

Abstract
In addition to transmission involving extracellular free particles, a generally accepted model of virus propagation is one wherein virus replicates in one cell, producing infectious particles that transmit to the next cell via cell junctions or induced polarized contacts. This mechanism of spread is especially important in the presence of neutralizing antibody, and the concept underpins analysis of virus spread, plaque size, viral and host functions, and general mechanisms of virus propagation. Here, we demonstrate a novel process involved in cell-to-cell transmission of herpes simplex virus (HSV) in human skin cells that has not previously been appreciated. Using time-lapse microscopy of fluorescent viruses, we show that HSV infection induces the polarized migration of skin cells into the site of infection. In the presence of neutralizing antibody, uninfected skin cells migrate to the initial site of infection and spread over infected cells to become infected in a spatially confined cluster containing hundreds of cells. The cells in this cluster do not undergo cytocidal cell lysis but harbor abundant enveloped particles within cells and cell-free virus within interstitial regions below the cluster surface. Cells at the base and outside the cluster were generally negative for virus immediate-early expression. We further show, using spatially separated monolayer assays, that at least one component of this induced migration is the paracrine stimulation of a cytotactic response from infected cells to uninfected cells. The existence of this process changes our concept of virus transmission and the potential functions, virus, and host factors involved.
INTRODUCTION
The mechanisms involved in the transmission of infectious viruses between cells are of fundamental importance for our overall understanding of virus replication, virulence, and pathogenesis and for long-term objectives in combating infection (1). Much of our understanding of virus cell-to-cell transmission has been underpinned by analysis of plaque formation in culture, which demonstrates for many viruses two main routes of transmission, i.e., the production of extracellular virions which infect new cells from without and intercellular infection where infectious particles transmit between cells via cell-to-cell contacts or specialized, sometimes induced connections, e.g., viral synapses (2, 3). Many viruses, including herpesviruses, spread by direct cell-to-cell transmission and, where examined, indeed as part of the definition, cell-to-cell spreading is generally resistant to neutralizing antibody that would otherwise block transmission by extracellular virus (3–6). Plaque formation is then widely analyzed, e.g., to determine the effect of mutation in virus-encoded genes on overall transmission efficiency, to identify distinct requirements for specific virus proteins during cell-to-cell transmission compared to those in extracellular entry (7–11), to analyze receptor usage and relocalization at cell-to-cell junctions (12), and to demonstrate host responses and interactions with incoming genomes (13), among many other types of investigation. However, while certain basic observations on cell-to-cell transmission have been established, novel features continue to emerge. For example, in HIV and human T-cell leukemia virus (HTLV) it has been demonstrated that infection induces polarized contacts, termed viral synapses, via which virus cell-to-cell transmission then occurs (14–16). In vaccinia virus, it appears that the transmission rate during plaque formation (in the absence of extracellular virus neutralization) is accelerated by virtue of a surfing mechanism, whereby virus emerging from an infected cell skips over and is specifically repelled by immediately adjacent cells, so that the virus infects naive cells further away, thus promoting a more rapid overall transmission and larger plaques (17).
In herpes simplex virus (HSV), the main route of transmission in human tissues is cell-to-cell spread, occurring during primary infection when progeny virus spreads from the primary infected cell to adjacent cells in the mucocutaneous tissue and then to axonal termini of sensory neurons (18). Cell-to-cell transmission also occurs upon reactivation from latency, when newly replicated virus spreads from the sensory neuron to the mucocutaneous tissue. The simplest model of cell-to-cell transmission is plaque formation in cell culture, which has been widely studied in monkey or rodent cell lines such as Vero and BHK cells, and results from a generally cytocidal infection producing progeny viruses which efficiently spread across the monolayer. While infection produces extracellular virus, the majority of HSV remains cell associated and transmits across cell junctions despite the presence of extracellular virus-neutralizing antibody. The precise mechanisms of intercellular transmission remain to be fully understood. It is generally assumed that transmission parameters, including, e.g., plaque diameter, represent many multiple cumulative processes, including, e.g., efficiency of virus replication and assembly of infectious particles, sorting to and potential reorganization of appropriate junctions, intercellular virus transport, and possible intrinsic immune effectors that may resist infection (1, 8, 10, 19).
We have now found that in human skin keratinocytes, an epithelial cell type of more physiological relevance for HSV infection processes, the outcome of infection was significantly different than in the other cells. In skin cells in the presence of neutralizing antibody, HSV infection did propagate from cell-to-cell but did not show the typical cytocidal plaque formation seen in other cells. Instead, infected cells formed distinct bunched clusters with a distinctive surrounding area in which uninfected cells aligned in an oriented fashion. The diameter of the plaques was smaller in skin keratinocytes than, for example, in Vero cells, but this did not reflect lower success of transmission. Rather, we show abundant virus cell-to-cell transmission. However, in contrast to the typical perception of HSV plaque propagation, the outcome of infection was due to an induced polarized migration of cells to the site of infection. In additional analysis, we show that was a consequence at least in part of paracrine signaling from the infected cells. Although relatively simple in concept, and with many aspects requiring further investigation, this work has potentially profound ramifications, revealing a new process that has not been previously appreciated, changing our perception of virus spread and of the potential mechanisms involved.
MATERIALS AND METHODS
Cells, virus, and infection.
Vero were grown in Dulbecco modified minimal essential medium (DMEM; Gibco) containing 10% newborn calf serum (NCS) and penicillin-streptomycin. RPE-1 cells, a human telomerase immortalized retinal pigment epithelial line, were grown in DMEM-F12 (Sigma) supplemented with 200 mM glutamine, 10% fetal bovine serum (FBS), and penicillin-streptomycin. Human epithelial keratinocytes were the HaCaT cell line (20) grown in DMEM containing 10% FBS and penicillin-streptomycin or primary keratinocytes which had been immortalized with telomerase (21). We have previously show that in single-step growth curves HSV normally replicates, as well in HaCaT cells as, e.g., Vero cells (22). The telomerase-immortalized keratinocytes (hTert-keratinocytes) were grown in Ham's/F-12 (PAA) supplemented with 10% FBS, penicillin-streptomycin, 10 ng of mouse epidermal growth factor/ml, 1 ng of cholera toxin/ml, 400 ng of hydrocortisone/ml, 5 μg of insulin/ml, 5 μg of transferrin/ml, and 13 ng of liothyronine/ml. All supplements were from Sigma except mouse epidermal growth factor which was from AbD Serotec, Oxford, United Kingdom. The virus strains were HSV-1[17] or HSV-1[17].ICP0-YFP, a derivative of HSV-1[17] that expresses the immediate-early protein ICP0 fused to YFP and has been previously characterized (23). Pooled neutralizing human serum (HS; Sigma) was used at 1%. Clinical-grade purified neutralizing human immunoglobulin (IVIg, Carimune NF, nanofiltered, human immune globulin; CSL Behring) was used at 5 mg/ml, having demonstrated complete neutralization of extracellular virus at this dose (>6-log reduction in virus titer).
Immunofluorescence studies.
Immunofluorescence analysis was performed exactly as described previously (24). Anti-ICP4 antibody (Virusys) was used at 1:400. For fluorescence readout of total cells, alongside the yellow fluorescent protein (YFP) fluorescence to identify infected cells, monolayers were stained with CellMask Orange (Invitrogen), a general probe for plasma membrane staining which can be used in live cells. For CellMask staining of live cells, the medium was replaced with fresh medium containing the probe (5 μg/ml) for 5 min, the cells then washed in fresh medium and then imaged. For fixed cells, samples were washed with phosphate-buffered saline (PBS), fixed with 3.7% paraformaldehyde in PBS, and blocked with PBS containing 10% NCS. Images were collected using Zeiss Axiovert 135TV microscope and Zeiss ×10 (NA 0.5) or ×40 (NA 0.6) LD lenses. Images were taken using a filter set XF66 (Glen Spectra, United Kingdom) using the green fluorescent protein (GFP) setting to image YFP-ICP0. Each channel was captured sequentially with a Retiga 2000R camera using Image Pro plus software. Composite illustrations were prepared using Adobe software. For more detailed comparative analysis of infection in HaCaT cells samples were examined by confocal microscopy using a Zeiss LSM510 confocal microscope. Images were collected using a ×10 objective (NA 0.6) and compiled using the Z sectioning module. The resulting images were analyzed using Axiovision Le software to construct orthogonal axial images for each channel. Example images are representative of numerous images gathered for each virus and condition.
Time-lapse microscopy.
Cells plated in 35-mm dishes were infected with 50 to 100 PFU per dish with either HSV-1[17] or HSV-1[17].ICP0-YFP. Infections were performed in the absence or presence of 1% pooled HS or clinical grade purified immunoglobulin (IVIg, 5 mg/ml) in media containing 2% NCS. Dishes were placed on a heated chamber at 37°C in an environmentally controlled mini-incubator (PeCon GmBH) maintained at 5% CO2. Images were captured on a Zeiss Axiovert 135TV microscope using ×10 or ×40LD lenses and Image Pro plus software capture suit for sequential collection of fluorescence and phase channels for each time point. Routinely single isolated small clusters of infected cells were identified by the positive YFP signal (ca. 8 to 12 h postinfection) and time lapse collection then initiated using the AFA module of the software. Animation of the time series was carried out after merging the fluorescence and phase channels using Image Pro Plus and compressed with ImageJ software. Image Pro Plus cell tracking module was performed by manually tracking individual cells on zoomed versions of each image series and overlaying each track on the video animation. Total distance from the origin of the track was then calculated by the automated software and plotted.
Cell migration in gap-filling assays.
Cells were plated within 35-mm μ-Dishes (Ibidi) that contain two chambers separated by a removable silicone insert with a specific gap size of 500 μm. Cells were plated in the adjoining chambers and once the monolayers were formed (24 h), the silicone inserts were removed leaving the defined gap. Fresh medium containing 2% NBS and 1% pooled HS was added, and the chambers were placed in an environmentally controlled mini-incubator. Time-lapse examination of gap closure and cell migration was then initiated, capturing phase images every 5 min over the subsequent 18-h period. Images were compiled and compressed as described above.
Cell migration in response to paracrine signaling.
Cells were plated on the bottom wells (2 × 105 cells/well) of 24 multiwell plates (Falcon) into which FluoroBlock cell culture inserts (BD Biosciences) were to be inserted (see Fig. 5). These inserts are designed for the plating of cells on a membrane which contains pores of defined size. The base of the membrane blocks all fluorescence transmission, such that when using live cell fluorescence analysis with an inverted microscope, any fluorescence signal originates only from cells that migrated through the pores onto the bottom side of the membrane. Pores of different diameter (3 and 8 μm) were examined. The bottom cell monolayers in the 24-well plates were mock infected or infected with 100 PFU or 1,000 PFU of HSV-1[17].ICP0-YFP/well. After 1 h of adsorption in serum-free media, the inoculum was removed, and medium containing 2% FBS and 1% pooled neutralizing HS was added. The inserts, which had been separately plated with HaCaT cells at a density of 2 × 104 cells/insert, were then placed into the 24-well plates containing the mock-infected or infected monolayers. At different times after initial infection, the inserts were removed, incubated in a fresh dish in medium with 2 μM Calcein AM (Invitrogen) for 30 min at room temperature prior to live imaging microscopy. In separate assays, we confirmed that the test migratory cells from the upper inserts remained uninfected, as expected from the presence of the neutralizing antibody. Images of cell migration through the pores in response to medium from uninfected or infected lower chambers were then collected. At least three random fields using a low-power ×10 objective lens were obtained for each condition. Representative images are shown and were quantitated based on the total pixel density (Calcein AM staining) of the test cells for each condition.
Fig 5.

Infected HaCaT cell clustering. Monolayers of Vero, HaCaT, and RPE cells in 35-mm dishes were infected with 100 PFU of HSV-1[17]/well, stained at 24 h postinfection with CellMask, and then fixed with paraformaldehyde. Phase images (lower panels) are included to illustrate the morphological changes used to identify the viral plaques. Entire plaques were then imaged by z-sectioning (a total of ∼116 μm, with intervals of 7 μm/section) from the lower part of the plaque at the substratum to the most elevated point. The upper panels show the staining in a conventional x,y perspective, with green lines indicating the orthogonal axis applied for the z-sectioning. The resulting stacks (middle panel) shows the vertical perspective through the center of the plaque for each cell type.
Transmission electron microscopy (TEM).
Cells were fixed in 0.5% glutaraldehyde in 200 mM sodium cacodylate buffer for 30 min, washed in buffer, and secondarily fixed in reduced 1% osmium tetroxide–1.5% potassium ferricyanide for 60 min. The samples were washed in distilled water and stained overnight at 4°C in 0.5% magnesium uranyl acetate, washed in distilled water, and dehydrated in graded ethanol. The samples were then embedded flat in the dish in Epon resin. Resin filled stubs were placed on embedded cell monolayers and polymerized. The dish was removed, and the sample was reorientated for transverse vertical sectioning. Semithin survey sections 500 to 1,000 nm were first cut and collected onto glass slides and then stained with crystal violet. Images were collected on a Zeiss Axiovert 200M using Axiovision software. The polished block face and embedded sample were observed through a stereo microscope with a charge-coupled device (CCD) camera attached to the eyepiece, and images were collected. The sample was sectioned again when appropriate transverse sections were observed, and ultrathin sections (typically 50 to 70 nm) were cut and collected onto slot grids, stained with Reynold's lead citrate examined in a FEI Tecnai electron microscope with CCD camera image acquisition.
RESULTS
Altered plaque formation by HSV in human skin cells.
We compared plaque formation in Vero cells, a monkey kidney line widely used for study of growth and replication of HSV, to that in a human skin keratinocyte line (HaCaT) and a human retinal pigment cell line (RPE). Monolayers in 35-mm dishes were infected with ∼200 PFU of HSV-1[17] and then incubated in the presence of 1% HS to neutralize extracellular virus and limit plaque formation to primary plaques formed by cell-to-cell transmission. Plaques were observed after 60 h by phase-contrast microscopy or crystal violet staining (Fig. 1). Typical plaques 2 to 3 mm in diameter and with a central clear area resulting from cytocidal cell lysis were observed in Vero cells, with generally similar results and somewhat larger plaques in RPE cells (Fig. 1a). In contrast, in HaCaT cells, while overall plaque numbers were the same (data not shown), plaque morphology was distinctly different, lacking a clear central area with, instead, clustered, piled-up cells and a more uniform circular appearance of the plaque. Of note was the observation of a distinct zone around the plaques in HaCaT cells. This reflected the reorientation of the cells in the zone from a more random shape and organization within the body of the monolayers to a more elongated shape and uniform alignment oriented toward the plaque center (Fig. 1a, see also Fig. 1b and c and Fig. 4). This type of plaque morphology was surprisingly long-lived, with regular piled up clusters being observed for at least 3 to 4 days after initiation without overt central lysis.
Fig 1.
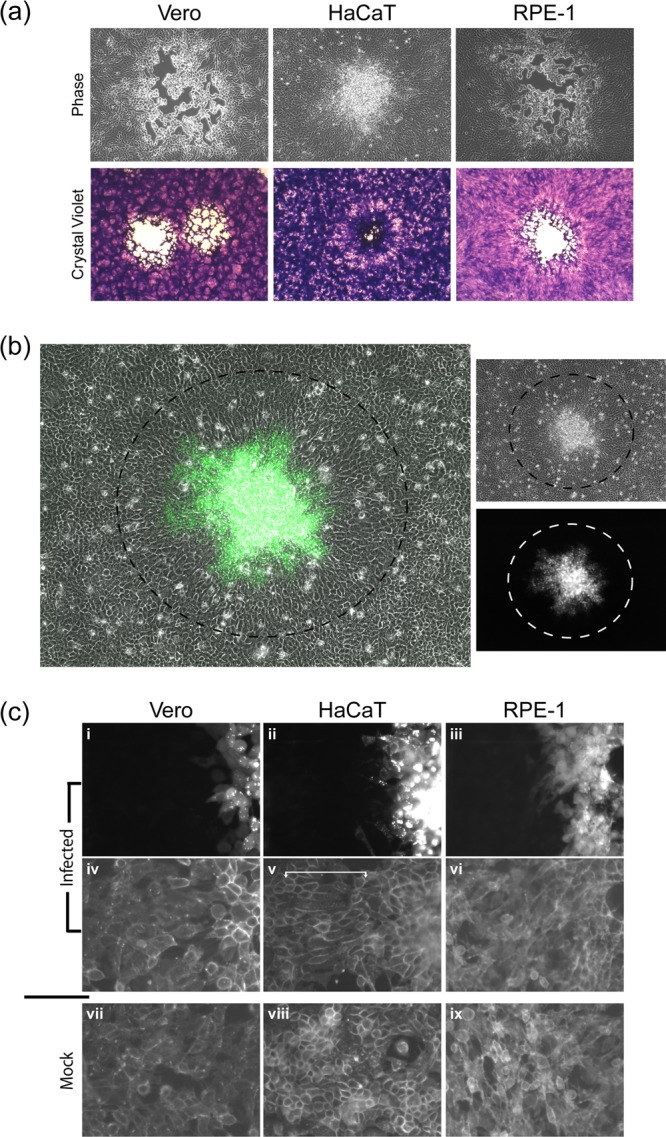
HSV spread comparison in different cell lines. Monolayers of Vero, HaCaT, and RPE cells were infected with ∼100 PFU of HSV-1[17]/well in the presence of HS. (a) Bright-field images (×10 objective lens) of plaques at 72 h postinfection by live microscopy (upper panels) prior to fixation and staining with crystal violet (lower panel). (b) Typical plaque formed in HaCaT cells by HSV-1[17].ICP0-YFP, showing a merged image of phase and YFP (48 h postinfection, ×10 objective lens). The dashed black circle indicates the area of YFP+ cells showing an elongated and oriented morphology compared to the surrounding cells (outside the circle). The images on the right show each channel independently, with the corresponding circles indicating the area of morphologically altered, YFP+ cells. (c) HSV-1[17].ICP0-YFP plaques in each of the cell types indicated (48 h postinfection, ×40 objective lens) showing plaque perimeters (panels i to vi) compared to mock monolayers (panels vii to ix). Live monolayers were stained with general plasma membrane stain CellMask (panels iv to ix) compared to the ICP0-YFP signal for virus infection (panels i to iii). The area of morphologically elongated GFP+ cells is indicated with a white bracket in the CellMask image (panel v).
Fig 4.

Polarized migration of HaCaT during plaque progression. Monolayers of HaCaT cells (a and b) or Vero cells (c and d) in 35-mm dishes were infected with ∼100 PFU of HSV-1[17].ICP0-YFP/dish in the presence of neutralizing antibody (1% HS) and incubated in an environmentally controlled microscope stage. Developing plaques were identified between 16 and 24 h and then recorded for both phase and YFP fluorescence for ∼18 h (1 frame every 5 min). The still images (a and c) show the last frame, merged for phase and YFP, of each time-lapse series. The time-lapse series upon which the quantitation was based (1 frame/25 min) are shown in accompanying movies (Fig. S4 and S5 in the supplemental material [https://www.dropbox.com/sh/mhj8p3qhswl4nyt/zk2VmGFjun]). The altered zone of cells surrounding the clustered plaque in HaCaT cells was readily observed. Random cells from outside this zone (blue tracks) and inside (red tracks) were individually tracked during the time lapse, and the distance from origin was plotted as a function of time (b and d).
To investigate the altered plaque formation in HaCaT cells, we used HSV-1[17].ICP0-YFP, a version of HSV-1[17] that expresses the immediate-early protein ICP0 fused to the autofluorescent protein YFP. After infection with ∼100 PFU per dish, individual developing plaques could be identified, and infected and uninfected surrounding cells were simultaneously imaged. Typical results again demonstrated the absence of a clear plaque and instead a turbid plaque with numerous cells in a piled-up cluster surrounded by a zone (indicated by a dotted black circle) wherein uninfected cells could be seen to reorient in a more uniform manner (Fig. 1b). The boundary of this zone from the more proximal oriented cells to the more distant randomly organized cells was readily observed (Fig. 1b, circle). The cells within the surrounding zone did not exhibit any detectable immediate-early ICP0-YFP expression (see the example with separate channels; there is a zone indicated by a black circle in the top phase panel, and the same zone is indicated by a white circle in the GFP channel). Although these analyses were performed in live cells, we also did not detect immediate-early proteins (or indeed any of a number of antigens tested) by immunostaining (data not shown), indicating that these surrounding cells were as yet uninfected.
We further compared the zones around the boundaries of live plaques in the three cell types using YFP in one channel (Fig. 1c, panels i to iii) and a total plasma membrane stain (Cellmask) in the second channel (Fig. 1c, panels iv to vi). For both Vero and RPE cells, cells beyond the YFP boundary did not exhibit any specific realignment, and no distinct differences from the general mock-infected picture could be observed (panels vii and ix). In contrast, in HaCaT cells, a distinct zone of more elongated and aligned cells could be seen outside the YFP+ boundary ((Fig. 1c, panel v, indicated by a white bracket). Similar results were obtained with other HSV-1 strains, e.g., KOS, and with HSV-2 (data not shown).
We interpret these results to indicate a qualitative difference in the response to HSV infection in HaCaT keratinocyte cells surrounding a developing plaque. One possible explanation was that this reflected not just a morphological reorganization but rather was a consequence of an active directional migration of the HaCaT cells with the consequential appearance of aligned, elongated cells. To pursue this, we first investigated basic growth features and motility properties in uninfected cells of the three types.
Comparison of cell migration.
Prior to analysis of cell motility, broad features of cell growth and plating were examined (Fig. 2). Each of the lines grew with approximately similar doubling times (data not shown) and formed confluent monolayers with a typical morphology seen in many previous studies (Fig. 2, panels i to iii). We noted that while Vero and RPE cells attached and flattened on the substratum rapidly after low density seeding (Fig. 2, panels iv and vi), HaCaT cells attached with similar efficiency, but flattened less well on the substratum until small islands formed (Fig. 2, panel v). To assess cell motility, we initially performed time-lapse microscopy in confluent and subconfluent monolayers of each of the cell types. In initial time-lapse analysis of confluent monolayers, relatively little migration was observed, although certain differences between the cell types were found, particularly in subconfluent monolayers. The key features and conclusions of these analyses were also observed in another type of assay, that of a “gap-filling” assay, typically used to assess motility and cell migration in a surrogate wound healing assay. Each of the cell types was plated in a two-chamber insert surrounded by a removable gasket, which when detached reveals a 500-μm gap in the substratum over which the cells then migrate with time. The results of these time-lapse assays in each line is summarized in Fig. 3 with accompanying movies showing the primary data included elsewhere (https://www.dropbox.com/sh/mhj8p3qhswl4nyt/zk2VmGFjun). These movies, together with others mentioned below, are supplied as avi or mp4 animations available for viewing online or download at the link specified at various places in this study. To aid viewing, the files were further compressed, which, although reducing overall image quality, maintains the key qualitative features. File names are accessed using “list view” rather than “grid view.”
Fig 2.
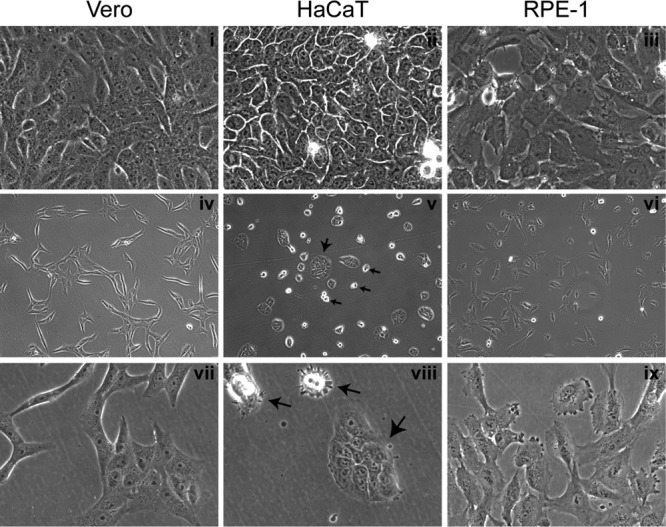
Morphological features of cell plating and growth. Monolayers of Vero, HaCaT, and RPE cells were plated at different densities. Typical images of live confluent monolayers (panels i to iii) or subconfluent (panels iv to ix) monolayers are shown. Images were recorded using a ×40 lens (panels i to iii and vii to ix) or ×10 objective lens (panels iv to vi). For the HaCaT cell panels, large arrows indicate small islands of adherent and flattened cells, whereas small arrows indicate individual rounded cells that have not flattened completely. For Vero and RPE cells, the individual cells flattened readily and then divided into islands, whereas for HaCaT cells, the individual cells remained rounded and poorly spread until present with neighboring cells to form islands.
Fig 3.

Motility characteristics of the cell lines. Vero, HaCaT, or RPE cell monolayers were plated in a two-chamber system with a removable gasket. As described in Materials and Methods, the subsequent gap filling assay was analyzed by time-lapse microscopy after removal of the gasket. (a) Individual snapshots at different times (0 to 18 h) after start of the assay showing gap filling by each of the cell lines. The frontline boundary of moving cells is outlined (black) on one boundary. Regions termed A within the denser internal area and B at the cell boundary are indicated by brackets. Boxes within the 18-h time point indicate typical AOI used to calculate cell density as described in the text. (b) Zoomed image (12-h time point) of the boundaries showing the distinct morphology of the migrating cells at the front. (c) Three independent AOI (black squares) within the defined regions A (original plated area) and B (just behind boundary) were quantified. The average cell density of an AOI in region A was standardized as 100% and then compared to that in the average AOI from just at the boundary. (d) Quantification of migration for 10 independent cells within area A for each cell line. Each cell was identified at the start of the time lapse, tracked (one point/5 min) for 18 h, and positioned from the origin plotted against time. The scales are identical for all cells.
Several notable conclusions were apparent from these analyses. First, in terms of simple motility into the gap and gap-filling, all of the lines tested exhibited this activity and with approximately similar kinetics, completing gap closure by ∼18 h (Fig. 3a, 0 to 18 h [see also supplementary material the URL above]). However, notwithstanding the similarity in this simple measure of motility, all three lines showed quite distinct properties. First and most obvious was the fact that, while at the beginning of time course all monolayers had a uniform starting boundary at the edge of the gap (Fig. 3, 0 h), both Vero and RPE cells then moved into the gap with broad spreading cell edges, probing the surroundings and proceeding in a nonuniform uncoordinated manner with an irregular front (Fig. 3a; see Fig. S1 and S2). For ease of reference, this is indicated by a black line on one boundary of the still images (Fig. 3a). In marked contrast, HaCaT cells migrated with a much more regular boundary, which moved in a unified front rate and did not exhibit the broad spreading lamellipodia seen with Vero and RPE cells (Fig. 3a and see Fig. S3). The time-lapse movies demonstrate the clear qualitative differences between the cell types in this regard. The differences in the leading edge boundaries are also shown at a higher magnification in a still image (Fig. 3b).
A second difference between the lines, likely as a consequence of the difference in migration properties, was that the areas just behind the leading edge movement in both Vero and RPE cells exhibited a much lower cell density than the area toward the interior of the cell sheet. This can be clearly seen in the accompanying movies but was also quantitated by enumerating cells in defined areas of interest (AOI) within region A (interior) or B (leading edge). Example AOI are marked with boxes (Fig. 3a, 18 h). In HaCaT cells, density within region B, the AOI were similar to those in region A AOI, whereas for both Vero and RPE cells the region B densities were noticeably reduced (Fig. 3c). Finally, and likely the explanation for these differences, was clearly evident from the time lapse movies of the gap filling assays (see Fig. S1 to S3). In Vero cells, it was mainly the cells at the leading edge that migrated into the gap, with cells at the origin (region A) remaining largely static. RPE cells also exhibited increased spreading of individual cells at the leading edge of the gap, together with some increased migration into the gap, although these cells were comparatively motile even within the denser origin region. However, HaCaT cells moved at the boundary and the interior at the same overall rates, migrating as a uniform field, resulting in a homogeneous density and a comparatively uniform regular leading edge. This difference in migration rates between the cell types, especially in the internal denser areas, was quantitated by tracking individual cells within region A's and plotting these as the distance from the origin versus time (Fig. 3c). Vero cells within the denser region away from the gap essentially did not move. RPE cells exhibited distinct uncoordinated motility of individual cells, while HaCaT cells were clearly more motile, independent of density. Together, these data indicated that in Vero cells, gap filling was mainly due to significant cell spreading (and cell enlargement), together with some selective migration of leading edge cells. RPE cells spread into the gap but were clearly more motile, migrating as individual cells at the gap edge but also in denser interior areas. HaCaT cells were distinct, and yet again in that gap filling was less due to cell spreading and enlargement but rather reflected migration in a uniform coordinated manner both at the leading edge and in the interior, more dense areas of the monolayer.
HSV infection induces polarized migration of keratinocytes.
We next examined the potential for cell migration during plaque development in HaCaT cells and compared this to Vero cells. Cell monolayers were infected with ∼100 PFU of HSV-1[17].ICP0-YFP/dish to allow the identification and monitoring of isolated developing plaques. Plaque progression was then recorded by time-lapse microscopy for both fluorescence and phase images and the merged channels animated as movies (Fig. 4 and see Fig. S4 and S5 at https://www.dropbox.com/sh/mhj8p3qhswl4nyt/zk2VmGFjun). At the initiation of the animations (24 h), there was a clear contrast in plaque morphology. In Vero cells (Fig. 4a and b and see also the accompanying movie in Fig. S4), the pattern of infection (YFP fluorescence) spread out from the center, which already showed a central cytolytic effect, with no overt reorganization of surrounding cells. In contrast, in HaCaT cells, infection was in a distinct central focus with a much more defined boundary, surrounded by a zone of cells aligned toward the focus (Fig. 4c). The accompanying movie (see Fig. S5) shows the striking result wherein this surrounding area reflects a zone of very active cell migration into the developing focus, with cells becoming YFP+ only within the cluster. Individual cells were tracked throughout the animated series and the results plotted in Fig. 4d. Cells exhibiting the realigned morphology (red tracks) showed a distinct directional migration into the focus (see Fig. S4 in the online material, and this is also plotted Fig. 4d), whereas more distant randomly ordered cells in the monolayer exhibited very little migration. In a further series of experiments, we examined plaque migration by time lapse microscopy in a different human skin system, using telomerase-immortalized keratinocytes (21) and observed virtually identical results (data not shown). In contrast, in infected Vero cells, although some individual cell migration appeared, this was then rapidly obscured due to the outwardly progressing plaque (Fig. 4a and b). By comparison of both the visual animations (see Fig. S4 and S5 in the online material [https://www.dropbox.com/sh/mhj8p3qhswl4nyt/zk2VmGFjun]) and the behavior and lack of orientation of surrounding uninfected Vero cells, it was clear that infection progressed in a qualitatively distinct manner in the two cell types.
The observations of induced cell migration were made under conditions which restrict transmission by extracellular virus, i.e., the presence of neutralizing antibody. In additional studies (data not shown), we considered whether the induced migration was dependent upon the presence of neutralizing antibody by conducting time-lapse analysis of plaque progression in semisolid media. The results demonstrated cell migration independent of the presence of antibody. However, in the complete absence of antibody and restricted diffusion, extensive plaque progression occurred, generally also with increased cytolysis at the center (see discussion).
Structural analysis of infection in skin cells.
We next pursued the distinct morphological aspects of the focus of infected HaCaT cells. Live developing plaques were stained with Cellmask at 24 h in each of the cell types, the plaques identified in phase and then imaged through the z dimension (Fig. 5). The resulting stacks (Cellmask, middle panel) show the vertical perspective through the center of the plaque (green line, top panels). In contrast to Vero and RPE cells, in HaCaT cells even by 24 h, the distinct piling of the cells in the vertical dimension could be observed. To increase the resolution and gain additional insight into the process, we next performed TEM through the z-dimension of the focus, as described in Materials and Methods. By first imaging the embedded block (before sectioning) at low magnification on a tilting stage, the vertical clusters of cells could be readily observed (Fig. 6a). Sequential sections were then cut, stained, and aligned, allowing a higher-level resolution across a single focus (Fig. 6b). The extremity of the image represents the surrounding zone of cells, just a single layer deep, that are migrating into the focus. Proceeding to the interior, e.g., the boxed areas in Fig. 6c, panels 3 and 4, cells crawling over the bottom layers can be now readily seen, with many cells at the peak in the center (probably under-representing the total due to processing). Finally, aligning the sequential sections from the block, the exact areas corresponding to those in panel B (numbers 1 to 4) could be identified and analyzed at high resolution (Fig. 6c, panels 1 to 4 [the lower panels display enlargements of the panel insets]). This analysis revealed several features of virus transmission in these cells.
Fig 6.

Ultrastructural characterization of HSV plaques in HaCaT cells. Monolayers of HaCaT cells in 35-mm dishes were infected with 100 PFU of HSV-1[17]/well and processed at 24 h postinfection for TEM as described in Materials and Methods. (a) Low-power binocular image of the vertical face of the embedded resin block. This image gives a three-dimensional view along the horizontal surface of the plastic support. Arrows indicate the plaques of clustered piled-up cells, with the size of the arrow indicating the distance from the block front. (b) The block was then sectioned into semithin survey sections (500 to 1,000 nm) and stained with crystal violet. Images were collected on a Zeiss Axiovert 200M using a ×40 objective lens, and the panels were assembled using Adobe Photoshop. (c) Sequential ultrathin sections (50 to 70 nm) of the same sample were then obtained and collected onto slot grids, stained, and examined by TEM. Because the sections analyzed are immediately adjacent to the section stained with crystal violet, corresponding areas could be matched. The TEM images of the sections in panel c, sections 1 to 4, therefore correspond to the boxes in panel b, sections 1 to 4. Upper panels (lower magnification) and lower panels (higher magnification on inset area) show different features of infection within the plaque as discussed in the text. In the lower panels, the features include extracellular virions (black arrows with white outline), nucleocapsids (short black arrows), and cell tight junctions (white arrows). In the upper part of panels 3 and 4, cells with no distinct features of infection (e.g., no early chromatin marginalization) are indicated by long black arrows. Box 5 in panel b shows flat uninfected cells that correspond to the elongated cells (TEM data not shown).
In the central “older” areas (Fig. 6c, regions 1 and 2), while there was a clear cytopathic effect, including marginated chromatin, the cells remained relatively intact, with numerous enveloped virions in the interstitial space between cells (panel 1 [lower panel, white outlined arrows]). It is known that skin cells form tight junctions, and numerous tight junctions could be observed in the vertical section between cells that had piled up over one another (Fig. 6c, panel 2 [lower panel, white arrow]). Considering the cells have moved over one another, these junctions may somehow be maintained or continuously formed as the cells migrate in the cluster. Numerous assembled virions were present between these connections (Fig. 6c, panel 2 [lower inset]). Few extracellular virions were present on the outermost surface to the focus. However, we cannot conclude (and in our view less likely) that there may be selective virion release on the internal aspect of the clusters, since surface material may have been lost during processing. Toward the perimeter to the focus, cells just beginning to migrate over the underlying cells could be observed (Fig. 6c, panels 3 and 4). The cell in panel 3, shows a migrating elongated cell with a tight junction (lower inset, white arrow) between the uppermost cell (which did not yet show overt chromatin margination) and underlying infected cells (upper panel, arrowed capsids in the nucleus). At the extreme perimeter of the focus, the monolayer comprised single flat cells with, as expected, no overt cytopathic effect.
HSV infection induces a paracrine mediator of skin cell migration.
Using both time-lapse microscopy and TEM analysis of vertical sections, we provide robust evidence for the induction of HaCaT cell migration and crawling over underlying cells, resulting in the focus of infection. Using a well-established cell migration assay system (see Materials and Methods), we next addressed whether there was evidence that these effects could be due to a paracrine mediator (Fig. 7a). Target HaCaT cells were plated in the upper chamber insert on a membrane support with defined 8-μm pores (Fig. 7b). The insert was then placed in a dish of test cells (lower chamber) that were either mock infected or infected with 100 or 1,000 PFU/dish. All cultures were in the presence of neutralizing antibody to prevent infectious extracellular virus. We confirmed that the upper target cells remained uninfected. Cell migration was then monitored by the presence of cells on the lower side of the membrane support by live cell staining. Only cells on the lower side are detected because the membrane is impervious to the excitation wavelengths. The results demonstrated a very clear increase in target cell migration with infected test cells compared to uninfected test cells and that this increase showed a dose response with increasing virus dose. This could be readily seen in the images of target cell migration taken at low magnification to avoid sample bias (Fig. 7c) and in the quantitation of the data (Fig. 7d). Identical results were obtained in repeat experiments, and in experiments using inserts with a 3-μm pore size instead of 8 μm. In some ways, this system is a more stringent test for paracrine effects than that occurring during focus formation, since the cells are not in direct contact with one another. We believe that the results indicate that HSV-infected skin cells release a soluble mediator that can stimulate cell migration. This conclusion is consistent with the observations during focus formation and indicates that a paracrine effect, at least in part, accounts for the induced migration of skin cells during virus infection and transmission.
Fig 7.

Paracrine-induced migration of skin cells by HSV infection. (a) Schematic diagram of culture system used to analyze HaCaT cell migration. The upper chamber (the insert) contains target cells plated onto a support with defined 8-μm pores. The support is impervious to light transmission. The lower chamber (a conventional 35-mm dish) contains a monolayer of test cells that would be mock infected or infected with 100 or 1,000 PFU of HSV-1[17].ICP0-YFP/dish. The cluster of red cells represents a developing plaque. The shading of the media indicates potential secretion of components that might induce cellular migration through the pores onto the bottom side of the membrane, where they can be detected by live-cell fluorescence staining (Calcein AM). (b) Image of the insert base support system showing Calcein AM signal through the pores of the membrane of the upper chamber. This represents the background signal. (c) Lower-chamber cells were mock infected or HSV infected as indicated and incubated with the upper-chamber cells for 72 h; representative low-power images were then taken of the bottom side of the upper-chamber membrane after staining with Calcein AM. The results show images of the target cells (I to III) for each of the test conditions (indicated on the left-hand side). (d) Average total Calcein AM staining (pixel density) of target cells after thresholding to remove the defined background signal (image in panel b). Values were obtained from three random low-power fields for each condition in each of the duplicated samples. The experiment was repeated three times with similar results.
DISCUSSION
Analysis of plaque formation in tissue culture models of infection represents one of the most fundamental methods used to study virus replication and cell-to-cell transmission. Notwithstanding the relatively straightforward concept, the precise mechanisms of intercellular transmission during plaque formation remain to be fully understood. Furthermore, recent results show that there are processes involved in cell-to-cell transmission that had previously been unrecognized, such as virus surfing in vaccinia virus plaque formation (17), or the induction of polarized cell-to-cell connections (14–16), or possible intercellular transmission via nanotubes (25). HSV plaque formation and cell-to-cell transmission has been widely studied in several types of cell lines, generally resulting in a cytocidal infection producing progeny viruses that efficiently spread across the monolayer. Infection produces extracellular virus which disseminates to surrounding cells, but the majority of HSV remains cell associated and transmits across cell junctions despite the presence of extracellular virus-neutralizing antibody, which otherwise neutralizes extracellular virus. Cell-to-cell transmission clearly represents many cumulative processes, including, for example, efficiency of virus replication and assembly of infectious particles, transport to the cell surface and potential reorganization of cell-to-cell junctions, intercellular virus transport, and possible restriction mechanisms that may resist infection (1, 8, 10, 19). However, it has been generally assumed that such transmission involves the propagation outward from the initially infected single cell to adjacent surrounding cells and progressively onward to form the resulting plaque in any particular situation. The results of the findings reported here indicate that we must add another hitherto-unrecognized factor in describing and accounting for the overall mechanisms of virus transmission, namely, the induced migration of uninfected cells to the site of infection. Although relatively simple in concept, our observations have significant implications (Fig. 8).
Fig 8.

Model of plaque progression and cell-to-cell transmission in HaCaT cells. The panels a to c represent the temporal development of the plaque. After initial infection, gray cell (circled 1), virus spreads intercellularly (circled 2). Additional signaling events, including a paracrine mechanism (circled 3), induce cell migration (circled 4) to the developing focus of infection. It is possible that intercellular communication (circled 5) contributes to migration, in addition to the paracrine mechanism. Uninfected cells migrate into and over uninfected cells, and cell-to-cell transmission occurs in reorganized clusters (circled 6 and 7) and, at least initially, continue to amplify (circled 6) the paracrine signal. The duration of migration (circled 8) and of the paracrine stimulus (circled 9) may be temporally regulated. This and further aspects of this process are discussed more fully in the text.
First, we noted an obvious difference in plaque formation that generally results in a pronounced cytopathic response, cell lysis, and a clear plaque center. In contrast, in HaCaT cells (and in an independent telomerase-immortalized keratinocyte line, data not shown), migrating cells crawled over the developing plaque, resulting in a piled cluster and a “turbid” plaque. Although the transverse electron miscroscopic analysis demonstrated obvious cytopathic effects, the cells retained structure without overt lysis and with abundant virus particles between the cells. It is noteworthy that previous studies have shown that with vaccinia virus, in certain cell types, productively infected cells exhibit a general increase in motility (26). Our results pertain to a paracrine stimulation by infected cells of migration in uninfected cells to the site of infection, although certain features of the underlying mechanism could be similar. based on the time-lapse microscopy findings, it would be generally expected that the base and outside layers of the plaque would be cells in earlier stages of infection as the uninfected cells migrated inward. HSV employs several mechanisms in the attempt to combat apoptosis pathways that are initiated by the cell (27–31), although, notwithstanding such virus countermeasures, a cytocidal lysed plaque phenotype normally results. The induced migration of skin cells that we observed here may be unrelated to the relative longevity of the infected cells and formation of cell clusters, being more related to intrinsic properties of the cells, e.g., lower capacity for apoptosis or more efficient antiapoptosis mechanisms effected by HSV. However, it is tempting to speculate that the progressive migration of uninfected cells into and over the infected cells could in some way be directly linked to prolonged survival (see below). It could be that the association between incoming uninfected skin cells migrating over infected cells signals events which reduce cytopathic effects or promote survival. Using markers for cell death that measure membrane permeability such as Calcein AM/EthD-1 or trypan blue, we detected little evidence of overt death in the HaCaT cell infected clusters (data not shown). Clearly, there may be several explanations, and the precise nature of the outcome of infection and relationship to cell migration will be the subject of further study.
Second, the observations of induced cell migration were made under conditions which restrict transmission by extracellular virus, i.e., the presence of neutralizing antibody. In additional work (data not shown), we demonstrated that induced migration occurred independently of the presence of antibody. We also demonstrated in the assay for a paracrine mediator in the two-chamber system that infected cells, in the presence of purified immunoglobulin, released a mediator of migration, whereas uninfected cells did not. Nevertheless, in the absence of antibody and without semisolid media, the production and diffusion of extracellular infectious virus overran inward cell migration, resulting in extensive primary plaque formation generally with increased destruction in the center. Thus, while neither neutralizing antibody (or other serum components) are required for cell migration, the presence of neutralizing antibody restricting transmission to cell-to-cell spread revealed the process of migration. However, we believe that antibody could also somehow contribute to the more prolonged survival or the reduced cytolytic events that we observed in the skin cell clusters. Keratinocytes are migratory cells and important players in immune mechanisms, secrete a large number of other proinflammatory cytokines and chemokines, and are involved in skin dendritic cell maturation (32–34). It is possible therefore that skin cells carry out immune effector mechanisms or interact with antibody or antibody-virus complexes in different ways from cells such as Vero or BHK cells. Indeed, it has recently been shown that antibody-virus complexes can be taken up by cells and effect intracellular antiviral responses (35, 36). It will be interesting in future work to examine the contribution of antibody in skin cells to HSV restriction and whether there is any contribution by mechanisms other than neutralization of extracellular virus to the phenotype we now observe in infected skin cells, cell-to-cell transmission, and cell migration.
Finally, we consider two of the most important questions which these findings raise, i.e., the mechanisms involved and the importance of the process to virus transmission. With regard to the mechanism, the two most straightforward explanations concern either (i) a paracrine effect, wherein a soluble mediator secreted from infected cells signaled migration toward the highest concentration at the developing plaque center, or (ii) some form of structural or mechanochemical signal transmitted via intrinsic cell-to-cell communication. We certainly do not rule out this latter possibility, but its investigation is currently beyond the scope of this work. However, using a recognized method to explore paracrine stimulation of migration, our results provide evidence that such a mechanism contributes to the HSV-induced cell migration observed in this work. Several paracrine mediators, including epidermal growth factor, transforming growth factor α, insulin-like growth factor 1, and platelet-derived growth factor, have each been shown to induce chemotaxis in keratinocytes (37–40). Another mediator, keratinocyte growth factor (KGF or FGF7), a member of the fibroblast growth factors family, is also a known mediator of keratinocyte migration (41–43) and is a good candidate for playing a role in orchestrating the activation of proteins involved in keratinocyte cell migration. In terms of other candidates, we have found no evidence for interferon in conditioned medium from infected HaCaT cells (data not shown). However, it may also be that HSV-encoded proteins are involved. Other herpesviruses of the beta- and gammaherpesvirus classes are known to encode soluble excreted proteins that bind to cellular chemokines to reduce chemotaxis (44, 45). However, they also encode chemokine homologues, which, while generally proinflammatory, are thought to act to aid dissemination via hemopoietic cell recruitment (44, 46–48). Moreover, very recent study has demonstrated that HSV gG (US4), either on the cell surface or as a secreted product, can act as a potent inducer of chemokine activity (49). It is therefore possible that HSV-encoded proteins are involved in the skin cell migration that we observed here. Future investigation will include a candidate approach to examine stimulation of cell and virus factors, as well as an unbiased approach to characterize the secretome of HSV-infected keratinocytes to identify and ultimately isolate the factor(s) responsible.
The most straightforward explanation for our results is that induced cell migration represents one of several countermeasures promoted by the cells to combat infection. It is possible that the paracrine effectors observed here act on additional cells in the skin in vivo, e.g., dendritic cells and that combined extrinsic and cell-intrinsic mechanisms related to migration act to restrict infection. However, it is possible, although less likely in our view, that virus induced migration represents some way to increase the pool of susceptible cells in the surroundings of neutralizing antibody that otherwise effectively limits extracellular spread. In conclusion, just as with many processes elucidated from tissue culture studies, especially since our observations were obtained in cells of direct physiological relevance, we propose that induced cell migration will play an important role in in vivo virus transmission and virus-host interactions. Although our results raise many questions, they reveal a hitherto-unrecognized process that must now be taken into account and investigated.
ACKNOWLEDGMENTS
We are grateful to Roger Everett for the supply of HSV-1[17].ICP0-YFP. We thank Marianne Bolstad, Sonia Barbosa, and Thomas Hennig for comments and assistance. We are grateful to Gill Elliott and Julianna Stylianos for the hTert-keratinocyte cells and for assistance.
This study was funded by Marie Curie Cancer Care.
Footnotes
Published ahead of print 8 May 2013
REFERENCES
- 1. Flint SJ, Enquist LW, Krug RM, Racaniello VR, Skalka AM. 2009. Principles of virology. ASM Press, Washington, DC [Google Scholar]
- 2. Mothes W, Sherer NM, Jin J, Zhong P. 2010. Virus cell-to-cell transmission. J. Virol. 84:8360–8368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sattentau Q. 2008. Avoiding the void: cell-to-cell spread of human viruses. Nat. Rev. Microbiol. 6:815–826 [DOI] [PubMed] [Google Scholar]
- 4. Black FL, Melnick JL. 1955. Microepidemiology of poliomyelitis and herpes-B infections: spread of the viruses within tissue cultures. J. Immunol. 74:236–242 [PubMed] [Google Scholar]
- 5. Christian RT, Ludovici PP, Jeter WS. 1971. Cell-to-cell transmission of herpes simplex virus in primary human amnion cells. Proc. Soc. Exp. Biol. Med. 138:1109–1115 [DOI] [PubMed] [Google Scholar]
- 6. Wheeler CE. 1960. Further studies on the effect of neutralizing antibody upon the course of herpes simplex infections in tissue culture. J. Immunol. 84:394–403 [PubMed] [Google Scholar]
- 7. Campadelli-Fiume G, Roizman B. 2006. The egress of herpesviruses from cells: the unanswered questions. J. Virol. 80:6716–6719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dingwell KS, Brunetti CR, Hendricks RL, Tang Q, Tang M, Rainbow AJ, Johnson DC. 1994. Herpes simplex virus glycoproteins E and I facilitate cell-to-cell spread in vivo and across junctions of cultured cells. J. Virol. 68:834–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Farnsworth A, Johnson DC. 2006. Herpes simplex virus gE/gI must accumulate in the trans-Golgi network at early times and then redistribute to cell junctions to promote cell-cell spread. J. Virol. 80:3167–3179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Johnson DC, Webb M, Wisner TW, Brunetti C. 2001. Herpes simplex virus gE/gI sorts nascent virions to epithelial cell junctions, promoting virus spread. J. Virol. 75:821–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Klupp B, Altenschmidt J, Granzow H, Fuchs W, Mettenleiter TC. 2008. Glycoproteins required for entry are not necessary for egress of pseudorabies virus. J. Virol. 82:6299–6309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Krummenacher C, Baribaud I, Eisenberg RJ, Cohen GH. 2003. Cellular localization of nectin-1 and glycoprotein D during herpes simplex virus infection. J. Virol. 77:8985–8999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sourvinos G, Everett RD. 2002. Visualization of parental HSV-1 genomes and replication compartments in association with ND10 in live infected cells. EMBO J. 21:4989–4997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Igakura T, Stinchcombe JC, Goon PK, Taylor GP, Weber JN, Griffiths GM, Tanaka Y, Osame M, Bangham CR. 2003. Spread of HTLV-1 between lymphocytes by virus-induced polarization of the cytoskeleton. Science 299:1713–1716 [DOI] [PubMed] [Google Scholar]
- 15. Jolly C, Sattentau QJ. 2004. Retroviral spread by induction of virological synapses. Traffic 5:643–650 [DOI] [PubMed] [Google Scholar]
- 16. McDonald D, Wu L, Bohks SM, KewalRamani VN, Unutmaz D, Hope TJ. 2003. Recruitment of HIV and its receptors to dendritic cell-T cell junctions. Science 300:1295–1297 [DOI] [PubMed] [Google Scholar]
- 17. Doceul V, Hollinshead M, van der Linden L, Smith GL. 2010. Repulsion of superinfecting virions: a mechanism for rapid virus spread. Science 327:873–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K. 2007. Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom: [PubMed] [Google Scholar]
- 19. Mettenleiter TC, Klupp BG, Granzow H. 2009. Herpesvirus assembly: an update. Virus Res. 143:222–234 [DOI] [PubMed] [Google Scholar]
- 20. Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. 1988. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 106:761–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dickson MA, Hahn WC, Ino Y, Ronfard V, Wu JY, Weinberg RA, Louis DN, Li FP, Rheinwald JG. 2000. Human keratinocytes that express hTERT and also bypass a p16INK4a-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol. Cell. Biol. 20:1436–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Abaitua F, Hollinshead M, Bolstad M, Crump CM, O'Hare P. 2012. A Nuclear localization signal in herpesvirus protein VP1-2 is essential for infection via capsid routing to the nuclear pore. J. Virol. 86:8998–9014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Everett RD, Sourvinos G, Orr A. 2003. Recruitment of herpes simplex virus type 1 transcriptional regulatory protein ICP4 into foci juxtaposed to ND10 in live, infected cells. J. Virol. 77:3680–3689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Abaitua F, Souto RN, Browne H, Daikoku T, O'Hare P. 2009. Characterization of the herpes simplex virus (HSV)-1 tegument protein VP1-2 during infection with the HSV temperature-sensitive mutant tsB7. J. Gen. Virol. 90:2353–2363 [DOI] [PubMed] [Google Scholar]
- 25. Eugenin EA, Gaskill PJ, Berman JW. 2009. Tunneling nanotubes (TNT) are induced by HIV-infection of macrophages: a potential mechanism for intercellular HIV trafficking. Cell. Immunol. 254:142–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sanderson CM, Way M, Smith GL. 1998. Virus-induced cell motility. J. Virol. 72:1235–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aubert M, Blaho JA. 1999. The herpes simplex virus type 1 regulatory protein ICP27 is required for the prevention of apoptosis in infected human cells. J. Virol. 73:2803–2813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Leopardi R, Van Sant C, Roizman B. 1997. The herpes simplex virus 1 protein kinase US3 is required for protection from apoptosis induced by the virus. Proc. Natl. Acad. Sci. U. S. A. 94:7891–7896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nguyen ML, Blaho JA. 2009. Cellular players in the herpes simplex virus-dependent apoptosis balancing act. Viruses 1:965–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nishiyama Y, Murata T. 2002. Anti-apoptotic protein kinase of herpes simplex virus. Trends Microbiol. 10:105–107 [DOI] [PubMed] [Google Scholar]
- 31. Ogg PD, McDonell PJ, Ryckman BJ, Knudson CM, Roller RJ. 2004. The HSV-1 Us3 protein kinase is sufficient to block apoptosis induced by overexpression of a variety of Bcl-2 family members. Virology 319:212–224 [DOI] [PubMed] [Google Scholar]
- 32. Kaplan DH, Igyarto BZ, Gaspari AA. 2012. Early immune events in the induction of allergic contact dermatitis. Nat. Rev. Immunol. 12:114–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li W, Henry G, Fan J, Bandyopadhyay B, Pang K, Garner W, Chen M, Woodley DT. 2004. Signals that initiate, augment, and provide directionality for human keratinocyte motility. J. Investig. Dermatol. 123:622–633 [DOI] [PubMed] [Google Scholar]
- 34. Mutou Y, Tsukimoto M, Homma T, Kojima S. 2010. Immune response pathways in human keratinocyte (HaCaT) cells are induced by ultraviolet B via p38 mitogen-activated protein kinase activation. J. Health Sci. 56:675–683 [Google Scholar]
- 35. Mallery DL, McEwan WA, Bidgood SR, Towers GJ, Johnson CM, James LC. 2010. Antibodies mediate intracellular immunity through tripartite motif-containing 21 (TRIM21). Proc. Natl. Acad. Sci. U. S. A. 107:19985–19990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McEwan WA, Tam JC, Watkinson RE, Bidgood SR, Mallery DL, James LC. 2013. Intracellular antibody-bound pathogens stimulate immune signaling via the Fc receptor TRIM21. Nat. Immunol. 14:327–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ando Y, Jensen PJ. 1993. Epidermal growth factor and insulin-like growth factor I enhance keratinocyte migration. J. Investig. Dermatol. 100:633–639 [DOI] [PubMed] [Google Scholar]
- 38. Barrandon Y, Green H. 1987. Cell migration is essential for sustained growth of keratinocyte colonies: the roles of transforming growth factor-alpha and epidermal growth factor. Cell 50:1131–1137 [DOI] [PubMed] [Google Scholar]
- 39. Cha D, O'Brien P, O'Toole EA, Woodley DT, Hudson LG. 1996. Enhanced modulation of keratinocyte motility by transforming growth factor-alpha (TGF-α) relative to epidermal growth factor (EGF). J. Investig. Dermatol. 106:590–597 [DOI] [PubMed] [Google Scholar]
- 40. Schultz G, Rotatori DS, Clark W. 1991. EGF and TGF-alpha in wound healing and repair. J. Cell. Biochem. 45:346–352 [DOI] [PubMed] [Google Scholar]
- 41. Ceccarelli S, Cardinali G, Aspite N, Picardo M, Marchese C, Torrisi MR, Mancini P. 2007. Cortactin involvement in the keratinocyte growth factor and fibroblast growth factor 10 promotion of migration and cortical actin assembly in human keratinocytes. Exp. Cell Res. 313:1758–1777 [DOI] [PubMed] [Google Scholar]
- 42. Sato C, Tsuboi R, Shi CM, Rubin JS, Ogawa H. 1995. Comparative study of hepatocyte growth factor/scatter factor and keratinocyte growth factor effects on human keratinocytes. J. Investig. Dermatol. 104:958–963 [DOI] [PubMed] [Google Scholar]
- 43. Tsuboi R, Sato C, Kurita Y, Ron D, Rubin JS, Ogawa H. 1993. Keratinocyte growth factor (FGF-7) stimulates migration and plasminogen activator activity of normal human keratinocytes. J. Investig. Dermatol. 101:49–53 [DOI] [PubMed] [Google Scholar]
- 44. Boomker JM, de Leij LF, The TH, Harmsen MC. 2005. Viral chemokine-modulatory proteins: tools and targets. Cytokine Growth Factor Rev. 16:91–103 [DOI] [PubMed] [Google Scholar]
- 45. Vink C, Smit MJ, Leurs R, Bruggeman CA. 2001. The role of cytomegalovirus-encoded homologs of G protein-coupled receptors and chemokines in manipulation of and evasion from the immune system. J. Clin. Virol. 23:43–55 [DOI] [PubMed] [Google Scholar]
- 46. Penfold ME, Dairaghi DJ, Duke GM, Saederup N, Mocarski ES, Kemble GW, Schall TJ. 1999. Cytomegalovirus encodes a potent alpha chemokine. Proc. Natl. Acad. Sci. U. S. A. 96:9839–9844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Saederup N, Lin YC, Dairaghi DJ, Schall TJ, Mocarski ES. 1999. Cytomegalovirus-encoded beta chemokine promotes monocyte-associated viremia in the host. Proc. Natl. Acad. Sci. U. S. A. 96:10881–10886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang D, Bresnahan W, Shenk T. 2004. Human cytomegalovirus encodes a highly specific RANTES decoy receptor. Proc. Natl. Acad. Sci. U. S. A. 101:16642–16647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Viejo-Borbolla A, Martinez-Martin N, Nel HJ, Rueda P, Martin R, Blanco S, Arenzana-Seisdedos F, Thelen M, Fallon PG, Alcami A. 2012. Enhancement of chemokine function as an immunomodulatory strategy employed by human herpesviruses. PLoS Pathog. 8:e1002497. 10.1371/journal.ppat.1002497 [DOI] [PMC free article] [PubMed] [Google Scholar]