

Abstract
Net1 is a RhoA guanine nucleotide exchange factor (GEF) that is overexpressed in a subset of human cancers and contributes to cancer cell motility and invasion in vitro. However, the molecular mechanism accounting for its role in cell motility and invasion has not been described. In the present work, we show that expression of both Net1 isoforms in breast cancer cells is required for efficient cell motility. Although loss of Net1 isoform expression only partially blocks RhoA activation, it inhibits lysophosphatidic acid (LPA)-stimulated migration as efficiently as knockdown of RhoA itself. However, we demonstrate that the Net1A isoform predominantly controls myosin light-chain phosphorylation and is required for trailing edge retraction during migration. Net1A interacts with focal adhesion kinase (FAK), localizes to focal adhesions, and is necessary for FAK activation and focal adhesion maturation during cell spreading. Net1A expression is also required for efficient invasion through a Matrigel matrix. Analysis of invading cells demonstrates that Net1A is required for amoeboid invasion, and loss of Net1A expression causes cells to shift to a mesenchymal phenotype characterized by high β1-integrin activity and membrane type 1 matrix metalloproteinase (MT1-MMP) expression. These results demonstrate a previously unrecognized role for the Net1A isoform in controlling FAK activation during planar cell movement and amoeboid motility during extracellular matrix (ECM) invasion.
INTRODUCTION
The small G protein RhoA is aberrantly expressed in many human cancers, including breast cancer, and its activation is essential for cancer cell migration and invasion (1–4). Localized activation of RhoA during cell migration contributes to diverse processes, including leading edge consolidation and trailing edge retraction. It accomplishes these tasks by controlling cortical actin polymerization and retrograde actin flow at the leading edge and by generating the actomyosin contraction that is necessary for both focal adhesion maturation and dissolution (5–7). During cell movement through an extracellular matrix (ECM), RhoA also controls a spatially localized actomyosin contraction that is crucial for efficient ECM invasion (8, 9). Not surprisingly, activation of RhoA is critical for metastasis in mouse models of human cancer (10). However, regulatory mechanisms controlling aberrant RhoA activation in human cancer are not well defined.
RhoA activation is controlled by a family of proteins called Rho guanine nucleotide exchange factors (Rho GEFs), which catalyze the release of GDP and thereby allow Rho proteins to bind GTP. More than 70 Rho GEFs exist in the human genome, a subset of which target RhoA (11, 12). The neuroepithelioma transforming gene 1 (Net1) is a Rho GEF whose gene was originally cloned as a transforming gene in NIH 3T3 focus formation assays (13). It is specific for RhoA and RhoB in vitro and stimulates RhoA-dependent actin stress fiber formation when overexpressed in cells (14, 15). Net1 is unusual among Rho GEFs in that it localizes to the nucleus due to the presence of multiple nuclear localization signal (NLS) sequences present in its amino terminus. Nuclear localization of Net1 is an important negative regulatory mechanism, since RhoA activation and actin stress fiber formation only occur when Net1 is redistributed to the cytoplasm (16, 17). Two isoforms of Net1 exist in cells, Net1 and Net1A, which are identical except for the presence of alternative amino-terminal regulatory domains. The Net1A isoform contains a shorter amino terminus that lacks two of the NLS sequences present in Net1, thereby conferring a less stringent regulation of nuclear localization. Thus, cells are more sensitive to Net1A overexpression, which tends to result in enhanced cytoplasmic localization and elevated RhoA activation and F-actin polymerization (16).
Recent findings support the notion that cells utilize the Net1A isoform, rather than Net1, to control actin cytoskeletal organization. For example, Net1A is specifically required for actin cytoskeletal rearrangement in fibroblasts and keratinocytes in response to transforming growth factor β (TGF-β) stimulation (18, 19). Moreover, we have observed that Net1A is relocalized from the nucleus to the plasma membrane in response to Rac1 activation and is required for focal adhesion maturation (20). Additionally, knockdown of the Net1 isoform in MCF7 breast cancer cells but not Net1A reduces estrogen-driven proliferation (21). Thus, it may be that the Net1 isoform is more important for cell proliferation, while Net1A controls aspects of cell motility.
A number of studies indicate that Net1 proteins may contribute to cancer initiation and progression. For example, overexpression of N-terminally truncated Net1 is transforming in cultured fibroblasts (13, 14, 16), and Net1 transcripts have been found to be overexpressed in human gastric cancers, hepatocellular carcinomas, and gliomas (22–24). In addition, we have shown that coexpression of Net1 and β4-integrin in node-positive breast cancer patients is associated with a high risk for distant metastasis (25), and others have found that overexpression of Net1 isoform mRNA correlates with reduced metastasis-free survival in estrogen receptor-positive breast cancer patients (21). Furthermore, small interfering RNA (siRNA)-mediated knockdown of both Net1 isoforms together inhibits gastric cancer cell motility and invasion (22, 26). These studies suggest that one or both Net1 isoforms may play a role in metastatic cancer progression.
In the present work, we explored the mechanistic basis for control of cell motility and invasion by Net1 isoforms. We show that expression of both Net1 isoforms is required for cell motility in multiple human breast cancer cell lines and for RhoA activation and peripheral myosin light-chain (MLC) phosphorylation in MDA-MB-231 cells. However, the Net1A isoform preferentially stimulated myosin light-chain phosphorylation, localized to focal adhesions, and was required for FAK activation, focal adhesion maturation, and trailing edge retraction. Similarly, expression of Net1A was necessary for amoeboid ECM invasion. In each of these assays, inhibition of Net1A expression blocked cell movement and invasion as potently as inhibition of RhoA expression, and siRNA-mediated knockdown of both Net1 isoforms could only be rescued by reexpression of catalytically active Net1A. These results indicate that both Net1 isoforms contribute to planar cell motility. However, the Net1A isoform is primarily required for control of FAK activity and focal adhesion dynamics during planar movement and for amoeboid motility in an extracellular matrix environment.
MATERIALS AND METHODS
Cells, tissues, and reagents.
MDA-MB-231 and MDA-MB-435 human breast cancer cells were grown in Dulbecco's modified Eagle's medium (DMEM)–Ham's F-12 (1:1) (HyClone) plus 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). HeLa cells were grown in modified Eagle's medium (HyClone) plus 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. SUM149 cells were grown in Ham's F-12 medium (HyClone) plus 5% FBS, 2 mM glutamine, 1 μg/ml hydrocortisone, 5 μg/ml insulin, 5 μg/ml transferrin, and 50 μM selenium. BT549 cells were grown in Roswell Park Memorial Institute medium (RPMI) plus 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. All cells were grown in a humidified 5% CO2 incubator, except for HeLa cells, which were cultured in a 10% incubator.
Rabbit anti-Net1 was previously described (25) and was utilized for the Western blot shown in Fig. 7A. The following commercial antibodies were used: mouse anti-GAPDH (anti-glyceraldehyde-3-phosphate dehydrogenase), mouse anti-Src, mouse anti-Net1 (sc-271207 and sc-271941), mouse anti-FAK (sc-1688), and nonspecific rabbit IgG from Santa Cruz Biotechnology, Santa Cruz, CA; rabbit anti-phospho-S19 MLC2, mouse anti-phospho-S19 MLC2, rabbit anti-phospho-Y418-Src, rabbit anti-FAK, rabbit anti-phospho-Y397 FAK, and rabbit anti-β1-integrin from Cell Signaling Technology, Danvers, MA; mouse antipaxillin from BD Biosciences, San Diego, CA; mouse anti-β1-integrin (4B4) from Coulter, Fullerton, CA; mouse anti-MLC2 and rabbit anti-membrane type 1 matrix metalloproteinase (anti-MT1-MMP) from Abcam, Cambridge, MA; mouse anti-RhoA from Cytoskeleton, Denver, CO; and Alexa Fluor 647-phalloidin, Alexa Fluor 488-phalloidin, anti-mouse antibody–Alexa Fluor 647, and anti-rabbit antibody–Alexa Fluor 594 from Invitrogen, Grand Island, NY. Cy2- and Cy3-conjugated anti-mouse and anti-rabbit antibodies were from Jackson ImmunoResearch. Bosutinib, Y-27632, ML-7, PF-562271, and blebbistatin were from Fisher Scientific.
Fig 7.
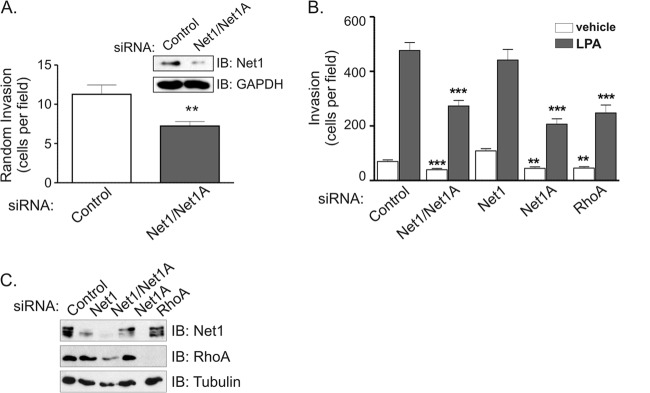
Net1A is specifically required for invasion of a Matrigel extracellular matrix. (A) MDA-MB-231 cells were transfected with control- or Net1 dual isoform-specific siRNAs and then allowed to invade overnight through a Matrigel-coated Transwell apparatus. Both wells contained 10% FBS. The inset shows a representative Western blot using an antibody that primarily recognizes Net1. Shown is the average from four independent experiments. Errors are SEM. **, P < 0.01. (B) MDA-MB-231 cells were transfected with the siRNAs indicated. Two days later, the cells were serum starved and allowed to migrate toward vehicle or LPA overnight through Matrigel-coated Transwells. Shown is the average from three independent experiments. Errors are SEM. **, P < 0.01; ***, P < 0.001. (C) Representative Western blot of siRNA-transfected MDA-MB-231 cells.
Mouse Net1 and Net1A in pEF-HA (hemagglutinin) were described previously (16). Net1A containing Myc and Flag epitopes was created by PCR and subcloned into pcDNA3.1 (Invitrogen). Catalytically inactive Net1A L267E in pEF-HA was made by PCR using Pfu Turbo polymerase (Agilent). Net1AΔC4 in pEF-HA was described previously (27). HA-epitope-tagged mouse FAK was a kind gift from Melanie Cobb. Y397F and L1034S mutants in HA-FAK were created by PCR. Flag epitope-tagged FAK was created by PCR and subcloned into pFlag-CMV (cytomegalovirus) (Sigma-Aldrich). All gene inserts created by PCR were fully sequenced to confirm the correct amplification. pCMV5M-L63RhoA was described previously (28). Nucleus-localized Myc-tagged β-galactosidase (Myc–β-gal) was described previously (20).
Migration and invasion assays.
MDA-MB-231 cells were transfected with control, Net1, or RhoA siRNAs using RNAiMax (Invitrogen, Grand Island, NY) or INTERFERin (Polyplus, New York, NY) according to the manufacturers' protocols. The final concentration of siRNAs in all transfections was 10 nM, except in the inverse invasion experiments using Matrigel (BD Biosciences), where the concentration was 20 nM. The siRNA sequences were from Sigma Genosys (St. Louis, MO) and consisted of the following: control AUUGUAUGCGAUCGCAGACdTdT (corresponding to a scrambled Net1 target sequence; used only for the experiments shown in Fig. 3); control GAUCAUACGUGCGAUCAGAdTdT (corresponding to a scrambled sequence targeting p21CIP1; used for all other siRNA transfection experiments); Net1/Net1A#1, GAGUGGACAUAAACUUUACdTdT; Net1/Net1A#2, GGAGGAUGCUAUAUUGAUAdTdT; RhoA, CAGAUACCGAUGUUAUACUdTdT; Net1#1, GAAAACGCAGAGAGAAAGAUU; Net1#2, AACGCAGAGAGAAAGAUGAUU; Net1A#1, AGGUUUGAGGGAGUACUUGUU; and Net1A#2, GGACCAUACGAGUCCUAGAUU. In all experiments after Fig. 3, Net1A#2 and Net1#1 siRNAs were used. For random migration, 2 days after siRNA transfection the cells were trypsinized and 2.5 × 104 cells were placed in the upper chamber of a 24-well Transwell plate (BD Biosciences). Membranes contained 8-μm pores and were either uncoated or coated with Matrigel. After 6 h (migration assays) or 24 h (Matrigel invasion assays), the cells in the upper well were removed using a cotton swab, and the cells on the bottom were fixed and stained with 4′,6-diamidino-2-phenylindole (DAPI) (1 μg/ml; Sigma-Aldrich). Cells that had traversed the membrane were counted in 10 random fields using the 20× objective of a Zeiss Axiophot microscope. For chemotactic migration and invasion, cells were starved 16 h prior to trypsinization in media without FBS. For these assays, 2.5 × 105 cells were placed in the upper well of the chamber. Media in the bottom well contained lysophosphatidic acid (LPA) (2 μM unless otherwise stated) (Sigma-Aldrich) or FBS (10%). Cells were allowed 6 h for migration assays and 24 h for invasion assays.
Fig 3.

Both Net1 isoforms are required for breast cancer cell migration toward LPA or serum. (A) MDA-MB-231 cells were transfected with a control siRNA, an siRNA specific for both Net1 isoforms (Net1/Net1A#1), or siRNAs specific for individual Net1 isoforms (Net1#1 and -#2 and Net1A#1 and -#2). Cells were then starved overnight and tested for migration in Transwell chambers toward LPA or 10% FBS gradients. Shown is the average from five independent experiments. Errors are SEM. The P values shown are compared to the control siRNA under the same migration conditions. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (B) Quantitative real-time PCR analysis of Net1 isoform mRNA expression in MDA-MB-231 cells transfected with control- and Net1 isoform-specific siRNAs. Net1 isoform expression was assessed 3 days after transfection. Shown is a representative experiment from three independent experiments. Errors are standard deviations. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (C) Representative Western blot of MDA-MB-231 cells transfected with the denoted siRNAs. The upper band corresponds to Net1; the lower band is Net1A.
Inverse invasion assays.
MDA-MB-231 cell invasion of a Matrigel plug was analyzed by inverse invasion assays (29, 30). Three days after transfection with control- or Net1-specific siRNAs, parental MDA-MB-231 cells or MDA-MB-231 cells stably expressing green fluorescent protein (GFP) were trypsinized and counted. A total of 1 × 104 viable cells were allowed to adhere to the membrane beneath a 100-μl plug of 1:1 Matrigel–phosphate-buffered saline (PBS) mixture. After 4 h, unattached cells were washed away, and medium lacking serum was placed in the bottom of the well. Serum-containing medium was placed in the upper chamber. Cells were allowed to migrate for 3 or 4 days before being fixed with 3.7% formaldehyde for 30 min at room temperature. Cells were permeabilized with 0.2% Triton X-100 in PBS and blocked with 1% donkey serum in PBS-Tween 20 (0.1%) (PBST) before undergoing staining with primary antibody for 6 h at 37°C. After being washed three times with PBST, samples were incubated with Cy3-conjugated secondary antibody (2 μg/ml), DAPI (2 μg/ml), and Alexa Fluor 647-phalloidin (according to the manufacturer) in 1% bovine serum albumin (BSA) in PBST overnight at 4°C. Samples were washed and stored in PBS. Imaging was performed within 1 week of sample generation using the 40× air objective on a Nikon A1 confocal microscope controlled by NIS Elements software. To quantify the distance migrated, images were acquired at 5-μm intervals starting from the surface of the membrane and moving into the matrix. To determine cell shape, 0.25- or 0.3-μm-interval stacks of the cell-containing layers were collected and observed using the “volume view” option. Single, rounded cells were classified as amoeboid and elongated cells as mesenchymal; a population of contiguous cells was considered “cohesive” and not included in the single-cell analysis. For rescue of the inverse invasion phenotype, cells were first transfected with Net1A siRNA. Forty-eight hours later, they were transfected with plasmids bearing Myc-tagged β-galactosidase, HA-Net1A, or HA-Net1A L267E using Lipofectamine and the Plus reagent (Invitrogen) according to the manufacturer's instructions. Twenty-four hours posttransfection, the inverse invasion was performed as described, with the cells invading for only 1 day before undergoing fixation and staining.
RhoA activation assays.
RhoA activation in control and Net1 knockdown cells was measured using an absorbance-based G-LISA kit, according to the manufacturer's directions (Cytoskeleton, Denver, CO). Briefly, siRNA-transfected cells were allowed to grow for 3 days posttransfection and then starved overnight before treatment with vehicle (phosphate-buffered saline) or 20 μM LPA for 5 min. Cells were harvested in G-LISA lysis buffer supplemented with protease and phosphatase inhibitors (80 mM β-glycerophosphate, 50 mM NaF, 1 mM sodium orthovanadate, 1 mM EDTA, 10 μg/ml leupeptin, 10 μg/ml pepstatin A, 2 μg/ml aprotinin, 1 mM phenylmethylsulfonyl fluoride [PMSF]), clarified by centrifugation, and immediately snap-frozen. The protein concentration was determined by measurement with Precision Red, and the RhoA activity of equal amounts of total protein was analyzed in duplicate for each sample. Samples were also analyzed by Western blotting to verify that Net1 knockdown was efficient and did not have secondary effects on RhoA expression. The results of three independent experiments were combined after normalization of the results to the absorbance of control samples.
Immunofluorescence microscopy and quantification of images.
For immunofluorescence microscopy, MDA-MB-231 cells were plated on glass coverslips and then transfected with the appropriate siRNAs. After 3 days, an area within the cells was cleared using a pipette tip and the cells were allowed to migrate into the cleared area for 6 h. The cells were then fixed for 5 min at 37°C with 3.7% formaldehyde in PBS and then permeabilized with 0.2% Triton X-100 in PBS for 5 min at room temperature. The cells were then incubated for 1 h at 37°C with the appropriate primary antibodies diluted in PBST plus 1% BSA (Sigma-Aldrich). After being washed in PBST, the cells were incubated for 1 h at 37°C with Cy2- or Cy3-conjugated anti-mouse or anti-rabbit antibodies diluted to 2 μg/ml in PBST plus 1% BSA. After being washed with PBST, the cells were mounted on glass slides with Fluormount reagent (EMD4 Biosciences, San Diego, CA). Cells were visualized with a Zeiss Axiophot epifluorescence microscope, and micrographs were acquired using Axiovision software. Staining for phospho-Y397 FAK required incubation of primary antibody overnight at 4°C for optimal signal. For pMLC staining, cells were fixed in 2% paraformaldehyde for 15 min at room temperature before permeabilizations in 0.2% Triton in PBS for 5 min. Coverslips were blocked in 3% BSA in PBS for 30 min at room temperature before incubation in primary antibody, also in 3% BSA, for 1 h at 37°C. Coverslips were washed with PBS before incubation at room temperature with secondary antibody and then further washed with PBS and rinsed with deionized water before mounting. Images were acquired using a 60× oil objective on a Nikon TiE wide-field confocal microscope controlled by NIS Elements software using constant photomultiplier tube (PMT) detector and laser settings. For rescue of pMLC staining, cells were allowed to recover for 24 h from siRNA transfection before subsequent transfection of the indicated vector. Twenty-four hours posttransfection, the cells were fixed and stained as described above. Images were acquired using a 60× oil objective on a Nikon TiE confocal microscope and deconvoluted for 10 iterations in Autoquant software.
To calculate elongation indices, the length of the cells from the center of the nucleus to the farthest point on the cell was calculated using Axiovision software. This value was then divided by the width of the cell in the nucleus, perpendicular to the first axis (31). For analysis of FAK activation after wounding or replating, images were acquired with a constant exposure time, and the background subtraction was performed uniformly for all samples and time points.
Replating assays.
To assess the contribution of Net1 to FAK activation and adherence, MDA-MB-231 cells were starved for 16 h and then trypsinized, washed, and resuspended in 0.5% lipid-free BSA (Sigma-Aldrich) in DMEM–F-12 at 37°C for 30 min before being replated in serum-free medium on collagen IV (BD Biosciences)-coated coverslips (for indirect immunofluorescence) or tissue culture dishes (for immunoprecipitation) and harvested at the time points indicated.
HA-Net1 and HA-Net1A localization.
To visualize Net1 isoform localization, MDA-MB-231 cells were plated on collagen-coated coverslips and transfected with HA-Net1, HA-Net1A, or empty vector, using Lipofectamine and the Plus reagent (Invitrogen) or TrueFect-Lipo reagent (United Biosystems), according to the manufacturer's instructions. Images were acquired using a 60× oil objective on a Nikon TiE wide-field fluorescence microscope with a CoolSnap HQ2 camera set at a 10-MHz readout speed controlled by NIS Elements software and were deconvoluted for 10 iterations using AutoQuant software.
Immunoprecipitation and Western blotting.
For analysis of proteins coprecipitating with Net1, actively growing MDA-MB-231 cells were lysed in radioimmunoprecipitation assay (RIPA) buffer without SDS (1.0% Triton X-100, 0.5% sodium deoxycholate, 50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 80 mM β-glycerophosphate, 50 mM NaF, 1 mM sodium orthovanadate, 1 mM EDTA, 10 μg/ml leupeptin, 10 μg/ml pepstatin A, 2 μg/ml aprotinin, 1 mM phenylmethylsulfonyl fluoride), and incubated on ice for 10 min, and insoluble proteins were pelleted by centrifugation (16,000 × g, 10 min, 4°C). Equal amounts of soluble lysate were precleared by incubation for 30 min at 4°C with 2 μg of nonspecific mouse IgG plus protein A-Sepharose (Rockland Immunochemicals, Gilbersville, PA). Clarified lysates were then incubated with 2 μg of control IgG or mouse anti-Net1 plus protein A-Sepharose for 2 h at 4°C. Precipitates were washed with a mixture of 20 mM Tris-HCl (pH 8.0), 125 mM NaCl, 5 mM MgCl2, and 0.5% Triton X-100, resuspended in 2× Laemmli sample buffer, and resolved by SDS-PAGE. Proteins were transferred to polyvinylidene difluoride (PVDF) membrane and analyzed by Western blotting as described previously (27).
For analysis of proteins coprecipitating with FAK or Net1 mutants, plasmids were transfected (Lipofectamine and the Plus reagent; Invitrogen) into HeLa cells for 48 h before lysis in Triton lysis buffer (0.5% Triton X-100, 20 mM Tris-HCl [pH 8.0], 100 mM NaCl, 80 mM β-glycerophosphate, 50 mM NaF, 1 mM sodium orthovanadate, 1 mM EDTA, 10 μg/ml leupeptin, 10 μg/ml pepstatin A, 2 μg/ml aprotinin, 1 mM PMSF) or RIPA buffer lacking SDS. Samples were incubated on ice for 10 min before insoluble proteins were pelleted by centrifugation. Soluble lysates were precleared with nonspecific IgG as described above before immunoprecipitation with the relevant antibody.
For analysis of RNA interference efficiency, siRNA-transfected cells were lysed in SDS lysis buffer (2.0% SDS, 20 mM Tris-HCl [pH 8.0], 100 mM NaCl, 80 mM β-glycerophosphate, 1 mM Na2VO3, 20 mM NaF, 10 μg/ml leupeptin, 10 μg/ml pepstatin A, 10 μg/ml aprotinin, 1 mM PMSF). DNA within the lysates was sheared using a sonicator or by 10 passages through a 23-gauge needle. Protein concentrations were determined by bicinchoninic acid assay (Pierce Biotechnology, Rockford, IL), and equal amounts of total protein were resolved by SDS-PAGE. Efficiency of knockdown was confirmed by Western blotting.
RESULTS
Net1 expression is required for random and LPA-stimulated breast cancer cell motility.
Net1 expression has previously been observed to be important for gastric cancer cell motility (22, 26). To determine whether Net1 is also required for breast cancer cell motility, MDA-MB-231 cells were transfected with siRNAs targeting both Net1 isoforms, and effects of Net1 knockdown were compared to those of siRNA-mediated knockdown of RhoA. Random or LPA-stimulated cell migration was monitored using Transwell chambers. In these assays, we observed that knockdown of Net1 resulted in a 40 to 60% reduction in random or LPA-stimulated cell migration, respectively (Fig. 1A and B). Importantly, the magnitude of this effect was not significantly different from that caused by inhibition of RhoA expression. A representative Western blot showing Net1 and RhoA knockdown is shown in Fig. 1C. To determine whether this effect was applicable to other invasive breast cancer cell lines, we assessed the effects of Net1 or RhoA knockdown in BT549, SUM149, and MDA-MB-435 cells. In these assays, we observed that both SUM149 and MDA-MB-435 cells required Net1 expression for motility. Net1 knockdown in BT549 cells did not produce a statistically significant change in motility, even though RhoA knockdown did have an effect (Fig. 1D). Thus, three of the four invasive breast cancer cell lines that we tested required Net1 expression for motility, and this requirement was similar to that for RhoA itself.
Fig 1.

Net1 is required for breast cancer cell motility. (A) MDA-MB-231 cells were transfected with control-, Net1 and Net1A dual targeting-, or RhoA-specific siRNAs. Three days later, the cells were tested for random migration in Transwell chambers. Both upper and lower chambers contained 10% FBS. Shown is the average from five independent experiments. Errors are standard errors of the mean (SEM). Statistical significance was calculated using Student's t test. ***, P < 0.001. (B) Effect of Net1 or RhoA knockdown on LPA-stimulated MDA-MB-231 cell migration in Transwell assays. Shown is the average from three independent experiments. Errors are SEM. ***, P < 0.001. (C) Representative Western immunoblots (IB) of total cell lysates from siRNA-transfected cells used for migration assays. (D) Effect of Net1 dual isoform or RhoA knockdown in BT549, SUM149, and MDA-MB-435 human breast cancer cells. Shown is the average from at least three independent experiments. Errors are SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Because Net1 knockdown was nearly as efficient as RhoA knockdown at inhibiting cell migration, we tested the extent to which Net1 controlled LPA-stimulated RhoA activation. MDA-MB-231 cells were starved overnight and then stimulated with LPA for 5 min, which is the peak time for LPA-stimulated RhoA activation in these cells (not shown). The cells were then lysed, and RhoA activation was measured. We observed that inhibition of Net1 expression partially blocked RhoA activation following LPA stimulation but did not significantly reduce basal levels of RhoA activity (Fig. 2A). A major effect of RhoA activation is to stimulate actomyosin contraction by promoting ROCK-mediated myosin light-chain (MLC) phosphorylation (32, 33). Thus, we examined whether Net1 expression was required for LPA-stimulated MLC2 phosphorylation on its activating site, serine 19. We observed that Net1 knockdown and RhoA knockdown inhibited MLC2 phosphorylation to similar degrees (Fig. 2B). To determine whether this effect was confined to specific areas within the cell, we assessed pS19-MLC2 levels by immunofluorescence. These assays showed that there was a general decrease in pS19-MLC2 staining in Net1 knockdown cells, with the greatest loss in the cell periphery. Similar results were observed with RhoA knockdown (Fig. 2C and D). Thus, these data suggest that Net1 controls localized RhoA activation within the cell periphery and explain why Net1 knockdown only partially inhibited RhoA activation when measured in lysed cells (Fig. 2A). The similar efficacies of Net1 and RhoA knockdown at blocking MLC2 phosphorylation also may explain why both proteins were similarly required for cell motility, as MLC phosphorylation is necessary for generation of actomyosin contractility.
Fig 2.

Contribution of Net1 isoforms to LPA-stimulated RhoA activation and peripheral myosin light-chain phosphorylation. (A) MDA-MB-231 cells were transfected with control-, Net1 dual isoform-, or RhoA-specific siRNAs, starved, and then stimulated with LPA for 5 min. RhoA activation was tested by G-LISA assay. Shown is the average from three independent experiments. Errors are SEM. **, P < 0.01; ***, P < 0.001. (B) Representative Western blot of siRNA-transfected cells. (C) Immunofluorescence analysis of myosin light-chain phosphorylation 2 on serine 19 (pMLC2) in siRNA-transfected cells. Cells were starved and then stimulated with LPA for 5 min. Shown are representative maximum-intensity z-plane images. Green, pMLC2; blue, DNA. Bar, 10 μm. (D) Quantification of pMLC2 staining in the cell periphery in siRNA-transfected cells. Shown is the average from three independent experiments. Errors are SEM. *, P < 0.05; **, P < 0.01. (E) Rescue of pMLC2 in Net/Net1A siRNA-transfected cells. Shown are representative maximum-intensity z-plane images. Green, pMLC2; blue, DNA; red, transfected protein. Bar, 21 μm. (F) Quantification of pMLC2 rescue in siRNA-transfected cells. Shown is the average from three independent experiments. Errors are SEM. **, P < 0.01.
Recent work indicates that Net1 isoforms play divergent roles in the cell. For example, Net1A expression is selectively stimulated by TGF-β in keratinocytes and is required for actin cytoskeletal rearrangement (18), and we have shown that Net1A is preferentially exported from the nucleus following Rac1 activation in MCF7 breast cancer cells and is required for focal adhesion maturation (20). Moreover, expression of the Net1 isoform is stimulated by estrogen in MCF7 cells and is preferentially required for estrogen-driven proliferation (21). To determine which Net1 isoform accounted for effects on MLC2 phosphorylation, MDA-MB-231 cells were transfected with control or Net1/Net1A siRNAs and then retransfected with a control plasmid expressing nucleus-localized β-galactosidase (Myc–β-gal), or siRNA-resistant expression vectors for HA-Net1, HA-Net1A, or catalytically inactive Net1A (HA-Net1A L267E) (16, 34). The cells were then fixed and stained for DNA, pMLC2, and transfected protein (Myc or HA epitope). We observed that expression of Myc–β-gal or HA-Net1 did not rescue pMLC2 staining, while expression of wild-type HA-Net1A did rescue it. Importantly, expression of catalytically inactive Net1A did not restore pMLC2 staining (Fig. 2E). Effects on pMLC staining were quantified in Fig. 2F. These data indicate that expression of the Net1A isoform is specifically required for peripheral MLC2 phosphorylation in MDA-MB-231 cells and that the ability to activate RhoA is a key aspect of Net1A function.
Both isoforms of Net1 contribute to LPA- and serum-stimulated chemotaxis.
Given the exclusive role of Net1A in controlling peripheral MLC2 phosphorylation in MDA-MB-231 cells, we examined whether there was a specific requirement for Net1A in controlling breast cancer cell motility. Small interfering RNAs were designed against the unique regions of Net1 or Net1A and then tested for effects on LPA- or FBS-stimulated cell motility in Transwell assays. As expected, concurrent knockdown of both Net1 isoforms efficiently blocked cell migration toward LPA or serum (Fig. 3A). Surprisingly, blocking individual Net1 isoform expression also fully inhibited cell migration in response to LPA or FBS. Quantitative PCR and Western blot analysis showed that each siRNA was specific for its intended target (Fig. 3B and C). Thus, although Net1A appears to be the major regulator of MLC2 phosphorylation, these data indicate that both Net1 isoforms contribute to MDA-MB-231 cell motility.
Net1A, but not Net1, is required for trailing edge retraction in migrating breast cancer cells.
To explore why MDA-MB-231 cells require expression of both Net1 isoforms for overall motility, we assessed their roles in controlling mechanistic aspects of cell movement. RhoA drives cell migration in part by stimulating actomyosin contraction, which in the rear of the cell results in disassembly of focal adhesions and retraction of the trailing edge. Lack of trailing edge retraction classically results in long extensions in the rear of the migrating cell (31, 35, 36). To monitor the contribution of each Net1 isoform to trailing edge retraction, we assessed the effects of their knockdown on cells migrating into a cleared area. Effects on trailing edge retraction were quantified as an elongation index, which reflects the length versus width of a cell. Increases in the elongation index have been correlated previously with an inability to retract the trailing edge (31). In these experiments, we observed that knockdown of either Net1A alone or both Net1 isoforms together caused cells to assume a spindly morphology with short extensions in the front and long extensions in the rear of the cell (Fig. 4A). However, knockdown of the Net1 isoform did not affect cell morphology, although it did elicit a small reduction in MLC2 phosphorylation when tested by Western blotting (Fig. 4B). This suggests that the Net1 isoform may play a minor role in generating actomyosin contractility that was not apparent in our previous pMLC2 rescue experiments (Fig. 2E and F). Importantly, the morphology of Net1A knockdown cells was indistinguishable from that observed in RhoA knockdown cells. This change in morphology was reflected in statistically significant increases in the elongation index that were identical for both Net1A and RhoA knockdown cells (Fig. 4C).
Fig 4.

Net1A is required for trailing edge retraction in MDA-MB-231 cells. (A) MDA-MB-231 cells were transfected with the siRNAs shown. Three days later, the monolayer was scratched and cells were allowed to migrate back into the cleared area. After 6 h, the cells were fixed and stained for DNA (blue) and F-actin (red). Arrows show the direction of migration. Shown are representative images from three independent experiments. Bar, 20 μm. (B) Representative Western blot of siRNA-transfected cells. (C) Quantification of elongation index in migrating MDA-MB-231 cells transfected with the siRNAs shown. The elongation index was measured as the distance from the furthest tip of the cell to the middle of the nucleus divided by the width of the cell at that portion of the nucleus. Shown are the combined results from three independent experiments. Bars denote median values. ***, P < 0.001. (D) Reexpression of Net1 proteins in control or Net1 dual isoform siRNA-transfected cells. Two days after siRNA transfection, cells were retransfected with expression vectors for Myc epitope-tagged, nucleus-localized β-galactosidase or the siRNA-resistant, HA-tagged Net1 isoforms shown. Net1A L/E, catalytically inactive Net1A L267E. Cells were allowed to migrate into a wounded area, fixed, and stained for the antigens shown. Arrows denote the direction of migration. Shown are representative images from three independent experiments. Bar, 20 μm. (E) Representative Western blot of siRNA-transfected cells. (F) Quantification of elongation indices from three independent rescue experiments. Bars denote median values. ***, P < 0.001. (G) Representative Western blot of transfected cells from elongation index rescue experiments.
To determine whether expression of either isoform alone was sufficient for rear end retraction, we knocked down expression of both Net1 isoforms and then transfected the cells with Myc–β-gal, HA-Net1, HA-Net1A, or HA-Net1A L267E. The cells were then allowed to migrate into a cleared area, and elongation indices were quantified. In these experiments, we observed that expression of the control protein β-galactosidase did not rescue cell morphology. Similarly, expression of the Net1 isoform did not rescue the ability of knockdown cells to retract their trailing edges (Fig. 4D to G). However, wild-type Net1A expression effectively rescued the morphology of the migrating cells and completely restored the elongation index to control values. Moreover, expression of catalytically inactive Net1A L267E did not rescue trailing edge retraction (Fig. 4D to G). Thus, these experiments demonstrate that Net1A uniquely controls the morphology of migrating MDA-MB-231 cells and that it must be able to activate RhoA to regulate rear end retraction.
Net1A interacts with FAK in a RhoA-dependent manner.
Since expression of both Net1 isoforms was necessary for efficient cell motility, we examined their subcellular distribution in MDA-MB-231 cells. Previously, we and others have shown that Net1 isoforms are localized in the nucleus and that extranuclear localization is required to stimulate RhoA activation and actin polymerization (16, 17). Thus, export of Net1 isoforms outside the nucleus would be predicted to occur if they controlled RhoA-dependent cell motility.
MDA-MB-231 cells were plated on collagen to activate integrin signaling and then transfected with expression vectors for HA-Net1 or HA-Net1A. HA epitope-tagged Net1 isoforms were expressed because antibodies suitable for immunofluorescent detection of endogenous Net1 isoforms are not available. Two days later, the cells were fixed and stained for the HA epitope, as well as for active FAK (pY397-FAK). FAK activation was assessed because it is required for focal adhesion assembly and disassembly at the leading and trailing edges of a cell, respectively (37, 38). Localization of Net1 isoforms was examined by confocal microscopy. We observed that HA-Net1 localized exclusively to the nucleus in >85% of cells (Fig. 5A). The few cells that did show extranuclear HA-Net1 exhibited diffuse cytoplasmic staining (not shown). On the other hand, HA-Net1A was commonly localized both in the nucleus and within punctate areas in the leading and trailing edges. Within the leading and trailing edges, these spots colocalized with active FAK, indicating that they were focal adhesions being actively remodeled (Fig. 5A). Localization of Net1A to focal adhesions was also observed when catalytically inactive Net1A was transfected, although this was less prominent (not shown). This indicates that ability to activate RhoA was not strictly required for focal adhesion localization of Net1A. Although the majority of cells overexpressing HA-Net1A exhibited an elongated, migratory phenotype, the population of HA-Net1-expressing cells was equally distributed among rounded stationary and elongated motile cells (not shown). Interestingly, HA-Net1, HA-Net1A, and active FAK also showed significant colocalization in the nucleus. Nuclear localization of FAK has been observed by others (39). Taken together, the results from this experiment demonstrate that a subpopulation of Net1A but not Net1 localizes outside the nucleus to phospho-FAK-positive focal adhesions concentrated within the leading edge and trailing segments of the cell.
Fig 5.
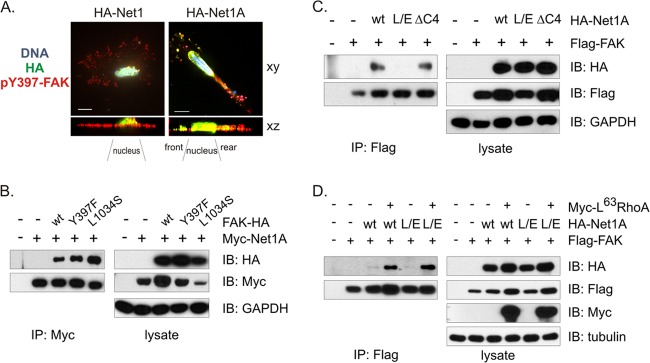
Net1A colocalizes with active FAK and interacts with FAK in a RhoA-regulated manner. (A) MDA-MB-231 cells were plated on collagen and transfected with expression vectors for HA-Net1 or HA-Net1A. The cells were then fixed and stained for HA (green), pY397-FAK (red), and DNA (blue). Shown are single x-y views of representative HA-Net1- or HA-Net1A-transfected cells, and the sum of the intensities for each cell in the x-z direction. Bar, 10 μm. (B) HeLa cells were transfected with HA epitope-tagged wild-type or mutant FAK, and wild-type Myc epitope-tagged Net1A. Myc-Net1A was then immunoprecipitated and tested for coprecipitation of FAK proteins by Western blotting. Shown is a representative experiment from three independent experiments. (C) HeLa cells were transfected with Flag epitope-tagged wild-type FAK and HA epitope-tagged wild-type Net1A, catalytically inactive Net1A L267E (L/E), or Net1A lacking the C-terminal PDZ domain binding site (ΔC4). Flag-tagged FAK was immunoprecipitated and tested for coprecipitation of Net1A proteins by Western blotting. Shown is a representative experiment from three independent experiments. (D) HeLa cells were transfected with wild-type (wt) or catalytically inactive (L/E) HA-Net1A and wild-type Flag-FAK, minus or plus constitutively active, Myc epitope-tagged L63 RhoA. Flag-FAK was immunoprecipitated and tested for coprecipitation of Myc-tagged proteins by Western blotting. Shown is a representative experiment from three independent experiments.
FAK is recruited to nascent focal adhesions through its C-terminal focal adhesion targeting (FAT) domain and is subsequently activated by intermolecular phosphorylation on tyrosine 397 (Y397) (40–42). Phosphorylation at this site provides a docking site for Src, which then further phosphorylates FAK to initiate cell signaling (37, 43). Thus, we tested whether Y397 phosphorylation of FAK or the functionality of its FAT domain was necessary for interaction with Net1A. HeLa cells were transfected with Myc-epitope-tagged Net1A and HA epitope-tagged wild-type, Y397F, or L1034S FAK. The L1034S mutation blocks interaction between FAK and integrins, as well as between FAK and paxillin (44, 45). Myc-Net1A was then immunoprecipitated and tested for coprecipitation of FAK by Western blotting. As shown in Fig. 5B, all three FAK proteins coprecipitated effectively with Net1A, with a weak preference for the FAT domain mutant. Thus, activation of FAK is not required for interaction with Net1A.
We then examined whether Net1A required catalytic activity or the presence of its C-terminal PDZ domain binding site for interaction with wild type FAK. The PDZ binding site within Net1A mediates interaction with the PDZ domain-containing proteins Magi1b, Dlg1, and CARMA1/3 (27, 34, 46, 47). HeLa cells were transfected with Flag epitope-tagged FAK and HA epitope-tagged wild-type, L267E, or ΔC4 Net1A. FAK was then immunoprecipitated and tested for coprecipitation of Net1A. In these experiments, we did not observe a requirement for the C-terminal PDZ domain binding site of Net1A, as the ΔC4 mutant coprecipitated with FAK as efficiently as wild-type Net1A. However, coprecipitation of catalytically inactive Net1A L267E was significantly impaired (Fig. 5C). This suggests that RhoA activation may stimulate the interaction between Net1A and FAK. To test this directly, we examined the ability of wild-type and catalytically inactive Net1A to coimmunoprecipitate with wild-type FAK in the presence or absence of coexpressed, constitutively active L63RhoA. We observed that coexpression of L63RhoA potently stimulated the coprecipitation of transfected Net1A and FAK (Fig. 5D). This indicates that RhoA activation potentiates the interaction between FAK and Net1A.
Net1A expression is required for FAK activation and focal adhesion maturation.
Since FAK controls focal adhesion formation and disassembly, we then tested whether inhibition of Net1A expression affected focal adhesion formation following integrin engagement. MDA-MB-231 cells were transfected with control-, Net1A-, or RhoA-specific siRNAs and then trypsinized and replated on a collagen matrix. After 1 h, the cells were fixed and stained for paxillin, pY397-FAK, and DNA. In these experiments, we observed that control siRNA-transfected cells exhibited strong pY397-FAK-positive focal adhesions on the cell edges and extending deep within the cell. However, Net1A and RhoA siRNA-transfected cells were largely devoid of these focal adhesions, with only a few small, peripheral pY397-FAK-positive focal adhesions remaining (Fig. 6A). Quantification of the number of focal adhesions per square micrometer or mean focal adhesion size throughout the cell indicated that Net1A and RhoA knockdown cells were equally impaired at promoting focal adhesion initiation and maturation (Fig. 6B and C).
Fig 6.

Net1A is required for focal adhesion maturation during cell adhesion and interacts with FAK. (A) MDA-MB-231 cells were transfected with control-, Net1A-, or RhoA-specific siRNAs and then replated on collagen-coated coverslips. After 1 h, the cells were fixed and stained for pY397-FAK (red) and DNA (blue). Shown are representative images. Bar, 10 μm. (B) Quantification of focal adhesion (FA) number per μm2 in cells 1 h after replating. Focal adhesions were identified by pY397-FAK and paxillin colocalization. Shown are data from three independent experiments. Bars are median values. ***, P < 0.001. (C) Quantification of focal adhesion size. Shown is the average from three independent experiments. Errors are SEM. ***, P < 0.001. (D) Coimmunoprecipitation of endogenous FAK with endogenous Net1 proteins is stimulated by replating MDA-MB-231 cells on collagen. Shown is a representative experiment from three independent experiments. (E) Inhibitors of MLCK (ML-7; 5 μM), ROCK (Y-27632; 10 μM), FAK (PF-562271; 8 ng/ml), and Src (bosutinib; 2 μM) block coprecipitation of endogenous Net1 proteins and FAK. Cells were treated with inhibitors overnight prior to lysis. Shown is a representative experiment from three independent experiments. DMSO, dimethyl sulfoxide. (F) Coimmunoprecipitation of endogenous FAK with Net1 proteins is sensitive to the myosin ATPase inhibitor blebbistatin (30 min, 20 μM). Shown is a representative experiment from three independent experiments.
We then examined whether replating cells on collagen stimulated the interaction between endogenous Net1 isoforms and FAK. MDA-MB-231 cells were grown on plastic or trypsinized and replated on collagen for 1 h. Endogenous Net1 proteins were immunoprecipitated with an antibody that recognizes both Net1 isoforms and tested for the coprecipitation of endogenous FAK. In these experiments, we observed that coprecipitation of FAK was strongly enhanced by replating cells on collagen, indicating that integrin engagement stimulates FAK interaction with Net1 proteins (Fig. 6D).
To identify the integrin-stimulated signaling pathways that are required for interaction between endogenous Net1 and FAK, MDA-MB-231 cells were pretreated overnight with inhibitors of myosin light-chain kinase (MLCK) (ML-7), ROCK (Y-27632), FAK (PF-562271), or Src (bosutinib). Endogenous Net1 proteins were immunoprecipitated and tested for coprecipitation of endogenous FAK by Western blotting. We observed that MLCK inhibition completely eliminated the interaction between Net1 and FAK, while inhibition of ROCK, FAK, and Src was somewhat less effective (Fig. 6E). Since both MLCK inhibition and ROCK inhibition reduce actomyosin contraction and RhoA activation stimulated interaction between Net1A and FAK (Fig. 5C and D), we examined whether directly inhibiting actomyosin contraction by using blebbistatin also blocked Net1 interaction with FAK. We observed that blebbistatin treatment was at least as effective as ML-7 at inhibiting FAK coprecipitation with Net1 (Fig. 6F). Taken together, these data indicate that the interaction between Net1 isoforms and FAK is driven by actomyosin contraction and that this interaction contributes to focal adhesion maturation.
Net1A expression is required for extracellular matrix invasion.
Because Net1A played such a profound role in controlling cell motility and spreading, we then examined whether Net1 isoforms were required for MDA-MB-231 cell invasion through an extracellular matrix. Cells were transfected with control siRNA or siRNA targeting both Net1 isoforms, and the cells were allowed to invade Matrigel-coated Transwells overnight in the absence of a ligand gradient. We observed that knockdown of both Net1 isoforms blocked unstimulated Matrigel invasion by 40% (Fig. 7A). We then assessed the requirement for individual Net1 isoforms in LPA-stimulated invasion. In these experiments, we observed that Net1A knockdown was as effective as knockdown of both Net1 isoforms and was as effective as knockdown of RhoA itself. Moreover, knockdown of the Net1 isoform did not significantly affect Matrigel invasion (Fig. 7B). A representative Western blot of siRNA-transfected cells is shown in Fig. 7C. These results indicate that Net1A, but not Net1, is required for efficient Matrigel invasion in MDA-MB-231 cells. This is distinct from LPA-stimulated cell motility in the absence of an ECM, which required expression of both Net1 isoforms (Fig. 3).
Net1A controls amoeboid movement in invading cells.
Individual cancer cells invade an extracellular matrix (ECM) by two major forms of movement, termed amoeboid and mesenchymal (9, 48, 49). In amoeboid movement, cells assume a rounded, nonpolarized morphology and squeeze between matrix fibers, propelled by actomyosin contraction in the rear of the cell. Mesenchymal movement is characterized by a polarized, fibroblast-like morphology and relies on protease-dependent degradation of the ECM and the formation of strong integrin-dependent contacts. It is thought to rely on Rac-driven lamellipodial protrusion at the front of the cell, as well as on RhoA-driven actomyosin contraction in the rear. Cancer cells can generally switch between these forms of movement as they invade an ECM (49, 50), although particular cancer cell lines often exhibit a preference for one form of movement over another (29). Amoeboid movement is considered a fast form of locomotion through a matrix, while mesenchymal invasion is considerably slower (8).
Currently, it is not known whether particular RhoA GEFs control specific forms of ECM invasion. However, because we observed that MDA-MB-231 cell invasion of a Matrigel matrix was partly dependent on Net1A activity, we examined whether Net1A contributed to a specific form of invasive movement. MDA-MB-231 cells were transfected with control- or Net1A-specific siRNAs, plated on a thick Matrigel plug, and allowed to invade toward a serum gradient. After 3 days, the cells were fixed and stained for F-actin, active β1-integrin, and MT1-MMP. MT1-MMP is the major metalloproteinase used by MDA-MB-231 cells during ECM invasion (49, 51, 52). In control siRNA-transfected cells, we observed that singly invading cells did so as a mixed population, with approximately equal numbers of amoeboid and mesenchymal morphologies. Importantly, Net1A or RhoA knockdown significantly changed this ratio, such that there were many fewer amoeboid invading cells. The increase in cells with the mesenchymal morphology was accompanied by a large increase in MT1-MMP and active β1-integrin staining (Fig. 8A). The change in morphology of invading cells is quantified in Fig. 8B.
Fig 8.

Net1A is required for amoeboid invasive movement. (A) MDA-MB-231 cells were transfected with control-, Net1A-, or RhoA-specific siRNAs. Three days later, the cells were plated on a thick Matrigel plug and allowed to invade for another 3 days. The cells were then fixed and stained for active β1-integrin (purple), MT1-MMP (red), F-actin (green), and DNA (blue) and visualized by confocal microscopy. Shown are maximum-intensity projections of representative x-y fields. Bar, 10 μm. (B) Quantification of cell morphologies from control and Net1A and RhoA siRNA-transfected cells during Matrigel invasion. Shown is the average from three independent experiments. Errors are SEM. *, P < 0.05; **, P < 0.01. (C) Median distance invaded by GFP-expressing MDA-MB-231 cells transfected with control, Net1A, or RhoA siRNAs. The extent of cell invasion was assessed by monitoring the GFP signal of at least six separate fields for each siRNA in four separate experiments. Shown is a representative experiment. (D) Representative Western blot of siRNA-transfected cells. (E) Reexpression of wild type but not catalytically inactive Net1A rescues amoeboid Matrigel invasion. MDA-MB-231 cells were transfected with control- or Net1A-specific siRNAs. Two days later, the cells were retransfected with Myc-tagged β-galactosidase (Myc-βgal), siRNA-resistant wild-type Net1A, or catalytically inactive Net1A L267E. One day after that, the cells were fixed and stained for HA or Myc epitope-tagged proteins, F-actin, and DNA. The top panels show F-actin (green) and DNA (blue). The bottom panels show expression of transfected proteins. Shown are representative images. Bar, 10 μm. (F) Quantification of cell morphology in transfected cells from three independent experiments. Errors are SEM. ***, P < 0.001.
We then examined whether this change in morphology was accompanied by a decrease in invasion efficiency. MDA-MB-231 cells stably expressing GFP were transfected with control-, Net1A-, or RhoA-specific siRNAs and allowed to invade a Matrigel plug for 3 days, and the distance invaded by the cells was assessed. As shown in Fig. 8C, knockdown of Net1A or RhoA reduced the distances invaded by similar degrees. Western blotting of cells prior to plating on Matrigel showed that the siRNAs were specific for their intended targets and that Net1A and RhoA knockdowns similarly inhibited MLC2 phosphorylation (Fig. 8D). Comparable results were obtained with parental MDA-MB-231 cells stained for F-actin (not shown). These data indicate that Net1A controls amoeboid movement in an ECM environment and is required for efficient invasion.
We then examined whether reexpression of wild-type or catalytically inactive Net1A could rescue amoeboid movement in invading Net1A knockdown cells. MDA-MB-231 cells were transfected with control- or Net1A-specific siRNAs and then retransfected with the control protein β-galactosidase, wild-type HA-Net1A, or HA-Net1A L267E. Cells were allowed to invade the Matrigel for 1 day and fixed and stained for epitope-tagged proteins, F-actin, and DNA. In these experiments, we observed that expression of wild-type Net1A efficiently restored amoeboid movement, but that β-galactosidase or Net1A L267E expression did not rescue this form of motility (Fig. 8E and F). These results indicate that the ability of Net1A to control amoeboid invasion requires its catalytic activity toward RhoA.
DISCUSSION
In the present work, we show that Net1 isoform expression is required for motility in multiple human breast cancer cell lines. Depletion of both Net1 isoforms reduces RhoA activation and diminishes myosin light-chain phosphorylation, particularly in the cell periphery. Moreover, depletion of the Net1A isoform yields a cellular phenotype that is consistent with an inability of migrating cells to retract their trailing edge. We also show that Net1A interacts with FAK in a RhoA-regulated manner and is required for FAK activation during cell spreading. Finally, we demonstrate that Net1A expression is required for efficient invasion and controls the ability of cells to move through the ECM by amoeboid movement. These data indicate that Net1 isoforms, especially Net1A, may be important mediators of breast cancer cell motility and invasion.
It is noteworthy that the majority of phenotypes we examined only required Net1A expression. These included rear edge retraction, FAK activation, focal adhesion maturation, and amoeboid invasive activity. These findings fit with previous studies of other cell types showing that Net1A was preferentially required for cell adhesion, TGF-β-stimulated actin cytoskeletal reorganization, and focal adhesion maturation (18, 20, 21). Given these findings, it is not clear why both Net1 isoforms are required for MDA-MB-231 cell motility in our study (Fig. 3). In this regard, we did observe that a minority of cells exhibited Net1 localization outside the nucleus, and we have shown recently in MCF7 cells that active Rac1 can occasionally stimulate relocalization of Net1 outside the nucleus (20). Moreover, we observed that knockdown of Net1 alone also caused a modest reduction in myosin light-chain phosphorylation (Fig. 4B). Even so, it is difficult to reconcile the apparently large contribution of Net1 to MDA-MB-231 cell motility in Transwell assays with these relatively minor effects. This suggests that Net1 may contribute to cell motility in a way that we did not measure. Future work will be required to identify this contribution.
When assessing the role of Net1 isoforms in controlling RhoA activation and MLC2 phosphorylation, we observed that knockdown of both Net1 isoforms only partially inhibited LPA-stimulated RhoA activation and preferentially blocked MLC2 phosphorylation in the cell periphery (Fig. 2). This suggests that Net1 function is localized in the cell, an idea that is supported by our previous work with MCF7 cells showing that Net1A is relocalized to the plasma membrane by active Rac1 (20). In addition, we observed here that Net1A was preferentially localized to focal adhesions and the leading and trailing edges, further supporting the idea of localized function (Fig. 5). The stronger effect of Net1 knockdown on MLC2 phosphorylation than on RhoA activation is unlikely to be due to its potential GEF activity toward RhoB, as these cells express very little RhoB and Net1 knockdown produced nearly equivalent effects as RhoA knockdown. Thus, we favor the interpretation that Net1 is one of multiple RhoA GEFs responding to LPA or other motility stimuli in MDA-MB-231 cells and that its function is localized. Importantly, others have shown that p115-RhoGEF, PDZ-Rho GEF, and LARG are LPA-regulated RhoA GEFs (53, 54), and we have observed that MDA-MB-231 cells express PDZ-Rho GEF (not shown). Thus, it is likely that multiple RhoA GEFs are responding to LPA stimulation in MDA-MB-231 cells to control different aspects of cell signaling.
An important finding of this work is that Net1A interacts with FAK and controls its activation (Fig. 5 and 6). Since FAK activation is necessary for the formation of new focal adhesions at the leading edge and dissolution of focal adhesions at the trailing edge (37, 38), it is likely that interaction with FAK is an important aspect of how Net1A controls cell motility. Interestingly, we observed that this interaction was stimulated by RhoA activation and reduced by small molecule inhibitors of MLCK, ROCK, FAK, Src, and myosin. The simplest explanation for these findings is that actomyosin contraction stimulates Net1A interaction with FAK. This may produce a positive-feedback loop whereby RhoA activation downstream of Net1A stimulates increased actomyosin contraction, which in turn would result in additional Net1A recruitment and enhanced FAK activation. Actomyosin contraction is generally accepted to stimulate FAK activation by aggregating integrins (55), which in turn promotes juxtaposition of FAK molecules to allow for their intermolecular phosphorylation on Y397 (41, 42). Thus, we favor a model whereby Net1A controls FAK activation indirectly, downstream of RhoA activation and actomyosin contraction. We propose that Net1A is part of an amplification mechanism to accelerate actomyosin contraction and focal adhesion maturation. In this regard, other Rho GEFs have been shown to interact with FAK. For example, transfected PDZ-Rho GEF has been shown to coimmunoprecipitate with transfected FAK and is localized to focal adhesions in rat fibroblasts plated on fibronectin (31). Similarly, p190RhoGEF interacts with FAK in Neuro-2A cells replated on fibronectin (56, 57). Thus, it may be that additional RhoA GEFs that we have not tested also contribute to FAK activation in MDA-MB-231 cells.
Another significant finding of this work is the requirement for Net1A expression for efficient invasion of a Matrigel extracellular matrix (Fig. 7 and 8). Importantly, we observed that Net1A knockdown blocked Matrigel invasion as efficiently as knockdown of RhoA itself. Single cells generally invade an extracellular matrix by two distinct forms of movement, amoeboid and mesenchymal motility. Amoeboid movement is characterized by rounded cells that squeeze between matrix fibers by generating actomyosin contraction in the rear of the cell. Mesenchymal movement is characterized by fibroblast-like cells that utilize strong integrin-dependent contacts and secrete various proteases to create a path for travel (48, 49). In our experiments, we observed that MDA-MB-231 cells invade Matrigel by equal use of amoeboid and mesenchymal mechanisms. However, knockdown of Net1A expression largely precluded amoeboid invasion and shifted the cells to the slower mesenchymal movement. This was characterized by a large increase in β1-integrin activation and MT1-MMP expression (Fig. 8). To our knowledge, this is the first time that a particular Rho GEF has been shown to control a class of invasive movement.
On the surface, this phenotype may seem difficult to reconcile with the interaction of Net1A and FAK we identified for 2D motility. However, cell movement through an extracellular matrix is qualitatively distinct from that on a planar surface, and many of the signaling mechanisms that apply to 2D motility are altered during 3D movement (8, 9). For example, cells moving in 3D matrices do not exhibit readily discernible focal adhesions (58). Instead, in this environment key focal adhesion proteins, such as FAK, contribute to the formation of cell protrusions between matrix fibers and are required to maintain the speed and directional persistence of cell movement (58).
It is generally accepted that invading cancer cell morphology is plastic, switching between amoeboid and mesenchymal movement as needed (49, 50). For example, mesenchymal invasion is usually required for cells to pass through a dense region of the extracellular matrix or a basement membrane (59). Our findings indicate that Net1A activity is an essential factor in determining the mode of cell movement. Importantly, it has been observed previously that long-term treatment with TGF-β, which in many cell types stimulates an epithelial-to-mesenchymal transition, leads to a decrease in expression of both Net1 isoforms (18). Similarly, Net1A expression falls off drastically when cells invade a basement membrane during chicken gastrulation (60). Thus, it is possible that cells generally downregulate Net1A expression when mesenchymal movement is necessary. Our results also suggest the interesting possibility that Net1A expression actively suppresses the mesenchymal phenotype. In the future it will be important to assess the role of Net1 isoforms in classical models of epithelial to mesenchymal transition, as well as in animal models of metastasis.
ACKNOWLEDGMENTS
We thank Sarita Menon for help with confocal microscopy and the Clark, Dessauer, and Denicourt lab meeting groups for critical analysis of this work.
This work was supported by Public Health Service grant CA116356 from the National Cancer Institute, by grant BCTR123806 from Susan G. Komen for the Cure, and by grant RP100502 from the Cancer Prevention and Research Institute of Texas.
Footnotes
Published ahead of print 20 May 2013
REFERENCES
- 1. Fritz G, Brachetti C, Bahlmann F, Schmidt M, Kaina B. 2002. Rho GTPases in human breast tumours: expression and mutation analyses and correlation with clinical parameters. Br. J. Cancer 87: 635– 644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fritz G, Just I, Kaina B. 1999. Rho GTPases are over-expressed in human tumors. Int. J. Cancer 81: 682– 687 [DOI] [PubMed] [Google Scholar]
- 3. Jaffe AB, Hall A. 2005. Rho GTPases: biochemistry and biology. Annu. Rev. Cell Dev. Biol. 21: 247– 269 [DOI] [PubMed] [Google Scholar]
- 4. Wheeler AP, Ridley AJ. 2004. Why three Rho proteins? RhoA, RhoB, RhoC, and cell motility. Exp. Cell Res. 301: 43– 49 [DOI] [PubMed] [Google Scholar]
- 5. Burridge K, Wennerberg K. 2004. Rho and Rac take center stage. Cell 116: 167– 179 [DOI] [PubMed] [Google Scholar]
- 6. Olson MF, Sahai E. 2009. The actin cytoskeleton in cancer cell motility. Clin. Exp. Metastasis 26: 273– 287 [DOI] [PubMed] [Google Scholar]
- 7. Ridley AJ. 2011. Life at the leading edge. Cell 145: 1012– 1022 [DOI] [PubMed] [Google Scholar]
- 8. Friedl P, Wolf K. 2010. Plasticity of cell migration: a multiscale tuning model. J. Cell Biol. 188: 11– 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sanz-Moreno V, Marshall CJ. 2010. The plasticity of cytoskeletal dynamics underlying neoplastic cell migration. Curr. Opin. Cell Biol. 22: 690– 696 [DOI] [PubMed] [Google Scholar]
- 10. Narumiya S, Tanji M, Ishizaki T. 2009. Rho signaling, ROCK and mDia1, in transformation, metastasis and invasion. Cancer Metastasis Rev. 28: 65– 76 [DOI] [PubMed] [Google Scholar]
- 11. Meller N, Merlot S, Guda C. 2005. CZH proteins: a new family of Rho-GEFs. J. Cell Sci. 118: 4937– 4946 [DOI] [PubMed] [Google Scholar]
- 12. Rossman KL, Der CJ, Sondek J. 2005. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 6: 167– 180 [DOI] [PubMed] [Google Scholar]
- 13. Chan AM, Takai S, Yamada K, Miki T. 1996. Isolation of a novel oncogene, NET1, from neuroepithelioma cells by expression cDNA cloning. Oncogene 12: 1259– 1266 [PubMed] [Google Scholar]
- 14. Alberts AS, Treisman R. 1998. Activation of RhoA and SAPK/JNK signalling pathways by the RhoA-specific exchange factor mNET1. EMBO J. 17: 4075– 4085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Srougi MC, Burridge K. 2011. The nuclear guanine nucleotide exchange factors Ect2 and Net1 regulate RhoB-mediated cell death after DNA damage. PLoS One 6: e17108. 10.1371/journal.pone.0017108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Qin H, Carr HS, Wu X, Muallem D, Tran NH, Frost JA. 2005. Characterization of the biochemical and transforming properties of the neuroepithelial transforming protein 1. J. Biol. Chem. 280:7603– 7613 [DOI] [PubMed] [Google Scholar]
- 17. Schmidt A, Hall A. 2002. The Rho exchange factor Net1 is regulated by nuclear sequestration. J. Biol. Chem. 277: 14581– 14588 [DOI] [PubMed] [Google Scholar]
- 18. Papadimitriou E, Vasilaki E, Vorvis C, Iliopoulos D, Moustakas A, Kardassis D, Stournaras C. 2012. Differential regulation of the two RhoA-specific GEF isoforms Net1/Net1A by TGF-beta and miR-24: role in epithelial-to-mesenchymal transition. Oncogene 31: 2862– 2875 [DOI] [PubMed] [Google Scholar]
- 19. Shen X, Li J, Hu PP, Waddell D, Zhang J, Wang XF. 2001. The activity of guanine exchange factor NET1 is essential for transforming growth factor-beta-mediated stress fiber formation. J. Biol. Chem. 276: 15362– 15368 [DOI] [PubMed] [Google Scholar]
- 20. Carr HS, Morris CA, Menon S, Song EH, Frost JA. 2013. Rac1 controls the subcellular localization of the Rho guanine nucleotide exchange factor Net1A to regulate focal adhesion formation and cell spreading. Mol. Cell. Biol. 33: 622– 634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dutertre M, Gratadou L, Dardenne E, Germann S, Samaan S, Lidereau R, Driouch K, de la Grange P, Auboeuf D. 2010. Estrogen regulation and physiopathologic significance of alternative promoters in breast cancer. Cancer Res. 70: 3760– 3770 [DOI] [PubMed] [Google Scholar]
- 22. Leyden J, Murray D, Moss A, Arumuguma M, Doyle E, McEntee G, O'Keane C, Doran P, MacMathuna P. 2006. Net1 and Myeov: computationally identified mediators of gastric cancer. Br. J. Cancer 94: 1204– 1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shen SQ, Li K, Zhu N, Nakao A. 2008. Expression and clinical significance of NET-1 and PCNA in hepatocellular carcinoma. Med. Oncol. 25: 341– 345 [DOI] [PubMed] [Google Scholar]
- 24. Tu Y, Lu J, Fu J, Cao Y, Fu G, Kang R, Tian X, Wang B. 2010. Over-expression of neuroepithelial-transforming protein 1 confers poor prognosis of patients with gliomas. Jpn. J. Clin. Oncol. 40: 388– 394 [DOI] [PubMed] [Google Scholar]
- 25. Gilcrease MZ, Kilpatrick SK, Woodward WA, Zhou X, Nicolas MM, Corley LJ, Fuller GN, Tucker SL, Diaz LK, Buchholz TA, Frost JA. 2009. Coexpression of alpha6beta4 integrin and guanine nucleotide exchange factor Net1 identifies node-positive breast cancer patients at high risk for distant metastasis. Cancer Epidemiol. Biomarkers Prev. 18: 80– 86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Murray D, Horgan G, MacMathuna P, Doran P. 2008. NET1-mediated RhoA activation facilitates lysophosphatidic acid-induced cell migration and invasion in gastric cancer. Br. J. Cancer 99: 1322– 1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Carr HS, Cai C, Keinanen K, Frost JA. 2009. Interaction of the RhoA exchange factor Net1 with Discs Large Homolog 1 protects it from proteasome mediated degradation and potentiates Net1 activity. J. Biol. Chem. 284: 24269– 24280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Frost JA, Xu S, Hutchison MR, Marcus S, Cobb MH. 1996. Actions of Rho family small G proteins and p21-activated protein kinases on mitogen-activated protein kinase family members. Mol. Cell. Biol. 16: 3707– 3713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hooper S, Marshall JF, Sahai E. 2006. Tumor cell migration in three dimensions. Methods Enzymol. 406: 625– 643 [DOI] [PubMed] [Google Scholar]
- 30. Scott RW, Hooper S, Crighton D, Li A, Konig I, Munro J, Trivier E, Wickman G, Morin P, Croft DR, Dawson J, Machesky L, Anderson KI, Sahai EA, Olson MF. 2010. LIM kinases are required for invasive path generation by tumor and tumor-associated stromal cells. J. Cell Biol. 191: 169– 185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Iwanicki MP, Vomastek T, Tilghman RW, Martin KH, Banerjee J, Wedegaertner PB, Parsons JT. 2008. FAK, PDZ-RhoGEF and ROCKII cooperate to regulate adhesion movement and trailing-edge retraction in fibroblasts. J. Cell Sci. 121: 895– 905 [DOI] [PubMed] [Google Scholar]
- 32. Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, Matsuura Y, Kaibuchi K. 1996. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J. Biol. Chem. 271: 20246– 20249 [DOI] [PubMed] [Google Scholar]
- 33. Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. 1996. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science 273: 245– 248 [DOI] [PubMed] [Google Scholar]
- 34. Garcia-Mata R, Dubash AD, Sharek L, Carr HS, Frost JA, Burridge K. 2007. The nuclear RhoA exchange factor Net1 interacts with proteins of the Dlg family, affects their localization, and influences their tumor suppressor activity. Mol. Cell. Biol. 27: 8683– 8697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Worthylake RA, Lemoine S, Watson JM, Burridge K. 2001. RhoA is required for monocyte tail retraction during transendothelial migration. J. Cell Biol. 154: 147– 160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yoshinaga-Ohara N, Takahashi A, Uchiyama T, Sasada M. 2002. Spatiotemporal regulation of moesin phosphorylation and rear release by Rho and serine/threonine phosphatase during neutrophil migration. Exp. Cell Res. 278: 112– 122 [DOI] [PubMed] [Google Scholar]
- 37. Cohen LA, Guan JL. 2005. Mechanisms of focal adhesion kinase regulation. Curr. Cancer Drug Targets 5: 629– 643 [DOI] [PubMed] [Google Scholar]
- 38. Mitra SK, Schlaepfer DD. 2006. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr. Opin. Cell Biol. 18: 516– 523 [DOI] [PubMed] [Google Scholar]
- 39. Lim ST, Chen XL, Lim Y, Hanson DA, Vo TT, Howerton K, Larocque N, Fisher SJ, Schlaepfer DD, Ilic D. 2008. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol. Cell 29: 9– 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hildebrand JD, Schaller MD, Parsons JT. 1993. Identification of sequences required for the efficient localization of the focal adhesion kinase, pp125FAK, to cellular focal adhesions. J. Cell Biol. 123: 993– 1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Katz BZ, Miyamoto S, Teramoto H, Zohar M, Krylov D, Vinson C, Gutkind JS, Yamada KM. 2002. Direct transmembrane clustering and cytoplasmic dimerization of focal adhesion kinase initiates its tyrosine phosphorylation. Biochim. Biophys. Acta 1592: 141– 152 [DOI] [PubMed] [Google Scholar]
- 42. Toutant M, Costa A, Studler JM, Kadare G, Carnaud M, Girault JA. 2002. Alternative splicing controls the mechanisms of FAK autophosphorylation. Mol. Cell. Biol. 22: 7731– 7743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schaller MD, Hildebrand JD, Shannon JD, Fox JW, Vines RR, Parsons JT. 1994. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol. Cell. Biol. 14: 1680– 1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sieg DJ, Hauck CR, Schlaepfer DD. 1999. Required role of focal adhesion kinase (FAK) for integrin-stimulated cell migration. J. Cell Sci. 112: 2677– 2691 [DOI] [PubMed] [Google Scholar]
- 45. Tachibana K, Sato T, D'Avirro N, Morimoto C. 1995. Direct association of pp125FAK with paxillin, the focal adhesion-targeting mechanism of pp125FAK. J. Exp. Med. 182: 1089– 1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dobrosotskaya IY. 2001. Identification of mNET1 as a candidate ligand for the first PDZ domain of MAGI-1. Biochem. Biophys. Res. Commun. 283: 969– 975 [DOI] [PubMed] [Google Scholar]
- 47. Vessichelli M, Ferravante A, Zotti T, Reale C, Scudiero I, Picariello G, Vito P, Stilo R. 2012. Neuroepithelial transforming gene 1 (Net1) binds to caspase activation and recruitment domain (CARD)- and membrane-associated guanylate kinase-like domain-containing (CARMA) proteins and regulates nuclear factor kappaB activation. J. Biol. Chem. 287: 13722– 13730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sahai E, Marshall CJ. 2003. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat. Cell Biol. 5: 711– 719 [DOI] [PubMed] [Google Scholar]
- 49. Wolf K, Mazo I, Leung H, Engelke K, von Andrian UH, Deryugina EI, Strongin AY, Brocker EB, Friedl P. 2003. Compensation mechanism in tumor cell migration: mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J. Cell Biol. 160: 267– 277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sanz-Moreno V, Gadea G, Ahn J, Paterson H, Marra P, Pinner S, Sahai E, Marshall CJ. 2008. Rac activation and inactivation control plasticity of tumor cell movement. Cell 135: 510– 523 [DOI] [PubMed] [Google Scholar]
- 51. Jiang WG, Davies G, Martin TA, Parr C, Watkins G, Mason MD, Mansel RE. 2006. Expression of membrane type-1 matrix metalloproteinase, MT1-MMP in human breast cancer and its impact on invasiveness of breast cancer cells. Int. J. Mol. Med. 17: 583– 590 [PubMed] [Google Scholar]
- 52. Wolf K, Wu YI, Liu Y, Geiger J, Tam E, Overall C, Stack MS, Friedl P. 2007. Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat. Cell Biol. 9: 893– 904 [DOI] [PubMed] [Google Scholar]
- 53. Wang Q, Liu M, Kozasa T, Rothstein JD, Sternweis PC, Neubig RR. 2004. Thrombin and lysophosphatidic acid receptors utilize distinct rhoGEFs in prostate cancer cells. J. Biol. Chem. 279: 28831– 28834 [DOI] [PubMed] [Google Scholar]
- 54. Yamada T, Ohoka Y, Kogo M, Inagaki S. 2005. Physical and functional interactions of the lysophosphatidic acid receptors with PDZ domain-containing Rho guanine nucleotide exchange factors (RhoGEFs). J. Biol. Chem. 280: 19358– 19363 [DOI] [PubMed] [Google Scholar]
- 55. Chrzanowska-Wodnicka M, Burridge K. 1996. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J. Cell Biol. 133: 1403– 1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lim Y, Lim ST, Tomar A, Gardel M, Bernard-Trifilo JA, Chen XL, Uryu SA, Canete-Soler R, Zhai J, Lin H, Schlaepfer WW, Nalbant P, Bokoch G, Ilic D, Waterman-Storer C, Schlaepfer DD. 2008. PyK2 and FAK connections to p190Rho guanine nucleotide exchange factor regulate RhoA activity, focal adhesion formation, and cell motility. J. Cell Biol. 180: 187– 203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhai J, Lin H, Nie Z, Wu J, Canete-Soler R, Schlaepfer WW, Schlaepfer DD. 2003. Direct interaction of focal adhesion kinase with p190RhoGEF. J. Biol. Chem. 278: 24865– 24873 [DOI] [PubMed] [Google Scholar]
- 58. Fraley SI, Feng Y, Krishnamurthy R, Kim DH, Celedon A, Longmore GD, Wirtz D. 2010. A distinctive role for focal adhesion proteins in three-dimensional cell motility. Nat. Cell Biol. 12: 598– 604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sabeh F, Shimizu-Hirota R, Weiss SJ. 2009. Protease-dependent versus -independent cancer cell invasion programs: three-dimensional amoeboid movement revisited. J. Cell Biol. 185: 11– 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nakaya Y, Sukowati EW, Wu Y, Sheng G. 2008. RhoA and microtubule dynamics control cell-basement membrane interaction in EMT during gastrulation. Nat. Cell Biol. 10: 765– 775 [DOI] [PubMed] [Google Scholar]