

Abstract
Complex I deficiency is commonly associated with mitochondrial oxidative phosphorylation diseases. Mutations in nuclear genes encoding structural subunits or assembly factors of complex I have been increasingly identified as the cause of the diseases. One such factor, NDUFAF2, is a paralog of the NDUFA12 structural subunit of the enzyme, but the mechanism by which it exerts its function remains unknown. Herein, we demonstrate that the Neurospora crassa NDUFAF2 homologue, the 13.4L protein, is a late assembly factor that associates with complex I assembly intermediates containing the membrane arm and the connecting part but lacking the N module of the enzyme. Furthermore, we provide evidence that dissociation of the assembly factor is dependent on the incorporation of the putative regulatory module composed of the subunits of 13.4 (NDUFA12), 18.4 (NDUFS6), and 21 (NDUFS4) kDa. Our results demonstrate that the 13.4L protein is a complex I assembly factor functionally conserved from fungi to mammals.
INTRODUCTION
Mitochondrial complex I (NADH:ubiquinone oxidoreductase, EC 1.6.5.3) is an L-shaped structure embedded in the inner mitochondrial membrane that transfers electrons from NADH to ubiquinone coupled to the translocation of protons to the intermembrane space (1). In mammals, it is composed of 45 dissimilar polypeptides encoded by both the mitochondrial and the nuclear DNA (2) These proteins, together with an FMN molecule and eight iron sulfur clusters, are organized in three functional modules: (i) the N module, responsible for the binding and oxidation of NADH; (ii) the Q module, the final acceptor of the complex, which transfers the electrons to ubiquinone; and (iii) the P module, involved in proton translocation by a conformational-driven mechanism (1). The N and Q modules are located in the peripheral arm protruding into the matrix and comprise all known cofactors, while the P module forms the membrane part of the enzyme and contains all the mitochondrial DNA (mtDNA)-encoded subunits.
In the membrane, complex I is usually associated with complex III and IV in supramolecular structures called supercomplexes, whose biosynthesis remain unsolved (3). These supercomplexes are regarded as relevant for reducing the diffusion distance of the substrates, improving electron transfer, reducing the formation of reactive oxygen species, and stabilizing the individual respiratory complexes (3–6).
Complex I dysfunction is the most frequent oxidative phosphorylation (OXPHOS) disorder in humans where defects in enzyme function and/or assembly have been associated with the development of clinically heterogeneous diseases (7). To date, mutations in at least 20 subunit genes and nine genes encoding assembly factors have been described, with a myriad of affected patients still waiting for a genetic diagnosis (for reviews, see references 8 and 9).
Given its huge complexity, the assembly of complex I is a multistep process, in which different subunits combine into assembly intermediates that subsequently join together to form the mature and functional enzyme (10). This process is aided by several assembly factors, proteins that do not belong to the mature enzyme but rather associate with assembly intermediates during biogenesis of complex I (11).
In recent years, several models for complex I assembly have being proposed, all of which imply that assembly intermediates join to form the holo-complex in a sequential pathway. However, some controversy exists regarding assembly subcomplexes between the different model systems (12, 13).
The fungus Neurospora crassa has been an important model system, providing remarkable insight into complex I assembly. The Neurospora enzyme is composed of 43 different polypeptides (14), all of them displaying homologues in the mammalian complex (15). The characterization of mutant strains harboring disrupted complex I genes led to the outline of an assembly model, in which three membrane arm intermediates are assembled independently, two of them combining to originate the large membrane arm intermediate that joins with the small intermediate forming the membrane arm. The peripheral domain can be assembled separately and upon combination with the membrane domain produces a mature enzyme (16). However, intermediates of assembly lacking part of the N module (nuo24 and nuo51) have been described (17, 18), demonstrating that the membrane arm can associate with an “incomplete” hydrophilic domain (17).
A slightly different pathway was described for mammalian complex I assembly in which peripheral and membrane subunits associate in the early steps of assembly. The most recent model proposes that an early assembly intermediate is anchored to the membrane prior to its extension by addition of membrane and peripheral subunits (12). A peripheral subcomplex containing some of the core subunits expands by incorporation of a small membrane complex holding ND1, thus forming a 400-kDa intermediate. This associates with a 460-kDa membrane complex containing ND2, ND3, ND4L, and ND6 and then with another membrane subcomplex containing ND4 and ND5, leading to an 830-kDa intermediate. With the addition of the N module and some remaining subunits, mature complex I is subsequently fully assembled (10). In parallel to this de novo synthesis of complex I, Lazarou and colleagues proposed a regeneration process in which the exchange of preexisting subunits with newly imported ones could maintain complex I homeostasis (19).
Assembly factors are associated with all of these assembly intermediates, although the precise function of most of them remains elusive. One such assembly factor, NDUFAF2, was found to be associated with the 830-kDa intermediate, which accumulates in patients who are mitochondrion deficient in some nucleus-located complex I genes, namely, NDUFV1 (20), NDUFS4 (21), and NDUFS6 (22). This intermediate is a stalled subassembly, since import of the missing subunit restores complex I assembly, and furthermore, the 830-kDa intermediate is present in small amounts in control cells (19), whereas most of the NDUFAF2 protein is found as a monomer (20). The absence of NDUFAF2 due to null mutations does not entirely prevent assembly or activity of complex I, although it leads to the development of a progressive encephalopathy (20, 23).
A better understanding of the specific involvement of NDUFAF2 in complex I assembly will provide important insights into its mechanism of action while identifying specific stages of assembly not previously uncovered. For these purposes, we investigated the effects of disruption of the N. crassa homologue of NDUFAF2 (13.4L) and of its paralog protein, the 13.4-kDa complex I subunit (NDUFA12), in complex I assembly and function. Moreover, we characterized the 13.4L-containing structures in specific complex I mutants and propose a model for the final steps of complex I assembly. Herein, we show that N. crassa 13.4L is associated with assembly intermediates of complex I that contain both nucleus- and mtDNA-encoded subunits and are able to form supercomplexes through association with dimeric complex III.
MATERIALS AND METHODS
Strains and plasmids.
Manipulation of N. crassa wild type strains 74-OR23-1A and 74-OR8-1a as well as of the complex I mutant strains nuo9.8 (24), nuo12.3 (25), nuo14 (14), nuo21 (26), nuo21.3a (27), nuo21.3c (28), nuo24 (17), nuo51 (18), and nuo78 (29) was performed according to standard procedures (30). The nuo13.4 (FGSC 13054a and FGSC 13055A), nuo18.4 (FGSC 15972a), and 13.4L (FGSC 11150a) mutant strains were obtained from the FGSC. The plasmid vectors pCRII-TOPO (Invitrogen) and pQE-31 (Qiagen) were used for intermediate cloning and protein expression in the bacterial systems DH5α and M15, respectively.
Molecular cloning and protein expression.
The 13.4L C-terminal encoding region was amplified from genomic DNA by PCR using primers 5′-CTGCAGCTCGACCTAGCTGGCAATGCC-3′ and 5′-CTGC AGGGAAGCATCTAAATCACCCCTTC-3′ that generated the restriction sites for PstI (in bold). The amplified 745-bp fragment was cloned in pCRII-TOPO, sequenced, excised with PstI, and subcloned into pQE-31 for protein expression. The cDNA encoding the 13.4-kDa protein was amplified from the N. crassa Sachs library by PCR using primers 5′-GAGCTCAATGAACTATATCGGCGACACC-3′ and 5′-GAGCTCTTAG TGTCTAGGAGCAGCAACAGG-3′ that introduced the restriction sites for SacI (in bold). The amplified 361-bp fragment was cloned in pCRII-TOPO, sequenced, excised with SacI, and subcloned in pQE-31 for protein expression. Bacterial strains containing pQE-31 recombinant plasmids were subsequently induced with IPTG in order to express the fungal proteins. In this way, the 13.4-kDa polypeptide and a 26.5-kDa polypeptide were separately expressed as fusion proteins containing 17 and 24 extra residues at the N terminus, respectively, including a six-histidine tag. The fusion proteins were purified in nickel columns and used to immunize rabbits and raise specific polyclonal antisera against the polypeptides as previously described (31).
Electrophoretic techniques.
Crude mitochondria (32) were thawed on ice and centrifuged at 10,000 × g for 10 min. The pellet was suspended in solubilization buffer containing 50 mM NaCl, 50 mM imidazole-HCl (pH 7.0), 1 mM EDTA, 2 mM phenylmethylsulfonyl fluoride (PMSF), and 5 mM 6-aminocaproic acid. Mitochondria were solubilized with digitonin (DIG) using a detergent/protein ratio of 4 g/g. The samples were incubated 15 min on ice followed by centrifugation at 10,000 × g for 30 min, and each lane was loaded with mitochondria extracts containing 150 μg of protein prior to solubilization. For blue native (BN) PAGE, different linear gradient gels overlaid with a 3% stacking gel were used. Lanes from the first dimension BN PAGE were subsequently excised and resolved in a second-dimension 12% SDS-PAGE upon incubation in a 1% SDS–1% β-mercaptoethanol solution for 2 h at room temperature (33). In-gel activity assays were performed as described elsewhere (34).
Oxygen consumption and enzymatic activities.
Oxygen uptake in mitochondria was measured polarographically at 25°C with a Clark-type oxygen electrode (Hansatech) in a total volume of 1 ml. The assays were started by the addition of 1 mM NADH to a mitochondrial suspension (0.1 to 1 mg) in 0.3 M sucrose, 10 mM potassium phosphate (pH 7.2), 5 mM MgCl2, 1 mM EGTA, 10 mM KCl, 4 μM carbonyl cyanide m-chlorophenylhydrazone, 0.02% (wt/vol) bovine serum albumin (BSA), which had been incubated with 20 μg/ml alamethicin for 2 min. Rotenone and antimycin A were added to final concentrations of 15 μM and 0.2 μg/ml, respectively.
NADH:hexaammineruthenium III (HAR) reductase activity was measured photometrically (ε340 = 6.22 mM−1 cm−1) in 50 mM Tris HCl, pH 8.0, in the presence of 1 mM KCN, 120 μM NADH, and 2 mM HAR.
Immunoprecipitation.
Mitochondria (1 to 2 mg) were resuspended in 30 mM Tris HCl (pH 7.5), 300 mM NaCl, and 1 mM PMSF and solubilized with 4 g digitonin (DIG)/g protein for 30 min on ice. Insoluble material was discarded in a preclearing step by centrifugation at 10,000 × g for 30 min. The supernatant was then incubated with specific antiserum overnight at 4°C in an orbital shaker. Antibody-antigen complexes were captured by incubation with protein A-Sepharose beads (Sigma-Aldrich) for 1 h at 4°C and recovered by centrifugation. Following six washings with 30 mM Tris HCl (pH 7.5), 300 mM NaCl, and 0.1% Triton X-100 and four with 30 mM Tris HCl (pH 7.5), the immunoprecipitates were analyzed by SDS-PAGE.
Miscellaneous techniques.
PCR and general cloning procedures were carried out according to standard protocols (35). The techniques used for protein determination (36), SDS-PAGE (37), blotting and incubation of blots with antisera (38), detection of anti-rabbit alkaline phosphatase-conjugated secondary antibodies (39), sucrose-gradient centrifugation analysis of detergent-solubilized mitochondrial proteins (27), and carbonate extraction (40) have been described and were performed accordingly.
RESULTS
Identification of the fungal NDUFAF2 homologue.
In silico analysis of the N. crassa genomic database (Neurospora Sequencing Project) led to the identification of the gene (NCU00278.5) encoding the 13.4L protein, which is a homologue of the described mammalian complex I assembly chaperone NDUFAF2 (20) and a paralog of the 13.4-kDa structural subunit of complex I (14). The single-copy gene is located on linkage group III of the fungal genome and comprises 2 exons. The corresponding polypeptide, predicted to be mitochondrial according to Mitoprot software (41), has 271 amino acid residues with a molecular mass of 30,408 Da and an isoelectric point of 9.69. Alignment of both Neurospora proteins revealed that they are 23% identical and 33% similar (Fig. 1), whereas a multiple sequence alignment revealed that the fungal 13.4L protein is not highly homologous to its mammalian equivalents.
Fig 1.

13.4L is the paralog of the 13.4-kDa structural subunit of complex I. Identical amino acid residues are shown on a gray background. Accession numbers are XP_961049 and XP_957749 for the 13.4-kDa subunit and 13.4L, respectively.
13.4L strongly interacts with the mitochondrial membrane.
To determine the mitochondrial localization of the 13.4-kDa subunit and 13.4L, we raised specific antibodies against each of the proteins. They recognized proteins with apparent molecular masses of approximately 14 kDa and 35 kDa, respectively, in mitochondrial extracts from the wild-type strain that were absent in mitochondria from the corresponding knockout mutants (Fig. 2A).
Fig 2.

The 13.4L protein is a mitochondrial protein that strongly interacts with the membrane. (A) Steady-state levels of the 13.4-kDa subunit of complex I and the paralog 13.4L in several N. crassa strains determined by Western blotting. (B) The 13.4-kDa subunit of complex I is a peripheral arm component. Mitochondria from the wild-type (wt) and nuo12.3 strains were centrifuged in sucrose gradients (7.5 to 25%) upon digitonin solubilization. Fractions (labeled 1 to 11 from top to bottom) from the gradients were collected and analyzed by Western blotting with individual antisera against the complex I subunits 21.3c, 18.4, and 13.4 kDa, as well as against the complex III CORE II subunit. (C) Membrane localization of the 13.4L protein. Mitochondria (T) were incubated under alkaline conditions and resolved by centrifugation into supernatant (S) and pellet (P) fractions and subsequently analyzed by Western blotting. Mitochondria from the wild type (wt) (M) were also sonicated and separated into membrane structures (SMP) and soluble fraction (S).
The steady-state levels of both proteins were subsequently analyzed in N. crassa mutants bearing different complex I assembly phenotypes (Table 1). As depicted in Fig. 2A, the levels of the 13.4-kDa subunit are decreased in complex I mutants with a disturbed membrane arm (nuo9.8 and nuo14) and are almost undetectable in the nuo21.3c mutant, which lacks the peripheral arm. In mutants that assemble an almost intact complex I (such as nuo21 and nuo21.3a), the 13.4-kDa subunit is present in wild-type amounts. Upon sucrose gradient analysis of mitochondrial proteins, we also observed that the 13.4-kDa subunit sediments with peripheral arm subunits in the complex I mutant nuo12.3 (25), which accumulates the peripheral intermediate (Fig. 2B).
Table 1.
Proteins and corresponding N. crassa mutant strains employed
| Subunit and strain | Protein in Homo sapiens | Property(ies) | Assembly and intermediatea | Activity | Reference |
|---|---|---|---|---|---|
| 78 kDa; nuo78 | NDUFS1 | N4, N5, N1b | No; a | No | 29 |
| 51 kDa; nuo51 | NDUFV1 | N3, NADH, FMN | Yes | No | 18 |
| 24 kDa; nuo24 | NDUFV2 | N1a | Yes | No | 17 |
| 21.3a kDa; nuo21.3a | NDUFA7 | Yes | Yes | 27 | |
| 21.3c kDa; nuo21.3c | NDUFS8 | N6a, N6b | No; MA | No | 28 |
| 21 kDa; nuo21 | NDUFS4 | Phosphorylation | Yes | Yes | 26 |
| 20.8 kDa; nuo20.8 | NDUFA8 | No; PA, small | No | 42 | |
| 18.4 kDa; nuo18.4 | NDUFS6 | Zinc binding | Yes | Yes | This work |
| 14 kDa; nuo14 | NDUFA13 | Proapoptotic | No; PA, small | No | 14 |
| 13.4 kDa; nuo13.4 | NDUFA12 | Nitrosylated | Yes | Yes | This work |
| 12.3 kDa; nuo12.3 | NDUFB10 | No; PA, large | No | 25 | |
| 9.8 kDa; nuo9.8 | NDUFA1 | No; PA, small | No | 24 | |
| 13.4L; 13.4L | NDUFAF2 | Assembly factor | Yes | Yes | This work |
a, intermediate that contains the P and Q modules of complex I; PA, peripheral arm intermediate; MA, membrane arm intermediate; small, small intermediate of the membrane arm; large, large intermediate of the membrane arm.
Overall, our results clearly demonstrate that the 13.4-kDa protein is part of the peripheral arm of complex I from N. crassa consistent with its localization in the mammalian enzyme (43).
In contrast, steady-state levels of the 13.4L protein (Fig. 2A) remain similar upon complex I disruption, suggesting that this protein is not a structural component of the mature enzyme.
We next addressed the localization of the 13.4L protein through alkaline extraction of mitochondria from different Neurospora strains. As illustrated in Fig. 2C, the 13.4L protein was found to be distributed between the pellet and supernatant fractions in all the tested strains, although more abundant in the pellet of the wild type. As controls, we analyzed the behavior of the Rieske iron sulfur protein (RIS-complex III) (44) and the 20.8-kDa subunit of complex I (45). As shown, the RIS protein was solubilized by the alkaline treatment whereas the 20.8-kDa protein remained in the pellet fraction, in accordance with previous reports (46). Moreover, we also separated mitochondria into mitochondrial membranes and soluble fraction by sonication and verified that the 13.4L was found mainly in the membranes with only a small portion in the supernatant (Fig. 2C). Taken together, our results suggest that the hydrophilic protein 13.4L strongly interacts with the mitochondrial membrane.
Complex I assembly and function is independent of the presence of 13.4L.
The mammalian homologue of the 13.4L protein has been suggested to be a complex I assembly factor, although its specific function remains unknown (20). To uncover its functional relevance, we set out to characterize the knockout mutants 13.4L and nuo13.4, obtained from the Fungal Genetics Stock Centre (FGSC) (47), in terms of steady-state levels of complex I subunits. Protein levels of the tested complex I subunits were unchanged between the wild type and the mutant strains (Fig. 3). Furthermore, several subunits from either the peripheral domain or the membrane arm were analyzed, and the results implied that disruption of either of these proteins does not dramatically interfere with complex I proteins. To assess whether the 13.4-kDa and the 13.4L proteins could be compensating for one another, given their homology, we produced a double mutant strain devoid of both the 13.4-kDa and the 13.4L proteins (nuo13.4×L), which did not display significantly altered steady-state levels of complex I subunits (Fig. 3).
Fig 3.
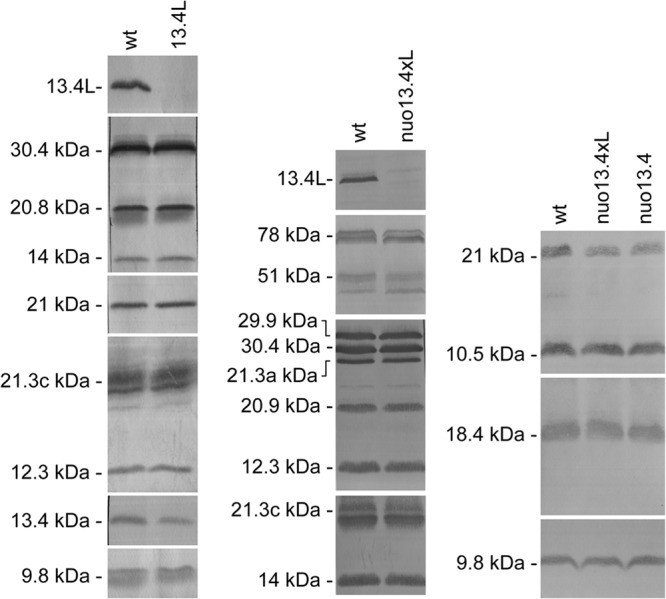
Steady-state levels of complex I subunits in the 13.4L and the nuo13.4×L mutants determined by Western blotting. Subunits 20.9, 20.8, 14, 12.3, and 9.8 kDa are components of the membrane arm whereas subunits 78, 51, 30.4, 29.9, 21.3a, 21.3c, 21, 18.4, 13.4, and 10.5 kDa are present in the peripheral domain of the enzyme.
We next analyzed mitochondria from the single and double mutants by BN PAGE and observed that all the mutant strains displayed complex I bands, similar to those of the wild-type strain (Fig. 4A, B, and C). Moreover, the patterns of complexes and supercomplexes within the respiratory chain were comparable in all strains, indicating that neither the 13.4-kDa nor the 13.4L protein is required for the assembly of the respiratory chain.
Fig 4.
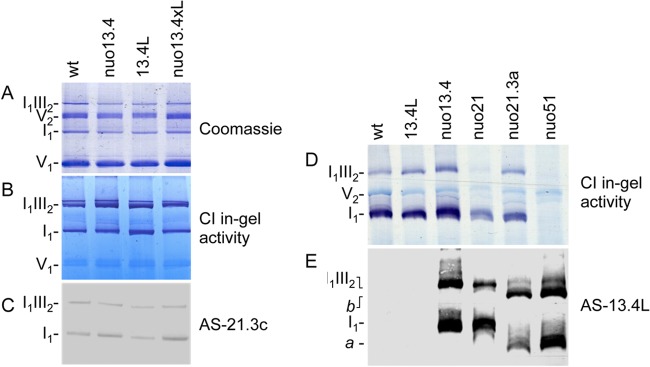
Effects of 13.4L disruption in respiratory chain organization. Mitochondrial proteins were separated in a 4-to-13% BN PAGE (A, B, and C) or 4-to-9% BN PAGE (D and E). The gels were stained with Coomassie blue (A), stained for NADH dehydrogenase activity (B and D), or analyzed by Western blotting with antibodies against complex I subunit 21.3c kDa (C) and the 13.4L protein (E). a and b are high-molecular-weight structures recognized by AS-13.4L.
To ascertain the function of the 13.4L protein in N. crassa, we examined its distribution in mitochondrial proteins from a group of complex I mutant strains, which assembled an almost intact enzyme, using nondenaturing gels (Fig. 4D and E). Complex I and its supercomplex I1III2 were identified by NADH:NBT (nitroblue tetrazolium) reductase in-gel activity (Fig. 4D) and the presence of the 13.4L protein was evaluated through Western blot analysis (Fig. 4E). The anti-13.4L antibody did not recognize the holoenzyme in the wild type but specifically recognized high-molecular-weight complexes in several complex I mutants, such as nuo13.4, nuo21, and nuo51 (Fig. 4E). We observed that this protein associates with different structures in different complex I mutants that do not correlate with active or inactive complex I. Moreover, complex I mutants which accumulate only early assembly intermediates do not display these 13.4L-containing high-molecular-weight complexes (data not shown). Overall, our data provide evidence that the N. crassa 13.4L protein is involved in a late step of complex I assembly, as previously described for mammals (20), conveying a conserved role for the protein from fungi to humans.
To assess the effects of nuo-13.4 and 13.4L deletions in oxidative phosphorylation, we measured rates of oxygen consumption by wild-type and mutant intact mitochondria, namely, through NADH oxidation in the presence of the hydrophobic peptide alamethicin (Table 2). Oxygen consumption by wild-type mitochondria was inhibited around 64% with rotenone, indicating that NADH enters the mitochondria and is oxidized by complex I. Likewise, NADH oxidation was inhibited upon addition of rotenone to mitochondria of the single and double mutants, confirming the presence of functional complex I in all the mutant strains. However, the percentage of rotenone inhibition was lower in the mutants, although both the nuo13.4 and the double mutant presented slightly increased NADH oxidase activity. We observed a clear decrease in complex I activity in the double mutant as determined by rotenone sensitive NADH oxidation (Table 2). The amount of complex I was further evaluated by measuring NADH:HAR(III) reductase activity in nuo13.4×L mitochondria, which exhibited a decreased amount of complex I, consistent with the observed decreased complex I activity. This decrease in complex I was not observed through BN PAGE analysis of the double mutant (Fig. 4), probably due to the lower sensitivity of the method compared to that of enzymatic assays. Antimycin A completely blocked the respiratory activities, indicating the exclusive use of the canonical respiratory chain by all strains.
Table 2.
Enzymatic activities of wild-type and mutant mitochondria
| Strain | Enzymatic activitya |
||
|---|---|---|---|
| NADH:HAR (arbitrary units) | NADH oxidaseb (nmol O2 min−1 mg−1) (% rotenone sensitivity) | Complex I (nmol O2 min−1 mg−1) (% of wt) | |
| Wild type | 0.434 ± 0.036 | 146.9 ± 14.0 (64) | 94.0 (100) |
| nuo13.4 | 0.334 ± 0.020 | 232.8 ± 20.2 (32) | 74.5 (79.2) |
| 13.4L | 0.360 ± 0.034 | 135.6 ± 14.4 (50) | 67.8 (72.1) |
| nuo13.4×L | 0.226 ± 0.016 | 190.2 ± 20.7 (21) | 39.9 (42.4) |
Values are means and standard errors from at least 4 independent mitochondrial preparations.
Activity was measured in the presence of 20 μg/ml alamethicin. Respiratory activities were fully sensitive to 0.4 μM antimycin A.
The 13.4L protein interacts with complex I subunits.
Given the demonstration that 13.4L is not required for complex I assembly and function although it associates with high-molecular-weight structures in specific complex I mutants, we decided to investigate which proteins are present in such 13.4L recognized structures. We purified the complexes present in the wild type, nuo13.4, and nuo51 using the antibody AS-13.4L which, as expected, immunoprecipitated the 13.4L protein in all strains. Moreover, as depicted in Fig. 5A, a subset of complex I subunits were coimmunoprecipitated by AS-13.4L in nuo13.4 and nuo51 but not in the wild-type strain. Likewise, the 13.4L protein was also coimmunoprecipitated from nuo21 and nuo51 with an antibody against the whole complex I (Fig. 5B). The complex III subunit COREII was not immunoprecipitated in any of the strains (data not shown), probably due to the low stability of the supercomplex under the experimental conditions used, further supported by the fact that the ratio of I1III2 supercomplex and monomeric I1 is known to be much lower in N. crassa than in mammalian mitochondria under the same solubilization conditions (6). Our results indicate that the 13.4L-recognized high-molecular-weight structures are enriched in complex I subunits and most certainly represent complex I assembly intermediates.
Fig 5.

13.4L interacts with complex I assembly intermediates. (A) Immunoprecipitation of complex I subunits with 13.4L antiserum. Immunoprecipitates were analyzed by Western blotting with a mixture of antisera against the proteins indicated on the left (subunits 30.4, 29.9, and 21.3c kDa belong to the Q module; subunits 20.8, 14, and 12.3 kDa are part of the P module). (B) Immunoprecipitation of the 13.4L protein by antiserum against complex I. Mitochondria from nuo21 (a) and nuo51 (b) were separately solubilized and incubated with either an antiserum to the whole complex I (1) or an antibody to the 13.4L protein (2). The immunoprecipitates were analyzed by Western blotting with a mixture of antisera against the proteins indicated on the left. (C and D) Association of the 13.4L to complex I assembly intermediates. Mitochondrial proteins were resolved by BN PAGE (4 to 13%) and subsequently analyzed by Western blotting with antisera against the 13.4L protein (C) and the 21.3c kDa subunit of complex I (D).
The 13.4L protein associates with complex I assembly intermediates.
Having established that the 13.4L protein interacts with complex I subunits in specific complex I mutants (Fig. 4E and 5A), we next set out to determine whether this interaction was mutant specific or occurred overall upon complex I disruption. A thorough analysis of Fig. 4E revealed high-molecular-weight complexes observed in mutants nuo13.4 and nuo21 different from those visualized in nuo21.3a and nuo51 mutants. Mitochondria from a plethora of complex I mutants were thus solubilized with digitonin, resolved by BN PAGE followed by Western blotting, and subsequently immunodecorated with antibodies against the 13.4L and the 21.3c complex I subunit. As depicted in Fig. 5C and 6A, and according to the pattern of the bands observed upon detection with AS-13.4L, the complex I mutants can be divided into 3 groups: group 1, encompassing mutants nuo21.3a, nuo24, nuo51, and nuo78, in which two bands are recognized; group 2, which includes mutants nuo13.4, nuo18.4, and nuo21 displaying two different bands; and group 3, including mutant nuo21.3c and those assembling early complex I intermediates, where no bands are detected. A similar analysis, performed with specific antiserum against the complex I subunit 21.3c, detected complex I in its monomeric form and associated in supercomplexes in all the strains except nuo21.3c and nuo78 (Fig. 5D). The 21.3c subunit was also identified in the 13.4L-containing band (a) in group 1 mutants (Fig. 5D). The observed 13.4L-containing structures in group 1 are smaller than complex I and the supercomplex I1III2, whereas bands detected in group 2 display molecular weights similar to those of monomeric complex I and its supercomplex with dimeric complex III. Interestingly, all of the mutants exhibiting association of 13.4L in high-molecular-weight structures are devoid of peripheral arm subunits of complex I accumulating late assembly intermediates of the enzyme. More so, mutants with mutations in peripheral arm subunits that do not accumulate late assembly intermediates (mutants in Q-module subunits) are devoid of 13.4L-containing subassemblies. Overall, our results indicate that 13.4L is a late assembly factor in N. crassa.
Fig 6.

Intermediate a is the smallest structure containing the 13.4L protein. (A) Association of 13.4L with complex I assembly intermediates. Mitochondrial proteins from the indicated strains were analyzed by BN PAGE (4 to 10%), and the gel was blotted and probed with an antibody against either the 13.4L protein or the 21.3c kDa subunit of complex I. Identified complexes are indicated on the left (a is the smallest structure containing the 13.4L protein). (B) Immunoprecipitation of complex I subunits from nuo78 with 13.4L antiserum. Mitochondria from wild-type (wt) and nuo78 strains were solubilized and incubated with antiserum against the 13.4L protein. Immunoprecipitates were resolved by SDS-PAGE, blotted onto nitrocellulose, and subsequently incubated with a mixture of antisera against the proteins indicated on the left. (C) Intermediate a does not contain the 18.4- or the 13.4-kDa subunit (nuo78 lane in both gels). Mitochondrial proteins from the indicated strains were analyzed by BN PAGE (4 to 10%) followed by Western blotting with antibodies against either the 18.4-kDa or the 13.4-kDa subunit of complex I. Identified complexes are indicated on the left. (D) Complex III is fully assembled in supercomplexes I1III2 and a1III2. Mitochondrial proteins from the indicated strains were analyzed by BN PAGE (4 to 10%), and the gel was blotted and probed with an antibody against the complex III Rieske iron sulfur (RIS) protein.
The assembly intermediate a identified by anti-13.4L has the same molecular weight as the complex I intermediate accumulated in nuo78 mutant (48). To confirm whether they are indeed the same structure, we immunoprecipitated nuo78 mitochondria with AS-13.4L, yielding the 13.4L protein as well as several complex I subunits from both the peripheral arm and the membrane arm (Fig. 6B). However, subunits belonging to the N module of complex I (13.4-, 18.4-, 21-, 24-, 51-, and 78-kDa subunits) were not coprecipitated with 13.4L. Moreover, antibodies against either the 13.4-kDa or the 18.4-kDa subunit do not recognize intermediate a or its supercomplex with the dimer of complex III (Fig. 6C). These results indicate that the nuo78 complex I intermediate is the smaller structure (a in Fig. 4E, 5, and 6) with which the 13.4L assembly factor interacts and that the higher-molecular-weight complex (molecular weight equal to that of I1) results from the addition of other complex I subunits to this intermediate. Both of these complexes are able to associate with fully assembled dimeric complex III, forming the supercomplexes a1III2 and I1III2, respectively (Fig. 6D).
More so, our results indicate that the 13.4L-containing intermediate a present in group 1 mutants is able to incorporate complex I subunits, yielding an “incomplete” complex I to which the assembly factor is no longer bound. In group 2 mutants, however, the 13.4L protein remains associated until the last step of the complex I assembly pathway is accomplished.
13.4L interaction with complex I assembly intermediates is dependent on their membrane integration.
We observed that the 13.4L protein associates with intermediates of complex I despite the fact that the functional relevance of this interaction remained unclear. To address this issue, we analyzed complex I-deficient mutants with different intermediate phenotypes by one-dimensional BN PAGE followed by two-dimensional (2D) BN SDS-PAGE (Fig. 7). The lack of association between 13.4L and the peripheral arm accumulated in nuo21.3a suggests that this assembly factor associates only with complex I intermediates in a late stage of assembly, upon joining of membrane and peripheral components. Moreover, we suggest that the 13.4L assembly factor is specifically recruited to complex I intermediates in group 1 mutants for stabilization purposes, given that, upon further incorporation of complex I subunits yielding an almost mature enzyme, 13.4L dissociates. However, we cannot discard the possibility that the peripheral arm accumulated in nuo21.3a results from instability of “incomplete” complex I, in which case 13.4L would not associate with it.
Fig 7.
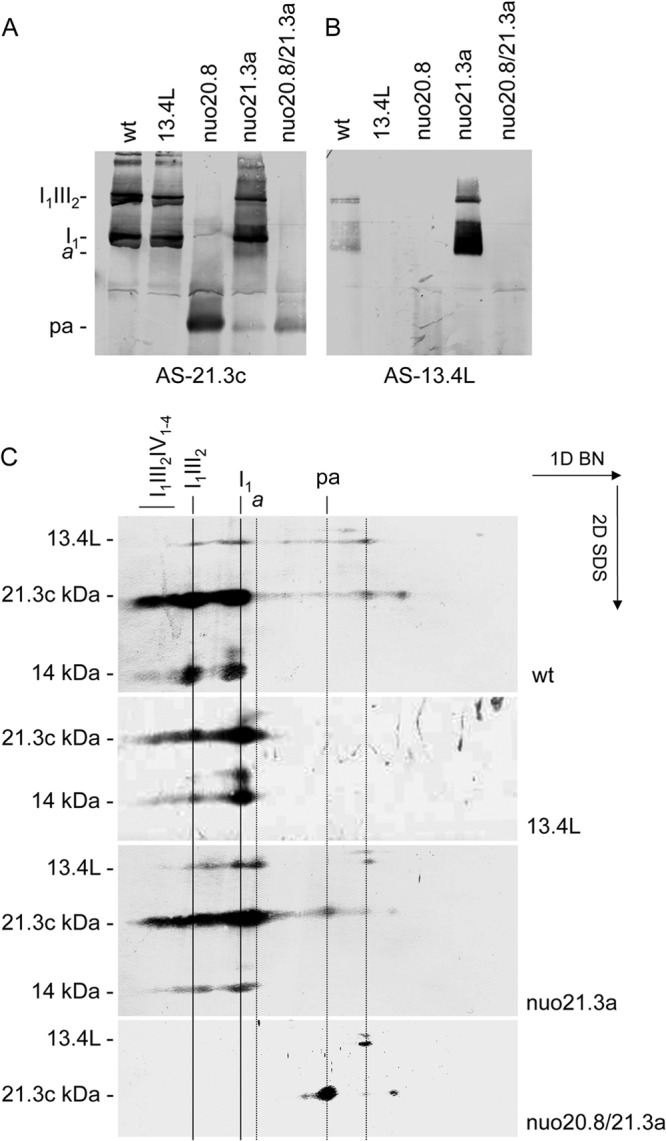
The 13.4L protein does not associate with the peripheral arm. Mitochondrial proteins were resolved in a BN PAGE and either blotted onto a membrane (A and B) or resolved by 2D SDS-PAGE (C). The filters were probed with antisera against the 21.3c kDa subunit of complex I (A), the 13.4L protein (B), and the proteins indicated on the left (C). Relevant OXPHOS complexes and supercomplexes are indicated. a, the smallest intermediate to which the 13.4L associates; pa, the peripheral arm of complex I.
Accordingly, 13.4L was not found to be associated with the peripheral domain of complex I in the nuo20.8 mutant. However, the double mutant nuo20.8/21.3a accumulates only an “incomplete” peripheral arm that was recognized by an antibody against the 21.3c kDa subunit (subunits are designated according to their molecular masses; subunit 21.3c kDa is the third identified subunit with a molecular mass of 21.3 kDa) of complex I but not by the 13.4L antibody, indicating that the assembly factor does not associate with this “incomplete” early assembly intermediate (Fig. 7).
As such, we conclude that the 13.4L assembly factor associates only with late assembly intermediates of complex I.
The 13.4L protein associates with genuine assembly intermediates.
Surprisingly, 13.4L could also be detected in wild type mitochondria associated with high-molecular-weight structures similar to those found in group 1 mutants under normal vegetative growth conditions, although in minute amounts (Fig. 7). We set out to determine under which conditions these intermediates accumulate in the wild type. We found that upon transfer to 37°C for 3 h of a wild-type culture growing at 26°C, the accumulation of assembly intermediates was easily detected by AS-13.4L in BN PAGE Western blots (Fig. 8A). Furthermore, this accumulation could be blocked by the addition of either cycloheximide or potassium cyanide (Fig. 8B), suggesting that the intermediates accumulate due to an accelerated growth rate that is prevented by protein synthesis or respiratory chain inhibitors. Accordingly, the shift in growth temperature also resulted in accumulation of assembly intermediates in nuo13.4, which already displayed these structures under normal growth conditions (Fig. 8A, AS-13.4L antibody).
Fig 8.

Effect of temperature (A and B), and oxidative stress (B) in complex I assembly intermediates. Indicated strains were grown at 26°C for 16 h followed by 3-h incubation either at 26°C or at 37°C in the presence of 1 mg/ml cycloheximide (CHM), 1 mM KCN, 10 mM N-acetylcysteine (NAC), 10 mM H2O2 (H2O2), or 10 mM H2O2 plus cycloheximide (CHM+H2O2). Mitochondrial extracts were analyzed by Western blotting with antibodies against the 13.4L protein, the complex I subunit 21.3c kDa, and the COREII complex III subunit.
Strikingly, we also observed that oxidative stress exposure resulting from the addition of hydrogen peroxide to an exponentially growing culture led to accumulation of 13.4L-containing assembly intermediates in the wild type (Fig. 8C, AS-13.4L antibody). We suggest that hydrogen peroxide exposure leads to the accumulation of complex I intermediates that probably result from damage/instability of the mature enzyme, which may be more pronounced in the nuo13.4 mutant than in the wild-type strain.
Additionally, we observed that nuo13.4 assembly intermediates appeared as smaller 13.4L-recognized structures upon oxidative stress exposure. To assess the conversion of nuo13.4 complex I assembly intermediates into smaller intermediates, we resolved mitochondrial extracts from the wild type, nuo13.4, and nuo51 upon growth in the presence or absence of hydrogen peroxide by BN PAGE. As depicted in Fig. 9A, H2O2 exposure leads to the conversion of nuo13.4 intermediates into structures that migrate exactly in the same position as those present in nuo51. Moreover, exposure of nuo51 to oxidative stress does not alter the AS-13.4L band pattern, suggesting that intermediate a is the smaller structure with which the 13.4L protein associates. The conversion of the 13.4L-recognized intermediates from nuo13.4 was also addressed by 2D BN SDS-PAGE analysis (Fig. 9B). It is clear that 13.4L and complex I subunits appear in lower-molecular-weight structures upon H2O2 exposure that do not contain the 78-kDa subunit, providing further evidence that intermediate a is that present in the nuo78 mutant.
Fig 9.

The 13.4L protein stabilizes complex I assembly intermediates. (A and B) Conversion of complex I from nuo13.4 into intermediate a upon oxidative stress induction. The nuo13.4 mutant was grown at 26°C in the presence or absence of 10 mM H2O2 for the last 3 h of growth. Mitochondria were then analyzed by BN PAGE (A) followed by two-dimensional BN SDS-PAGE (B). The 1D gel was probed with AS-13.4L, and the 2D gels were analyzed with antisera as indicated on the left. (C) Schematic representation of the final steps of complex I assembly. Late assembly intermediates of complex I present in the different mutant strains and mature complex I (gray), incomplete subdomains (white), and 13.4L assembly factor (black). The mutant strains indicated represent those in which such intermediates accumulate. N, Q, and P are distinct functional domains of the mature enzyme.
DISCUSSION
Complex I dysfunction is a major cause of OXPHOS disorders in humans usually associated with defects in enzyme function and/or assembly (7). Here, we used N. crassa as a model system to investigate the functional relevance of the complex I assembly factor 13.4L (homologue of the human NDUFAF2 protein), and we provide evidence that this protein is functionally conserved from fungi to humans. Despite the fact that we observed an association of this assembly factor to complex I assembly intermediates that accumulate in specific complex I mutant strains, our results indicate that this late-stage assembly factor is not essential for complex I assembly or function.
In view of our evidence, we propose a model (Fig. 9C) in which the 13.4L protein is involved in the stabilization of a complex I assembly intermediate, which accumulates in specific complex I mutants, as well as during accelerated growth and upon oxidative stress exposure. In the latter case, the stabilization of the assembly intermediate could subsequently lead to the repair of the damaged molecules by incorporation of newly imported peripheral arm subunits. In this model, it can be envisaged that repair of damaged complex I molecules can be accomplished without the need for total substitution of the enzyme, as has been suggested for the mammalian enzyme (19).
Our data for a cohort of complex I-deficient mutants indicates that the smallest intermediate with which the 13.4L associates is the one accumulated in nuo78, which lacks all the N-module components (13.4, 18.4, 21, 21.3a, 24, 51, and 78 kDa). This structure also accumulates in nuo21.3a, nuo24, and nuo51 but upon incorporation of the remaining N-module subunits leads 13.4L to dissociate from the final assembled structure. A similar interaction of the human NDUFAF2 with an 830-kDa intermediate that accumulates in patients' mitochondria deficient in some nuclear complex I genes, namely, NDUFV1 (20, 21), NDUFS1 (21), NDUFS4 (21), and NDUFS6 (22), was previously described. We suggest that intermediate a is the equivalent of this 830-kDa subcomplex, which also lacks the N module (19, 49, 50).
Furthermore, in group 2 mutants (nuo13.4, nuo18.4, and nuo21) the assembly factor associates with an intermediate that has the same molecular weight as mature complex I.
These mutants do not accumulate intermediate a, and 13.4L is probably stalled while waiting for the missing subunit. These findings suggest that in these mutants the assembly factor dissociates only upon incorporation of the 13.4-, 18.4-, and 21-kDa subunits. Indeed, these 3 subunits are present in the “incomplete” complex I assembled in mutants nuo21.3a and nuo51. In contrast, a previous study by Lazarou et al. reported NDUFAF2 to be associated only with the 830-kDa subassembly, not with the 970-kDa complex I lacking NDUFS6, in patient's mitochondria (19).
Remarkably, the 3 subunits (13.4, 18.4, and 21 kDa) have been described as a possible regulatory module, already present in complex I from some proteobacteria (51). It remains to be established whether these subunits interact with each other and form a subdomain within the complex. Our results indicate that they can be incorporated independently from each other, since the mutant in one subunit assembled the other two into complex I, although in these mutants the 13.4L assembly factor is found associated with the enzyme. We propose for the first time that 13.4L is involved in the regulation/assembly/repair of this modular part of complex I with a consequent role in the regulation of the enzyme.
Interestingly, all the 13.4L-containing intermediates are able to form supercomplexes with the dimer of complex III, indicating that the membrane part of the enzyme is already assembled. The assembly of “incomplete” complex I, with or without activity, into supercomplexes was described previously (3, 6); however, we observed here that intermediate a is already able to assemble into supercomplexes, making it the first supercomplex assembly intermediate in agreement with what was proposed for human respirasome biogenesis (3, 19). Indeed, Moreno-Lastres and coworkers have proposed a model of human supercomplex biogenesis where complex I N module is only incorporated upon complexes III and IV association (3).
How the N module is incorporated into intermediate a is yet to be established, although our results point to a sequential integration of the different subunits. However, we cannot discard the possibility that some subunits associate before incorporation. For instance, the interaction between the 24- and 51-kDa subunits was suggested previously (17, 52). In contrast, three of the N-module subunits (NDUFV2, NDUFV1, and NDUFA12) have been described in their free form upon fluorescence studies (53), consistent with their late incorporation into complex I.
Our results for mutant nuo21 are discordant with those described for mitochondria from patients or mice harboring NDUFS4 mutations. Herein, we illustrate the association of the 13.4L protein to the functional complex I present in the Neurospora nuo21 mutant (26). Mitochondria from patients and mice deficient in NDUFS4 were described as devoid of complex I activity, with accumulation of the 830-kDa intermediate associated with the NDUFAF2 assembly factor (20, 21, 49, 50). However, it was recently reported that the absence of NDUFS4 results in increased instability of complex I leading to the accumulation of two subcomplexes, the N module and the 830-kDa intermediate, which has been found to be associated with two assembly factors (49, 50). Furthermore, complex I activity was detected in different tissues of homozygous ndufs4 mutant mice, consistent with our results from nuo21 mutant. Given that two assembly factors have been found to be associated with the hampered complex I present in homozygous ndufs4 mutant mice, we would like to speculate that our 13.4L (NDUFAF2)-containing complexes could also contain CIA30 (NDUFAF1).
A patient suffering from Leigh syndrome caused by nonsense mutations in NDUFA12 (nuo-13.4) and presenting isolated complex I deficiency with reduced amounts of fully active enzyme was recently described (54). Likewise, our results showed that disruption of the homologous subunit in N. crassa is compatible with the assembly of a functional complex I that appears to be more susceptible to oxidative damage.
It was previously described that N. crassa complex I assembly occurs through a modular pathway in which the membrane and the peripheral domains of the enzyme assemble independently and then join together to form the mature complex I (55). This assembly pathway was established with assays in complex I mutants where both intermediates were observed. However, this pathway is not entirely compatible with the assembly pathway proposed for human complex I, where the peripheral arm was not detected as an assembly intermediate. For the first time, we propose that the late steps of N. crassa complex I assembly are identical to those of mammalian complex I, indicating a conserved pathway for enzyme assembly. Accordingly, the intermediate present in nuo78 is equivalent to a subcomplex that accumulates in specific complex I-deficient patients (21). This intermediate contains membrane and peripheral subunits of complex I and indicates that also in the fungus, peripheral arm subunits can be incorporated into membrane arm intermediates independently of the assembly of the peripheral arm, raising the prospect of more than one assembly pathway. Indeed, the accumulation of the peripheral domain was also observed in Chinese hamster fibroblasts upon disruption of NDUFA1 (9.8 kDa) (56). This result suggests that, as in N. crassa, disruption of a membrane arm subunit in mammals could lead to the accumulation of the peripheral domain of complex I. The formation of this intermediate in the absence of the membrane domain can be advantageous for the stability of the peripheral arm subunits that would otherwise be prone to degradation. In such cases, a nonphysiological assembly pathway can be “activated” due to the presence of stalled intermediates.
Herein, we provide evidence that 13.4L is a complex I assembly factor and that mitochondrial complex I biogenesis appears to be evolutionarily conserved. Moreover, the use of N. crassa validates it as an excellent model to characterize the assembly pathway of complex I and the proteins involved, providing important insights into mammalian mitochondrial complex I biogenesis.
ACKNOWLEDGMENTS
We thank Patrícia Carneiro for helpful discussions and for help preparing the manuscript.
This work was supported by the Fundação para a Ciência e a Tecnologia from Portugal through research grants coparticipated in by Fundo Europeu de Desenvolvimento Regional (FEDER) through COMPETE and by Fundos Nacionais under the project FCOMP-01-0124-FEDER-022718 (PEst-C/SAU/LA0002/2011).
Footnotes
Published ahead of print 6 May 2013
REFERENCES
- 1. Brandt U. 2006. Energy converting NADH:quinone oxidoreductase (complex I). Annu. Rev. Biochem. 75:69–92 [DOI] [PubMed] [Google Scholar]
- 2. Carroll J, Fearnley IM, Skehel JM, Shannon RJ, Hirst J, Walker JE. 2006. Bovine complex I is a complex of 45 different subunits. J. Biol. Chem. 281:32724–32727 [DOI] [PubMed] [Google Scholar]
- 3. Moreno-Lastres D, Fontanesi F, Garcia-Consuegra I, Martin MA, Arenas J, Barrientos A, Ugalde C. 2012. Mitochondrial complex I plays an essential role in human respirasome assembly. Cell Metab. 15:324–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Acin-Perez R, Fernandez-Silva P, Peleato ML, Perez-Martos A, Enriquez JA. 2008. Respiratory active mitochondrial supercomplexes. Mol. Cell 32:529–539 [DOI] [PubMed] [Google Scholar]
- 5. Schagger H, Pfeiffer K. 2000. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 19:1777–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marques I, Dencher NA, Videira A, Krause F. 2007. Supramolecular organization of the respiratory chain in Neurospora crassa mitochondria. Eukaryot. Cell 6:2391–2405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bugiani M, Invernizzi F, Alberio S, Briem E, Lamantea E, Carrara F, Moroni I, Farina L, Spada M, Donati MA, Uziel G, Zeviani M. 2004. Clinical and molecular findings in children with complex I deficiency. Biochim. Biophys. Acta 1659:136–147 [DOI] [PubMed] [Google Scholar]
- 8. Pagniez-Mammeri H, Loublier S, Legrand A, Benit P, Rustin P, Slama A. 2012. Mitochondrial complex I deficiency of nuclear origin I. Structural genes Mol. Genet. Metab. 105:163–172 [DOI] [PubMed] [Google Scholar]
- 9. Pagniez-Mammeri H, Rak M, Legrand A, Benit P, Rustin P, Slama A. 2012. Mitochondrial complex I deficiency of nuclear origin II. Non-structural genes. Mol. Genet. Metab. 105:173–179 [DOI] [PubMed] [Google Scholar]
- 10. Mimaki M, Wang X, McKenzie M, Thorburn DR, Ryan MT. 2012. Understanding mitochondrial complex I assembly in health and disease. Biochim. Biophys. Acta 1817:851–862 [DOI] [PubMed] [Google Scholar]
- 11. McKenzie M, Ryan MT. 2010. Assembly factors of human mitochondrial complex I and their defects in disease. IUBMB Life 62:497–502 [DOI] [PubMed] [Google Scholar]
- 12. Lazarou M, Thorburn DR, Ryan MT, McKenzie M. 2009. Assembly of mitochondrial complex I and defects in disease. Biochim. Biophys. Acta 1793:78–88 [DOI] [PubMed] [Google Scholar]
- 13. Remacle C, Barbieri MR, Cardol P, Hamel PP. 2008. Eukaryotic complex I: functional diversity and experimental systems to unravel the assembly process. Mol. Genet. Genomics 280:93–110 [DOI] [PubMed] [Google Scholar]
- 14. Marques I, Duarte M, Assuncao J, Ushakova AV, Videira A. 2005. Composition of complex I from Neurospora crassa and disruption of two “accessory” subunits. Biochim. Biophys. Acta 1707:211–220 [DOI] [PubMed] [Google Scholar]
- 15. Cardol P. 2011. Mitochondrial NADH:ubiquinone oxidoreductase (complex I) in eukaryotes: a highly conserved subunit composition highlighted by mining of protein databases. Biochim. Biophys. Acta 1807:1390–1397 [DOI] [PubMed] [Google Scholar]
- 16. Duarte M, Videira A. 2007. Mitochondrial NAD(P)H dehydrogenases in filamentous fungi, p 55–68 In González Siso M. (ed), Complex I and alternative dehydrogenases. Transworld Research Network, Trivandrum, Kerala, India [Google Scholar]
- 17. Almeida T, Duarte M, Melo AM, Videira A. 1999. The 24-kDa iron-sulphur subunit of complex I is required for enzyme activity. Eur. J. Biochem. 265:86–93 [DOI] [PubMed] [Google Scholar]
- 18. Fecke W, Sled VD, Ohnishi T, Weiss H. 1994. Disruption of the gene encoding the NADH-binding subunit of NADH: ubiquinone oxidoreductase in Neurospora crassa. Formation of a partially assembled enzyme without FMN and the iron-sulphur cluster N-3. Eur. J. Biochem. 220:551–558 [DOI] [PubMed] [Google Scholar]
- 19. Lazarou M, McKenzie M, Ohtake A, Thorburn DR, Ryan MT. 2007. Analysis of the assembly profiles for mitochondrial- and nuclear-DNA-encoded subunits into complex I. Mol. Cell. Biol. 27:4228–4237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ogilvie I, Kennaway NG, Shoubridge EA. 2005. A molecular chaperone for mitochondrial complex I assembly is mutated in a progressive encephalopathy. J. Clin. Invest. 115:2784–2792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vogel RO, van den Brand MA, Rodenburg RJ, van den Heuvel LP, Tsuneoka M, Smeitink JA, Nijtmans LG. 2007. Investigation of the complex I assembly chaperones B17.2L and NDUFAF1 in a cohort of CI deficient patients. Mol. Genet. Metab. 91:176–182 [DOI] [PubMed] [Google Scholar]
- 22. Kirby DM, Salemi R, Sugiana C, Ohtake A, Parry L, Bell KM, Kirk EP, Boneh A, Taylor RW, Dahl HH, Ryan MT, Thorburn DR. 2004. NDUFS6 mutations are a novel cause of lethal neonatal mitochondrial complex I deficiency. J. Clin. Invest. 114:837–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hoefs SJ, Dieteren CE, Rodenburg RJ, Naess K, Bruhn H, Wibom R, Wagena E, Willems PH, Smeitink JA, Nijtmans LG, van den Heuvel LP. 2009. Baculovirus complementation restores a novel NDUFAF2 mutation causing complex I deficiency. Hum. Mutat. 30:E728–E736 [DOI] [PubMed] [Google Scholar]
- 24. Marques I, Duarte M, Videira A. 2003. The 9.8 kDa subunit of complex I, related to bacterial Na(+)-translocating NADH dehydrogenases, is required for enzyme assembly and function in Neurospora crassa. J. Mol. Biol. 329:283–290 [DOI] [PubMed] [Google Scholar]
- 25. Duarte M, Sousa R, Videira A. 1995. Inactivation of genes encoding subunits of the peripheral and membrane arms of neurospora mitochondrial complex I and effects on enzyme assembly. Genetics 139:1211–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ferreirinha F, Duarte M, Melo AM, Videira A. 1999. Effects of disrupting the 21 kDa subunit of complex I from Neurospora crassa. Biochem. J. 342:551–554 [PMC free article] [PubMed] [Google Scholar]
- 27. Alves PC, Videira A. 1994. Disruption of the gene coding for the 21.3-kDa subunit of the peripheral arm of complex I from Neurospora crassa. J. Biol. Chem. 269:7777–7784 [PubMed] [Google Scholar]
- 28. Duarte M, Videira A. 2000. Respiratory chain complex I is essential for sexual development in Neurospora and binding of iron sulfur clusters are required for enzyme assembly. Genetics 156:607–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Harkness TA, Rothery RA, Weiner JH, Werner S, Azevedo JE, Videira A, Nargang FE. 1995. Disruption of the gene encoding the 78-kilodalton subunit of the peripheral arm of complex I in Neurospora crassa by repeat induced point mutation (RIP). Curr. Genet. 27:339–350 [DOI] [PubMed] [Google Scholar]
- 30. Davis RH, de Serres FJ. 1970. Genetic and microbiological research techniques for Neurospora crassa. Methods Enzymol. 17A:79–143 [Google Scholar]
- 31. Videira A, Werner S. 1989. Assembly kinetics and identification of precursor proteins of complex I from Neurospora crassa. Eur. J. Biochem. 181:493–502 [DOI] [PubMed] [Google Scholar]
- 32. Melo AM, Duarte M, Moller IM, Prokisch H, Dolan PL, Pinto L, Nelson MA, Videira A. 2001. The external calcium-dependent NADPH dehydrogenase from Neurospora crassa mitochondria. J. Biol. Chem. 276:3947–3951 [DOI] [PubMed] [Google Scholar]
- 33. Nijtmans LG, Henderson NS, Holt IJ. 2002. Blue native electrophoresis to study mitochondrial and other protein complexes. Methods 26:327–334 [DOI] [PubMed] [Google Scholar]
- 34. Krause F, Scheckhuber CQ, Werner A, Rexroth S, Reifschneider NH, Dencher NA, Osiewacz HD. 2004. Supramolecular organization of cytochrome c oxidase- and alternative oxidase-dependent respiratory chains in the filamentous fungus Podospora anserina. J. Biol. Chem. 279:26453–26461 [DOI] [PubMed] [Google Scholar]
- 35. Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 36. Bradford M. 1976. A rapid and sensitive method for the quantification of microgram quantities of protein using the principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 37. Zauner R, Christner J, Jung G, Borchart U, Machleidt W, Videira A, Werner S. 1985. Identification of the polypeptide encoded by the URF-1 gene of Neurospora crassa mtDNA. Eur. J. Biochem. 150:447–454 [DOI] [PubMed] [Google Scholar]
- 38. Towbin H, Staehelin T, Gordon J. 1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. U. S. A. 76:4350–4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Blake MS, Johnston KH, Russell-Jones GJ, Gotschlich EC. 1984. A rapid, sensitive method for detection of alkaline phosphatase-conjugated anti-antibody on Western blots. Anal. Biochem. 136:175–179 [DOI] [PubMed] [Google Scholar]
- 40. Azevedo JE, Nehls U, Eckerskorn C, Heinrich H, Rothe H, Weiss H, Werner S. 1992. Primary structure and mitochondrial import in vitro of the 20.9 kDa subunit of complex I from Neurospora crassa. Biochem. J. 288:29–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Claros MG, Vincens P. 1996. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur. J. Biochem. 241:779–786 [DOI] [PubMed] [Google Scholar]
- 42. da Silva MV, Alves PC, Duarte M, Mota N, Lobo-da-Cunha A, Harkness TA, Nargang FE, Videira A. 1996. Disruption of the nuclear gene encoding the 20.8-kDa subunit of NADH: ubiquinone reductase of Neurospora mitochondria. Mol. Gen. Genet. 252:177–183 [DOI] [PubMed] [Google Scholar]
- 43. Ugalde C, Vogel R, Huijbens R, Van Den Heuvel B, Smeitink J, Nijtmans L. 2004. Human mitochondrial complex I assembles through the combination of evolutionary conserved modules: a framework to interpret complex I deficiencies. Hum. Mol. Genet. 13:2461–2472 [DOI] [PubMed] [Google Scholar]
- 44. Hartl FU, Schmidt B, Wachter E, Weiss H, Neupert W. 1986. Transport into mitochondria and intramitochondrial sorting of the Fe/S protein of ubiquinol-cytochrome c reductase. Cell 47:939–951 [DOI] [PubMed] [Google Scholar]
- 45. Videira A, Tropschug M, Wachter E, Schneider H, Werner S. 1990. Molecular cloning of subunits of complex I from Neurospora crassa. Primary structure and in vitro expression of a 22-kDa polypeptide. J. Biol. Chem. 265:13060–13065 [PubMed] [Google Scholar]
- 46. Azevedo JE, Videira A. 1994. Characterization of a membrane fragment of respiratory chain complex I from Neurospora crassa. Insights on the topology of the ubiquinone-binding site. Int. J. Biochem. 26:505–510 [DOI] [PubMed] [Google Scholar]
- 47. McCluskey K. 2003. The Fungal Genetics Stock Center: from molds to molecules. Adv. Appl. Microbiol. 52:245–262 [DOI] [PubMed] [Google Scholar]
- 48. Ferreira M, Torraco A, Rizza T, Fattori F, Meschini MC, Castana C, Go NE, Nargang FE, Duarte M, Piemonte F, Dionisi-Vici C, Videira A, Vilarinho L, Santorelli FM, Carrozzo R, Bertini E. 2011. Progressive cavitating leukoencephalopathy associated with respiratory chain complex I deficiency and a novel mutation in NDUFS1. Neurogenetics 12:9–17 [DOI] [PubMed] [Google Scholar]
- 49. Calvaruso MA, Willems P, van den Brand M, Valsecchi F, Kruse S, Palmiter R, Smeitink J, Nijtmans L. 2012. Mitochondrial complex III stabilizes complex I in the absence of NDUFS4 to provide partial activity. Hum. Mol. Genet. 21:115–120 [DOI] [PubMed] [Google Scholar]
- 50. Leong DW, Komen JC, Hewitt CA, Arnaud E, McKenzie M, Phipson B, Bahlo M, Laskowski A, Kinkel SA, Davey GM, Heath WR, Voss AK, Zahedi RP, Pitt JJ, Chrast R, Sickmann A, Ryan MT, Smyth GK, Thorburn DR, Scott HS. 2012. Proteomic and metabolomic analyses of mitochondrial complex I-deficient mouse model generated by spontaneous B2 short interspersed nuclear element (SINE) insertion into NADH dehydrogenase (ubiquinone) Fe-S protein 4 (Ndufs4) gene. J. Biol. Chem. 287:20652–20663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yip CY, Harbour ME, Jayawardena K, Fearnley IM, Sazanov LA. 2011. Evolution of respiratory complex I: “supernumerary” subunits are present in the alpha-proteobacterial enzyme. J. Biol. Chem. 286:5023–5033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yano T, Sled VD, Ohnishi T, Yagi T. 1996. Expression and characterization of the flavoprotein subcomplex composed of 50-kDa (NQO1) and 25-kDa (NQO2) subunits of the proton-translocating NADH-quinone oxidoreductase of Paracoccus denitrificans. J. Biol. Chem. 271:5907–5913 [DOI] [PubMed] [Google Scholar]
- 53. Dieteren CE, Willems PH, Vogel RO, Swarts HG, Fransen J, Roepman R, Crienen G, Smeitink JA, Nijtmans LG, Koopman WJ. 2008. Subunits of mitochondrial complex I exist as part of matrix- and membrane-associated subcomplexes in living cells. J. Biol. Chem. 283:34753–34761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ostergaard E, Rodenburg RJ, van den Brand M, Thomsen LL, Duno M, Batbayli M, Wibrand F, Nijtmans L. 2011. Respiratory chain complex I deficiency due to NDUFA12 mutations as a new cause of Leigh syndrome. J. Med. Genet. 48:737–740 [DOI] [PubMed] [Google Scholar]
- 55. Videira A, Duarte M. 2002. From NADH to ubiquinone in Neurospora mitochondria. Biochim. Biophys. Acta 1555:187–191 [DOI] [PubMed] [Google Scholar]
- 56. Scheffler IE, Yadava N, Potluri P. 2004. Molecular genetics of complex I-deficient Chinese hamster cell lines. Biochim. Biophys. Acta 1659:160–171 [DOI] [PubMed] [Google Scholar]