

Abstract
The involvement of nuclear factor kappa B (NF-κB) in several processes in the postnatal and adult brain, ranging from neuronal survival to synaptogenesis and plasticity, has been documented. In contrast, little is known about the functions of NF-κB during embryonic brain development. It is shown here that NF-κB is selectively activated in neocortical neural progenitor cells in the developing mouse telencephalon. Blockade of NF-κB activity leads to premature cortical neuronal differentiation and depletion of the progenitor cell pool. Conversely, NF-κB activation causes decreased cortical neurogenesis and expansion of the progenitor cell compartment. These effects are antagonized by the proneuronal transcription factor Hes6, which physically and functionally interacts with RelA-containing NF-κB complexes in cortical progenitor cells. In turn, NF-κB exerts an inhibitory effect on the ability of Hes6 to promote cortical neuronal differentiation. These results reveal previously uncharacterized functions and modes of regulation for NF-κB and Hes6 during cortical neurogenesis.
INTRODUCTION
Development of the mammalian cerebral cortex initiates with mitotic neural progenitor cells located in the ventricular zone (VZ) of the embryonic neocortex. These cells initially undergo symmetric divisions to generate two new undifferentiated progenitors. At the onset of neurogenesis, certain neural progenitors begin to undergo asymmetric divisions that generate a new progenitor and a neuron. Regulation of the balance between progenitor maintenance and neuronal differentiation is essential for correct cortical development, but the mechanisms regulating these processes remain incompletely characterized (1, 2).
Hairy/enhancer of split (Hes) transcription factors, including Hes1 and Hes5, are important regulators of the balance between proliferation and differentiation during cortical neurogenesis. Hes1 and Hes5 act downstream of the Notch receptors to inhibit neuronal differentiation and promote maintenance of the undifferentiated progenitor state (3–5). The antineurogenic role of Notch-induced Hes proteins is antagonized, at least in part, by a related Hes family member, Hes6, which is not activated by Notch signaling and instead functions downstream of proteins that promote neuronal differentiation (6–8).
Functions of the Notch pathway are often modulated by cross talk with the transcription factor, nuclear factor kappa B (NF-κB) (9, 10). NF-κB is a heterodimer that controls the expression of many genes involved in regulating cell proliferation, survival, and differentiation during numerous physiological processes (11, 12). NF-κB dimers are ubiquitously expressed but are usually maintained in an inactive state in the cytosol by mechanisms that prevent and/or revert their nuclear translocation, including the association with factors generally referred to as inhibitors of NF-κB (IκBs). A variety of different stimuli lead to the activation of NF-κB by causing the phosphorylation and degradation of IκBs (13, 14).
NF-κB is important for nervous system processes like neuronal survival, dendrite development, synaptic plasticity, and learning (15–18). Little is known, however, about the involvement of NF-κB in early stages of brain development. Here, we show that mouse NF-κB is selectively activated in neocortical neural progenitor cells, where it inhibits neuronal differentiation and maintains the undifferentiated progenitor state. This effect is antagonized by Hes6, which interacts physically and functionally with NF-κB. This antagonism is bidirectional, since NF-κB suppresses Hes6-mediated promotion of cortical neurogenesis. Together, these results reveal a previously uncharacterized function for NF-κB during cortical neurogenesis and provide evidence that mutually repressive interactions between NF-κB and Hes6 drive signaling events that are critical for normal cerebral cortex development.
MATERIALS AND METHODS
DNA plasmids.
The following expression and reporter plasmids have been described previously: pCAGGS-GFP (where GFP is green fluorescent protein) (19); pcDNA3-IκBαM (20); pAd-Track-IKKβ-DN (where IKKβ-DN is dominant negative form of IκB kinase subunit beta) (21, 22); pCMV2-FLAG-Hes6WT, pCMV2-FLAG-Hes6Δ55-95, pCMV2-FLAG-Hes6ΔWRPW, pCMV2-FLAG-Hes1WT, and pEGFP (23, 24); pCMV-p65/RelA (pCMV-RelA) and pNF-κB-luciferase (25); and pRSV-β-gal (7, 24). The IκBαM coding sequence was subcloned as an EcoRI fragment into the EcoRI site of the pCIG2 vector, which contains a cDNA-internal ribosome entry site (IRES)-enhanced GFP (EGFP) expression cassette under the control of a cytomegalovirus (CMV) enhancer and chicken β-actin promoter (26). The IKKβ-DN coding sequence was subcloned as a blunt-ended NotI/HindIII fragment into the SmaI site of pCIG2.
Animals.
Animal procedures were conducted in accordance with the guidelines of the Canadian Council on Animal Care and were approved by the Montreal Neurological Institute Animal Care Committee. NF-κBLacZ reporter mice were generated and genotyped as described previously (20). For analysis of staged embryos, NF-κB male mice were crossed to C57BL/6 females to obtain heterozygous embryos containing a single transgenic allele. For staging of mouse embryos, the day of the appearance of the vaginal plug was considered embryonic day 0.5 (E0.5).
Analysis of NF-κB reporter mouse embryos.
Heterozygous NF-κBLacZ embryos containing a single transgenic allele were recovered, fixed, and cryostat sectioned as described previously (27). β-Galactosidase (β-Gal) activity in NF-κBLacZ embryos was detected histochemically by rinsing sections from staged embryos three times in solution A (80 mM Na2HPO4, 20 mM NaH2PO4 [pH 7.4], 2 mM MgCl2, 0.2% IGEPAL [octylphenoxypolyethoxyethanol], 0.1% sodium deoxycholate), followed by incubation overnight at 37°C in solution A containing 5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, and 1 mg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) (Invitrogen). After this, sections were extensively rinsed in phosphate-buffered saline (PBS) and counterstained with eosin before being mounted with Fluoromount-G (SouthernBiotech). For immunohistochemical analysis, sections from staged embryos were rinsed twice in HEPES-buffered saline (HBS) and then preincubated for 1 h in blocking solution. With primary antibodies raised in mice, the blocking solution was provided by the “mouse-on-mouse” (MOM) kit purchased from Vector Laboratories Inc. With primary antibodies raised in other species, the blocking solution consisted of 5% normal donkey serum (Jackson ImmunoResearch Laboratories), 0.1% Triton X-100, and 0.5 mg/ml bovine serum albumin (Sigma) in HBS. Sections were then incubated for 1 to 2 h at room temperature in blocking solution containing the following primary antibodies: rabbit anti-β-Gal (1/2,000; Cappel), goat anti-β-Gal (1/1,000; Biogenesis), mouse anti-β-Gal (1/50; Developmental Studies Hybridoma Bank), rabbit anti-Hes6 (1/1,000; Abcam), rabbit anti-FoxG1 (1/500; Abcam), rabbit anti-Pax6 (1/500; Covance), mouse anti-intermediate filament protein nestin (1/10; Developmental Studies Hybridoma Bank), guinea pig anti-Hes1 (1/500) (28), or mouse anti-type III β-tubulin (βIII-tubulin) (1/300; Promega) antibody. Sections were then extensively rinsed in blocking solution, followed by incubation with the appropriate secondary antibodies for 1 h at room temperature. Secondary antibodies against primary reagents raised in various species were conjugated to Alexa Fluor 555 or Alexa Fluor 488 (1/1,000; Invitrogen). Sections were then rinsed twice with blocking solution and several times with PBS, counterstained with Hoechst 33258 (1/5,000; Sigma) for 5 min, rinsed twice with PBS, mounted with Fluoromount-G, and examined by fluorescence microscopy. Images were acquired using either a digital video camera mounted on a Zeiss Axioskop 2 microscope or a Retiga EXi camera (Qimaging) on a Zeiss Axio Imager.M1 microscope. Images were digitally assigned to the appropriate red, green, or blue channels using Northern Eclipse image acquisition software (Empix).
In utero electroporation.
Pregnant CD1 mice at gestational stage E13.5 were obtained from Charles River Laboratories. Mice were anesthetized with isoflurane, and uterine horns were exposed as described previously (29). Embryos were injected into the lateral ventricles with an empty pCIG2 plasmid, a pCIG2-IκBαM construct, or a pCIG2-IKKβ-DN construct (2 μg/μl in each case) using an Eppendorf Femtojet microinjector (30). The head of each embryo was held between tweezer-type circular electrodes (Harvard Apparatus) across the uterus wall, and five electrical pulses (amplitude, 50 V; duration, 50 ms; intervals, 950 ms) were delivered using an ECM830 square wave electroporator (BTX Harvard Apparatus). Gestation was then allowed to continue for 2 days, after which embryos were collected (E15.5) and fixed in 4% paraformaldehyde. Double-labeling immunofluorescence analysis of the expression of GFP and markers of undifferentiated neural progenitors or postmitotic neurons was performed as described for embryonic brain sections. The following primary antibodies were used: mouse anti-intermediate filament protein nestin (1/10), mouse anti-βIII-tubulin (1/1,000), rabbit anti-GFP (1/500; Invitrogen), and goat anti-Sox2 (1/500; R&D Systems) antibodies.
Organotypic forebrain slice culture and electroporation.
E13.5 embryos from staged CD1 pregnant mice were collected, and their brains were recovered in ice-cold Hanks' balanced salt solution (HBSS) supplemented with 0.7% glucose and 2.5 mM HEPES (pH 7.3). Brains were embedded in 4% low-melting-point agarose (Invitrogen) in HBSS, followed by preparation of 250-μm coronal slices of the forebrain by using a Leica VT1000S vibratome. Slices were collected on Nuclepore Track-Etched polyvinylpyrrolidone-free polycarbonate membranes (8.0-μm pores; Whatman Schleicher & Schuell) floating in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum, 1% glucose, and 1% penicillin-streptomycin. Slices were incubated at 37°C for 2 h prior to nucleic acid injection and electroporation. DNA plasmids (1 μg/μl) or small interfering RNA (siRNA) reagents (10 μM; Ambion) were mixed with Fast green dye and injected into the neocortical VZ. The ratio of effector nucleic acids (i.e., plasmids encoding IκBαM, IKKβ-DN, RelA, or FLAG-Hes6 or RelA or scrambled siRNA reagents) to GFP expression plasmid was kept at 4:1 to ensure that GFP-expressing cells also expressed the effector protein. This approach normally results in overlapping delivery of the two plasmids in greater than 90% of the electroporated cells (19). Electroporations were performed using a TSS20 Ovodyne electroporator (Intracel) as described previously (19), with the following settings: three pulses (25 V, 5-ms width), 500-ms interval between pulses, zero resistance, boost option selected. Electroporated slices on polycarbonate membranes were transferred to neurobasal medium supplemented with 1% B27, 1% glucose, 1% penicillin-streptomycin, and 1% glutamine. Electroporated slices were incubated at 37°C for either 48 h (loss-of-function experiments) or 96 h (RelA overexpression experiments), followed by fixation in 4% paraformaldehyde for 20 min, cryoprotection, and embedding in OCT compound (Tissue-Tek) for sectioning on a cryostat (16 μm). Double-labeling immunofluorescence analysis of the expression of GFP and markers of undifferentiated neural progenitors or postmitotic neurons was performed as described above for embryonic brain sections. The following primary antibodies were used: rabbit anti-GFP (1/500), rabbit anti-Pax6 (1/500), rabbit anti-activated caspase-3 (1/200; BD Pharmingen), mouse anti-Ki-67 (1/200; BD Pharmingen), mouse anti-intermediate filament protein nestin (1/10), mouse anti-βIII-tubulin (1/1,000), mouse anti-MAP2 (1/500; Sigma), mouse anti-NeuN (1/400; Millipore), mouse anti-glial fibrillary acidic protein (1/250; Sigma), and goat anti-Sox2 (1/500) antibodies. Two to four days after electroporation, sections were subjected to double-label immunofluorescence and quantitation of the percentage of GFP-positive cells that were also positive for the expression of Ki-67, nestin, MAP2, or NeuN. In each experiment, an average of 5 or 6 slices were electroporated and analyzed for expression of the different markers mentioned above.
Primary cortical neural progenitor cells.
Primary neural progenitor cells were established from dissociated dorsal telencephalic cortices obtained from CD1 mouse embryos collected between E12.5 and E13.5, as described previously (24, 27, 31). In transfection experiments, cells were transfected with a GFP expression plasmid (pEGFP; 200 ng/transfection) either alone or in combination with pCMV2-FLAG-Hes6 (500 ng/transfection), pCMV2-FLAG-Hes6 (500 ng/transfection) and pCMV-RelA (800 ng/transfection), or pCMV-RelA (800 ng/transfection). Three days after transfection, cells were fixed and subjected to double-label immunofluorescence and quantitation of the percentage of GFP-positive cells that were also positive for the expression of Ki-67, nestin, MAP2, or βIII-tubulin. At least 5 separate experiments were conducted in duplicate. In pharmacological inhibition studies, cortical progenitor cells were established from E13.5 CD1 embryos. Prior to treatment with the NF-κB inhibitor SN50, half of the culture medium was removed on day 1 in vitro, kept as conditioned medium, and replaced with fresh medium together with 2.5 μM SN50 peptide (Enzo Life Sciences), 2.5 μM mutated peptide (SN50M), or no peptide. Six hours later, the medium was replaced with a 1:1 mix of conditioned medium and fresh medium and cells were cultured for 18 h, followed by a second round of pharmacological treatment as described above. Cells were fixed on day 3 and subjected to immunocytochemistry for Ki-67, nestin, MAP2, or βIII-tubulin. For each condition and each antibody, 5 different fields were photographed using a 20× objective, with an average of about 300 cells per field. Cells were identified manually in Photoshop (Adobe Systems) and quantitated in ImageJ (National Institutes of Health). In transduction experiments, primary cortical progenitor cells were established from E13.5 NF-κBLacZ embryos. After 2 days in vitro, half of the culture medium was removed and replaced with fresh medium containing no adenovirus or adenovirus expressing either RelA and GFP (20), IKKβ-DN and GFP (21), or GFP alone (27). Cells were collected and subjected to determination of β-Gal activity as described previously (7, 24). RelA overexpression and IKKβ-DN expression were detected by Western blotting with rabbit anti-RelA antibody (1/5,000; Santa Cruz Biotechnology) or mouse anti-IKKβ antibody (1/1,000; Millipore). For expression studies, cortical progenitor cells obtained from E13.5 NF-κBLacZ litters were subjected to immunocytochemistry with anti-β-Gal antibody and either anti-intermediate filament protein nestin or anti-Hes6 (1/1,000; Abcam) antibody. In the latter case (Hes6), cultures were established from NF-κBLacZ embryos only, while in the former case (nestin), cultures were derived from mixed NF-κBLacZ and wild-type embryos.
Immunoprecipitation and Western blotting.
Dissected alar telencephalon from E14.5 CD1 mouse embryos was quickly rinsed in ice-cold HBSS and then incubated in 10 packed-tissue volumes of hypotonic buffer (20 mM Tris-HCl [pH 7.8], 10 mM KCl, 1.5 mM MgCl2) supplemented with 20 mM iodoacetamide, 10 μM MG132 (Boston Biochem), Complete protease inhibitor cocktail (Roche Applied Science), and 1 mM phenylmethylsulfonyl fluoride (PMSF) (32). Tissue was mechanically triturated and incubated for 15 min on ice, followed by the addition of 0.5 packed-tissue volume of hypertonic buffer (20 mM Tris-HCl [pH 7.8], 1 M KCl, 30 mM MgCl2, 20 mM iodoacetamide) supplemented with protease inhibitors. Tissue suspension was thoroughly mixed and centrifuged at 1,500 × g for 10 min. The resulting supernatant (postnuclear supernatant) and pellet (crude nuclear fraction) were collected. The crude nuclear pellet was rinsed and then resuspended in 2 packed-tissue volumes of lysis buffer (30 mM HEPES, pH 7.6, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 20 mM iodoacetamide) supplemented with protease inhibitors, followed by mechanical trituration and a brief sonication. The ensuing suspension was centrifuged at 14,000 × g for 15 min, and the supernatant was recovered (nuclear extract). This fraction was subjected to immunoprecipitation of endogenous RelA using rabbit anti-RelA or control rabbit antibodies using protein A/G-Plus agarose beads (Santa Cruz Biotechnology). Collected and rinsed immunoprecipitates were resuspended in 60 mM Tris-HCl (pH 6.8), 10% glycerol, and 2% SDS, followed by incubation at room temperature for 10 min and recovery of the eluted material by quick centrifugation. Eluates, together with 1/10 of input lysate, were incubated in the presence of 5% β-mercaptoethanol, followed by SDS-polyacrylamide gel electrophoresis on 12% gels, transfer to nitrocellulose, and Western blotting with rabbit anti-Hes6 (1/1,000; Novus Biologicals) or rabbit anti-RelA antibody. Human embryonic kidney 293 (HEK293) cells were transfected with plasmids encoding FLAG epitope-tagged Hes1 or Hes6 proteins (200 ng/transfection) using the SuperFect reagent (Qiagen). Lysates from transfected cells were subjected to immunoprecipitation with either anti-FLAG (Sigma) or anti-RelA antibody as described previously (23, 31). This step was followed by Western blotting of immunoprecipitates, together with 1/10 of each input lysate, with anti-FLAG (1/5,000) or anti-RelA antibody. Western blotting of p50 was performed using a mouse anti-p50 antibody (1/500; Santa Cruz Biotechnology).
Transcription assays.
To examine the effect of Hes6 on RelA-mediated transactivation, HEK293 cells were transfected with the pNF-κB-luciferase reporter plasmid (500 ng/transfection) in the absence or presence of pCMV-RelA (50 ng/transfection) and increasing amounts of pCMV2-FLAG-Hes6WT (100, 500, and 1,000 ng/transfection). Assays performed to determine the effect of RelA on the ability of Hes6 to suppress Hes1-mediated transcriptional repression were performed by transfecting HEK293 cells with the pFOX-neurogenin3 promoter-luciferase plasmid (7) in the absence or presence of pCMV2-FLAG-Hes1 (100 ng/transfection), pCMV2-FLAG-Hes6WT (750 ng/transfection), and increasing amounts of pCMV-RelA (500 and 1,000 ng/transfection). In each case, a pRSV-β-gal plasmid (250 ng/transfection) was used to normalize for transfection efficiency. Twenty-four hours after transfection, cell lysates were subjected to determination of luciferase and β-Gal activities as described previously (7, 23, 24).
RESULTS
Spatiotemporal activation of NF-κB signaling during embryonic brain development.
Little is known about the spatial and temporal pattern of NF-κB activation in the developing mammalian brain. To address this lack of information, we took advantage of previously characterized NF-κBLacZ reporter mice in which an NF-κB-responsive promoter drives transcription of a LacZ gene expressing a nuclear β-Gal isoform. These mice were shown to provide a reliable means of monitoring NF-κB activation in the nervous system (16, 20). The first detectable expression of β-Gal in the brain of NF-κBLacZ embryos was observed in the ventral and lateral telencephalon between E10.5 and E11.0, near the onset of telencephalic neurogenesis (Fig. 1A). NF-κB continued to be preferentially activated in the telencephalon at E13.5, a period of active telencephalic neurogenesis, with little or no expression detected in other brain regions (Fig. 1B to D). In the pallial (alar) region of the telencephalon, NF-κB activation was particularly robust in the VZ of the lateral pallium (neocortex), with no or undetectable activation in the VZ of the ventromedial sector. This situation occurred at various rostrocaudal pallial levels (Fig. 1C and D). Robust NF-κB activation was also observed in the lateral and medial ganglionic eminences in the subpallial (basal) region of the telencephalon of E13.5 NF-κBLacZ embryos, where β-Gal-expressing cells were present in both the VZ and the mantle zone (MZ; where postmitotic neurons are located) (Fig. 1B to D). NF-κB continued to be preferentially activated in the neocortical region of the pallium at E17.5, near the end of cortical neurogenesis, when activation was robust in the germinative zone (GZ), decreased in the intermediate zone (IZ; where migrating neurons are located), and again robust in the cortical plate (CP; where more developmentally mature neurons reside) (see Fig. S1A and B in the supplemental material). Activation of NF-κB in postmitotic cortical neurons has been described previously (20). Similar to earlier embryonic stages, little or no NF-κB activation was observed in the GZ of the ventromedial pallium, including the developing hippocampus (see Fig. S1A and B). NF-κB activation persisted in the subpallium at this stage and was also detected in a region likely corresponding to the preoptic area (see Fig. S1A). Western blotting of dissected telencephalon showed endogenous expression of the NF-κB subunits RelA and p50, suggesting that NF-κB complexes of these proteins are present in the developing neocortex (see Fig. S1C). These results provide evidence for a selective activation of NF-κB signaling in the dorsolateral pallium during cortical neurogenesis.
Fig 1.
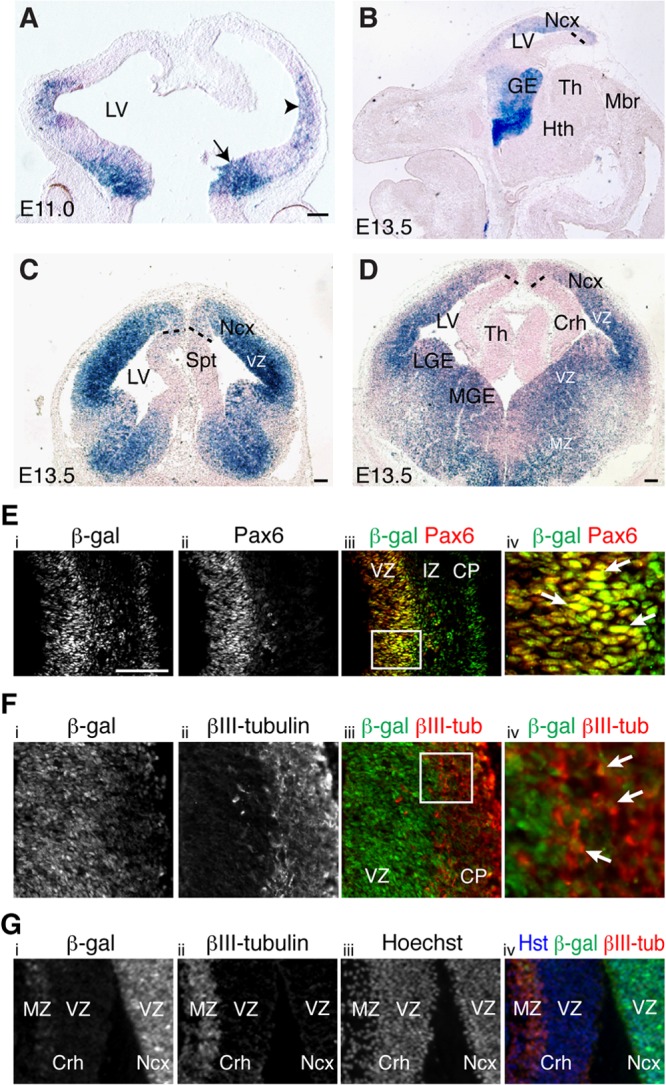
Activation of NF-κB signaling in neocortical neural progenitor cells during neurogenesis. (A) Coronal view of β-Gal activity (blue staining) in the telencephalon of E11.0 NF-κBLacZ embryos. The arrow and arrowhead point to the first detectable β-Gal activity in ventral and lateral telencephalon, respectively. Dorsal is at the top and ventral is at the bottom. (B) Sagittal view of β-Gal activity in the brain of E13.5 NF-κBLacZ embryos showing preferential activation in the telencephalon, with little or no expression in other brain regions. (C and D) Coronal views of β-Gal activity in the brain of E13.5 NF-κBLacZ embryos showing robust activation in the VZ of the lateral pallium (neocortex) but little or no activation in the ventromedial pallial VZ (cortical hem). Strong β-Gal expression is also observed in both lateral and medial ganglionic eminences in the subpallium. The section in panel C is rostral to the section in panel D. Dotted lines roughly demarcate the boundary of the β-Gal+ territory in the VZ of the medial pallium. Abbreviations: Crh, cortical hem; GE, ganglionic eminence; Hth, hypothalamus; LGE, lateral ganglionic eminence; LV, lateral ventricle; Mbr, midbrain; MGE, medial ganglionic eminence; MZ, mantle zone; Ncx, neocortex; Spt, septum; Th, thalamus; VZ, ventricular zone. Scale bars, 100 μm. (E to G) Double-label immunofluorescence analysis of β-Gal and Pax6 (E) or βIII-tubulin (βIII-tub) (F and G) expression in the pallium of E13.5 NF-κBLacZ embryos showing β-Gal expression in neural progenitor cells in the neocortical VZ (E) and in postmitotic neurons in the pallial MZ, both laterally (F) and medially (G). The iv panels represent high-magnification views of the boxed areas in the iii panels. Arrows point to examples of double-labeled cells. Dorsal is at the top and lateral is to the right in all panels. CP, cortical plate; IZ, intermediate zone. Scale bar, 100 μm.
Activation of NF-κB signaling in neocortical neural progenitor cells.
In agreement with the activation of NF-κB in the VZ, we observed that β-Gal was expressed in neocortical cells that also expressed the neural progenitor marker protein Pax6 in NF-κBLacZ embryos (Fig. 1E). Neocortical cells expressing β-Gal also expressed the neural progenitor markers FoxG1, Hes1, and nestin at this stage (see Fig. S1D to F in the supplemental material). β-Gal immunoreactivity also overlapped with nestin expression in primary cultures of cortical neural progenitor cells from NF-κBLacZ embryos (see Fig. S1G). In contrast, the majority of β-Gal+ cells in the lateral pallium of NF-κBLacZ embryos did not express the neuronal protein βIII-tubulin at E13.5; a partial overlap of β-Gal and βIII-tubulin was observed only in the MZ at this stage (Fig. 1F). A converse situation was observed in the ventromedial pallium, where the few detectable β-Gal+ cells were absent from the VZ and were instead located in the MZ, where they coexpressed βIII-tubulin (Fig. 1G). These findings provide evidence that NF-κB is activated in neocortical neural progenitor cells during telencephalic neurogenesis.
Premature differentiation of cortical neurons by inhibition of endogenous NF-κB signaling.
To study the involvement of NF-κB in the developing neocortex, we first examined the effect of inhibiting endogenous NF-κB signaling in neural progenitor cells. In one approach, we utilized a previously described mutated form of IκBα, IκBαM, to prevent IκBα-dependent NF-κB activation (33). In IκBα-dependent pathways, phosphorylation of IκBα by the beta subunit of the IκB kinase complex (IKKβ) (in canonical and some atypical NF-κB pathways), or independently of the IKK complex (in most atypical pathways), triggers the polyubiquitylation and degradation of IκB through the proteasome, allowing p50-RelA NF-κB complexes to translocate to the nucleus and regulate transcription. IκBαM, which contains S32A and S36A mutations that prevent its phosphorylation by IKKs, is not degraded in response to signaling pathways that target IκBα, thereby causing the retention of p50-RelA in the cytosol (33).
A bicistronic DNA plasmid driving the coexpression of IκBαM and GFP, or a control DNA expressing GFP alone, was injected into the lateral ventricles of E13.5 mouse embryonic brains followed by in utero electroporation. In this procedure, DNA is taken up by VZ progenitor cells, which subsequently differentiate into neurons that migrate toward the pial surface. Analysis of electroporated embryos collected at E15.5 revealed that expression of IκBαM caused a significant decrease in the number of electroporated cells expressing the neural progenitor marker proteins, nestin and Sox2 (Fig. 2A, B, and D). Concomitantly, expression of IκBαM caused a significant increase in the number of cells expressing the neuronal marker protein βIII-tubulin (Fig. 2C and D). Remarkably, embryos expressing exogenous IκBαM exhibited many ectopic βIII-tubulin+ cells in the neocortical VZ, where little or no expression of this neuronal protein is normally detected. The relative distributions of electroporated cells across the VZ, IZ, and CP were essentially equivalent in embryos expressing GFP alone or together with IκBαM, which is suggestive of no detectable cell displacement or perturbation of postmitotic neuronal migration (see Fig. S2 in the supplemental material). The same results were obtained in a complementary experimental approach in which endogenous NF-κB activity was inhibited using a previously described dominant negative form of IKKβ (IKKβ-DN; which is catalytically inactive) to inhibit canonical but not most forms of atypical or noncanonical NF-κB signaling (34, 35) (Fig. 2E).
Fig 2.

NF-κB signaling is important to prevent premature neuronal differentiation in the developing neocortex in vivo. (A to C) Double-label immunofluorescence analysis of GFP and either nestin (A), Sox2 (B), or βIII-tubulin (βIII-tub) (C) expression 48 h after in utero electroporation of E13.5 mouse embryos with either a bicistronic plasmid expressing both IκBαM and GFP or the empty vector expressing GFP alone. Bottom rows, high-magnification views of the areas indicated by rectangles in the right-hand panels. Where shown (when IκBαM was used), dotted lines roughly demarcate the GFP+ electroporated area in the VZ. Hst, Hoechst. Scale bar, 100 μm. (D) Quantification of the fraction of GFP+ cells coexpressing nestin, Sox2, or βIII-tubulin. Results are shown as the means ± standard errors of the means (SEM) (*, P < 0.05; **, P < 0.01; n = 5 electroporated embryos per condition; t test). (E) Quantification of the fraction of GFP+ cells coexpressing nestin, Sox2, or βIII-tubulin 48 h after in utero electroporation of E13.5 embryos with either a bicistronic plasmid expressing both IKKβ-DN and GFP or the empty vector expressing GFP alone. Results are shown as the means ± SEM (***, P < 0.001; n = 3 electroporated embryos per condition; t test).
Virtually identical results were observed when the expression of IκBαM or IKKβ-DN was forced in organotypic forebrain slices from E13.5 mouse embryos by focal DNA injection in the neocortical VZ, followed by ex vivo electroporation. As in in utero electroporation studies, expression of IκBαM caused a significant decrease in the number of cells expressing the cell proliferation marker Ki-67 (Fig. 3A and C) or nestin (see Fig. S3A in the supplemental material). This effect was paralleled by a remarkable increase in the number of cells expressing neuronal marker proteins like MAP2 (Fig. 3B and C), NeuN (see Fig. S3B), or βIII-tubulin (see Fig. S3C). In these experiments, a GFP-expressing plasmid was electroporated alone or together with an IκBαM-expressing vector, with the amount of the latter being 4 times the amount of the GFP vector, to ensure that most, if not all, GFP+ cells would coexpress IκBαM (19). This situation most likely explains why the ensuing phenotypes were observed over an area larger than the domain containing GFP+ cells, as a number of cells probably expressed only IκBαM and not GFP. We observed no detectable changes in the number of cells expressing the apoptotic marker, activated caspase-3, in the CP of control and IκBαM-expressing cells (GFP, 4.7% ± 2.8%; GFP and IκBαM, 9.6% ± 7.1%; n = 5 electroporated slices per condition from each of 5 separate electroporations). Moreover, no changes in the expression of glial fibrillary acidic protein were detected following expression of IκBαM, suggesting no alterations of astrocyte differentiation (see Fig. S3D). Exogenous expression of IKKβ-DN also promoted neuronal differentiation and resulted in reduced numbers of mitotic progenitor cells (Fig. 4A to C).
Fig 3.
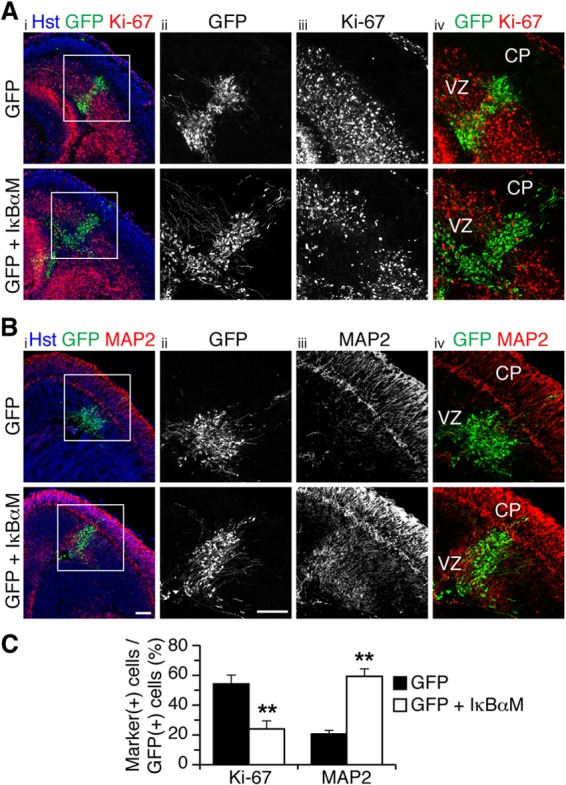
NF-κB signaling is important to prevent premature neuronal differentiation in the embryonic neocortex ex vivo. (A and B) Double-label immunofluorescence analysis of GFP and either Ki-67 (A) or MAP2 (B) expression in organotypic slice cultures from E13.5 mouse forebrain, 48 h after electroporation of plasmids encoding the indicated proteins. Columns ii to iv, high-magnifications from boxed areas in column i. Scale bars, 100 μm. (C) Quantification of the fraction of GFP+ cells coexpressing either Ki-67 or MAP2 (means ± SEM; **, P < 0.01; n = 16 electroporated slices per condition; t test).
Fig 4.

NF-κB signaling is important to prevent premature cortical neuronal differentiation ex vivo and in vitro. (A and B) Double-label immunofluorescence analysis of GFP and either Ki-67 (A) or MAP2 (B) expression in organotypic slice cultures from E13.5 mouse forebrain, 48 h after electroporation of plasmids encoding the indicated proteins. Scale bar, 100 μm. (C) Quantification of the fraction of GFP+ cells coexpressing Ki-67 or MAP2 (means ± SEM; ***, P < 0.001; n = 21 electroporated slices per condition; t test). (D and E) Double-label immunofluorescence analysis of GFP and either Ki-67 (D) or MAP2 (E) expression in organotypic slice cultures from E13.5 mouse forebrain, 48 h after electroporation of the indicated siRNA reagents together with GFP. Scr., control scrambled. Scale bar, 100 μm. (F) Quantification of the fraction of GFP+ cells coexpressing Ki-67 or MAP2 (means ± SEM; *, P < 0.05; n = 23 electroporated slices per condition; one-way analysis of variance [ANOVA]). (G) Double-label immunofluorescence analysis of cortical progenitor cells incubated in the absence (control) or presence of either SN50 or SN50M peptides, as indicated. Nestin and Ki-67 were used as progenitor cell markers, while βIII-tubulin (βIII-tub) and MAP2 were used as neuronal markers. Scale bar, 100 μm. (H) Quantification of the percentage of Hoechst-positive cells expressing either Ki-67, nestin, βIII-tubulin, or MAP2 under the three experimental conditions (means ± SEM; >500 cells were counted in each case; *, P < 0.05; **, P < 0.01; n = 4 separate experiments; one-way ANOVA).
To confirm these results, additional experiments were performed based on RNA interference and pharmacological NF-κB inhibition approaches. Electroporation of organotypic slices with RelA siRNA caused a remarkable decrease in the number of GFP+ cells expressing neural progenitor markers compared to control conditions, with a concomitant increase in the number of GFP+ cells expressing neuronal markers (Fig. 4D to F). Similarly, treatment of primary cultures of cortical neural progenitor cells with the SN50 peptide, a robust inhibitor of p50-RelA nuclear translocation (36), resulted in increased neuronal differentiation and reduced numbers of progenitor cells, whereas a mutated control peptide had no effect (Fig. 4G and H). Together, these results provide evidence that NF-κB signaling is required for maintenance of the undifferentiated neocortical neural progenitor state by inhibiting/delaying the neocortical progenitor-to-neuron transition. They suggest further that canonical NF-κB activation is involved in this function.
Inhibition of cortical neurogenesis by activation of NF-κB signaling.
We next examined whether NF-κB activation was sufficient to inhibit/delay neuronal differentiation in cortical progenitor cells. Forced overexpression of RelA caused increased β-Gal activity in primary cultures of cortical progenitor cells from NF-κBLacZ embryos (see Fig. S4C in the supplemental material), in agreement with previous evidence that forced RelA overexpression is sufficient to cause NF-κB activation (20, 37). Organotypic forebrain slices were subjected to focal electroporation of GFP alone or together with RelA. Four days later, the number of cells expressing GFP was quantitated in each of 5 radial sectors emanating from the GZ (sectors 1 and 2) toward the CP (sector 5) (Fig. 5A). Compared to the expression of GFP alone, exogenous RelA resulted in increased numbers of electroporated cells in sectors 1 and 2, where neural progenitor cells are mostly located, and a concomitant decrease in electroporated cells in sector 5, where migrated neurons are positioned (Fig. 5B). Expression of RelA caused neither a detectable alteration of the thickness of the neocortex (GFP, 484.1 ± 30.5 μm; GFP and RelA, 471.9 ± 10.2 μm; n = 20 slices from 4 separate electroporation experiments) nor a significant change in the number of GFP+ cells present 4 days after electroporation (GFP, 119.4 ± 27.2; GFP and RelA, 105.3 ± 9.5; n = 25 slices from 5 separate electroporation experiments), suggesting that the observed changes were not due to morphological alterations of the tissue or differential survival of the electroporated cells.
Fig 5.
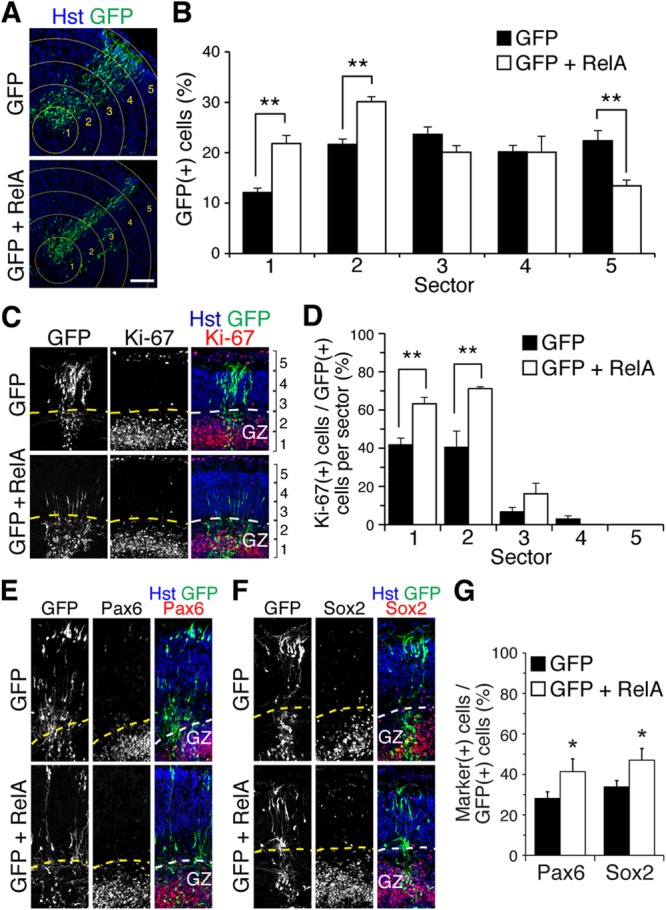
NF-κB is sufficient to inhibit/delay the progenitor-to-neuron transition during cortical neurogenesis ex vivo. (A) Depiction of GFP+ cell distribution in slice cultures from E13.5 mouse forebrains, 96 h after electroporation of plasmids encoding GFP or GFP-RelA. Neocortex was operationally divided into 5 sectors; sector 1 is in the GZ, sector 5 is close to the pial surface. Scale bar, 100 μm. (B) Quantification of the fraction of GFP+ cells (percentage of total) in each of the 5 different sectors (means ± SEM; **, P < 0.01; n = 25 electroporated slices per condition; two-way ANOVA). (C) Double-label immunofluorescence analysis of GFP and Ki-67 expression in organotypic slice cultures from E13.5 mouse forebrain, 96 h after electroporation of plasmids encoding GFP or GFP plus RelA. (D) Quantification of the fraction of GFP+ cells coexpressing Ki-67 in each of the 5 different sectors (means ± SEM; **, P < 0.01; n = 13 electroporated slices per condition; two-way ANOVA). (E and F) Double-label immunofluorescence analysis of GFP and either Pax6 (E) or Sox2 (F) expression in organotypic slice cultures from E13.5 mouse forebrain, 96 h after electroporation of plasmids encoding GFP or GFP plus RelA. (G) Quantification of the fraction of GFP+ cells coexpressing either Pax6 or Sox2 (means ± SEM; *, P < 0.05; n = 12 electroporated slices per condition; t test).
To determine whether the observed accumulation of exogenous RelA-expressing cells in inner sectors of the neocortex was the result of arrested/delayed neuronal differentiation resulting in increased numbers of progenitor cells, we first compared the numbers of GFP+ cells expressing the mitotic marker Ki-67 in each of the five radial sectors. This analysis revealed a significant increase in GFP+/Ki-67+ cells in sectors 1 and 2 when RelA was expressed compared to the number of cells with GFP alone (Fig. 5C and D). Consistent with this phenotype, we also observed that exogenous RelA expression led to increased numbers of electroporated cells expressing the progenitor markers Pax6 and Sox2 in the same sectors (Fig. 5E to G). Analysis of electroporated cells expressing the neuronal marker protein MAP2 revealed no evidence of increased numbers of neuronal cells at, or near, the GZ following exogenous RelA expression; instead, these numbers were decreased (GFP, 16.4% ± 1.7%; GFP and RelA, 9.1% ± 0.3%; P = 0.014; n = 15 electroporated slices per condition; t test) (see Fig. S5 in the supplemental material). This observation argues against a block/delay of neuronal migration, a phenotype that could be predicted to result in an accumulation of postmitotic neurons outside the GZ. Taken together, these results show that the NF-κB activity that is present within cortical progenitors is necessary and sufficient to inhibit neuronal differentiation and promote maintenance of the undifferentiated progenitor state.
Suppression of NF-κB-mediated inhibition of cortical neuronal differentiation by Hes6.
If cortical progenitors are to differentiate, the NF-κB signaling that is present during phases of active neurogenesis must be regulated in some manner. We observed that most β-Gal+ neocortical progenitor cells in E13.5 NF-κBLacZ embryos coexpressed the proneuronal basic helix-loop-helix (bHLH) protein Hes6 in vivo (Fig. 6A). The same situation was observed in primary cultures of cortical neural progenitors established from NF-κBLacZ embryos (see Fig. S6A in the supplemental material). More importantly, immunoprecipitation studies using dissected cortices from E13.5 mouse embryos showed that precipitation of endogenous RelA resulted in the coprecipitation of an endogenous protein of ∼28 kDa that was recognized by anti-Hes6 antibodies (Fig. 6B; see also Fig. S6B in the supplemental material). The 28-kDa band recognized by these antibodies was abolished after transduction of primary cortical progenitor cells with a lentivirus expressing an Hes6 short hairpin RNA (shRNA) reagent, indicating that it corresponded to endogenous Hes6 (see Fig. S6C and D). Additional coimmunoprecipitation studies in HEK293 cells expressing FLAG epitope-tagged Hes6 also demonstrated that Hes6 could be coprecipitated with endogenous RelA (Fig. 6C and D). This association was specific, because RelA did not coprecipitate with the related Hes family member Hes1, which performs an antineurogenic function that is antagonistic to Hes6 activity. Because both Hes6 and RelA can interact with the transcriptional corepressor Groucho/TLE (7, 25), we tested whether Groucho/TLE might act as a bridge between RelA and Hes6. A mutated form of Hes6 lacking the C-terminal WRPW tetrapeptide required for Groucho/TLE binding was competent to interact with RelA, indicating that the ability of Hes6 to form a complex with the latter did not require the Groucho/TLE association (Fig. 6D). In contrast, deletion of helix 2 of the Hes6 bHLH domain completely abolished the coimmunoprecipitation of Hes6 with RelA, suggesting that this interaction requires an intact Hes6 bHLH domain (Fig. 6D).
Fig 6.
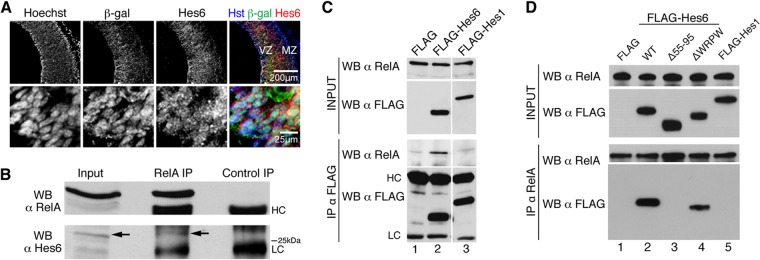
Hes6 is expressed in neocortical progenitor cells in which NF-κB is activated and forms complexes with NF-κB subunit RelA. (A) Double-label immunofluorescence analysis of β-Gal and Hes6 expression in the neocortex of E13.5 NF-κBLacZ embryos. The bottom row depicts high-magnification views of β-Gal and Hes6 coexpression in VZ cells. MZ, mantle zone. Scale bars, 200 μm or 25 μm, as indicated. (B) Nuclear extracts from dissected cortices from E14.5 CD1 mouse embryos were subjected to immunoprecipitation (IP) with anti-RelA or control antibody. Immunoprecipitates were analyzed with each input lysate by Western blotting (WB) with anti-RelA (top) or anti-Hes6 (bottom) antibody. The arrow points to the position of the Hes6 immunoreactive band. HC, immunoglobulin heavy chain; LC, immunoglobulin light chain. (C) Protein extracts from HEK293 cells transfected with empty FLAG vector (FLAG), FLAG-tagged Hes6 (FLAG-Hes6), or FLAG-tagged Hes1 (FLAG-Hes1) were subjected to immunoprecipitation with anti-FLAG antibody. Immunoprecipitates (bottom two panels) were analyzed together with each input lysate (top two panels) by Western blotting with anti-RelA or anti-FLAG antibodies. (D) Protein extracts from HEK293 cells transfected with empty FLAG vector (FLAG), FLAG-tagged wild-type Hes6 (WT), FLAG-Hes6 lacking most of helix 2 (i.e., amino acids 55 to 95) (Δ55-95), FLAG-Hes6 lacking the WRPW motif required for Gro/TLE binding (ΔWRPW), or FLAG-Hes1 were subjected to immunoprecipitation with anti-RelA antibody. Immunoprecipitates (bottom two panels) were analyzed together with each input lysate (top two panels) by Western blotting with anti-RelA or anti-FLAG antibody.
To determine the functional significance of the Hes6-RelA interaction, we examined whether Hes6 could modulate the ability of RelA to activate transcription. In transient-transfection/transcription assays, the expression of RelA alone led to a significant activation of a luciferase reporter gene under the control of tandem NF-κB binding sites (Fig. 7A). Hes6 inhibited RelA-mediated transactivation in a dose-dependent manner (Fig. 7A, second to fifth bars) but had no significant effect on reporter gene expression in the absence of RelA (Fig. 7A, sixth to eighth bars). This result suggests that Hes6 can negatively regulate the transactivating ability of RelA-containing NF-κB complexes. Based on this finding, we next tested if Hes6 could antagonize the inhibition of neuronal differentiation and expansion of the progenitor pool caused by forced RelA overexpression (Fig. 7B and C). Compared to expression of GFP alone, expression of Hes6 in organotypic slice cultures caused a detectable decrease in the number of electroporated cells located in the inner sectors (Fig. 7C, sector 2, first and fourth bars), with a concomitant increase in the number of cells in the outer region (Fig. 7C, sector 5, first and fourth bars). More importantly, the coexpression of Hes6 with RelA abolished both the RelA-mediated accumulation of electroporated cells in sectors 1 and 2 and the concomitant decrease of electroporated cells in sector 5 (Fig. 7C, sectors 1 and 5, second and third bars). Furthermore, exogenous Hes6 antagonized both the increase in Ki-67+ progenitor cells and the decrease in MAP2+ neuronal cells caused by RelA (Fig. 7D and E). Taken together, these results identify Hes6 as a previously uncharacterized modulator of NF-κB biochemical activity and suggest that Hes6 can antagonize the antineurogenic effect of RelA-containing NF-κB complexes during cortical neurogenesis.
Fig 7.
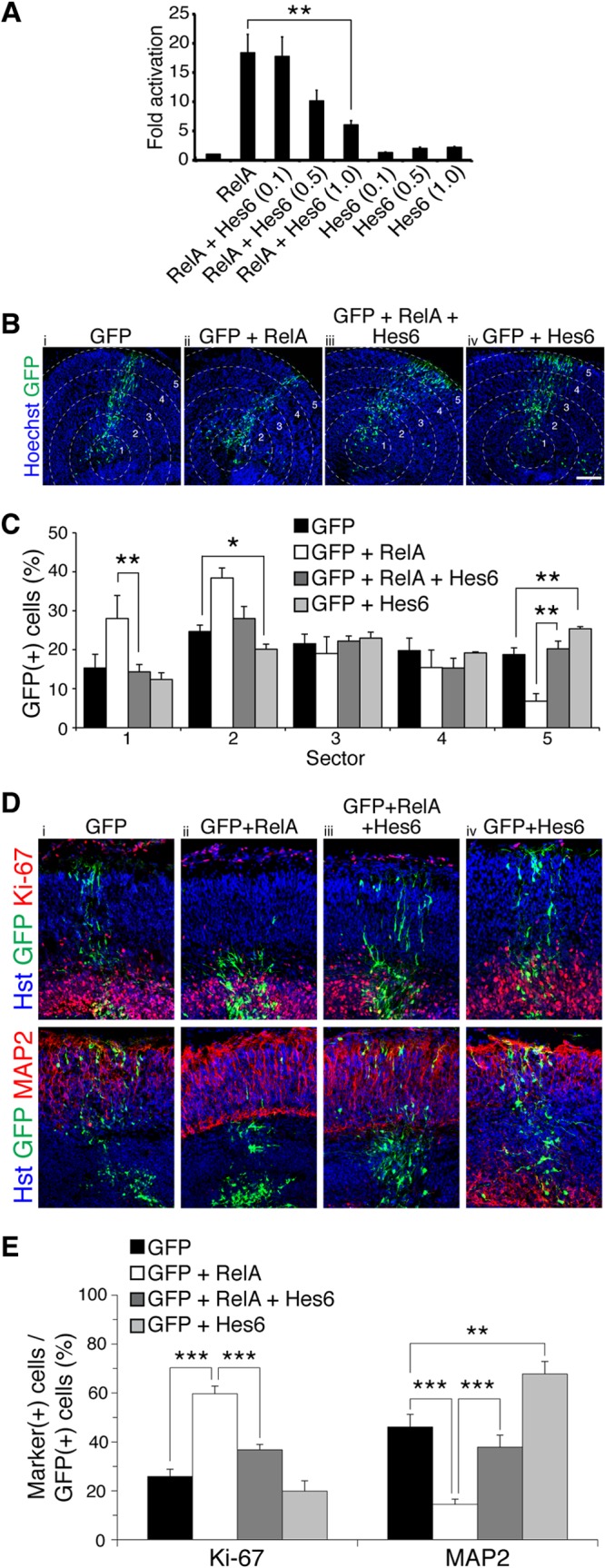
Hes6 antagonizes RelA-mediated transactivation and inhibition of cortical neuronal differentiation. (A) Quantification of luciferase activity in HEK293 cells transfected with a reporter plasmid in which luciferase expression is under the control of NF-κB binding sites. Reporter was transfected alone or together with plasmids encoding RelA (0.05 μg/transfection) or Hes6 (0.1, 0.5, or 1.0 μg/transfection). Results are shown as the means ± standard deviations (SD) (**, P < 0.01; n = 4 separate experiments performed in duplicate; t test). (B) Distribution of GFP+ cells in slice cultures from E13.5 mouse forebrain, 96 h after electroporation of plasmids encoding the indicated combinations of proteins. Neocortex was operationally divided into 5 sectors. Scale bar, 100 μm. (C) Quantification of the fraction of GFP+ cells present in each of the 5 different sectors (means ± SEM; *, P < 0.05; **, P < 0.01; n = 19 electroporated slices per condition; t test). (D) Double-label immunofluorescence analysis of GFP and either Ki-67 or MAP2 expression in organotypic slice cultures from E13.5 mouse forebrain, 96 h after electroporation of plasmids encoding the indicated combinations of proteins. (E) Quantification of the fraction of GFP+ cells coexpressing either Ki-67 or MAP2 (means ± SEM; **, P < 0.01; ***, P < 0.001; n = 14 electroporated slices per condition; t test).
Suppression of Hes6-mediated promotion of cortical neuronal differentiation by NF-κB.
We next examined whether NF-κB activation could inhibit Hes6 biochemical and biological functions. Hes6 suppresses Hes1-mediated transcriptional repression, an effect thought to underlie at least some of the functions of Hes6 during cortical development (6, 7, 24). Hes6 suppressed Hes1-mediated transcriptional repression from the promoter of the neurogenin3 gene (Fig. 8A, first to third bars), as previously shown (7). The exogenous expression of Hes6 increased promoter activity above starting levels, due to promoter derepression through inhibition of endogenous Hes1 (7, 24). Coexpression of RelA antagonized the inhibitory effect of Hes6 on Hes1 transcription repression activity in a dose-dependent manner, causing decreased promoter activation; in contrast, RelA caused a modest increase in promoter activity in the absence of Hes6 (Fig. 8A, fourth to seventh bars). To extend these observations, primary cortical progenitor cell cultures were transfected with GFP in the absence or presence of Hes6 alone or Hes6 and RelA together, followed by quantification of the numbers of transfected cells displaying hallmarks of undifferentiated progenitors or postmitotic neurons. As shown (24), exogenous Hes6 expression led to decreased numbers of progenitor cells (expressing Ki-67 or nestin) and promoted an increase in the number of βIII-tubulin+ (or MAP2+) neurons (Fig. 8B, first and second bars). Both of these effects were blocked by the coexpression of RelA (Fig. 8B, second and third bars). Expression of RelA alone caused a modest but significant increase in the number of progenitor cells (Fig. 8B, fourth bar), with no detectable effect on neuronal cell number under these conditions. The latter observation may be due to the fact that any decrease in neurons caused by RelA-mediated inhibition/delay of differentiation might have been partly compensated by the ability of NF-κB to promote cortical neuronal survival in culture (20). Taken together, these results provide evidence that NF-κB and Hes6 are coexpressed in neocortical neural progenitor cells, physically and functionally interact, and exert cross-inhibitory actions on each other. These observations suggest that NF-κB and Hes6 are part of antagonistic developmental programs during cortical neurogenesis.
Fig 8.
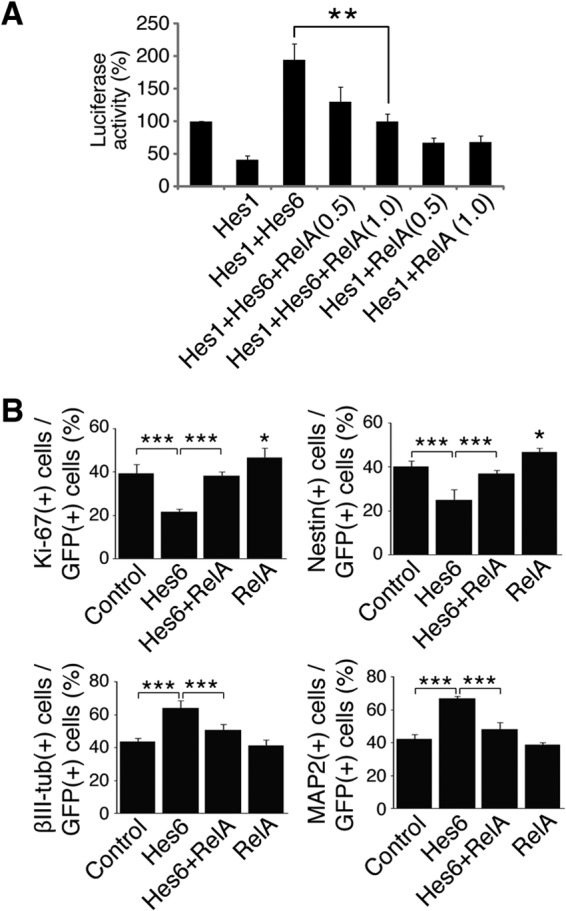
RelA antagonizes Hes6-mediated inhibition of Hes1 and promotion of cortical neuronal differentiation. (A) Quantification of luciferase activity in HEK293 cells transfected with a reporter plasmid in which luciferase expression is under the control of an ∼3.0-kb fragment of the neurogenin3 promoter, which contains multiple Hes1 binding sites. Reporter plasmid was transfected alone (luciferase activity is considered 100%) or in combination with plasmids encoding Hes1 in the absence or presence of Hes6 and increasing amounts of RelA (0.5 or 1.0 μg/transfection). Results are shown as the means ± SD (**, P < 0.01; n = 4 separate experiments performed in duplicate; t test). (B) Quantification of the percentage of GFP+ cells coexpressing Ki-67, nestin, βIII-tubulin (βIII-tub), or MAP2 72 h after transfection of E13.5 telencephalon-derived cortical progenitor cells using plasmids encoding GFP alone (control) or together with the indicated proteins. Results are shown as the means ± SD (>500 cells were counted in each case; *, P < 0.05; ***, P < 0.001; n = 5 separate experiments performed in duplicate; one-way ANOVA followed by Tukey's post hoc test).
DISCUSSION
Roles for NF-κB in the regulation of neuronal survival, synaptogenesis, learning, and memory have been demonstrated. In contrast, little is known about the involvement of NF-κB in the control of neuronal differentiation in the developing brain. We have demonstrated that NF-κB signaling becomes activated in neural progenitor cells in the dorsolateral pallium (precursor of the cerebral cortex) at developmental stages coinciding with the onset of neurogenesis in this region of the brain (2, 38). NF-κB activation then persists in neocortical progenitor cells throughout the period of cortical neurogenesis. More importantly, experimental strategies that either inhibited or activated NF-κB signaling in neocortical progenitor cells have shown that NF-κB signaling is necessary and sufficient to maintain the neural progenitor state and inhibit/delay neuronal differentiation during cortical development. Specifically, in vivo inhibition of NF-κB activity caused the differentiation of a significant number of supernumerary neurons, with a concomitant decrease in cells expressing markers typical of undifferentiated neural progenitors. A converse situation was observed after NF-κB activation through RelA overexpression. Our studies did not suggest a perturbation of postmitotic neuronal migration as a result of NF-κB inhibition, because the relative distribution of cells with impaired NF-κB activity was essentially equivalent to control cells across the VZ, IZ, and CP. Moreover, RelA overexpression studies provided no evidence for an accumulation of postmitotic neurons near the proliferative zone, as one would expect if neuronal migration were inhibited. Instead, these experiments showed a significant increase in the number of undifferentiated progenitors in response to NF-κB activation, consistent with a delayed/blocked progenitor-to-neuron transition. Based on these observations, we propose that NF-κB promotes maintenance of the undifferentiated cell compartment at the expense of neuronal differentiation, ensuring that the pool of undifferentiated cortical progenitors not be prematurely depleted by differentiative cell divisions (Fig. 9).
Fig 9.
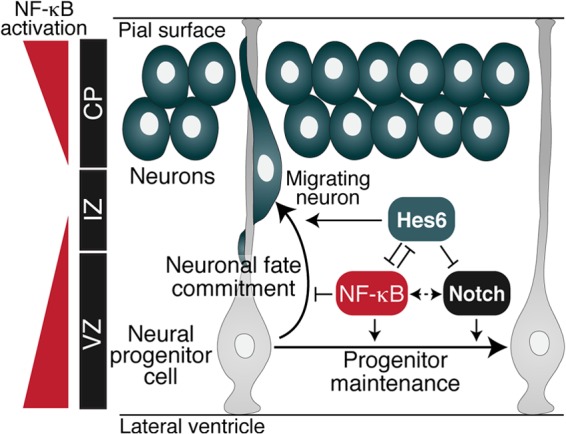
Proposed role for NF-κB during cortical neurogenesis. NF-κB signaling is robustly activated in neocortical VZ progenitors, transiently downregulated during early stages of neuronal differentiation, and then strongly activated in more developmentally mature neurons in the CP. NF-κB acts in undifferentiated progenitors to prevent/delay neuronal differentiation, thereby supporting progenitor cell maintenance. This role is similar to the function of the Notch signaling pathway (dotted double-pointed arrow). Hes6 antagonizes NF-κB in progenitor cells that are committed to undergo neuronal cell lineage progression. Conversely, NF-κB antagonizes the proneuronal activity of Hes6 to promote preservation of the neural progenitor pool. Hes6 also opposes Notch signaling-mediated inhibition of neuronal differentiation.
We have shown further that little or no NF-κB activation can be detected in the VZ of the ventromedial pallium (precursor of the hippocampus), in contrast to the robust activation observed in the dorsolateral pallium. Although it is conceivable that NF-κB signaling could be activated in the ventromedial pallium at below-detection levels, the remarkable difference in NF-κB activation between lateral and medial pallial progenitors suggests that NF-κB is important for the regulation of cortical but not hippocampal neurogenesis during embryonic development. In this regard, recent studies have shown that stress-induced activation of NF-κB signaling in the adult hippocampus causes impaired adult hippocampal neurogenesis (39), suggesting that neuronal differentiation in the ventromedial pallium might be incompatible with activation of NF-κB signaling. It should be noted, however, that both the present and previous (20) studies have shown that NF-κB is activated in postmitotic neurons in the developing cortical hem and adult hippocampus. Thus, it is reasonable to suggest that NF-κB has roles in both cortical and hippocampal postmitotic neurons, in contrast to its specific involvement in cortical but not hippocampal progenitors.
The role of NF-κB in cortical progenitor cell maintenance is analogous to the function of the Notch-Hes1/5 pathway, which is essential for maintenance of the undifferentiated state in neural stem/progenitor cells (28, 40). Because NF-κB and Notch-Hes1/5 pathways cross talk to enhance each other's functions in a variety of cellular contexts (9), it is reasonable that they might also cooperate in cortical progenitors during embryonic neurogenesis. This possibility is consistent with the demonstration that Notch signaling promotes adult neural stem cell self-renewal (41, 42) and that this function is enhanced by NF-κB, at least in part, through RelA-mediated potentiation of Notch-mediated Hes1 gene transcription (43). A positive effect of NF-κB on Notch-induced Hes1 expression was also observed in other cellular contexts (44).
Our present results suggest a previously uncharacterized mechanism through which NF-κB signaling could cooperate with the Notch-Hes1/5 pathway to promote maintenance of the undifferentiated cortical progenitor state. Earlier studies showed that the neurogenic Hes family member Hes6 acts antagonistically to the Notch pathway to promote cortical neuronal differentiation (6, 7, 24). This function could be mediated, at least in part, by the ability of Hes6 to suppress Hes1-mediated transcriptional repression (6, 7). Our results demonstrate that the NF-κB subunit, RelA, interacts physically and functionally with Hes6, which is expressed in cortical neural progenitor cells in which NF-κB signaling is activated. Activation of NF-κB through RelA overexpression suppresses the ability of Hes6 to both inhibit Hes1-mediated transcriptional repression and promote neuronal differentiation. These observations suggest that NF-κB might cooperate with Notch signaling in cortical neural progenitor cells by antagonizing Hes6 and therefore enhancing Hes1 protein activity. The latter effect would be particularly important in cortical progenitor cells in which the Hes1 expression level is low during its cyclical oscillation (28). The ensuing augmentation of Hes1 activity would result in an increased ability to repress neuronal differentiation programs, thereby contributing to maintenance of the undifferentiated state.
The functional interaction between NF-κB and Hes6 appears to be mutually inhibitory, because the latter can suppress both the transcriptional activity and the antineurogenic effect of RelA in cortical progenitor cells. It is therefore possible that the cortical progenitor-to-neuron transition is regulated in part by the balance of NF-κB and Hes6 activity (Fig. 9). NF-κB might enhance Notch-mediated promotion of the undifferentiated state, at least in part, by antagonizing Hes6 during early phases of cortical development, when mostly maintenance divisions are occurring. In contrast, Hes6 might inhibit NF-κB activity during cortical neuronal fate commitment and progression, concomitant with the persistent downregulation of Hes1 (and/or other antineurogenic genes) and the consolidation of neuronal differentiation programs. These scenarios are hypothesized to be specific to the developing neocortex and not the ventromedial pallium, where NF-κB is not activated and Hes6 functions might be modulated by alternative mechanisms. The cross talk of Notch and NF-κB pathways through Hes6 might therefore represent a specific regulatory process during cortical neurogenesis.
Recent studies have identified mutations associated with intellectual disability within two genes involved in NF-κB signaling. Specifically, mutations in the TRAPPC9 gene, encoding NIK and IKKβ binding protein (NIBP), were detected in four separate families with individuals affected by autosomal recessive nonsyndromic mental retardation (45–47). Cultured skin fibroblasts from individuals with homozygous mutations in TRAPPC9 show impaired NF-κB signaling (47). Importantly, NIBP is expressed throughout the embryonic cerebral cortex during neurogenesis and enhances canonical NF-κB signaling in neuronal cells (48). Mutations in the CC2D1A gene have also been linked to autosomal recessive nonsyndromic mental retardation (49, 50). CC2D1A is expressed in the VZ of the developing brain during neurogenesis (49), and the CC2D1A protein promotes activation of canonical NF-κB signaling (51). Taken together with the present findings, these results suggest that NF-κB signaling might be defective in these patients during brain development, resulting in an impaired generation and/or maturation of cortical neurons. This possibility is consistent with the microcephaly observed in some affected individuals carrying TRAPPC9 mutations. In the future, it will be important to determine whether disruption of the functions of the mouse orthologs of NIBP or CC2D1A might cause perturbations of cortical neurogenesis.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the Canadian Institutes of Health Research to P.A.B. and S.S. (MOP-13957 and MOP-42479). P.A.B. and S.S. held Chercheur National Awards from the Fonds de la Recherche en Santé du Québec.
We thank T. Tetsuka for plasmids, R. Kageyama for anti-Hes1 antibody, M. Maira for assistance during electroporation studies, N. Unsain for assistance with NF-κBLacZ mice, and M. Bouchard-Levasseur, S. Perrino, and I. Murillo for excellent animal care.
Footnotes
Published ahead of print 20 May 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01610-12.
REFERENCES
- 1.Farkas LM, Huttner WB. 2008. The cell biology of neural stem and progenitor cells and its significance for their proliferation versus differentiation during mammalian brain development. Curr. Opin. Cell Biol. 20:707–715 [DOI] [PubMed] [Google Scholar]
- 2.Kriegstein A, Alvarez-Buylla A. 2009. The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 32:149–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guillemot F. 2007. Cell fate specification in the mammalian telencephalon. Prog. Neurobiol. 83:37–52 [DOI] [PubMed] [Google Scholar]
- 4.Kageyama R, Ohtsuka T, Shimojo H, Imayoshi I. 2008. Dynamic Notch signaling in neural progenitor cells and a revised view of lateral inhibition. Nat. Neurosci. 11:1247–1251 [DOI] [PubMed] [Google Scholar]
- 5.Louvi A, Artavanis-Tsakonas S. 2006. Notch signaling in vertebrate neural development. Nat. Rev. Neurosci. 7:93–102 [DOI] [PubMed] [Google Scholar]
- 6.Bae SK, Bessho Y, Hojo M, Kageyama R. 2000. The bHLH gene Hes6, an inhibitor of Hes1, promotes neuronal differentiation. Development 127:2933–2943 [DOI] [PubMed] [Google Scholar]
- 7.Gratton MO, Torban E, Jasmin SB, Theriault FM, German MS, Stifani S. 2003. Hes6 promotes cortical neurogenesis and inhibits Hes1 transcription repression activity by multiple mechanisms. Mol. Cell. Biol. 23:6922–6935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koyano-Nakagawa N, Kim J, Anderson D, Kintner C. 2000. Hes6 acts in a positive feedback loop with the neurogenins to promote neuronal differentiation. Development 127:4203–4216 [DOI] [PubMed] [Google Scholar]
- 9.Ang HL, Tergaonkar V. 2007. Notch and NF-κB signaling pathways: do they collaborate in normal vertebrate brain development and function? Bioessays 29:1039–1047 [DOI] [PubMed] [Google Scholar]
- 10.Bonini SA, Ferrari-Toninelli G, Uberti D, Montinaro M, Buizza L, Lanni C, Grilli M, Memo M. 2011. Nuclear factor κB-dependent neurite remodeling is mediated by Notch pathway. J. Neurosci. 31:11697–11705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hayden MS, Ghosh S. 2008. Shared principles in NF-κB signaling. Cell 132:344–362 [DOI] [PubMed] [Google Scholar]
- 12.Perkins ND. 2007. Integrating cell-signaling pathways with NF-κB and IKK function. Nat. Rev. Mol. Cell Biol. 8:49–62 [DOI] [PubMed] [Google Scholar]
- 13.Baker RG, Hayden MS, Ghosh S. 2011. NF-κB, inflammation, and metabolic disease. Cell Metab. 13:11–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chariot A. 2009. The NF-κB-independent functions of IKK subunits in immunity and cancer. Trends Cell Biol. 19:404–413 [DOI] [PubMed] [Google Scholar]
- 15.Boersma MC, Dresselhaus EC, De Biase LM, Mihalas AB, Bergles DE, Meffert MK. 2011. A requirement for nuclear factor-kappaB in developmental and plasticity-associated synaptogenesis. J. Neurosci. 31:5414–5425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lebrun-Julien F, Duplan L, Pernet V, Osswald I, Sapieha P, Bourgeois P, Dickson K, Bowie D, Barker PA, Di Polo A. 2009. Excitotoxic death of retinal neurons in vivo occurs via a noncell-autonomous mechanism. J. Neurosci. 29:5536–5545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li J, Gu X, Ma Y, Calicchio ML, Kong D, Teng YD, Yu L, Crain AM, Vartanian TK, Pasqualini R, Arap W, Libermann TA, Snyder EY, Sidman RL. 2010. Nna1 mediates Purkinje cell dendritic development via lysyl oxidase propeptide and NF-κB signaling. Neuron 68:45–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meffert MK, Chang JM, Wiltgen BJ, Fanselow MS, Baltimore D. 2003. NF-kappa B functions in synaptic signaling and behavior. Nat. Neurosci. 6:1072–1078 [DOI] [PubMed] [Google Scholar]
- 19.Maira M, Long JE, Lee AY, Rubenstein JL, Stifani S. 2010. Role for TGF-beta superfamily signaling in telencephalic GABAergic neuron development. J. Neurodev. Disord. 2:48–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhakar AL, Tannis LL, Zeindler C, Russo MP, Jobin C, Park DS, MacPherson S, Barker PA. 2002. Constitutive nuclear factor-κB activity is required for central neuron survival. J. Neurosci. 22:8466–8475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ho WC, Dickson KM, Barker PA. 2005. Nuclear factor-kappaB induced by doxorubicin is deficient in phosphorylation and acetylation and represses nuclear factor-kappaB-dependent transcription in cancer cells. Cancer Res. 65:4273–4281 [DOI] [PubMed] [Google Scholar]
- 22.Russo SJ, Wilkinson MB, Mazei-Robison MS, Dietz DM, Maze I, Krishnan V, Renthal W, Graham A, Birnbaum SG, Green TA, Robison B, Lesselyong A, Perrotti LI, Bolaños CA, Kumar A, Clark MS, Neumaier JF, Neve RL, Bhakar AL, Barker PA, Nestler EJ. 2009. Nuclear factor kappa B signaling regulates neuronal morphology and cocaine reward. J. Neurosci. 29:3529–3537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Belanger-Jasmin S, Llamosas E, Tang Y, Joachim K, Osiceanu AM, Jhas S, Stifani S. 2007. Inhibition of cortical astrocyte differentiation by Hes6 requires amino- and carboxy-terminal motifs important for dimerization and phosphorylation. J. Neurochem. 103:2022–2034 [DOI] [PubMed] [Google Scholar]
- 24.Jhas S, Ciura S, Belanger-Jasmin S, Dong Z, Llamosas E, Theriault FM, Joachim K, Tang Y, Liu L, Liu J, Stifani S. 2006. Hes6 inhibits astrocyte differentiation and promotes neurogenesis through different mechanisms. J. Neurosci. 26:11061–11071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tetsuka T, Uranishi H, Imai H, Ono T, Sonta S, Takahashi N, Asamitsu K, Okamoto T. 2000. Inhibition of nuclear factor-κB-mediated transcription by association with the amino-terminal enhancer of split, a Groucho-related protein lacking WD40 repeats. J. Biol. Chem. 275:4383–4390 [DOI] [PubMed] [Google Scholar]
- 26.Hand R, Bortone D, Mattar P, Nguyen L, Heng JI, Guerrier S, Boutt E, Peters E, Barnes AP, Parras C, Schuurmans C, Guillemot F, Polleux F. 2005. Phosphorylation of neurogenin2 specifies the migration properties and the dendritic morphology of pyramidal neurons in the neocortex. Neuron 48:45–62 [DOI] [PubMed] [Google Scholar]
- 27.Theriault FM, Nuthall HN, Dong Z, Lo R, Barnabe-Heider F, Miller FD, Stifani S. 2005. Role for Runx1 in the proliferation and neuronal differentiation of selected progenitor cells in the mammalian nervous system. J. Neurosci. 25:2050–2061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shimojo H, Ohtsuka T, Kageyama R. 2008. Oscillations in Notch signaling regulate maintenance of neural progenitors. Neuron 58:52–64 [DOI] [PubMed] [Google Scholar]
- 29.Walantus W, Castaneda D, Elias L, Kriegstein A. 2007. In utero intraventricular injection and electroporation of E15 mouse embryos. J. Vis. Exp. 6:239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghanem N, Andrusiak MG, Svoboda D, Al Lafi SM, Julian LM, McClellan KA, De Repentigny Y, Kothary R, Ekker M, Blais A, Park DS, Slack RS. 2012. The Rb/E2F pathway modulates neurogenesis through direct regulation of the Dlx1/Dlx2 bigene cluster. J. Neurosci. 32:8219–8230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nuthall HN, Joachim K, Stifani S. 2004. Phosphorylation of serine 239 of Groucho/TLE1 by protein kinase CK2 is important for inhibition of neuronal differentiation. Mol. Cell. Biol. 24:8395–8407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Husain J, Lo R, Grbavec D, Stifani S. 1996. Affinity for the nuclear compartment and expression during cell differentiation implicate phosphorylated Groucho/TLE1 forms of higher molecular mass in nuclear functions. Biochem. J. 317:523–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. 1996. Suppression of TNFα-induced apoptosis by NF-κB. Science 274:787–789 [DOI] [PubMed] [Google Scholar]
- 34.Li Q, Van Antwerp D, Mercurio F, Lee KF, Verma IM. 1999. Severe liver degeneration in mice lacking the IkappaB kinase 2 gene. Science 284:321–325 [DOI] [PubMed] [Google Scholar]
- 35.Manning A, Rao A. 1997. IKK-1 and IKK-2: cytokine-activated IκB kinases essential for NF-κB activation. Science 278:860–866 [DOI] [PubMed] [Google Scholar]
- 36.Lin YZ, Yao SY, Veach RA, Torgerson TR, Hawiger J. 1995. Inhibition of nuclear translocation of transcription factor NF-kappa B by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. J. Biol. Chem. 16:14255–14258 [DOI] [PubMed] [Google Scholar]
- 37.Schmitz ML, Baeuerle PA. 1991. The p65 subunit is responsible for the strong transcription activating potential of NF-kappa B. EMBO J. 10:3805–3817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hébert JM, Fishell G. 2008. The genetics of early telencephalon patterning: some assembly required. Nat. Rev. Neurosci. 9:678–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koo JW, Russo SJ, Ferguson D, Nestler EJ, Duman RS. 2010. Nuclear factor-κB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc. Natl. Acad. Sci. U. S. A. 107:2669–2674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoon K, Gaiano N. 2005. Notch signaling in the mammalian central nervous system: insights from mouse mutants. Nat. Neurosci. 8:709–715 [DOI] [PubMed] [Google Scholar]
- 41.Androutsellis-Theotokis A, Leker RR, Soldner F, Hoeppner DJ, Ravin R, Poser SW, Rueger MA, Bae SK, Kittappa R, McKay RD. 2006. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature 442:823–826 [DOI] [PubMed] [Google Scholar]
- 42.Hitoshi S, Alexson T, Tropepe V, Donoviel D, Elia AJ, Nye JS, Conlon RA, Mak TW, Bernstein A, van der Kooy D. 2002. Notch pathway molecules are essential for the maintenance, but not the generation, of mammalian neural stem cells. Genes Dev. 16:846–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Andreu-Agullo C, Morante-Redolat JM, Delgado A, Farinas I. 2009. Vascular niche factor PEDF modulates Notch-dependent stemness in the adult subependymal zone. Nat. Neurosci. 12:1514–1523 [DOI] [PubMed] [Google Scholar]
- 44.Espinosa L, Santos S, Inglés-Esteve J, Muñoz-Canoves P, Bigas A. 2002. p65-NF-kappaB synergizes with Notch to activate transcription by triggering cytoplasmic translocation of the nuclear receptor corepressor N-CoR. J. Cell Sci. 115:1295–1303 [DOI] [PubMed] [Google Scholar]
- 45.Mir A, Kaufman L, Noor A, Motazacker MM, Jamil T, Azam M, Kahrizi K, Rafiq MA, Weksberg R, Nasr T, Naeem F, Tzschach A, Kuss AW, Ishak GE, Doherty D, Ropers HH, Barkovich AJ, Najmabadi H, Ayub M, Vincent JB. 2009. Identification of mutations in TRAPPC9, which encodes the NIK- and IKK-beta-binding protein, in nonsyndromic autosomal-recessive mental retardation. Am. J. Hum. Genet. 85:909–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mochida GH, Mahajnah M, Hill AD, Basel-Vanagaite L, Gleason D, Hill RS, Bodell A, Crosier M, Straussberg R, Walsh CA. 2009. A truncating mutation of TRAPPC9 is associated with autosomal-recessive intellectual disability and postnatal microcephaly. Am. J. Hum. Genet. 85:897–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Philippe O, Rio M, Carioux A, Plaza JM, Guigue P, Molinari F, Boddaert N, Bole-Feysot C, Nitschke P, Smahi A, Munnich A, Colleaux L. 2009. Combination of linkage mapping and microarray-expression analysis identifies NF-kappaB signaling defect as a cause of autosomal-recessive mental retardation. Am. J. Hum. Genet. 85:903–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu WH, Pendergast JS, Mo XM, Brambilla R, Bracchi-Ricard V, Li F, Walters WM, Blits B, He L, Schaal SM, Bethea JR. 2005. NIBP, a novel NIK and IKK(beta)-binding protein that enhances NF-(kappa)B activation. J. Biol. Chem. 280:29233–29241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Basel-Vanagaite L, Attia R, Yahav M, Ferland RJ, Anteki L, Walsh CA, Olender T, Straussberg R, Magal N, Taub E, Drasinover V, Alkelai A, Bercovich D, Rechavi G, Simon AJ, Shohat M. 2005. The CC2D1A, a member of a new gene family with C2 domains, is involved in autosomal recessive nonsyndromic mental retardation. J. Med. Genet. 43:203–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rogaeva A, Galaraga K, Albert PR. 2007. The Freud-1/CC2D1A family: transcriptional regulators implicated in mental retardation. J. Neurosci. Res. 85:2833–2838 [DOI] [PubMed] [Google Scholar]
- 51.Zhao M, Li XD, Chen Z. 2010. CC2D1A, a DM14 and C2 domain protein, activates NF-kappaB through the canonical pathway. J. Biol. Chem. 285:24372–24380 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.