors of IMP-1 MBL with Ki values as low as 11μM [54]. N-acylated thiosemicarbazide 52 has the maximum inhibitory activity showing mixed inhibition (Ki competitive = 11 ± 4 μM and Ki uncompetitive = 20 ± 5 μM) for the IMP-1 enzyme Figure 11.
Hetrocyclic Thiones and Disulfides
Tamilselvi and Mugesh [55] studied the hydrolysis of cephalosporins and found that cephalosporins having heterocyclic -SR side chains are less prone to MBL-mediated hydrolysis. This is partly due to the inhibitory activity of the thione moieties eliminated during hydrolysis. They carried out the enzymatic hydrolysis of oxacillin in the presence of hetrocyclic thiones and disulfides 53–58 (Figure 12) and found that the catalytic activity of the BcII enzyme was significantly reduced by these compounds. However, these inhibitors are not as potent as the aliphatic thiol 9[12].
Fig. 12.

Thiones and disulfides as BcII enzyme inhibitors
Pyrrole Derivatives
Mohamed et al., [56] recently synthesized pyrrole derivatives and tested for their inhibitory activity against the IMP-1 enzyme from P. aeruginosa and Klebsiella pneumonia. They reported some compounds showing micromolar inhibition constants for IMP-1 MBL. Compound 59 showed the maximum inhibition with a Ki value of 12 ± 4 μM, while compounds 60 and 61 (Figure 13) have Ki values of 15 ± 5 μM and 18 ± 9 μM, respectively.
Fig. 13.
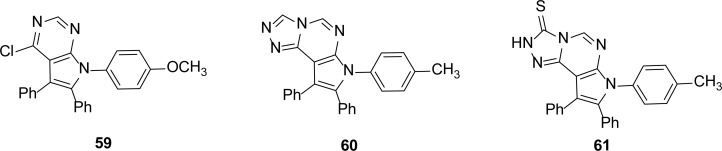
Pyrrole Based inhibitors of the IMP-1 enzyme
Single Stranded DNAs
Kim [57] and his colleagues identified single-stranded DNAs as rapid, reversible, and non-competitive inhibitors of Bacillus cereus 5 / B / 6 MBL, with Ki and K′i values in the nanomolar range. They assayed these ssDNA’s against other zinc-dependent metalloenzymes like porcine carboxypeptidase A, but there was no inhibitory effect on the catalytic activity of these enzymes. Hence, these inhibitors are highly specific to B. cereus 5 / B / 6 metallo-β-lactamase. They found 30 residue ssDNA (Ki = 0.92 nM and K′i = 11 nM) and 10 residue ssDNA (Ki = 0.31 nM and K′i = 1.5 nM), the most potent non-competitive inhibitors of B. cereus MBL.
Sulfonic Acid Derivatives
Siemann [58] and his coworkers reported N-arylsulfonyl hydrazones as novel inhibitors of the IMP-1 enzyme. The most potent inhibitor of IMP-1 MBL is compound 62 with an IC50 of 1.6 μM. 4-Morpholinoethanesulfonic acid 63 has also been reported as a competitive inhibitor of MBL from B. fragilis with a Ki of 23 ± 5 mM [59]. Simm [60] reported bulgecin A 64 as a novel inhibitor of binuclear MBLs. It competitively inhibits (Ki = 230 ± 10 μM) BcII enzyme in its two-zinc form, but fails to inhibit when the enzyme is in the single-zinc form, while it inhibits (Ki = 2.5 ± 0.3 μM) the L1 enzyme in a partially competitive mode (Fig. 14).
Fig. 14.

Sulfonated inhibitors of MBLs
Summary
To overcome the problem of increasing resistance of pathogenic bacteria by expressing metallo-β-lactamases to the presently known β-lactam antibacterial agents, several research groups are actively engaged in discovering broad spectrum potent inhibitors of metallo-β-lactamases. Several chemical classes of metallo-β-lactamases have been reported, but there is no such inhibitor to overcome this problem. The challenge for the medicinal chemists and pharmaceutical industries will be to continue to identify such a broad spectrum inhibitor to get rid of this clinical threat. In this review, currently known potent inhibitors of metallo-β-lactamases are presented and we are hopeful that this review could provide a platform for designing a broad spectrum potent inhibitor of all metallo-β-lactamases.
Authors’ Statement
Competing Interests
The authors declare no conflict of interests.
Footnotes
This article is available from: http://dx.doi.org/10.3797/scipharm.1302-08
References
- [1].Masuda N, Sakagawa E, Ohya S. Outer membrane proteins responsible for multiple drug resistance in pseudomonas aeruginosa. Antimicrob Agents Chemother. 1995;39:645–649. doi: 10.1128/AAC.39.3.645. http://dx.doi.org/10.1128/AAC.39.3.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Vella P, Hussein WM, Leung EWW, Clayton D, Ollis DL, Mitic N, McGeary PR. The identification of new metallo-β-lactamase inhibitor leads from fragment-based screening. Bioorg Med Chem Lett. 2011;21:3282–3285. doi: 10.1016/j.bmcl.2011.04.027. http://dx.doi.org/10.1016/j.bmcl.2011.04.027. [DOI] [PubMed] [Google Scholar]
- [3].Ambler RP. The structure of beta-lactamases. Philos Trans R Soc London B. 1980;289:321–331. doi: 10.1098/rstb.1980.0049. http://dx.doi.org/10.1098/rstb.1980.0049. [DOI] [PubMed] [Google Scholar]
- [4].Felici A, Amicosante G, Oratore A, Strom R, Ledent P, Joris B, Fanuel L, Frère JM. An overview of the kinetic parameters of class B beta-lactamases. Biochem J. 1993;291:151–155. doi: 10.1042/bj2910151. http://www.ncbi.nlm.nih.gov/pubmed/8471035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gilpin ML, Fluston M, Payne D, Cramp R, Hood I. Isolation and structure determination of two novel phenazines from a streptomyces with inhibitory activity against metallo-enzymes, including metallo-β-lactamase. J Antibiot. 1995;48:1081–1085. doi: 10.7164/antibiotics.48.1081. http://dx.doi.org/10.7164/antibiotics.48.1081. [DOI] [PubMed] [Google Scholar]
- [6].Payane DJ, Bateson JH, Gasson BC, Proctor D, Khushi T, Farmar TH, Tolson DA, Bell D, Skett PW, Marshall AC, Reid R, Ghosez L, Combret Y, Marchand-Brynaert J. Inhibition of metallo-beta-lactamases by a series of mercaptoacetic acid thiol ester derivatives. Antimicrob Agents Chemother. 1997;41:135–140. doi: 10.1128/aac.41.1.135. http://www.ncbi.nlm.nih.gov/pubmed/8980769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Greenlee ML, Laub JB, Balkovec JM, Hammond ML, Hammond GG, Pompliano DL, Epstein-Toney JH. Synthesis and SAR of thioester and thiol inhibitors of IMP-1 Metallo-β-Lactamase. Bioorg Med Chem Lett. 1999;9:2549–2554. doi: 10.1016/s0960-894x(99)00425-4. http://dx.doi.org/10.1016/S0960-894X(99)00425-4. [DOI] [PubMed] [Google Scholar]
- [8].Bounanga S, Laws AP, Galleni M, Page MI. The mechanism of catalysis and the inhibition of the Bacillus cereuszinc-dependent β-lactamase. Biochem J. 1998;331:703–711. doi: 10.1042/bj3310703. http://www.ncbi.nlm.nih.gov/pubmed/9560295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yang KW, Crowder MW. Inhibition Studies on the Metallo-β-lactamase L1 from Stenotrophomonas maltophilia. Arch Biochem Biophys. 1999;368:1–6. doi: 10.1006/abbi.1999.1293. http://dx.doi.org/10.1006/abbi.1999.1293. [DOI] [PubMed] [Google Scholar]
- [10].Mollard C, Moali C, Papamicael C, Christian Damblon C, Vessilier S, Amicosante G, Schofield CJ, Galleni M, Frere J-M, Roberts GCK. Thiomandelic Acid, a Broad Spectrum Inhibitor of Zinc_β-Lactamases. J Biol Chem. 2001;276:45015–45023. doi: 10.1074/jbc.M107054200. http://dx.doi.org/10.1074/jbc.M107054200. [DOI] [PubMed] [Google Scholar]
- [11].Kurosaki H, Yamaguchi Y, Higashi T, Soga K, Matsueda S, Yumoto H, Misumi S, Yamagata Y, Arakawa Y, Goto M. Irreversible Inhibition of Metallo-b-lactamase (IMP-1) by 3-(3-Mercaptopropionylsulfanyl)- propionic Acid Pentafluorophenyl Ester. Angew Chem Int Ed. 2005;44:3861–3864. doi: 10.1002/anie.200500835. http://dx.doi.org/10.1002/anie.200500835. [DOI] [PubMed] [Google Scholar]
- [12].Siemann S, Clarke AJ, Viswanatha T, Dmitrienko GI. Thiols as Classical and Slow-Binding Inhibitors of IMP-1 and Other Binuclear Metallo-β-lactamases. Biochemistry. 2003;42:1673–1683. doi: 10.1021/bi027072i. http://dx.doi.org/10.1021/bi027072i. [DOI] [PubMed] [Google Scholar]
- [13].Liénard BMR, Hüting R, Lassaux P, Galleni M, Frère J-M, Schofield CJ. Dynamic Combinatorial Mass Spectrometry Leads to Metallo-β-lactamase Inhibitors. J Med Chem. 2008;51:684–688. doi: 10.1021/jm070866g. http://dx.doi.org/10.1021/jm070866g. [DOI] [PubMed] [Google Scholar]
- [14].Heinz U, Bauer R, Wommer S, Meyer-Klaucke W, Papamichaels C, Bateson J, Adolph HW. Coordination Geometries of Metal Ions in D- or L-Captopril-inhibited Metallo-β-lactamases. J Biol Chem. 2003;278:20659–20666. doi: 10.1074/jbc.M212581200. http://dx.doi.org/10.1074/jbc.M212581200. [DOI] [PubMed] [Google Scholar]
- [15].Liénard BM, Garau G, Horsfall L, Karsisiotis AI, Damblon C, Lassaux P, Papamicael C, Roberts GC, Galleni M, Dideberg O, Frère JM, Schofield CJ. Structural basis for the broad-spectrum inhibition of metallo-beta-lactamases by thiols. Org Biomol Chem. 2008;6:2282–2294. doi: 10.1039/b802311e. http://dx.doi.org/10.1039/b802311e. [DOI] [PubMed] [Google Scholar]
- [16].Lassaux P, Hamel M, Gulea M, Delbrück H, Mercuri PS, Horsfall L, Dehareng D, Kupper M, Frère JM, Hoffmann K, Galleni M, Bebrone C. Mercaptophosphonate Compounds as Broad-Spectrum Inhibitors of the Metallo-β-lactamases. J Med Chem. 2010;53:4862–4876. doi: 10.1021/jm100213c. http://dx.doi.org/10.1021/jm100213c. [DOI] [PubMed] [Google Scholar]
- [17].Yamaguchi Y, Jin W, Matsunaga K, Ikemizu S, Yamagata Y, Wachino J, Shibata N, Arakawa Y, Kurosaki H. Crystallographic investigation of the inhibition mode of a VIM-2 metallo-beta-lactamase from Pseudomonas aeruginosa by a mercaptocarboxylate inhibitor. J Med Chem. 2007;50:6647–6653. doi: 10.1021/jm701031n. http://dx.doi.org/10.1021/jm701031n. [DOI] [PubMed] [Google Scholar]
- [18].Jin W, Arakawa Y, Yasuzawa H, Taki T, Hashiguchi R, Mitsutani K, Shoga A, Yamaguchi Y, Kurosaki H, Shibata N, Ohta M, Goto M. Comparative study of the inhibition of metallo--lactamases (IMP-1 and VIM-2) by thiol compounds that contain a hydrophobic group. Biol Pharm Bull. 2004;27:851–856. doi: 10.1248/bpb.27.851. http://dx.doi.org/10.1248/bpb.27.851. [DOI] [PubMed] [Google Scholar]
- [19].Concha NO, Janson CA, Rowling P, Pearson S, Cheever CA, Clarke BP, Lewis C, Galleni M, Frère JM, Payne DJ, Bateson JH, Abdel-Meguid SS. Crystal Structure of the IMP-1 Metallo β-Lactamase from Pseudomonas aeruginosa and Its Complex with a Mercaptocarboxylate Inhibitor: Binding Determinants of a Potent, Broad-Spectrum Inhibitor. Biochemistry. 2000;39:4288–4298. doi: 10.1021/bi992569m. http://dx.doi.org/10.1021/bi992569m. [DOI] [PubMed] [Google Scholar]
- [20].Gelb MH, Svaren JP, Abeles RH. Fluoro ketone inhibitors of hydrolytic enzymes. Biochemistry. 1985;24:1813–1817. doi: 10.1021/bi00329a001. http://dx.doi.org/10.1021/bi00329a001. [DOI] [PubMed] [Google Scholar]
- [21].Brady K, Wei A, Ringe D, Abeles RH. Structure of chymotrypsin-trifluoromethyl ketone inhibitor complexes: comparison of slowly and rapidly equilibrating inhibitors. Biochemistry. 1990;29:7600–7607. doi: 10.1021/bi00485a009. http://dx.doi.org/10.1021/bi00485a009. [DOI] [PubMed] [Google Scholar]
- [22].Christianson DW, Lipscomb WN. Complex between carboxypeptidase A and a possible transition-state analog: mechanistic inferences from high-resolution x-ray structures of enzyme-inhibitor complexes. J Am Chem Soc. 1986;108:4998–5003. http://dx.doi.org/10.1021/ja00276a048. [Google Scholar]
- [23].Walter MW, Felici A, Galleni M, Soto RP, Adlington RM, Baldwin JE, Frère J-M, Gololobov M, Schofield CJ. Trifluoromethyl alcohol and ketone inhibitors of metallo-β-lactamases. Bioorg Med Chem Lett. 1996;6:2455–2458. http://dx.doi.org/10.1016/0960-894X(96)00453-2. [Google Scholar]
- [24].Walter MW, Adlington RM, Baldwin JE, Schofield CJ. Synthesis of metallo-β-lactamase inhibitors. Tetrahedron. 1997;53:7275–7290. http://dx.doi.org/10.1016/S0040-4020(97)00404-3. [Google Scholar]
- [25].Toney JH, Fitzgerald PM, Grover-Sharma N, Olson SH, May WJ, Sundelof JG, Vanderwall DE, Cleary KA, Grant SK, Wu JK, Kozarich JW, Pompliano DL, Hammond GG. Antibiotic sensitization using biphenyl tetrazoles as potent inhibitors of Bacteroides fragilis metallo-beta-lactamase. Chem Biol. 1998;5:185–198. doi: 10.1016/s1074-5521(98)90632-9. http://dx.doi.org/10.1016/S1074-5521(98)90632-9. [DOI] [PubMed] [Google Scholar]
- [26].Toney JH, Cleary KA, Hammond GG, Yuan X, May WJ, Hutchins SM, Ashton WT, Vanderwall DE. Structure-activity relationships of biphenyl tetrazoles as metallo-β-lactamase inhibitors. Bioorg Med Chem Lett. 1999;9:2741–2746. doi: 10.1016/s0960-894x(99)00458-8. http://dx.doi.org/10.1016/S0960-894X(99)00458-8. [DOI] [PubMed] [Google Scholar]
- [27].Toney JH, Hammond GG, Fitzgerald PM, Sharma N, Balkovec JM, Rouen GP, Olson SH, Hammond ML, Greenlee ML, Gao YD. Succinic acids as potent inhibitors of plasmid-borne IMP-1 metallo-beta-lactamase. J Biol Chem. 2001;276:31913–31918. doi: 10.1074/jbc.M104742200. http://dx.doi.org/10.1074/jbc.M104742200. [DOI] [PubMed] [Google Scholar]
- [28].Moloughney JG, D Thomas J, Toney JH. Novel IMP-1 metallo-β-lactamase inhibitors can reverse meropenem resistance in Escherichia coli expressing IMP-1. FEMS Microbiol Lett. 2005;243:65–71. doi: 10.1016/j.femsle.2004.11.042. http://dx.doi.org/10.1016/j.femsle.2004.11.042. [DOI] [PubMed] [Google Scholar]
- [29].Olsen L, Jost S, Adolph H-W, Pettersson I, Hemmingsen L, Jørgensen FS. New leads of metallo-β-lactamase inhibitors from structure-based pharmacophore design. Bioorg Med Chem. 2006;14:2627–2635. doi: 10.1016/j.bmc.2005.11.046. http://dx.doi.org/10.1016/j.bmc.2005.11.046. [DOI] [PubMed] [Google Scholar]
- [30].Hiraiwa Y, Morinaka A, Fukushima T, Kudo T. Metallo-β-lactamase inhibitory activity of phthalic acid derivatives. Bioorg Med Chem Lett. 2009;19:5162–5165. doi: 10.1016/j.bmcl.2009.07.018. http://dx.doi.org/10.1016/j.bmcl.2009.07.018. [DOI] [PubMed] [Google Scholar]
- [31].Walter MW, Valladares MH, Adlington RM, Amicosante G, Baldwin JE, Frère J-M, Galleni M, Rossolini GR, Schofield CJ. Hydroxamate Inhibitors ofAeromonas hydrophilaAE036 Metallo-B-lactamase. Bioorg Chem. 1999;27:35–40. http://dx.doi.org/10.1006/bioo.1998.1111. [Google Scholar]
- [32].Liénard BMR, Horsfall LE, Galleni M, Frère J-M, Schofield CJ. Inhibitors of the FEZ-1 metallo-β-lactamase. Bioorg Med Chem Lett. 2007;17:964–968. doi: 10.1016/j.bmcl.2006.11.053. http://dx.doi.org/10.1016/j.bmcl.2006.11.053. [DOI] [PubMed] [Google Scholar]
- [33].Nagano R, Adachi Y, Imamura H, Yamada K, Hashizume T, Morishima H. Carbapenem derivatives as potential inhibitors of various β-lactamases, including class B metallo-β-lactamases. Antimicrob Agents Chemother. 1999;43:2497–2503. doi: 10.1128/aac.43.10.2497. http://www.ncbi.nlm.nih.gov/pubmed/10508031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Nagano R, Adachi Y, Hashizume T, Morishima H. In vitro antibacterial activity and mechanism of action of J.-111,225, a novel 1β-methylcarbapenem, against transferable IMP-1 metallo-β-lactamase producers. J Antimicrob Chemother. 2000;45:271–276. doi: 10.1093/jac/45.3.271. http://dx.doi.org/10.1093/jac/45.3.271. [DOI] [PubMed] [Google Scholar]
- [35].Hovel FV, Vanwetswinkell S, Marchand-Brynaertt J, FasWez J. Synthesis and rearrangment of potential zinc β-lactamase inhibitors. Tetrahedron Lett. 1995;36:9313–9316. http://dx.doi.org/10.1016/0040-4039(95)02028-N. [Google Scholar]
- [36].Buynak JD, Chen H, Vogeti L, Gadhachanda VR, Buchanan CA, Palzkill T, Shaw Rw, Spencer J, Walsh TR. Penicillin-derived inhibitors that simultaneously target both metallo- and serine-β-lactamases. Bioorg Med Chem Lett. 2004;14:1299–1304. doi: 10.1016/j.bmcl.2003.12.037. http://dx.doi.org/10.1016/j.bmcl.2003.12.037. [DOI] [PubMed] [Google Scholar]
- [37].Tsang WY, Dhanda A, Schofield CJ, Frère J-M, Galleni M, Page MI. The inhibition of metallo-β-lactamase by thioxo-cephalosporin derivatives. Bioorg Med Chem Lett. 2004;14:1737–1739. doi: 10.1016/j.bmcl.2004.01.047. http://dx.doi.org/10.1016/j.bmcl.2004.01.047. [DOI] [PubMed] [Google Scholar]
- [38].Badarau A, Llina's A, Laws AP, Damblon C, Page MI. Inhibitors of Metallo-β-lactamase Generated from β-Lactam Antibiotics. Biochemistry. 2005;44:8578–8589. doi: 10.1021/bi050302j. http://dx.doi.org/10.1021/bi050302j. [DOI] [PubMed] [Google Scholar]
- [39].Beharry Z, Chen H, Gadhachanda VR, Buynak JD, Palzkill T. Evaluation of penicillin-based inhibitors of the class A and B β-lactamases from Bacillus anthracis. Biochem Biophys Res Commun. 2004;313:541–545. doi: 10.1016/j.bbrc.2003.11.158. http://dx.doi.org/10.1016/j.bbrc.2003.11.158. [DOI] [PubMed] [Google Scholar]
- [40].Ganta SR, Perumal S, Pagadala SR, Samuelsen O, Spencer J, Pratt RF, Buynak JD. Approaches to the simultaneous inactivation of metallo- and serine-beta-lactamases. Bioorg Med Chem Lett. 2009;19:1618–1622. doi: 10.1016/j.bmcl.2009.02.018. http://dx.doi.org/10.1016/j.bmcl.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Johnson JW, Gretes M, Goodfellow VJ, Marrone L, Heynen ML, Strynadka NC, Dmitrienko GI. Cyclobutanone analogues of beta-lactams revisited: insights into conformational requirements for inhibition of serine- and metallo-beta-lactamases. J Am Chem Soc. 2010;132:2558–2560. doi: 10.1021/ja9086374. http://dx.doi.org/10.1021/ja9086374. [DOI] [PubMed] [Google Scholar]
- [42].Sanschagrin F, Levesque RC. A specific peptide inhibitor of the class B metallo-beta-lactamase L-1 from Stenotrophomonas maltophilia identified using phage display. J Antimicrob Chemother. 2005;55:252–255. doi: 10.1093/jac/dkh550. http://dx.doi.org/10.1093/jac/dkh550. [DOI] [PubMed] [Google Scholar]
- [43].Bounaga S, Galleni M, Laws AP, Page MI. Cysteinyl peptide Inhibitors of Bacillus cereus Zinc β-Lactamase. Bioorg Med Chem. 2001;9:503–510. doi: 10.1016/s0968-0896(00)00257-1. http://dx.doi.org/10.1016/S0968-0896(00)00257-1. [DOI] [PubMed] [Google Scholar]
- [44].Sun Q, Law A, Crowder MW, Geysen HM. Homo-cysteinyl peptide inhibitors of the L1 metallo-β-lactamase, and SAR as determined by combinatorial library synthesis. Bioorg Med Chem Lett. 2006;16:5169–5175. doi: 10.1016/j.bmcl.2006.07.001. http://dx.doi.org/10.1016/j.bmcl.2006.07.001. [DOI] [PubMed] [Google Scholar]
- [45].Horsfall LE, Garau G, Lie′nard BMR, Dideberg O, Schofield CJ. Competitive inhibitors of the CphA metallo-beta-lactamase from Aeromonas hydrophila. Antimicrob Agents Chemother. 2007;51:2136–2142. doi: 10.1128/AAC.00866-06. http://dx.doi.org/10.1128/AAC.00866-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Roll DM, Yang Y, Wildey MJ, Bush K, Lee MD. Inhibition of metallo-beta-lactamases by pyridine monothiocarboxylic acid analogs. J Antibiot. 2010;63:255–257. doi: 10.1038/ja.2010.20. http://dx.doi.org/10.1038/ja.2010.20. [DOI] [PubMed] [Google Scholar]
- [47].Payne DJ, Hueso-Rodríguez JA, Boyd H, Concha NO, Janson CA, Gilpin M, Bateson JH, Cheever C, Niconovich NL, Pearson S, Rittenhouse S, Tew D, Díez E, Pérez P, De La Fuente J, Rees M, Rivera-Sagredo A. Identification of a series of tricyclic natural products as potent broad-spectrum inhibitors of metallo-beta-lactamases. Antimicrob Agents Chemother. 2002;46:1880–1886. doi: 10.1128/AAC.46.6.1880-1886.2002. http://dx.doi.org/10.1128/AAC.46.6.1880-1886.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Denny BJ, Lambert PA, West PWJ. The flavonoid galangin inhibits the L1 metallo-beta-lactamase from Stenotrophomonas maltophilia. FEMS Microbiol Lett. 2002;208:21–24. doi: 10.1111/j.1574-6968.2002.tb11054.x. http://www.ncbi.nlm.nih.gov/pubmed/11934488. [DOI] [PubMed] [Google Scholar]
- [49].Bebrone C. Metallo-beta-lactamases (classification, activity, genetic organization, structure, zinc coordination) and their superfamily. Biochem Pharmacol. 2007;74:1686–1701. doi: 10.1016/j.bcp.2007.05.021. http://dx.doi.org/10.1016/j.bcp.2007.05.021. [DOI] [PubMed] [Google Scholar]
- [50].Walsh TR, Tolemman MA, Poirel L, Nordmann P. Metallo-beta-lactamases: the quiet before the storm? Clin Microb Rev. 2005;18:306–325. doi: 10.1128/CMR.18.2.306-325.2005. http://www.ncbi.nlm.nih.gov/pubmed/15831827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Docquier JD, Lamotte-Brasseur J, Galleni M, Amicosante G, Frere JM, Rossolini GM. On functional and structural heterogeneity of VIM-type metallo-beta-lactamases. J Antimicrob Chemother. 2003;51:257–266. doi: 10.1093/jac/dkg067. http://dx.doi.org/10.1093/jac/dkg067. [DOI] [PubMed] [Google Scholar]
- [52].Minond D, Saldanha SA, Subramaniam P, Spaargaren M, Spicer T, Fotsing JR, Weide T, Fokin VV, Sharpless KB, Galleni M, Bebrone C, Lassaux P, Hodder P. Inhibitors of VIM-2 by screening pharmacologically active and click-chemistry compound libraries. Bioorg Med Chem. 2009;17:5027–5037. doi: 10.1016/j.bmc.2009.05.070. http://dx.doi.org/10.1016/j.bmc.2009.05.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Weide T, Saldanha SA, Minond D, Spicer TP, Fotsing JR, Spaargaren M, Frère JM, Bebrone C, Sharpless KB, Hodder PS, Fokin VV. NH-1,2,3-Triazole-based Inhibitors of the VIM-2 Metallo-β-Lactamase: Synthesis and Structure-Activity Studies. ACS Med Chem Lett. 2010;1:150–154. doi: 10.1021/ml900022q. http://dx.doi.org/10.1021/ml900022q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Faridoon, Hussein WM, Vella P, Islam NU, Ollis DL, Schenk G, McGeary RP. 3-Mercapto-1,2,4-triazoles and N-acylated thiosemicarbazides as metallo-β-lactamase inhibitors. Bioorg Med Chem Lett. 2012;22:380–386. doi: 10.1016/j.bmcl.2011.10.116. http://dx.doi.org/10.1016/j.bmcl.2011.10.116. [DOI] [PubMed] [Google Scholar]
- [55].Tamilselvi A, Mugesh G. Metallo-β-lactamase-Catalyzed Hydrolysis of Cephalosporins: Some Mechanistic Insights into the Effect of Heterocyclic Thiones on Enzyme Activity. Inorg Chem. 2011;50:749–756. doi: 10.1021/ic100253k. http://dx.doi.org/10.1021/ic100253k. [DOI] [PubMed] [Google Scholar]
- [56].Mohamed MS, Hussein WM, McGeary RP, Vella P, Schenk G, Abd El-hameed RH. Synthesis and kinetic testing of new inhibitors for a metallo-β-lactamase from Klebsiella pneumonia and Pseudomonas aeruginosa. Eur J Med Chem. 2011;46:6075–6082. doi: 10.1016/j.ejmech.2011.10.030. http://dx.doi.org/10.1016/j.ejmech.2011.10.030. [DOI] [PubMed] [Google Scholar]
- [57].Kim S-K, Sims CL, Wozniak SE, Drude SH, Whitson D, Shaw RW. Antibiotic resistance in bacteria: novel metalloenzyme inhibitors. Chem Biol Drug Des. 2009;74:343–348. doi: 10.1111/j.1747-0285.2009.00879.x. http://dx.doi.org/10.1111/j.1747-0285.2009.00879.x. [DOI] [PubMed] [Google Scholar]
- [58].Siemann S, Evanoff DP, Marrone L, Clarke AJ, Viswanatha T, Dmitrienko GI. N-arylsulfonyl hydrazones as inhibitors of IMP-1 metallo-beta-lactamase. Antimicrob Agents Chemother. 2002;46:2450–2457. doi: 10.1128/AAC.46.8.2450-2457.2002. http://dx.doi.org/10.1128/AAC.46.8.2450-2457.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Fitzgerald PMD, Wu JK, Toney JH. Unanticipated inhibition of the metallo-beta-lactamase from Bacteroides fragilis by 4-morpholineethanesulfonic acid (MES): a crystallographic study at 1.85-A resolution. Biochemistry. 1998;37:6791–6800. doi: 10.1021/bi9730339. http://dx.doi.org/10.1021/bi9730339. [DOI] [PubMed] [Google Scholar]
- [60].Simm AM, Loveridge EJ, Crosby J, Avison MB, Walsh TR, Bennett PM. Bulgecin A: a novel inhibitor of binuclear metallo-beta-lactamases. Biochem J. 2005;387:585–590. doi: 10.1042/BJ20041542. http://dx.doi.org/10.1042/BJ20041542. [DOI] [PMC free article] [PubMed] [Google Scholar]