

Abstract
Calpains regulate a wide spectrum of biological functions, including migration, adhesion, apoptosis, secretion, and autophagy, through the modulating cleavage of specific substrates. Ubiquitous microcalpain (μ-calpain) and millicalpain (m-calpain) are heterodimers composed of catalytic subunits encoded, respectively, by CAPN1 and CAPN2 and a regulatory subunit encoded by CAPNS1. Here we show that calpain is required for the stability of the deubiquitinating enzyme USP1 in several cell lines. USP1 modulates DNA replication polymerase choice and repair by deubiquitinating PCNA. The ubiquitinated form of the USP1 substrate PCNA is stabilized in CAPNS1-depleted U2OS cells and mouse embryonic fibroblasts (MEFs), favoring polymerase-η loading on chromatin and increased mutagenesis. USP1 degradation directed by the cell cycle regulator APC/Ccdh1, which marks USP1 for destruction in the G1 phase, is upregulated in CAPNS1-depleted cells. USP1 stability can be rescued upon forced expression of calpain-activated Cdk5/p25, previously reported as a cdh1 repressor. These data suggest that calpain stabilizes USP1 by activating Cdk5, which in turn inhibits cdh1 and, consequently, USP1 degradation. Altogether these findings point to a connection between the calpain system and the ubiquitin pathway in the regulation of DNA damage response and place calpain at the interface between cell cycle modulation and DNA repair.
INTRODUCTION
Calpains regulate a wide spectrum of biological functions, including migration, adhesion, apoptosis, secretion, and autophagy through the modulated cleavage of specific substrates (reviewed in references 1–3). Ubiquitous microcalpain (μ-calpain) and millicalpain (m-calpain) are heterodimers composed of a catalytic subunit encoded, respectively, by CAPN1 and CAPN2 and a regulatory subunit encoded by CAPNS1. Both μ-calpain and m-calpain are negatively modulated by calpastatin. By a proteomic approach, we identified USP1 deubiquitinase as a CAPNS1-interacting protein. USP1 is a key modulator of DNA repair, partly through deubiquitination of its known targets FANCD2 (4, 5) and PCNA (6). Usp1 knockout (KO) mice have a severe phenotype and die soon after birth (7). Usp1−/− cells are defective in FANCD2 focus formation and are hypersensitive to DNA damage (8). PCNA ubiquitination is higher in USP1-depleted cells than in control cells, thus leading to recruitment of error-prone, translesion DNA synthesis (TLS) polymerases and the consequent increase in mutation rate (6). USP1 promotes inhibitor of DNA binding (ID) protein stability and stem cell-like characteristics in osteosarcoma and is required for normal skeletogenesis (9). Interestingly, mice lacking CAPNS1 in cells of the osteoblast lineage are defective in bone development and remodeling in vivo (10).
UV light irradiation activates hUSP1 autocleavage at glycines 670 and 671, inducing its subsequent proteasomal degradation (6). USP1 mutants with mutation in the catalytic cysteine 90 or in the autocleavage sites are more stable but can still be degraded in the cell (5), suggesting that alternative degradation pathways may be eventually activated. Indeed, USP1 is ubiquitinated in the G1 phase by the anaphase-promoting complex/cyclosome complexed to the substrate adaptor protein cdh1 (APC/Ccdh1) and consequently targeted to the proteasome for degradation (11). It is well established that APC/Ccdh1 ubiquitin ligase, by adding ubiquitin chains to cell cycle regulators, targets them to proteasomal degradation and modulates cell cycle progression and differentiation (12). In addition, the decrease of USP1 levels before S-phase entry allows PCNA ubiquitination and consequent recruitment of translesion DNA polymerases in response to UV to the sites of DNA damage (11). These data indicate that APC/Ccdh1 links cell cycle modulation to DNA repair pathway choice (11). USP1 stability and function require its interaction with UAF1/WDR48 (13), a WD repeat-containing protein, originally described as an endosomal regulator of vesicular traffic (14) that may alternatively bind and stabilize USP12 and USP46 (15).
Here we show that μ-calpain activity is required for USP1 protein stability in several cell lines. Accordingly, the USP1 substrate, ubiquitinated PCNA, is stabilized in CAPNS1-depleted U2OS cells and mouse embryonic fibroblasts (MEFs), favoring polymerase η (pol-η) loading on chromatin and increased mutagenesis. We characterize the calpain/USP1 axis and show the involvement of calpain-activated Cdk5/p25 kinase and cdh1 in this regulatory network. Altogether these findings highlight a function of calpain in the modulation of cell cycle and DNA damage response.
MATERIALS AND METHODS
Chemicals and reagents.
The protease inhibitors MG132 and Z-Leu-Leu-CHO were purchased from Sigma-Aldrich. Lipofectamine RNAiMAX and Lipofectamine 2000 were bought from Invitrogen, and Mirus TransIT-LT1 was purchased from MirusBio. USP1-specific small interfering RNA (siRNA) with the sequence AACCCUAUGUAUGAAGGAUAU was purchased from Eurofins Mwg/Operon. RNA interference (RNAi) oligonucleotides specific for Cdk5 and cdh1 were purchased from Santa Cruz Biotechnology. CAPNS1 siRNA was already described (16).
Plasmids and constructs.
Green fluorescent protein (GFP)-tagged USP1 and FLAG-tagged USP1 were kind gifts from Renè Bernards (Netherlands Cancer Institute), and myc-USP1 and mutant derivatives were kindly donated by Tony T. Huang (New York University [NYU]). FLAG-WDR48 and enhanced GFP (EGFP)-tagged pol-η were kindly provided by Jae Jung (University of Southern California) and Alan Lehmann (Sussex University), respectively. p25- and p35-expressing plasmids were kindly donated by Elena Agostoni and Francesca Persichetti (ISAS, Trieste, Italy). C-terminal FLAG-tagged USP1 was produced by subcloning PCR amplified cDNA into 3× FLAG-CMV14. Point mutants were obtained using the QuikChange site-directed mutagenesis kit from Stratagene (Agilent Technologies) following the procedure suggested by the manufacturer.
Cell culture and transfection.
Wild-type and Capns1−/− mouse embryonic fibroblasts were previously described (17) and kindly donated by P. Greer (Queen's University, Kingston, Ontario, Canada). 293T, Phoenix, U2OS, and BJ-ET cells and the mouse fibroblasts described above were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS). U2OS, 293T, and BJ-ET cells were transiently transfected using TransIT-LT1 (Mirus) or RNAiMAX (Invitrogen) according to the manufacturer's instructions.
Immunological procedures.
Standard protocols for immunoblotting and immunofluorescence were used. Antibodies against CAPNS1, CAPN2, actin, FLAG, and tubulin were purchased from Sigma-Aldrich. Antibodies against CAPN1, PCNA, CDK5, p35, p21, and ID2 were purchased from Santa Cruz Biotechnology, Inc., Santa Cruz, CA. Antibody against myc tag was purchased from BD-Pharmingen. Anti-cdh1 antibody was purchased from Calbiochem. Initial studies were performed using an anti-USP1 antibody provided by Fanconi Anemia Research Fund and one from T. Huang (NYU). Almost all of the blots of the present article are decorated with anti-USP1 antibody purchased from Bethyl Laboratories, Inc. (no. A301-698A). The only exceptions are the blots shown in Fig. 2, in which the anti-USP1 antibody is from T. Huang's laboratory.
Fig 2.

CAPNS1 depletion affects USP1 protein level. (a) CAPNS1-, LACZ-, or USP1-specific siRNAs were transfected in U2OS cells. Three days later, the cells were exposed to 30 J/m2 UV light irradiation or left untreated. Samples were collected 1, 3, and 5 h postirradiation and utilized to monitor USP1 protein by Western blotting. (b and c) Effect of CAPNS1 depletion in other osteosarcoma cell lines. MG63 cells (b) or SAOS cells (c) were transfected with the indicated siRNAs, and 72 h later, the cells were lysed and analyzed with the indicated antibodies. (d) Capns1 wild-type (w.t.) MEFs, Capns1 knockout (k.o.) MEFs, and Capns1-rescued MEFs (R) were irradiated or not with 30 J/m2 UV light, and the lysates were prepared 5 h after irradiation and utilized to monitor the USP1 protein level by Western blotting. (e) CAPNS1-, LACZ-, or USP1-specific siRNAs were transfected in WI38 fibroblasts. Three days later, the cells were exposed to 30 J/m2 UV light irradiation or left untreated. Samples were collected 1, 3, and 5 h postirradiation and utilized to monitor the USP1 protein level by Western blotting. (f) BJ-ET Rasv12-inducible fibroblasts were infected with an AAV vector expressing sh-CAPNS1 or sh-LUC. Three days later, tamoxifen (TMX) was added (or not) to the cells to induce Rasv12 expression. Lysates were collected 3 days after induction and utilized to monitor the USP1 protein level by Western blotting.
Immunoprecipitations and MS.
A protocol compatible with mass spectrometry (MS) was followed. True-blot beads (mouse TrueBlot; eBioscience) were washed 3 times with ice-cold lysis buffer (50 mM HEPES [pH 7.4], 150 mM NaCl, 50 mM NaF, 1 mM EDTA, 0.2% Triton X-100, 0.5 mM NaF, 1 mM dithiothreitol [DTT], 1 mM Na3VO4 protease inhibitors [protease inhibitor cocktail; Sigma]) and then resuspended in 0.5 ml of lysis buffer supplemented with 0.1 mg/ml dextran (catalog no. D1037; Sigma). Antibodies were added to the beads at a ratio of 1:100 and incubated on the rotator in the cold room for 45 min. Beads were then washed twice with lysis buffer. Cells were washed with ice-cold phosphate-buffered saline (PBS) and lysed by adding lysis buffer and dextran directly to the plate (on ice). Cells were scraped, and the lysates were spun for 10 min at high speed. At this point, 100 μl of lysates was removed into a fresh tube (input). Lysates were added to the washed antibody beads and incubated in the cold room for 2.5 h. Beads were washed 3 times and analyzed by mass spectrometry or run in SDS-PAGE. Mass spectrometric (MS) analysis was performed using an Applied Biosystems 4800 matrix-assisted laser desorption ionization–tandem time of flight (MALDI-TOF/TOF) instrument. Liquid chromatography (LC)-MALDI MS is similar in many respects to the more classical LC-MS/MS that is based on electrospray instruments. One of the key differences is that with LC-MALDI MS, the LC run is spotted and dried onto a sample plate, and this results in the LC run being “frozen in time.” Because of this property, each peak in the LC run is typically only fragmented a single time, at the apex of its elution. Therefore, it is more difficult to judge the relative abundance of a protein in a sample solely based on spectral counts (18). The data were searched using the MASCOT search engine (version 2.0) using a human proteome database (Swiss-Prot) and the reversed database as a decoy. Only those proteins with a false discovery rate of <5% were included for further analysis. Besides some well-known substrates, including calpain catalytic subunits, we identified USP1 deubiquitinating enzyme as a novel interacting partner. In the case of USP1, four peptides were identified that account for 7% protein coverage (taking into account the peptides that could not be identified because they were either too small or too big to be detected by the mass spectrometer).
Single-cell gel electrophoresis (comet assay).
An alkaline comet assay was performed according to the manufacturer's protocol (Trevigen kit; 4250-050-K) with some modifications. MEFs were UV irradiated (30 J/m2) or left untreated. Forty-eight or 72 h later, the cells were washed in PBS and then gently scraped. A total of 4 × 105 cells/ml were combined with molten LM agarose at a ratio of 1:10 and immediately pipetted onto CometSlide. Slides were treated with lysis and unwinding solution. After electrophoresis (21 V, 45 min), the slides were fixed in 70% ethanol, dried, and stained with SYBR green. Slides were observed by epifluorescence microscope, and 50 random comets per sample were analyzed using CometScore software (TriTek).
supF mutation assay.
U2OS cells were transfected with lacZ-, CAPNS1-, or USP1-specific siRNAs using Lipofectamine RNAiMAX (Invitrogen) according to manufacturer's instructions. Forty-eight hours after transfection, cells were subjected to a second-round transfection with siRNAs along with the either undamaged or UVC-irradiated (254 nm of radiation using a 2400 UV Stratalinker from Stratagene) pSP189 plasmid using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Forty-eight hours later, plasmids were retrieved using a plasmid miniprep kit (Sigma) and digested with DpnI to eliminate unreplicated plasmids. The plasmid DNA was then electroporated into the MBM7070 bacterial strain, which carries an amber mutation in the β-galactosidase gene. Transformed bacteria were plated on agar plates with 50 μg/ml ampicillin, 2 mM isopropyl-1-thiogalactopyranoside and 200 μg/ml X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside). Mutant (white) and wild-type (blue) colonies were counted to determine the mutation frequency (number of white colonies over total colonies).
Statistical analysis.
Results are expressed as means ± standard deviations of at least three independent experiments. Statistical analysis was performed using Student's t test with the level of significance set at P < 0.05.
RESULTS
USP1 interacts with CAPNS1.
A proteomic approach was followed for the identification of novel CAPNS1-interacting proteins. Preparative coimmunoprecipitation of endogenous proteins was achieved avoiding the use of overexpressed molecules to reduce the interference of artifacts linked to the forced accumulation of a protein in a cell. Crude extracts from HT-1080 fibroblasts were immunoprecipitated with a commercial monoclonal anti-CAPNS1 antibody, and the products were analyzed by mass spectrometry with an Applied Biosystems 4800 MALDI TOF/TOF instrument.
To verify the proteomic data, we transfected 293T cells with a FLAG-USP1-expressing construct or the unrelated FLAG-USP33 cDNA as the negative control. The cell lysates were immunoprecipitated with an antibody against CAPNS1 and analyzed by Western blotting to investigate the presence of the transfected deubiquitinases among the immunoprecipitation products. A representative experiment is shown in Fig. 1a: only USP1 was found in the CAPNS1 immunoprecipitates. Apparently, the central 341 amino acids (aa) of the protein are sufficient for USP1-CAPNS1 interaction (Fig. 1b and c). However, the production of a large collection of single, double, or multiple point mutants will be required to finely dissect the interaction. Indeed USP1 is not organized in adjacent domains specifying distinct functions. For instance, the catalytic triad involves the cysteine domain between 82 and 99, the aspartic acid domain between 197 and 213 and the histidine domain between 576 and the 776 (6) (see Fig. 7b). Actually, using the prediction software SliMPred (available at http://bioware.ucd.ie/), we found that USP1 contains several stretches of amino acids with a disordered structure (19), just like calpastatin, which recognizes calpain through the combined action of three distinct motifs (20).
Fig 1.

USP1 and CAPNS1 interact in U2OS cells. FLAG-USP1 and FLAG-USP33 (a) were transfected in 293T cells, and 24 h later, the cells were lysed. Total lysates were prepared and used for immunoprecipitation (IP) against anti-CAPNS1. Immunoprecipitation products and respective inputs were analyzed by Western immunoblotting (IB) to detect the tagged proteins and endogenous CAPNS1. (b and c) Mapping the USP1-CAPNS1 interaction. The myc-USP1 wild type (w.t.) and mutants were transfected in 293T cells, and 24 h later, the cells were lysed. The lysates were prepared and used for immunoprecipitation against CAPNS1. Immunoprecipitation products and respective inputs were analyzed by Western blotting to detect the tagged proteins and endogenous CAPNS1. Cells stably expressing GFP-USP1 were transfected with FLAG-CAPNS1 (d); alternatively, cells stably expressing HA-CAPNS1 were transfected with GFP-USP1 (e). Twenty-four hours later, the cells were fixed and decorated with anti-FLAG or anti-HA antibodies and analyzed by fluorescence microscopy. (f) In vitro calpain cleavage assay. USP1-Flag (left gel) or p50 NF-κB1 (right gel) was produced as [35S]methionine-labeled protein by in vitro transcription and translation and incubated for the indicated time intervals (minutes) with commercial microcalpain, as previously described (38). The reactions were then stopped in Laemmli buffer and analyzed by SDS-PAGE and autoradiography. Parallel reactions in the presence of 10 μM EGTA were carried out to prove the calcium dependency of the reactions. (g) Time course effect of MG132 on endogenous USP1 in U2OS cells. (h) U20S cells, preincubated for 12 h with MG132, were irradiated or not with 30 J UV light. Three hours later, the lysates were immunoprecipitated with either anti-CAPNS1 antibody or anti-FLAG antibody. The IP products were analyzed by Western blotting with the indicated antibodies.
Fig 7.

Calpain acts as a brake against APC/Ccdh1-dependent proteasomal degradation of USP1. (a) U2OS cells were transfected with the indicated siRNAs and 72 h later incubated for 16 h with mimosine. Afterwards the lysates were prepared and utilized for immunoprecipitation with anti-cdh1 antibody. The immunoprecipitation products were utilized for Western blotting to detect the indicated proteins. (b) The scheme reports the position of previously described relevant regions of USP1, including the catalytic triad Cys, Asp, and His (6), the autocleavage site (6), the cdh1 recognition region (11), and the UAF1/WDR48 interacting domain (33). In addition, we marked Ser16, which according to Scansite is a Cdk5 kinase or Cdc2 kinase target site (http://scansite3.mit.edu/), and the position of the predicted calpain target site at serine 13 (http://calpain.org). (c, left panel) Schematics of the USP1 wild type and point mutants utilized to evaluate the role of Cdk5 and calpain modifications on USP1 stability. (Right panel) U2OS cells were transfected with the indicated siRNAs and 48 h later with the indicated plasmids. Twenty-four hours after transfection, lysates were prepared and utilized for Western blot analysis with anti-FLAG antibody. The blots were also decorated with anti-CAPNS1 and antiactin antibodies to monitor silencing efficiency and as loading control. (d) Working hypothesis. When calpain is active, USP1 is cleaved at the very N terminus. We hypothesize that this cleavage product is less susceptible to APC/Ccdh1 interaction or ubiquitination. Calpain may activate Cdk5/p25, which in turn inhibits APC/Ccdh1, further preserving USP1 stability. When calpain is off, Cdk5 is inactive and USP1 is rapidly ubiquitinated by APC/Ccdh1 and directed to proteasomal degradation.
Since USP1 has been reported to localize mainly in the nucleus and calpain mainly in the cytoplasm, an alternative approach was followed to determine whether USP1 and CAPNS1 are present in the same cellular compartments. U2OS cells stably expressing GFP-USP1 were transiently transfected with FLAG-CAPNS1; alternatively, stably transfected U2OS cells expressing HA-CAPNS1 were transiently transfected with GFP-USP1, and 24 h later, the cells were analyzed by a fluorescence microscope. Two representative fields selected for Fig. 1d and e show that ectopic USP1 and CAPNS1 may both localize in the nucleus.
To further confirm the interaction of CAPNS1 and USP1 in vivo, we investigated whether endogenous USP1 is present in a complex with endogenous CAPNS1 before and after irradiation with UV light. Since we have found that calpain can rapidly destroy USP1 in vitro (Fig. 1f), the cells were incubated with MG132 for 12 h before processing for immunoprecipitation. As shown in Fig. 1h, endogenous USP1 is specifically immunoprecipitated by an anti-CAPNS1 antibody and not by an anti-FLAG antibody used as the negative control. Notably, autocleaved USP1 is more abundant in the CAPNS1-containing complex upon UV irradiation, possibly suggesting a transient increase in USP1 activity. Indeed, active USP1 autocleaves itself (6). Remarkably, MG132 treatment causes an accumulation of autocleaved USP1 since it prevents its proteasome-dependent degradation (6), besides inhibiting calpain (Fig. 1g).
CAPNS1 depletion is linked to a reduction in USP1 protein levels in several cell lines.
Knockout of both Usp1 and Capns1 during mouse development leads to severe defects in bone formation (9, 10). We therefore investigated the effect of calpain depletion on USP1 protein stability in human osteosarcoma U2OS cells using siRNA. We studied the effect of calpain depletion both under basal conditions and upon UV light irradiation, inducing USP1 proteosomal turnover (6). After siRNA transfection, the cells were left untreated or were exposed to 30 J/m2 UV light, and lysates were collected at different time points after irradiation. CAPNS1 depletion is coupled to a reduction in USP1 protein level in both untreated and irradiated cells (Fig. 2a). To maximize efficiency, an siRNA “smart-pool” was utilized to deplete CAPNS1. Each CAPNS1-specific siRNA of the pool is effective to silence CAPNS1 and to reduce USP1 protein levels (data not shown). Similar results (Fig. 2b and c) were obtained using other osteosarcoma cell lines.
Capns1 knockout mice die during embryonic development; therefore, in a first attempt to address whether USP1 is modulated by CAPNS1 also in the mouse, we utilized Capns1 knockout (KO) and derivative MEFs kindly provided by P. A. Greer (Queen's University, Canada) (17). As occurs in human osteosarcoma cells, a reduction in USP1 protein level was found in Capns1 KO MEFs (Fig. 2d). This phenotype can be effectively rescued in a KO MEF derivative cell line when Capns1 is reintroduced (Fig. 2d).
To investigate whether CAPNS1 is important for USP1 stability also in human fibroblasts, we analyzed the USP1 protein level in CAPNS1-depleted diploid fetal lung WI38 fibroblasts. Also in this cellular system, calpain appears to be required for USP1 stabilization under basal conditions, and the kinetics of degradation upon UV treatment are not affected (Fig. 2e).
The USP1 protein level is quite low in human diploid BJ fibroblasts immortalized with telomerase but can be induced upon Rasv12 overexpression, concomitantly with the rise in CAPNS1 levels (Fig. 2f). Interestingly, CAPNS1 depletion, obtained upon infection with an adeno-associated virus (AAV) vector expressing a short hairpin targeting CAPNS1 (16), is coupled to a reduction of the USP1 protein level also in this cellular model (Fig. 2f). Altogether, these data support a role of CAPNS1 in maintaining USP1 protein stability in the described cellular types.
Ubiquitinated PCNA is stabilized in CAPNS1-depleted U2OS cells.
To address whether the reduction of USP1 level observed upon CAPNS1 depletion has an impact on the prototype target of USP1, the PCNA protein clamp, we monitored its ubiquitination state and analyzed the activity of its downstream effectors. PCNA is ubiquitinated by Rad6/Rad18, leading to the recruitment of TLS DNA pol-η (21) to PCNA foci on chromatin, and subsequently deubiquitinated by USP1 to reestablish the interaction with high-fidelity DNA polymerases (6).
Since we have found that CAPNS1 depletion is linked to a reduction in USP1 protein, we expected to observe an overubiquitination of PCNA and an increase of PCNA foci in the absence of calpain. CAPNS1-depleted cells, and LACZ-depleted cells as a control, were UV light irradiated or left untreated, and chromatin-bound PCNA was detected by immunofluorescence 5 and 20 h after irradiation (Fig. 3a). Two hundred nuclei were analyzed in each sample, and the cells retaining Triton-resistant PCNA foci were scored as positive. Percentage averages of three independent replicas are reported in the graph in Fig. 3b. PCNA foci continue to accumulate in CAPNS1-depleted cells 20 h after UV light irradiation, while PCNA foci diminish after a transient increase in control cells.
Fig 3.

CAPNS1 perturbs PCNA ubiquitination. CAPNS1 or LACZ was depleted from U2OS cells using specific siRNAs, and 72 h later, the cells were either left untreated or irradiated with 30 J/m2 UV. The cells were fixed 1, 5, and 20 h later, and Triton-insoluble PCNA was detected by immunofluorescence. (a) Typical images of PCNA foci. (b) Average number of PCNA foci at different time points after irradiation. Two hundred nuclei were analyzed for each sample. The error bars represent standard deviations from three independent experiments. (c) HA-CAPNS1-expressing U2OS cells, U2OS cells infected with pRetro Super carrying sh-CAPNS1 or an empty pRetro Super vector were irradiated or not with 30 J/m2 UV light, and the respective lysates were prepared 1, 3, and 5 h postdamage to monitor the PCNA ubiquitination (Ub-PCNA) level. (d) U2OS cells infected with pRetro Super carrying sh-CAPNS1 or an empty pRetro Super vector were irradiated or not with 15, 30, and 60 J/m2 UV light, and the respective lysates were prepared 1, 5, and 20 h after irradiation to monitor PCNA ubiquitination by Western blotting. Longer and shorter exposures are shown. (e) U2OS cells, stably expressing hyperstable GFP-USP1 mutated in its autocleavage site GFP-USP1(GG670/671AA) or catalytically inactive GFP-USP1C90S (f) were transfected with LACZ- or CAPNS1-specific siRNA and 72 h later irradiated with 30 J/m2 UV or left untreated. The lysates were collected at various time points (1, 3, 5, and 20 h postdamage) and utilized to monitor endogenous PCNA ubiquitination by Western blotting. Longer and shorter exposures are shown.
Next, we compared PCNA ubiquitination by Western blotting during the first 5 h after UV light irradiation in cells expressing different levels of CAPNS1. In particular, stably transfected U2OS cells expressing hemagglutinin (HA)-tagged CAPNS1, U2OS cells expressing a short hairpin designed to deplete CAPNS1 (sh-CAPNS1), and p-Retro Super-transduced U2OS cells (pRs-control) were left untreated or irradiated with 30 J/m2 UV light. Cell lysates were collected 1, 3, and 5 h afterwards to monitor PCNA ubiquitination by Western blotting. The data presented in Fig. 3c show that PCNA ubiquitination is inversely correlated to CAPNS1 protein levels. Notably, PCNA ubiquitination kinetics monitored by Western blotting appears faster than the kinetics of PCNA focus formation, underscoring the existence of multiple levels of regulation governing protein-DNA architecture.
We also compared PCNA ubiquitination in CAPNS1-depleted cells and control cells upon exposure to 0, 15, 30, and 60 J/m2 UV light. Lysates were collected 1, 5, and 20 h after irradiation and analyzed by Western blotting to investigate PCNA ubiquitination dynamics (Fig. 3d). A general increase in PCNA ubiquitination is evident in CAPNS1-depleted samples. Moreover, while PCNA ubiquitination clearly decreases 20 h after damage in control cells exposed to 30 or 60 J/m2 UV light, this decline is milder in CAPNS1-depleted cells, in accordance with the observed reduction in the amount of USP1 protein.
To determine whether CAPNS1's effect on PCNA ubiquitination is USP1 dependent, we stably transfected U2OS cells with a plasmid coding for GFP-tagged USP1 mutated in its autocleavage site, GFP-USP1(GG670/671AA) (and therefore more stable), or GFP-tagged USP1 mutated in its catalytic site, GFP-USP1C90S. The levels of mutant GFP-USP1 remain high also 20 h after UV light irradiation (Fig. 3e and f). The hyperstable GFP-USP1, but not the catalytically inactive USP1 mutant, counteracts the increase in PCNA ubiquitination coupled to CAPNS1 depletion (Fig. 3e), indicating that the perturbation in PCNA deubiquitination occurring in CAPNS1-depleted cells is USP1 dependent. Notably, a cleavage product of GFP-USP1C90S appears upon UV light irradiation in control cells but not in CAPNS1-depleted cells. Apparently, calpain can cleave in the region of the autocleavage site when USP1 catalytic function is knocked down.
CAPNS1 depletion enhances pol-η loading on chromatin and increases mutagenesis.
PCNA monoubiquitination allows the proper localization of pol-η into DNA repair foci, and UV-induced degradation of USP1 is critical to maintain proper localization and translesion synthesis function of pol-η (6). To investigate whether the higher level of PCNA ubiquitination observed in CAPNS1-depleted cells was linked to an increase in pol-η loading on chromatin, CAPNS1-depleted and control cells were transiently transfected with a plasmid coding for GFP–pol-η and subsequently irradiated or not with UV light. GFP–pol-η uniformly stains the nuclei in untreated control cells. Five hours after irradiation, pol-η foci are visible in control cells (Fig. 4a), while a certain number of foci are already present prior to irradiation in CAPNS1-depleted cells (Fig. 4b). Focus formation in CAPNS1-depleted cells can be reduced upon overexpression of wild-type myc-USP1 but not by a catalytically inactive USP1 derivative, myc-USP11–746 (Fig. 4b).
Fig 4.

CAPNS1 depletion is linked to a defect in DNA repair. CAPNS1 (CS1)-depleted or LACZ-depleted U2OS cells were transfected with GFP7–pol-η and, where indicated, with wild-type USP1 (USP1 wt) or catalytically inactive USP1. Twenty hours later, the cells were irradiated with 30 J/m2 UV light or left untreated. Five hours later, the cells were fixed and analyzed by immunofluorescence. (a) Typical distribution of GFP–pol-η in control cells (left picture) and upon UV light-induced focus formation (right picture). (b) Percentage of GFP–pol-η focus-containing cells obtained in three independent experiments. (c) U2OS cells were transfected with LACZ, CAPNS1, or USP1 siRNA and subsequently with irradiated marker plasmid. The plasmid was recovered from the transfected cells 24 h after transfection and utilized to transform bacteria and measure repair efficiency in a standard supF mutagenesis assay. The graph reports the increase in mutagenesis occurring in CAPNS1- or USP1-depleted cells compared to control cells. (d) Wild-type, Capns1 KO, and Capns1-rescued (R) MEFs were irradiated with 30 J/m2 UV light or left untreated, and lysates were prepared 5 h later to monitor the PCNA ubiquitination (Ub-PCNA) level by Western blotting. (e) Wild-type and Capns1 KO MEFs were irradiated or not with 30 J/m2 UV light and utilized to perform a comet assay 48 and 65 h later; typical images are shown. (f) Quantification of parameters indicative of DNA damage and recovery: tail moment, % of DNA in the tail, and olive moment. CTRL, control. (g) CAPNS1 depletion is coupled to a decrease in USP1, and therefore the equilibrium between deubiquitinated and ubiquitinated PCNA is shifted toward the ubiquitinated form. Ubiquitinated PCNA recruits pol-η on DNA replication forks and may increase mutagenesis if it is not replaced by high-fidelity DNA polymerase once the lesion is bypassed. (h) Effect of CAPNS1 silencing on another USP1 substrate, ID2. Seventy-two hours after transfection with the indicated siRNAs, U2OS cells were lysed and analyzed with the indicated antibodies.
Since USP1 depletion is linked to increased mutation frequency (6, 8), we determined mutation frequency in CAPNS1-depleted U2OS cells employing a supF mutagenesis assay; USP1- and LACZ-depleted cells were utilized as controls. As shown in the graph of Fig. 4c, USP1 depletion is coupled to a sharp increase in mutation frequency, as already reported (6). Similarly, CAPNS1 depletion is linked to an increase in mutation frequency, in accordance with the increase of pol-η loading on chromatin occurring in CAPNS1-depleted cells relative to control ones.
UV damage response is altered in Capns1−/− MEFs.
We monitored PCNA ubiquitination levels before and after UV light irradiation in wild-type, Capns1−/−, and Capns1-rescued (R) MEFs (17). Cells were either left untreated or irradiated with 30 J/m2 UV light. Five hours later, protein extracts were prepared and the PCNA ubiquitination level was analyzed by Western blotting. As shown in Fig. 4d, Capns1 depletion is coupled to an accumulation of ubiquitinated PCNA also in mouse embryonic fibroblasts. Next, we monitored the effect of UV light on DNA integrity in wild-type and Capns1−/− MEFs by means of the alkaline comet assay (single-cell electrophoresis). This assay allows one to follow the induction of DNA damage and subsequent repair at the single-cell level. Single- and double-strand DNA breaks are visualized as a comet after electrophoresis and DNA staining with SYBR green. Cells were collected before and 48 or 65 h after irradiation and processed for the comet assay. Figure 4e shows some representative examples of single-cell DNA before and 48 h after UV light-induced DNA damage. The graphs reported in Fig. 4f show the mean values obtained in three independent replicas for three parameters indicative of DNA damage: tail moment, % DNA in tail, and “olive” moment. In wild-type cells, DNA repair intermediates increase 48 h after damage and then go back to the levels observed in untreated cells, indicating the activation of efficient DNA repair. On the other hand, both the induction of DNA repair intermediates and recovery appear impaired in Capns1−/− MEFs. Altogether, these data suggest that the hyperubiquitination of PCNA coupled to Capns1 depletion is linked to a defect in DNA repair triggered by UV light-induced DNA damage. This result is in accordance with a recent report that demonstrates the essential role of USP1 for homologous recombination (8). Overall, the data reported above demonstrate that calpain inhibition or depletion is linked to a decrease in USP1 level and downstream functionality as a consequence of a shift in the equilibrium between ubiquitinated and nonubiquitinated PCNA toward the ubiquitinated form. Overubiquitinated PCNA is linked to increased recruitment of translesion synthesis DNA polymerases, thus eventually impacting genome integrity (Fig. 4g). Remarkably, also ID2, a recently recognized substrate of USP1, is affected by calpain depletion. Indeed, CAPNS1 depletion in U2OS cells leads to ID2 destabilization and p21 accumulation, just like USP1 depletion (Fig. 4h), as recently reported (9).
Microcalpain activity is required for USP1 protein stability.
We found that USP1 protein levels are reduced upon CAPNS1 depletion in osteosarcoma cells and fibroblasts. To determine whether CAPNS1 affected USP1 at the mRNA level, quantitative reverse transcription-PCR (qRT-PCR) was performed using RNAs extracted from CAPNS1-depleted U2OS cells. RNAs extracted from USP1- and LACZ-depleted cells were included as controls. The graph in Fig. 5a shows that CAPNS1 depletion does not significantly affect USP1 mRNA.
Fig 5.
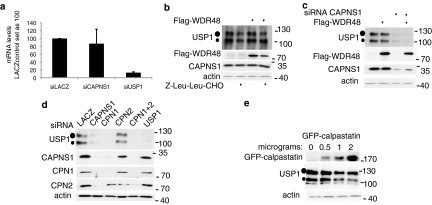
Microcalpain modulates USP1 stability. (a) U2OS cells were transfected with LACZ, CAPNS1, or USP1 siRNA. Seventy-two hours later, the cells were processed for qRT-PCR analysis. The graph shows the quantification of USP1 mRNA. For each mRNA, the control RNA extracted from LACZ-silenced cells is set as 100. (b) U2OS cells were transfected with Flag-WDR48 expression plasmid or an empty vector. Twenty-four hours later, the cells were incubated for 5 h with Z-Leu-Leu-CHO or left untreated and then utilized to monitor endogenous USP1 by Western blotting. (c) U2OS cells were transfected with CAPNS1 siRNAs and 48 h later were transfected with Flag-WDR48 expression plasmid. Twenty-four hours later, the cells were lysed and utilized for Western blots to monitor endogenous USP1. (d) LACZ, CAPNS1, CAPN1, CAPN2, CAPN1 and -2, or USP1 was depleted by siRNA from U2OS cells. Seventy-two hours later, the respective lysates were utilized for Western blots to monitor USP1 levels. (e) U2OS cells were transfected with increasing amounts of GFP-calpastatin, as indicated. Twenty-four hours later, the respective lysates were utilized for Western blots to monitor USP1 levels.
USP1 stability and function require its interaction with UAF1/WDR48 (13, 15), a WD repeat-containing protein, originally described as an endosomal regulator of vesicular traffic (14) that may alternatively bind and stabilize USP12 and USP46 (15). Therefore, we investigated the effect of calpain inhibition on UAF1/WDR48 stability. As shown in the blots of Fig. 5b and c, calpain inhibition by Z-Leu-Leu-CHO or by CAPNS1 silencing is coupled to a slight decrease in UAF1/WDR48 protein level, while the UAF1/WDR48 mRNA level remains unaffected (data not shown). UAF1/WDR48 overexpression, however, cannot rescue the stability of USP1 in the cells where calpain is inhibited (Fig. 5b) or depleted (Fig. 5c). We can therefore argue that the effect of calpain on USP1 signaling is not mediated by UAF1/WDR48.
To investigate whether CAPNS1 modulation of USP1 involves its proteolytic associated function, we transiently depleted the microcalpain (CAPN1) and millicalpain (CAPN2) catalytic subunits independently or simultaneously and checked USP1 protein levels. CAPN1 silencing like CAPNS1 silencing is coupled to a reduction in USP1 protein levels, whereas CAPN2 silencing does not affect USP1 (Fig. 5d). As previously described by others (17), CAPNS1 depletion is coupled to a decrease in CAPN1 and CAPN2 protein levels. Since we found that CAPN1 depletion leads to a sharp reduction in CAPNS1, we decided to further assess the requirement of the catalytic function of calpain in the modulation of USP1 by inhibiting calpain activity through calpastatin overexpression. Transfection of increasing amounts of a calpastatin-encoding plasmid into U2OS cells is coupled to a reduction of endogenous USP1 (Fig. 5e), just as in CAPNS1- and CAPN1-depleted cells, indicating that the catalytic activity of calpain is important to guarantee USP1 stability.
CAPNS1 modulates USP1 via APC/Ccdh1.
USP1 is a target of APC/Ccdh1, which marks it for proteasomal degradation in the G1 phase of the cell cycle (22). Therefore, we investigated whether the instability of USP1 occurring in CAPNS1-depleted cells could be linked to an increase in APC/Ccdh1 activity toward USP1. To this end, we knocked down cdh1 in CAPNS1-depleted cells and found that Cdh1 silencing rescues USP1 stability, both in an asynchronous population and in late G1 enriched cells, collected 16 h after mimosine addition (Fig. 6a). Calpain stabilizes USP1 specifically in late G1; indeed, USP1 is not affected by CAPNS1 depletion in G2/M-arrested cells, obtained by incubation with nocodazole for 16 h (Fig. 6b). Altogether, these results indicate that calpain specifically acts as a brake for USP1 degradation at the G1/S border.
Fig 6.

USP1 instability in CAPNS1-depleted cells can be rescued by cdh1 depletion or Cdk5/p25 overexpression. (a and b) U2OS cells were transfected with the indicated siRNA, and 48 h later, the cells were incubated or not with mimosine (a) to induce G1/S arrest or with nocodazole to achieve arrest in M phase (b). Sixteen hours later, cell lysates were collected and analyzed with the indicated antibodies. (c) U2OS cells were transfected with the indicated siRNAs, and 72 h later, the respective lysates were utilized for Western blots to monitor the USP1 level. (d and e) p35-overexpressing U2OS cells (d) or MCF10A cells (e) were transfected with the indicated siRNAs, and 48 h later, the cells were incubated with mimosine to induce G1/S arrest. The cells were then irradiated with 30 J/m2 UV light, and at the indicated time points, lysates were collected and analyzed by Western blotting with the indicated antibodies. (f and g) U2OS cells were transfected with the indicated plasmids and kept under selection for 2 weeks to increase the percentage of cells overexpressing p25 or p35. Afterwards, they were transfected with the indicated siRNAs, and 48 h later, the cells were left untreated (f) or were incubated with mimosine (g). Sixteen hours later, cell lysates were utilized for Western blots to monitor USP1 levels.
What is the mechanism behind calpain modulation of APC/Ccdh1? It is well established that calpain modulates p35/Cdk5 kinase by processing a p35 regulatory subunit to a p25 form (23) (24). Moreover, calpain-activated p25/Cdk5 can repress APC/Ccdh1 activity by triple phosphorylation of cdh1 (25). Accordingly, we found that CDK5 depletion determines a decrease in USP1 stability, just as CAPNS1 depletion does (Fig. 6c).
Endogenous Cdk5/p35 was shown to play an essential role in a number of cell types; in particular, in U2OS cells it is required for senescence induction (26). However, due to p35's short half-life in this cell line, endogenous p35 is barely detectable, unless the cells are treated with MG132 (26). To overcome this problem, we investigated calpain cleavage of p35 in U2OS cells overexpressing p35 in mimosine-treated cells, after UV light irradiation (Fig. 6d). The same time course experiment was carried out in MCF10A mammary cells (Fig. 6e).
In both cell lines, UV treatment is coupled to a reduction of p35 stability and an increase in the p25/p35 ratio. These events do not occur in CAPNS1-depleted cells, confirming published data on calpain-dependent processing of p35 to p25. Notably, cdc6, a well-established APC/Ccdh1 substrate, is more rapidly degraded (p35-U2OS) or is already less abundant in CAPNS1-depleted cells (MCF10A). These pieces of evidence further argue for an increase in APC/Ccdh1 activity in CAPNS1-depleted cells.
We hypothesized that under basal conditions calpain may stabilize USP1 by inhibiting cdh1 activity via activation of Cdk5. We found that transfection of Cdk5 activator p25, but not of its precursor p35, in CAPNS1-depleted cells can partially rescue USP1 stability (Fig. 6f). The rescue effect is more pronounced in late G1 enriched cells (Fig. 6g), confirming that calpain may act as a brake for APC/Ccdh1-dependent proteasomal degradation specifically in late G1. These data indicate that calpain may tune USP1 degradation acting on one of the key elements in the USP1 route for proteasomal degradation.
USP1 binds cdh1 more efficiently in CAPNS1-depleted cells.
We found that calpain stabilizes USP1 in a cdh1 (Fig. 6a)- and Cdk5/p25 (Fig. 6f and g)-dependent manner. These results are in accordance with published data on the role of calpain as an inhibitor of APC/Ccdh1-dependent proteasomal degradation, via activation of Cdk5/p25 (25). In this scenario, we expected that CAPNS1 depletion would enhance the engagement of USP1 in the APC/Ccdh1 complex. To verify this hypothesis, U2OS cells were transfected with CAPNS1 or control siRNA and subsequently incubated for 16 h to arrest them in late G1. Cell lysates were subjected to immunoprecipitation with an anti-cdh1 antibody, and the products were analyzed by Western blotting. As shown in Fig. 7a, the anti-cdh1 antibody can efficiently immunoprecipitate endogenous USP1 from protein extracts of CAPNS1-depleted cells. This result confirms that USP1/cdh1 complex formation is enhanced in CAPNS1-depleted cells in accordance with the increased APC/Ccdh1-dependent proteasomal clearance of USP1. Cdk5 is present in the USP1/cdh1 complex in CAPNS1-depleted cells, confirming previous studies that described a transient direct interaction between cdh1 and Cdk5 (25). In cells where calpain is active, the resulting complex may be transient and unstable given the reciprocal negative cross-regulation between APC/Ccdh1 and active Cdk5, as recently reported (27).
The stabilizing effect of calpain on USP1 could be due to the indirect effect on cdh1 but also to a direct stabilizing cleavage. In vitro transcribed/translated USP1 is a perfect target for calpain (Fig. 1f), confirming the predictions made by means of in silico search for calpain cleavage sites. Indeed, USP1 is rapidly degraded in vitro, suggesting that multiple calpain target sites are present which are probably masked in vivo by other proteins and/or by specific compartmentalization. Both N-terminus and C-terminus USP1 deletion mutants can be cleaved by calpain in vitro (data not shown), confirming the existence of multiple potential cleavage sites scattered throughout the protein.
Since we never detected a calpain-specific cleavage product of endogenous USP1 in vivo, we hypothesized that the cleavage may occur at the very N-terminal end of the protein, thus not affecting the apparent molecular weight on a regular SDS-PAGE. Alternatively, cleavage products may have a very short half-life and therefore may be difficult to detect. Interestingly, a published work on the function of various USPs utilizes a USP1 protein lacking the first 20 amino acids, most likely to increase the stability of the recombinant protein (28). Notably, this first stretch of amino acids is identical in human, mouse, and rat USP1. USP1 domains described in the literature, as well as a potential Cdk5 (Cdc2 kinase) target site at serine 16 and one potential N-terminal calpain cleavage site, are schematized in Fig. 7b. Indeed, using Scansite software, available at http://scansite3.mit.edu/, at high stringency, we found that serine 16 is located within a Cdk5 (or Cdc2 kinase) consensus sequences (Fig. 7b). Interestingly, Ser 16 has been found phosphorylated in human and mouse cells by means of mass spectrometry analysis (www.phosphosite.org/). In addition, using the software available at http://calpain.org, we found a potential calpain target at serine 13 (Fig. 7b). To investigate the importance of the potential Cdk phosphorylation site and of the potential calpain target site on USP1 stability, we produced specific point mutants. We mutated the potential Cdk target serine 16 (AGC) to an alanine (GCA) and the amino acid located at position P2 with respect to calpain predicted cleavage, leucine 12 (AGC) to a glycine (GCA), following the same strategy successfully used to pinpoint the calpain cleavage site on talin (29) (Fig. 7c, left panel). These constructs were transfected in CAPNS1-depleted cells and control cells to monitor their relative stability. As shown in the right panel of Fig. 7c, a C-terminally tagged USP1 is stabilized in the presence of calpain as endogenous USP1. On the other hand, a point mutant at position P2 with respect to a predicted calpain cleavage site (L12G) renders USP1 unstable, just like wild-type USP1 upon calpain inhibition. This result suggests that calpain cleavage of USP1 at the very N terminus protects it from proteasome-dependent degradation.
Calpain regulation of USP1 is quite complex and multifaceted. To recapitulate, in the present study we focused on the characterization of the calpain/Cdk5/APC/Ccdh1/USP1 axis in osteosarcoma cells under physiological conditions and upon UV damage.
Altogether the results presented in this study support a model schematized in Fig. 7d. Calpain cleaves USP1 at the very N terminus, resulting in its stabilization. In addition, calpain activates Cdk5/p25, which acts as a brake for APC/Ccdh1-dependent ubiquitination of USP1, further stabilizing the protein. In the absence of the stabilizing cleavage exerted by calpain, USP1 interacts with cdh1 (Fig. 7a) and is more prone to ubiquitination by APC/Ccdh1 (Fig. 6a). Other studies are ongoing to further dissect the complexity of calpain regulation of USP1 stability under physiological and pathological conditions.
DISCUSSION
Osteosarcoma, the most common primary bone tumor in young adults, is characterized by local invasion and distant metastasis. A recent report described USP1 as a “druggable” protein for differentiation therapy (30). Indeed, USP1 knockdown in osteosarcoma cells determined ID2 protein destabilization, cell cycle arrest, and osteogenic differentiation (9). Accordingly, USP1 is required for proper osteogenesis (7). Remarkably, silencing of calpain expression reduces the metastatic potential of the human osteosarcoma cell (31). Moreover, tissue-specific KO studies demonstrated that the calpain small subunit is essential for proper osteoblast activity and bone remodeling (10). We found that CAPNS1 depletion in osteosarcoma cells and fibroblasts is linked to USP1 destabilization in a cdh1/Cdk5-dependent manner. As expected, CAPNS1 silencing, just like USP1 depletion, is coupled to ID2 protein decrease and subsequent p21 accumulation. A number of clinical trials for different neural disorders and cardiovascular diseases and malignancies (3) targeting calpain are already going on. Since we found that calpain inhibition is coupled to USP1 instability, we may speculate that calpain inhibitors may represent convenient drugs for specific cancers where the calpain/Cdk5/cdh1/USP1 axis is active. It is certainly worthwhile planning to investigate the feasibility of a therapeutic approach for osteosarcoma based on calpain inhibition. In addition, since USP1 inhibitors have been proposed as potential drugs for non-small-cell lung carcinoma (30), also in this type of cancers, calpain inhibitors might be considered for combination therapy trials.
Certainly, caution must be observed due to the effects of USP1 depletion on genome stability. Indeed, we have demonstrated that calpain depletion, like USP1 depletion, is coupled to an increase in PCNA ubiquitination and subsequent overloading of translesion synthesis pol-η on DNA. Such a phenotype is linked to increased mutagenesis in U2OS cells, as previously described for USP1-depleted cells (6). Accordingly, a defect in DNA repair dynamics occurs in Capns1 KO MEFs. We showed for the first time that calpain plays an active role in the integrated signaling and genome maintenance that the cell activates to overcome DNA damage. Interestingly, the activation of the DNA repair enzyme poly(ADP-ribose) polymerase 1 (PARP1) in response to DNA damage was shown to trigger calcium overload and activation of mitochondrial calpain (32). Calpain activation resulted in apoptosis inducing factor (AIF) processing and apoptosis induction (32). This represents another example in which calpain inhibition after DNA damage could lead to accumulation of damaged DNA, in the absence of apoptosis. Calpain may indeed represent an important caretaker for the cell.
We have found that CAPNS1 interacts with USP1 in living cells. The region sufficient for this interaction encompasses the previously identified recognition site for UAF1/WDR48 (33). We therefore cannot exclude that the binding is mediated by this factor, raising the possibility that calpain also affects other signaling pathways involving this protein regulator. Interestingly, we showed that calpain depletion is coupled to a reduction in UAF1/WDR48 protein levels. UAF1/WDR48 is required not only for USP1 catalytic function and stability but also for USP46, an important modulator of GABA signaling and USP12 involved in histone H2A and H2B deubiquitination. It will therefore be worthwhile to investigate whether CAPNS1 plays any role in the signaling pathways governed by these deubiquitinating enzymes. In addition, we have demonstrated that calpain inhibits APC/Ccdh11 activity directed toward USP1. Accordingly, the stability of another cdh1 substrate, cdc6, decreases in CAPNS1-depleted cells. Besides the well-recognized cell cycle regulators and cyclins, this complex controls the proteasomal degradation of key components of the mitochondrial division machinery (34). cdh1 apparently acts as a link between cell cycle modulation and metabolism, and calpain supervision may contribute to the concerted modulation of such nodal points. Many studies have demonstrated the activating role of calpain on Cdk5 function. Calpain-mediated p35 cleavage appears to be crucial for the amplification of the Cdk5 activity that is required during differentiation (24). Triple phosphorylation of cdh1 by the calpain-activated Cdk5/p25 has a repressive function on the APC/Ccdh1 complex and results in cyclin B1 stabilization (25).
Our study confirms the regulatory role of Cdk5 on cdh1 and consequently on proteasomal degradation of USP1 and unveils a role of calpain at the interface between cell cycle regulation and DNA replication and/or repair. In addition, it opens up a number of questions about the effect of calpain on other Cdk5 substrates, such as p53 (35), erk (36), and related pathways and other APC/Ccdh1 substrates, such as rad17 (37), involved in DNA damage response.
To recapitulate, in this study we have unveiled a calpain/cdh1/USP1 axis. We detected a complex containing CAPNS1 and USP1 that leads to a transient USP1 stabilization and a complex containing USP1 and cdh1 in calpain-depleted cells, which is consistent with an increase in APC/Ccdh1-dependent USP1 clearance. In addition, we have found that a single Leu-to-Gly substitution at amino acid 12 of USP1, at the P2 position with regard to a predicted calpain cleavage site, determines a dramatic destabilization of USP1. Altogether, these data point to a stabilizing role of calpain for USP1 deubiquitinating enzyme in physiological conditions. Further studies are ongoing to dissect the CAPNS1-USP1-UAF1-cdh1 interaction and reciprocal regulation under pathological conditions linked to APC/Ccdh1 impairment and/or calcium overload. These studies will deepen our understanding of the molecular basis by which the calpain/calpastatin network can enhance cancer cell survival and may be instrumental for the design of novel drugs for specific diseases.
ACKNOWLEDGMENTS
We are grateful to Tony Huang (NYU) for constructive criticism and for providing myc-USP1 wild-type and mutant constructs, as well as anti-USP1 antibodies. We acknowledge Kenta Terai and Anindya Dutta (University of Virginia) for providing the supF assay protocol and reagents (pSP189 plasmid and competent bacteria). We thank René Bernards (NKI, Netherlands) for FLAG-USP1 and GFP-USP1, Alan Lehmann (University of Sussex, United Kingdom) for GFP–pol-η, and Jae U. Jung (University of Southern California) for FLAG-WDR48. We are grateful to Peter Greer (Queen's University, Canada) for Capns1−/− mouse embryonic fibroblasts and respective controls and Fanconi Anemia Research Fund for providing antibodies against USP1.
This work was supported by AIRC grant (G-2010) and FIRB grant (CINECA RBAP11LP2W) to C.S. F.C. was the recipient of a fellowship from Area Science Park (SHARM Project) and now is paid by a grant financed by Cross-Border Cooperation Programme Italy-Slovenia 2007–2013 (PROTEO) to F.D.
Footnotes
Published ahead of print 15 April 2013
REFERENCES
- 1. Sorimachi H, Hata S, Ono Y. 2010. Expanding members and roles of the calpain superfamily and their genetically modified animals. Exp. Anim. 59:549–566 [DOI] [PubMed] [Google Scholar]
- 2. Croall DE, Ersfeld K. 2007. The calpains: modular designs and functional diversity. Genome Biol. 8:218 doi:10.1186/gb-2007-8-6-218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Storr SJ, Carragher NO, Frame MC, Parr T, Martin SG. 2011. The calpain system and cancer. Nat. Rev. Cancer 11:364–374 [DOI] [PubMed] [Google Scholar]
- 4. Nijman SM, Huang TT, Dirac AM, Brummelkamp TR, Kerkhoven RM, D'Andrea AD, Bernards R. 2005. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol. Cell 17:331–339 [DOI] [PubMed] [Google Scholar]
- 5. Oestergaard VH, Langevin F, Kuiken HJ, Pace P, Niedzwiedz W, Simpson LJ, Ohzeki M, Takata M, Sale JE, Patel KJ. 2007. Deubiquitination of FANCD2 is required for DNA crosslink repair. Mol. Cell 28:798–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huang TT, Nijman SM, Mirchandani KD, Galardy PJ, Cohn MA, Haas W, Gygi SP, Ploegh HL, Bernards R, D'Andrea AD. 2006. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat. Cell Biol. 8:339–347 [DOI] [PubMed] [Google Scholar]
- 7. Kim JM, Parmar K, Huang M, Weinstock DM, Ruit CA, Kutok JL, D'Andrea AD. 2009. Inactivation of murine Usp1 results in genomic instability and a Fanconi anemia phenotype. Dev. Cell 16:314–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Murai J, Yang K, Dejsuphong D, Hirota K, Takeda S, D'Andrea AD. 2011. The USP1/UAF1 complex promotes double strand break repair through homologous recombination. Mol. Cell. Biol. 31:2462–2469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Williams SA, Maecker HL, French DM, Liu J, Gregg A, Silverstein LB, Cao TC, Carano RA, Dixit VM. 2011. USP1 deubiquitinates ID proteins to preserve a mesenchymal stem cell program in osteosarcoma. Cell 146:918–930 [DOI] [PubMed] [Google Scholar]
- 10. Shimada M, Greer PA, McMahon AP, Bouxsein ML, Schipani E. 2008. In vivo targeted deletion of calpain small subunit, Capn4, in cells of the osteoblast lineage impairs cell proliferation, differentiation, and bone formation. J. Biol. Chem. 283:21002–21010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cotto-Rios XM, Jones MJ, Busino L, Pagano M, Huang TT. 2011. APC/CCdh1-dependent proteolysis of USP1 regulates the response to UV-mediated DNA damage. J. Cell Biol. 194:177–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wasch R, Robbins JA, Cross FR. 2010. The emerging role of APC/CCdh1 in controlling differentiation, genomic stability and tumor suppression. Oncogene 29:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cohn MA, Kowal P, Yang K, Haas W, Huang TT, Gygi SP, et al. 2007. A UAF1-containing multisubunit protein complex regulates the Fanconi anemia pathway. Mol. Cell 28:786–797 [DOI] [PubMed] [Google Scholar]
- 14. Park J, Lee BS, Choi JK, Means RE, Choe J, Jung JU. 2002. Herpesviral protein targets a cellular WD repeat endosomal protein to downregulate T lymphocyte receptor expression. Immunity 17:221–233 [DOI] [PubMed] [Google Scholar]
- 15. Cohn MA, Kee Y, Haas W, Gygi SP, D'Andrea AD. 2009. UAF1 is a subunit of multiple deubiquitinating enzyme complexes. J. Biol. Chem. 284:5343–5351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Demarchi F, Cataldo F, Bertoli C, Schneider C. 2010. DNA damage response links calpain to cellular senescence. Cell Cycle 9:755–760 [DOI] [PubMed] [Google Scholar]
- 17. Arthur JS, Elce JS, Hegadorn C, Williams K, Greer PA. 2000. Disruption of the murine calpain small subunit gene, Capn4: calpain is essential for embryonic development but not for cell growth and division. Mol. Cell. Biol. 20:4474–4481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bish RA, Fregoso OI, Piccini A, Myers MP. 2008. Conjugation of complex polyubiquitin chains to WRNIP1. J. Proteome Res. 7:3481–3489 [DOI] [PubMed] [Google Scholar]
- 19. Tompa P. 2011. Unstructural biology coming of age. Curr. Opin. Struct. Biol. 21:419–425 [DOI] [PubMed] [Google Scholar]
- 20. Kiss R, Kovacs D, Tompa P, Perczel A. 2008. Local structural preferences of calpastatin, the intrinsically unstructured protein inhibitor of calpain. Biochemistry 47:6936–6945 [DOI] [PubMed] [Google Scholar]
- 21. Watanabe K, Tateishi S, Kawasuji M, Tsurimoto T, Inoue H, Yamaizumi M. 2004. Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 23:3886–3896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cotto-Rios XM, Jones MJ, Huang TT. 2011. Insights into phosphorylation-dependent mechanisms regulating USP1 protein stability during the cell cycle. Cell Cycle 10:4009–4016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kusakawa G, Saito T, Onuki R, Ishiguro K, Kishimoto T, Hisanaga S. 2000. Calpain-dependent proteolytic cleavage of the p35 cyclin-dependent kinase 5 activator to p25. J. Biol. Chem. 275:17166–17172 [DOI] [PubMed] [Google Scholar]
- 24. de Thonel A, Ferraris SE, Pallari HM, Imanishi SY, Kochin V, Hosokawa T, Hisanaga S, Sahlgren C, Eriksson JE. 2010. Protein kinase Czeta regulates Cdk5/p25 signaling during myogenesis. Mol. Biol. Cell 21:1423–1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Maestre C, Delgado-Esteban M, Gomez-Sanchez JC, Bolanos JP, Almeida A. 2008. Cdk5 phosphorylates Cdh1 and modulates cyclin B1 stability in excitotoxicity. EMBO J. 27:2736–2745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mao D, Hinds PW. 2010. p35 is required for CDK5 activation in cellular senescence. J. Biol. Chem. 285:14671–14680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang J, Li H, Zhou T, Zhou J, Herrup K. 2012. Cdk5 levels oscillate during the neuronal cell cycle: Cdh1 ubiquitination triggers proteosome-dependent degradation during S-phase. J. Biol. Chem. 287:25985–25994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Faesen AC, Luna-Vargas MP, Geurink PP, Clerici M, Merkx R, van Dijk WJ, Hameed DS, El Oualid F Ovaa H, Sixma TK. 2011. The differential modulation of USP activity by internal regulatory domains, interactors and eight ubiquitin chain types. Chem. Biol. 18:1550–1561 [DOI] [PubMed] [Google Scholar]
- 29. Franco SJ, Rodgers MA, Perrin BJ, Han J, Bennin DA, Critchley DR, Huttenlocher A. 2004. Calpain-mediated proteolysis of talin regulates adhesion dynamics. Nat. Cell Biol. 6:977–983 [DOI] [PubMed] [Google Scholar]
- 30. Chen J, Dexheimer TS, Ai Y, Liang Q, Villamil MA, Inglese J, Maloney DJ, Jadhav A, Simeonov A, Zhuang Z. 2011. Selective and cell-active inhibitors of the USP1/ UAF1 deubiquitinase complex reverse cisplatin resistance in non-small cell lung cancer cells. Chem. Biol. 18:1390–1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fan DG, Dai JY, Tang J, Wu MM, Sun SG, Jiang JL, Fan QY. 2009. Silencing of calpain expression reduces the metastatic potential of human osteosarcoma cells. Cell Biol. Int. 33:1263–1267 [DOI] [PubMed] [Google Scholar]
- 32. Vosler PS, Sun D, Wang S, Gao Y, Kintner DB, Signore AP, Cao G, Chen J. 2009. Calcium dysregulation induces apoptosis-inducing factor release: cross-talk between PARP-1- and calpain-signaling pathways. Exp. Neurol. 218:213–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Garcia-Santisteban I, Zorroza K, Rodriguez JA. 2012. Two nuclear localization signals in USP1 mediate nuclear import of the USP1/UAF1 complex. PLoS One 7:e38570 doi:10.1371/journal.pone.0038570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Horn SR, Thomenius MJ, Johnson ES, Freel CD, Wu JQ, Coloff JL, Yang CS, Tang W, An J, Ilkayeva OR, Rathmell JC, Newgard CB, Kornbluth S. 2011. Regulation of mitochondrial morphology by APC/CCdh1-mediated control of Drp1 stability. Mol. Biol. Cell 22:1207–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ajay AK, Upadhyay AK, Singh S, Vijayakumar MV, Kumari R, Pandey V, Boppana R, Bhat MK. 2010. Cdk5 phosphorylates non-genotoxically overexpressed p53 following inhibition of PP2A to induce cell cycle arrest/apoptosis and inhibits tumor progression. Mol. Cancer 9:204 doi:10.1186/1476-4598-9-204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brinkkoetter PT, Olivier P, Wu JS, Henderson S, Krofft RD, Pippin JW, Hockenberry D, Roberts JM, Shankland SJ. 2009. Cyclin I activates Cdk5 and regulates expression of Bcl-2 and Bcl-XL in postmitotic mouse cells. J. Clin. Invest. 119:3089–3101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang L, Park CH, Wu J, Kim H, Liu W, Fujita T, Balasubramani M, Schreiber EM, Wang XF, Wan Y. 2010. Proteolysis of Rad17 by Cdh1/APC regulates checkpoint termination and recovery from genotoxic stress. EMBO J. 29:1726–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Demarchi F, Bertoli C, Greer PA, Schneider C. 2005. Ceramide triggers an NF-kappaB-dependent survival pathway through calpain. Cell Death Differ. 12:512–522 [DOI] [PubMed] [Google Scholar]