

Abstract
Certain antigen-presenting cells (APCs) process and present extracellular antigen with major histocompatibility complex class I (MHC-I) molecules to activate naive CD8+ T cells in a process termed cross-presentation. We used insights gained from HIV immune evasion strategies to demonstrate that the clathrin adaptor protein adaptor protein 1 (AP-1) is necessary for cross-presentation by MHC-I molecules containing a cytoplasmic tail tyrosine signal (murine MHC-I molecules, human MHC-I HLA-A and HLA-B allotypes). In contrast, AP-1 activity was not needed for cross-presentation by MHC-I molecules containing a human MHC-I HLA-C cytoplasmic tail, which does not contain a tyrosine signal. AP-1 activity was also dispensable for presentation of endogenous antigens by MHC-I via the classical pathway. In APCs, we show that HIV Nef disrupts cross-presentation by MHC-I containing the tyrosine signal but does not affect cross-presentation by MHC-I containing the HLA-C cytoplasmic tail. Thus, we provide evidence for two separable cross-presentation pathways, only one of which is targeted by HIV.
INTRODUCTION
Human immunodeficiency virus (HIV) causes a persistent infection in which cytotoxic T lymphocytes (CTLs) control viremia, especially early in disease. However, CTLs fail to eradicate the virus and most untreated people ultimately develop AIDS and life-threatening opportunistic infections. HIV evades CTL recognition and lysis through the activity of the HIV-1 Nef protein (1), which disrupts major histocompatibility complex class I (MHC-I) antigen presentation (2) and the development of CTLs in vivo (3). Three amino acids in the cytoplasmic tail of MHC-I HLA-A and HLA-B allotypes (YXXXAXXD) are essential for responsiveness to Nef (4). In contrast, HLA-C allotypes, which lack two of these amino acids (CXXXAXXN), are not affected by Nef. HIV-infected people with elevated HLA-C expression have lower viral loads and an improved prognosis (reviewed in reference 5).
The HIV-1 Nef protein binds to HLA-A and HLA-B cytoplasmic tails and stabilizes an interaction between the cytoplasmic tail tyrosine and the clathrin adaptor protein 1 (AP-1) (6). AP-1 normally recognizes YXXϕ or (D/E)XXXLL trafficking signals in protein cargo and facilitates trafficking between the trans-Golgi network (TGN) and the endolysosomal pathway (7). The MHC-I YXXXAXXD AP-1 signal is cryptic and does not function in the absence of Nef in T cells (6). The crystal structure of this complex elegantly reveals how additional contacts provided by Nef stabilize the weak interaction between AP-1 and the MHC-I cytoplasmic tail (8). Nevertheless, the presence of this extended, highly conserved cryptic AP-1 binding site in the MHC-I cytoplasmic tail raises the possibility that AP-1 might naturally use this signal in some contexts.
In addition to the classical pathway for endogenous antigen presentation that all cells utilize, antigen-presenting cells (APCs), including macrophages and dendritic cells, present exogenous antigenic peptides in association with MHC-I using a process referred to as cross-presentation. The cross-presentation pathway is important for the activation of naive CD8+ T cells, a necessary first step for the development of effective immune responses to viral, bacterial, self-, and tumor-associated antigens (reviewed in reference 9).
The tyrosine residue in the MHC-I HLA-A and HLA-B cytoplasmic tails that forms the cryptic AP-1 signal is conserved in murine MHC-I allotypes. Transgenic mice expressing H-2Kb with a mutation in the cytoplasmic tail tyrosine mount inferior H-2Kb-restricted CTL responses against two immunodominant viral epitopes in vivo (10). However, the mechanism by which this tyrosine affects antigen presentation and the development of the CTL response is unknown.
Here, we demonstrate that in APCs, the cryptic AP-1 signal in MHC-I HLA-A and HLA-B cytoplasmic tails acquires the capacity to bind AP-1 and that this interaction is necessary for cross-presentation of exogenous antigens. Thus, we show that for HLA-A and HLA-B molecules, the cytoplasmic tail tyrosine is part of a cell-type-specific AP-1 signal that allows trafficking of MHC-I into cross-presentation compartments in APCs. We also demonstrate that this signal is needed for effective cross-priming of naive primary T lymphocytes. In contrast, MHC-I molecules containing HLA-C cytoplasmic tails, which naturally lack the conserved cytoplasmic tail tyrosine, do not require AP-1 to cross-present soluble antigen. Moreover, we show that the requirement for AP-1 is specific for cross-presentation and is not necessary for presentation of endogenous antigens via the classical MHC-I presentation pathway. Finally, we show that the HIV-1 Nef protein disrupts the natural AP-1-dependent MHC-I HLA-A and HLA-B cross-presentation and cross-priming pathways but does not affect cross-presentation by HLA-C. These results have important implications for understanding normal immune responses to viral antigens and mechanisms of viral immune evasion.
MATERIALS AND METHODS
DNA constructs.
The murine stem cell virus (MSCV) vector expressing hemagglutinin (HA) and HLA-A2 (MSCV HA-HLA-A2) (11), MSCV HA-HLA-A2-Y320A (6), the retroviral vector expressing the internal ribosome entry site (IRES) and placental alkaline phosphatase (PLAP) (MSCV IRES PLAP) (6), the retroviral vector in which AP-1 activity was inhibited by a dominant negative mutant that is unable to bind tyrosine signals (TBPM) and in which IRES and PLAP were expressed (MSCV AP-1 TBPM IRES PLAP) (6), and short hairpin RNA (shRNA) against an irrelevant sequence (negative-control shRNA [shNC]) and shRNA against the AP-1 μ1 subunit (shμ1) (12) have all been described previously.
MSCV Kb/A and Kb/C retroviral vectors were created by subcloning chimeric PCR products into XhoI and HpaI restriction sites of MSCV 2.1. The chimeras were created through a two-step PCR fusion protocol. The Kb template was pRSVH2-Kb, which was kindly provided by Yik Yeung Lawrence Yu. The HLA-A2 template was MSCV HLA-A2 (11), and the HLA-C template was HLA-Cw4 (13). Primer sequences are listed below. Step 1 primers were 5′ H2-Kb XhoI and 3′ overlap primers (3′ Kb-A2 overlap or 3′ Kb-C overlap) for amplification from pRSVH2-Kb and 5′ overlap primers (5′ Kb-A2 overlap or 5′ Kb-C overlap) and a primer (3′ HLA-A2 XhoI or 3′ HLA-C XhoI) for amplification from MSCV HLA-A2. Step 2 primers were 5′ H2-Kb BamHI and 3′ primers (3′ HLA-A2 XhoI or 3′ HLA-C XhoI) to create the chimeric PCR product. The H2-Kb sequence begins at amino acid position 1 and ends at amino acid position 331, just after the transmembrane domain. The HLA-A2 or HLA-Cw4 cytoplasmic tail sequence starts at amino acid position 332 of the chimera and for HLA-A2 continues for 31 amino acid residues to the stop codon (*) (RKSSDRKGGSYSQAASSDSAQGSDVSLTGL*) and for HLA-Cw4 continues for 34 amino acid residues to the stop codon (RKSSGGKGGSCSQAASSNSAQGSDESLIACKA*).
The HIV-7SF-green fluorescent protein (GFP) lentiviral vector was created and described previously (14) and was modified to express AP-1 TBPM upstream of the IRES GFP reporter. Briefly, linker primers (5′ AP-1 XbaI linker and 3′ AP-1 EcoRV linker) were designed to create XbaI and EcoRV restriction sites on the 5′ and 3′ ends of the AP-1 TBPM gene during amplification in a PCR using MSCV AP-1 TBPM IRES PLAP as a template. pHIV-7SF-GFP was digested with XbaI and HpaI, which cut at unique sites immediately upstream of the IRES GFP reporter. The digested plasmid was then ligated to the XbaI- and EcoRV-digested AP-1 TBPM PCR product. The resultant clones were confirmed through DNA sequencing.
The MSCV IRES GFP retroviral vector was created and described previously (15) and was modified to express ovalbumin (OVA) upstream of the IRES GFP reporter. Briefly, ovalbumin contains a secretion signal sequence beginning at amino acid 21 and ending at residue 47 (16). To assess the specific effect of TBPM on endogenous antigen presentation, linker primers (5′ OVA BglII-del 50 and 3′ OVA EcoRI) were designed to truncate the amino terminus and delete the secretion signal sequence from ovalbumin as well as to introduce an in-frame start codon at amino acid position 50 of the protein. Linker primers were engineered with BglII and EcoRI sites for cloning the deleted OVA into MSCV IRES GFP upstream of the IRES GFP reporter. The resultant clones from ligation of OVA into MSCV IRES GFP were confirmed through DNA sequencing.
Primer sequences.
The primer sequences were as follows: primer 5′ AP-1 XbaI linker, 5′-CGTCTAGAACCAGGATGTCCGCCAGCGCCGTCTACGTG-3′; primer 3′ AP-1 EcoRV linker, 5′-CGGATATCTCACTGGGTCCGGAGCTGGTAATC-3′; primer 5′ OVA BglII-del 50, 5′-GGAGATCTACCATGCAGGACACAGATAAATAAGG-3′; primer 3′ OVA EcoRI, 5′-GCGAATTCTTAAGGGGAAACACATCTGCCAAA-3′; primer 5′ H2-Kb XhoI, 5′-CACCTCGAGATGGTACCGTGCACG-3′; primer 3′ Kb-A2 overlap, 5′-ATCTGAGCTCTTCCTCCTCATCTTCATCACAAAAGCCAC-3′; primer 5′ Kb-C overlap, 5′-GTGGCTTTGTGATGAAGATGAGGAGGAAGAGCTCAGGT-3′; primer 5′ H2-Kb BamHI, 5′-CGCGGGATCCGCGACCATGGTACCGTGCACGCTGCTC-3′; primer 3′ HLA-A2 XhoI, 5′-CCGCTCGAGCGGTCACACTTTACAAGCTGTGAG-3′; and primer 3′ HLA-C XhoI, 5′-CCGCTCGAGTCAGGCTTTACAAGCGATGAGAGA-3′.
Cell lines and primary cell isolation.
(i) CEM-A2 cells. CEM T cells expressing HA-tagged HLA-A2 (CEM-A2 cells) were described previously (11). CEM-A2 cells were maintained in RPMI 1640 medium (GIBCO) formulated with 4.5 g/liter glucose, l-glutamine, and 100 mg/liter sodium pyruvate (CEM RPMI) supplemented with 10% fetal bovine serum (FBS), 10 mM HEPES, and 2 mM penicillin, streptomycin, and glutamine (R-10).
(ii) THP-1 cell lines.
The THP-1 (ATCC) monocytic cell line was maintained in RPMI 1640 medium (Gibco) with 2 mM l-glutamine that was modified to contain 10 mM HEPES, 1 mM sodium pyruvate, 4.5 g/liter glucose, and 1.5 g/liter sodium bicarbonate (THP-1 RPMI) and supplemented with 10% FBS and 0.05 mM 2-mercaptoethanol. For induction into macrophages, THP-1cells were stimulated with 100 ng/ml lipopolysaccharide (LPS) (day 1), followed 24 h later by the addition of 100 ng/ml phorbol 12-myristate 13-acetate (PMA) for 6 more days.
To create HA-A2, HA-A2-Y320A, Kb/A, and Kb/C stable lines, THP-1 monocytes were transduced with the corresponding retroviral vectors. Stable lines were selected and subsequently maintained in medium containing 1 mg/ml G418 (Geneticin; Gibco).
(iii) Primary human macrophages.
Leukopaks provided by the New York Blood Center were purified by Ficoll-Hypaque centrifugation, and CD14+ mononuclear cells were isolated using an EasySep human CD14+ selection kit (StemCell Technologies). To induce maturation, CD14+ cells were incubated in the presence of 50 ng/ml granulocyte-macrophage colony-stimulating factor (GM-CSF) and 50 ng/ml macrophage colony-stimulating factor in R-10 for 7 days.
(iv) Primary mouse dendritic cells.
C57BL/6 mouse bone marrow was obtained frozen from Astarte Biologics, thawed, and sorted using a negative-selection mouse hematopoietic progenitor cell enrichment kit (Stem Cell Technologies) following the manufacturer's instructions. Enriched progenitors were then used for lentiviral transduction and subsequently cultured for 5 days in R-10 supplemented with 50 ng/ml GM-CSF.
Virus preparation and transductions.
(i) Adenoviral vectors. An adenoviral vector expressing Nef was provided by the University of Michigan Gene Vector Core facility. Adenoviral transductions were performed in CEM-A2 cells as previously described (17). THP-1 and primary human macrophages were both transduced on day 4 of the induction protocol. Adherent macrophages were harvested at 72 h postransduction using cell dissociation solution (Invitrogen). The adenoviral vector was titrated, and Nef levels were assessed by Western blotting to achieve equal intracellular Nef concentrations between different cell types.
(ii) Retroviral vectors.
Retroviral supernatants were prepared as previously described (15, 18). Briefly, Bosc cells were transfected with MSCV constructs plus the retrovirus packaging vector pCL-Eco (19) and pseudotyped with pHCMV-G (20). Viral supernatants were harvested at 48 h after transfection. THP-1 cells were spin transduced in a tabletop centrifuge at a density of 5 × 105 cells per ml retroviral supernatants plus 8 μg/ml Polybrene at 2,500 rpm for 4 h at 25°C.
(iii) Lentivirus.
Lentiviral vectors were produced by transfection of 293 cells. pHIV-7SF-GFP vectors included the addition of helper plasmids pCMV-HIV-1 and pHCMV-G. shNC and shμ1 included the helper plasmids pRRE (21), pRSV-Rev (21), and pHCMV-G (22). Viral supernatants were harvested at 48 h after transfection. THP-1 cells were spin transduced in a tabletop centrifuge at 2,500 rpm for 4 h at 25°C at a density of 5 × 105 cells per ml lentiviral supernatants plus 8 μg/ml Polybrene. For transduction of bone marrow progenitors, lentiviral supernatants were harvested at 48 h posttransfection and then concentrated 10-fold by centrifugation at 10,000 × g for 4 h at 4°C. Cleared medium was removed by aspiration and viral pellets were resuspended in StemSpan serum-free medium (StemCell Technologies). The 10-fold-concentrated lentiviral supernatants were then used for spin transductions with freshly isolated hematopoietic progenitors in a tabletop centrifuge at 2,500 rpm for 2 h at 25°C at a density of 5 × 105 cells per ml plus 8 μg/ml Polybrene.
Flow cytometry.
For SIINFEKL peptide pulse experiments, cells were incubated in 1 mM SIINFEKL peptide (AnaSpec Inc.) for 1 h at 37°C prior to beginning the antibody staining. Stimulated cells were preincubated in biotin-free Fc receptor blocker (Accurate Chemical and Scientific Corp.) for 20 min on ice prior to antibody staining. Cells were stained with primary and secondary antibodies using standard techniques. Antibodies directed against the following proteins were used: PLAP (Serotec), HLA-A2 (BB7.2) (23) ascites (provided by the University of Michigan Hybridoma Core Facility; antibody was purified as previously described [24]), HA (HA.11; Covance), H-2Kb/H-2Db (BD Pharmingen), 25.D1.16 (eBioscience), and IgG2b isotype control (BD Biosciences). Secondary antibodies used were Alexa Fluor 488 goat mouse IgG1, R-phycoerythrin goat anti-mouse IgG2b (Invitrogen) for BB7.2, Alexa Fluor 488 goat mouse IgG1 for PLAP, R-phycoerythrin goat anti-mouse IgG (Invitrogen), and streptavidin R-phycoerythrin (Invitrogen). For all flow cytometry analyses, stained cells were analyzed on a Becton Dickinson FACScan or FACSCanto cytometer. Analysis was performed using FlowJo software (Tree Star Inc.).
MHC-I internalization and recycling assays.
The MHC-I internalization assay was performed as previously described using stimulated THP-1 cells and harvesting of the cells at the time points indicated (12). THP-1 cells were stained for HA using the standard flow cytometry techniques described above. The MHC-I recycling assay was developed previously (24). Stimulated THP-1 cells were harvested at the time points indicated and stained with the BB7.2 MHC-I antibody.
Immunoprecipitation and Western blotting.
Whole-cell lysates (WCLs) were normalized for total protein prior to polyacrylamide gel electrophoresis. Western blot assays to examine AP-1 μ1 expression were performed using standard techniques with anti-AP-1 μ1 (RY/1) antibody (25). Tubulin (Sigma) was used as a loading control. The secondary antibody for anti-AP-1 μ1 was horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (Zymed), and HRP-conjugated rat anti-mouse IgG1 (Zymed) secondary antibody was used for tubulin. Immunoprecipitations were performed as previously described (6), except that where indicated anti-HA agarose (Sigma) was used. Immunoprecipitates were eluted and analyzed by Western blotting as previously described (17). Antibodies for Western blotting were anti-HA antibody HA.11 (Covance) and anti-AP-1 γ (BD Biosciences). Secondary antibody for HA.11 was HRP-conjugated rat anti-mouse IgG1 (Zymed), and secondary antibody for anti-AP-1 γ was HRP-conjugated rat anti-mouse IgG (Zymed).
Confocal microscopy.
For all confocal microscopy experiments, 8-chambered glass slides (Lab-Tek II chamber slide) were pretreated with poly-l-lysine (Sigma), washed with distilled H2O, and allowed to dry prior to addition of cells. For THP-1 cell experiments, cells were stimulated with LPS and PMA as described above at a density of 1 × 105 cells per chamber. At 7 days after stimulation, cells were fixed and stained. For primary dendritic cell experiments, cells were induced with GM-CSF as described above at a density of 1 × 105 cells per chamber and subsequently fixed and stained on day 5. For microscopy staining, cells were washed with phosphate-buffered saline (PBS) containing calcium and magnesium (PBS++) twice before fixing in 2% paraformaldehyde for 15 min at room temperature. Cells were then washed again with PBS++ twice before they were permeabilized in 0.4% Tween for 15 min at 37°C. After permeabilization, cells were washed with PBS++ twice before treatment with (Accurate) Fc receptor blocker in PBS++ containing 2% goat serum, 10 mM HEPES, and 1% NaN3 (goat serum wash) for 30 min on ice prior to antibody staining. All cells were stained with primary and secondary antibodies for 30 min on ice in goat serum wash, except for 25.D1.16 antibody, which was incubated overnight at 4°C. Cells were washed after each antibody staining by rinsing twice with goat serum wash, followed by 3 washes with 5-min incubations on ice. Antibodies to the following proteins were used for staining: SIINFEKL bound to Kb (25.D1.16; eBioscience), GFP (Invitrogen), PLAP (Thermo), HA (HA.11, Covance), and AP-1 γ (Sigma). Secondary antibodies were streptavidin 546 and Alexa Fluor 488 IgG2a, Alexa Fluor 546 IgG1, and Alexa Fluor 647 IgG2b. All secondary antibodies were obtained from Molecular Probes. All samples were treated with ProLong Gold antifade reagent with DAPI (4′,6-diamidino-2-phenylindole; Invitrogen) after antibody staining. Images were collected using a Zeiss LSM 510 confocal microscope and assembled using Adobe Photoshop software. During all image capture, settings were not adjusted, once the level of the background signal was determined for each antibody using isotype or other specified negative controls.
Cross-presentation assay.
(i) Primary bone marrow-derived APCs. Hematopoietic progenitor cells isolated from mouse bone marrow as described above were transiently transduced with the HIV-7F-GFP with or without TBPM as described above. Immediately after transduction, the viral supernatant was removed and cells were resuspended in R-10 supplemented with 50 ng/ml GM-CSF. Cells were stimulated in 8-chambered microscope slides at a density of 1 × 105 cells per chamber. At 5 days poststimulation, 5 mg/ml OVA was added for 4 h at 37°C. Where indicated, 5 mg/ml bovine serum albumin (BSA) was included as a negative control. The cells were subsequently washed three times with PBS++ prior to fixing and permeabilizing steps for confocal microscopy.
(ii) THP-1 cells.
On day 7 poststimulation, 5 mg/ml OVA or BSA was added for 6 h at 37°C. Positive-control cells were pulsed with 1 mg/ml SIINFEKL peptide for 1 h at 37°C. All cells were subsequently washed three times with PBS++ prior to fixing and permeabilizing steps for confocal microscopy.
OVA uptake assay.
One milligram of ovalbumin was labeled using an Alexa Fluor 647 protein labeling kit (Molecular Probes), and cells were incubated with 0.01 mg/ml of Alexa Fluor 647-tagged OVA protein (OVA-647) for 4 h at 37°C. After incubation, cells were washed three times with twice the volume of PBS++ and then harvested using cell dissociation solution (Sigma).
Mice.
OT-1 transgenic mice were purchased from The Jackson Laboratory. Mice were maintained in a pathogen-free mouse facility. The mice were cared for according to the guidelines set by the University of Michigan Committee on Use and Care of Animals (UCUCA).
Ethics statement.
This study was carried out in strict accordance with the recommendations in the guide for the care and use of laboratory animals of the National Institutes of Health (26). Experimental mouse protocols were approved by the UCUCA (UCUCA protocol 09549). The University of Michigan animal welfare number for the National Institutes of Health Office for Protection from Research Risks (OPRR) is A3114-01. Mouse euthanasia was performed using CO2 overdose prior to organ harvest, and all efforts were made to minimize any animal suffering.
Purification of CD8+ OT-1 mouse T cells.
Spleens were extracted from OT-1 mice. The red blood cells were lysed using red cell lysis buffer (Sigma), and the CD8+ cells were isolated by positive selection using anti-CD8a (Ly-2) antibody-coated magnetic beads (magnetically activated cell sorter; Miltenyi Biotec).
CD8+ T cell cross-priming assay.
On day 7 poststimulation, THP-1 cells were incubated with the indicated amount of ovalbumin (or SIINFEKL peptide) at 37°C for 3 h. The cells were then fixed with 1% formaldehyde for 7 min at room temperature and washed with 200 μl PBS, following a final wash with RPMI medium. Medium was aspirated from the wells, and a suspension of 0.5 × 105 to 1.5 × 105 OT-1 cells in 250 μl RPMI medium was added to the wells. At 24 h, interleukin-2 (IL-2) levels were measured by enzyme-linked immunosorbent assay (ELISA).
IL-2 ELISA.
Plates were coated with purified anti-mouse IL-2 (catalog no. 554424; BD Pharmingen) overnight at room temperature. After blocking with 10% calf serum for 3 h, cell culture supernatants were incubated at 37°C for 1 h. The plates were washed 3 times with 0.05% Tween 20 in PBS, and a biotinylated antibody directed against IL-2 (catalog no. 554426; BD Pharmingen) was added to the plates for an overnight incubation at room temperature. The plates were washed 3 times, and streptavidin conjugated to horseradish peroxidase (catalog no. 554066; BD Pharmingen) was added to the plates for 20 min at room temperature. The plates were again washed 3 times, and the assay was developed using a tetramethylbenzidine substrate reagent set (catalog no. 555214; OptEIA; BD Pharmingen). The colorimetric assay result was read on a microplate reader (Synergy HT; Bio-Tek).
Statistical analysis.
An unpaired t test was used in statistical analyses. Two-tailed P values are indicated in the text.
RESULTS
AP-1 coprecipitates with MHC-I in macrophages, and this interaction depends on the MHC-I cytoplasmic tail tyrosine.
In HIV-infected T cells, Nef promotes an interaction between AP-1 and a cryptic AP-1 signal in the MHC-I HLA-A and HLA-B cytoplasmic tails, which is normally inactive in T cells. Nevertheless, the presence of this extended, highly conserved cryptic AP-1 binding site in the MHC-I cytoplasmic tail (YXXXAXXD) raised the possibility that AP-1 might naturally use this signal in a different context (Fig. 1a). Indeed, we observed coprecipitation of wild-type MHC-I HLA-A2 with AP-1 in THP-1 macrophages (Fig. 1b). Mutation of the tyrosine residue in the MHC-I cytoplasmic tail (HA-A2-Y320A) disrupted this interaction (Fig. 1b). Thus, the MHC-I HLA-A and HLA-B AP-1 tyrosine signal is active in macrophages.
Fig 1.

AP-1 coprecipitates with MHC-I proteins containing a cytoplasmic tail tyrosine, and AP-1 activity is required for cross-presentation in murine primary bone marrow-derived APCs. (a) Model for an interaction between HA-tagged human MHC-I (black) and clathrin AP-1 (gray oval). Residues in the MHC-I cytoplasmic tail previously shown to be important for AP-1 complex formation in Nef expressing T lymphocytes are indicated (Y320XXXA324XXD327). PM, plasma membrane. (b) Coimmunoprecipitation (co-IP) and Western blot (WB) analysis of MHC-I and AP-1 in differentiated THP-1 cells expressing the indicated MHC-I. Parental THP-1 cells were used as a negative control. The input was the whole-cell lysate of the indicated sample. (c) Model for an association between murine H2-Kb (light blue) and AP-1 (gray oval). The conserved tyrosine residue in the cytoplasmic tail region is indicated. Arrowheads indicate the positions of AP-1 and HA-A2 on the blot. (d). Schematic of lentiviral vectors used to express the AP-1 dominant negative mutant (TBPM) and GFP or GFP alone. (e) Immunofluorescent, confocal microscopic analysis of cross-presentation by murine bone marrow progenitor cells transduced with the indicated lentiviral vector, differentiated with GM-CSF, pulsed with soluble OVA or BSA, and stained with an antibody recognizing the ova peptide antigen (SIINFEKL)-Kb complex (25D1.16) and GFP. (f) Relative amount of cross-presentation signal from cells transduced with the indicated lentiviral vector and scored by a panel of 5 blinded reviewers. The mean scores ± standard error of the mean are shown (n = 38 and 41 for vector and TBPM, respectively). **, P < 0.02.
AP-1 is required for effective cross-presentation in primary bone marrow-derived APCs.
To investigate whether the interaction between AP-1 and the MHC-I cytoplasmic tail tyrosine was functionally significant, we asked whether AP-1 activity was required for cross-presentation of exogenous antigens by primary murine APCs, which express MHC-I molecules (H2-Kb) containing the conserved cytoplasmic tail tyrosine (Fig. 1c). For these experiments, AP-1 activity was inhibited with a dominant negative mutant that is unable to bind tyrosine signals (TBPM) (6). This mutant was introduced into primary murine bone marrow hematopoietic progenitor cells using a lentiviral vector, which also contained an IRES-driven GFP reporter cassette (Fig. 1d). The transduced cells differentiated into APCs after culturing with GM-CSF, and cross-presentation was assayed. For this analysis, APCs were incubated with soluble OVA protein and then stained with a monoclonal antibody (25.D1.16) that specifically recognizes the processed OVA peptide SIINFEKL bound to the murine MHC-I molecule H2-Kb (27). Using this assay, we readily detected cross-presenting primary murine APCs in control samples (Fig. 1e and f). However, inhibition of AP-1 tyrosine-binding activity by expression of TBPM in primary murine APCs significantly reduced the ability of the APCs to cross-present (Fig. 1e and f; P < 0.02).
AP-1 is required for effective cross-presentation in human monocytic cell lines.
The partial effect of TBPM expression on cross-presentation (Fig. 1f) may indicate the presence of multiple trafficking pathways, only some of which are AP-1 dependent. Alternatively, the partial effect may be due to low transduction of primary cells. Therefore, to further explore the role of AP-1 in cross-presentation, we used cells that were easier to manipulate in vitro.
For these studies, we used the human THP-1 cell line, a human acute monocytic leukemia cell line. THP-1 cells can be induced to differentiate into mature macrophages through stimulation with exogenous LPS and PMA. In addition, we engineered a chimeric MHC-I molecule that contained the murine Kb extracellular domain and the human HLA-A2 cytoplasmic tail (Kb/A; Fig. 2a) and stably expressed this molecule in THP-1 cells.
Fig 2.

AP-1 is required for cross-presentation in human macrophages and for cross-priming of primary CD8+ T cells. (a) Schematic diagram of the Kb/A chimeric MHC-I molecule, which contains the extracellular and transmembrane domains of H2-Kb (light blue) and the human HLA-A cytoplasmic tail sequence (black). The association of AP-1 (gray) with the MHC-I cytoplasmic tail is shown. (b) Immunofluorescent, confocal microscopic analysis of THP-1 Kb/A cells transduced with the indicated lentiviral vector, sorted by flow cytometry for GFP expression, differentiated, pulsed with OVA or BSA, as indicated, and stained with 25.D1.16. After sorting, the cell populations were 95% (vector) and 92% (TBPM) GFP-positive cells. (c) Lower-power magnifications of the images from panel b. (d) Quantification of cross-presentation signal by a panel of 5 blinded reviewers scoring the presence (score, 10) or absence (score, 0) of the cross-presentation signal in each cell. The mean scores for each condition ± standard error of the mean are shown (n = 16, 27, and 36 for untransduced, vector-expressing, and TBPM-expressing cells, respectively). ***, P < 0.0001. (e) Flow cytometric analysis of THP-1 Kb/A cells treated as described for panel b, except that they were pulsed with synthetic SIINFEKL peptide prior to staining with 25.D1.16. The control was differentiated parental THP-1 cells treated in parallel. (f) IL-2 production by murine primary CD8+ OT-1 T lymphocytes cocultured in triplicate with cells from the experiments whose results are shown in panels a to d. Where indicated, cells were pulsed with synthetic SIINFEKL peptide instead of OVA as a positive control.**, P < 0.001.
The human macrophages expressing the chimeric molecule Kb/A readily cross-presented SIINFEKL after incubation with soluble OVA, and cross-presentation was not affected by transduction with vector alone (Fig. 2b). However, inhibition of AP-1 tyrosine-binding activity by the expression of TBPM resulted in a reduction in cross-presentation (Fig. 2b) that was readily apparent in lower-power-magnification images when contrasted with that for untransduced cells or vector controls (Fig. 2c). Quantitation revealed that the effect was a highly significant 4-fold reduction in cross-presentation signal (P < 0.001; Fig. 2d). In contrast, TBPM did not affect the amount of Kb/A that was available to bind synthetic SIINFEKL peptide (Fig. 2e).
AP-1 is required for cross-priming of naive CD8+ primary T cells.
We then asked whether the inhibition of cross-presentation that we observed by microscopy was functionally significant in a cross-priming assay system that measures the capacity of cross-presenting APCs to prime naive primary T cells. This system utilizes OVA-specific naive primary CD8+ T cells freshly isolated from OT-1 transgenic mice (28). These T cells express a transgenic receptor that specifically recognizes SIINFEKL when presented by the mouse H2-Kb molecule. Effectively primed OT-1 T cells release a cascade of cytokines, including IL-2. We found that untransduced and vector-transduced human macrophages expressing the chimeric molecule Kb/A readily primed OT-1 primary T cells following exposure to exogenous OVA (Fig. 2f). However, inhibition of AP-1 activity by expression of TBPM significantly reduced the ability of APCs to effectively cross-prime and activate naive primary T cells to secrete IL-2 (P < 0.001; Fig. 2f). The effect of TBPM on cross-priming was not due to a nonspecific reduction in total MHC-I expression, as APCs expressing TBPM could still prime cells if they were pulsed with synthetic SIINFEKL peptide (Fig. 2f). Thus, TBPM functioned by specifically inhibiting the ability of APCs to effectively process and present soluble exogenous antigen.
Silencing AP-1 inhibits cross-presentation in human macrophages.
To ensure that the effect of TBPM was specific, we also inhibited AP-1 activity by RNA silencing. This was accomplished using lentiviral vectors expressing shRNAs against either an irrelevant sequence (shNC) or the AP-1 μ1 subunit (shμ1 [12]) (Fig. 3a). THP-1 cells expressing Kb/A were transduced with these vectors, sorted by flow cytometry for GFP expression, and induced to differentiate into macrophages. Silencing was confirmed by Western blotting (Fig. 3b), and cross-presentation was assayed as described for Fig. 2. As expected, similar levels of GFP, a marker of transduction, were observed in shNC and shμ1 (data not shown and Fig. 3c, bottom). Similar to the results observed using TBPM, silencing of AP-1 with shμ1 significantly reduced cross-presentation (P < 0.0001; Fig. 3c, with quantification shown in Fig. 3d) without reducing the amount of Kb/A available to bind synthetic SIINFEKL peptide (Fig. 3e).
Fig 3.

Silencing of AP-1 inhibits cross-presentation by MHC-I containing the conserved cytoplasmic tail tyrosine; AP-1 is not required for cross-presentation by HLA-C, which lacks the conserved cytoplasmic tail tyrosine. (a) Schematic of lentiviral vector expressing shRNAs against AP-1 μ1 (shμ1) or a negative-control shRNA (shNC). (b) Western blot analysis of THP-1 Kb/A cells transduced with the indicated lentiviral vector, sorted by flow cytometry for GFP expression, and differentiated into the macrophage lineage. (c) Immunofluorescent, confocal microscopic analysis of THP-1 Kb/A cells transduced with the indicated lentiviral vector, sorted by flow cytometry for GFP expression, differentiated, pulsed with OVA, as indicated, and stained with 25.D1.16 and GFP. The control was parental THP-1 cells treated in parallel. (d). Quantification of cross-presentation signal by a panel of 5 blinded reviewers scoring the presence (score, 10) or absence (score, 0) of the 25.D1.16 signal in each cell. The mean scores for each condition ± standard error of the mean are shown (n = 9, 16, and 12 for untransduced, shNC-expressing, and shμ1-expressing cells, respectively). ***, P < 0.0001. (e) Flow cytometric analysis of THP-1 Kb/A cells treated as described for panel c, except that they were pulsed with synthetic SIINFEKL peptide prior to staining with 25.D1.16. (f) Sequence comparison of human HLA-A, -B, and -C cytoplasmic tail domains. The positions of conserved residues previously demonstrated to be required for AP-1 complex formation with the MHC-I HLA-A and -B alleles and HIV Nef in T lymphocytes are shaded in yellow. (g) Schematic of Kb/C chimeric MHC-I molecule. The Kb/C chimera contains the extracellular and transmembrane domain of H2-Kb (light blue) and the human HLA-C cytoplasmic tail sequence (dark blue). (h) Immunofluorescent, confocal microscopic analysis of THP-1 Kb/C cells transduced with the indicated lentiviral vector, differentiated, pulsed with OVA, as indicated, and stained with 25.D1.16 and GFP. The control was parental differentiated THP-1 cells treated in parallel. (i) Quantification of cross-presentation signal by a panel of 5 blinded reviewers scoring the presence (score, 10) or absence (score, 0) of the 25.D1.16 signal in each cell. The mean scores for each condition ± standard error of the mean are shown (n = 17, 6, and 11 for untransduced, vector-expressing, and TBPM-expressing cells, respectively). (j) Flow cytometric analysis of THP-1 Kb/C cells treated as described for panel h, except that they were pulsed with SIINFEKL peptide prior to staining with 25.D1.16. The GFP-positive population is shown for transduced cells.
AP-1 is not required for cross-presentation by MHC-I molecules containing HLA-C cytoplasmic tails.
While the highly conserved tyrosine in the AP-1 binding site in the human MHC-I cytoplasmic tail (YXXXAXXD) is conserved in all murine MHC-I molecules and in human HLA-A and HLA-B molecules, the more recently evolved human MHC-I HLA-C protein lacks two amino acids within this sequence (CXXXAXXN; Fig. 3f and g). Thus, if the HLA-C cytoplasmic tail domain supports cross-presentation, we expect that it must occur independently of AP-1. To test this, we engineered an H2-Kb molecule with a human HLA-C cytoplasmic tail (Kb/C; Fig. 3g) and created stable THP-1 cell lines expressing the chimeric molecule.
Remarkably, we found that the Kb/C chimera efficiently cross-presented ovalbumin antigen following incubation of differentiated THP1 cells with OVA (Fig. 3 h). Moreover, cross-presentation occurred in the presence of TBPM as well as untransduced or vector control cells (Fig. 3h, with quantification shown in Fig. 3i). As expected, there was no effect of TBPM on Kb/C binding to the synthetic SIINFEKL peptide in differentiated macrophages (Fig. 3j). Thus, the effect of TBPM on cross-presentation is highly specific to MHC-I molecules containing a cytoplasmic tail tyrosine signal. This rules out the possibility that TBPM has a nonspecific global effect on MHC-I trafficking. Moreover, these data indicate that HLA-C-dependent cross-presentation must occur by a mechanism different from that used by HLA-A and HLA-B.
AP-1 is not required for presentation of endogenous antigens by MHC-I molecules containing HLA-A and HLA-B cytoplasmic tails.
We also considered the possibility that AP-1 activity might be important for nonspecific trafficking of MHC-I molecules containing the cytoplasmic tail tyrosine rather than for trafficking that was uniquely important for cross-presentation. To examine this possibility, we expressed OVA endogenously in cells using a retroviral vector that expressed GFP while simultaneously expressing TBPM from a separate retroviral vector that had a distinct marker (PLAP) so that we could identify cells expressing both endogenous OVA and TBPM (Fig. 4a). We found that APCs expressing endogenous OVA (MSCV OVA IRES GFP) were able to process and present SIINFEKL peptide regardless of whether TBPM was expressed (Fig. 4b, bottom, with quantification shown in Fig. 4c), whereas control cells transduced with a vector expressing GFP (MSCV IRES GFP) alone did not stain positively in this assay (Fig. 4b, bottom).
Fig 4.
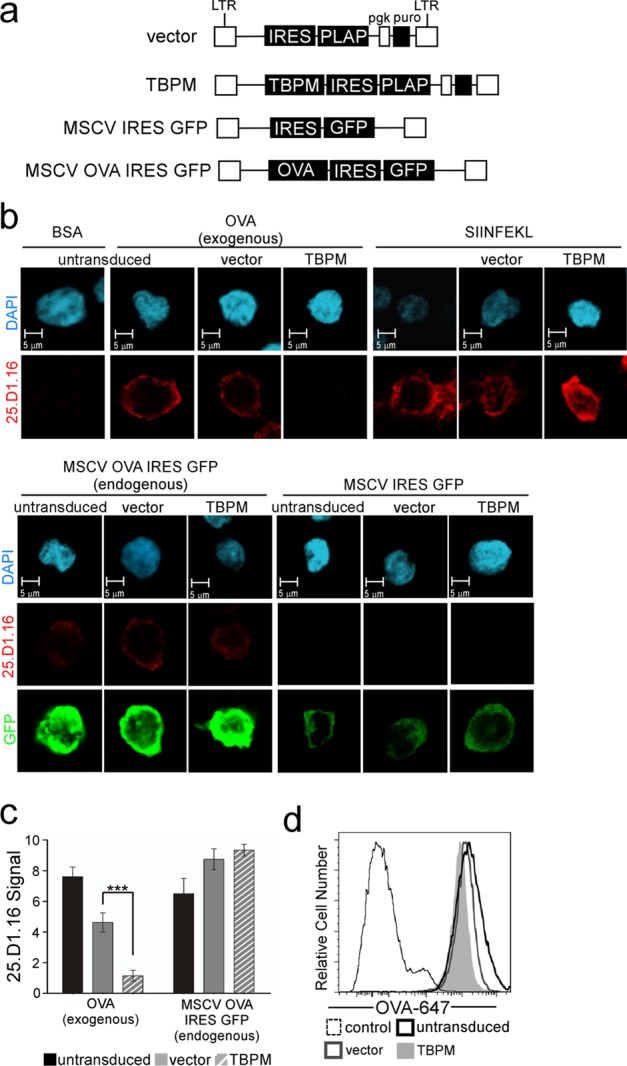
AP-1 activity is not necessary for presentation of endogenous antigens via the classical pathway. (a) Schematic map of retroviral vectors used to express TBPM and OVA with marker gene PLAP or GFP. LTR, long terminal repeat. (b) Immunofluorescent, confocal microscopic analysis of THP-1 Kb/A cells transduced with the indicated PLAP-expressing retroviral vector, sorted for PLAP expression, and transduced where indicated with a retroviral vector expressing OVA (MSCV OVA IRES GFP) or empty vector control (MSCV IRES GFP). The cell populations were 96% (vector) and 99% (TBPM) PLAP positive by flow cytometry analysis after sorting and stimulation. (Top) Cells were pulsed with BSA, OVA, or SIINFEKL peptide, as indicated, and stained with 25.D1.16; (bottom) unpulsed cells transduced with the indicated vector(s) were stained with an antibody against GFP and with 25.D1.16. (c) Quantification of antigen presentation measured by 25.D1.16 staining and scored by a panel of 5 blinded reviewers for the presence (score, 10) or absence (score, 0) of the 25.D1.16 signal in each cell. The numbers of images scored for exogenous or endogenous OVA presentation were 24 and 11, respectively, for untransduced cells; 27 and 8, respectively, for vector-expressing cells; and 35 and 13, respectively, for TBPM-expressing cells. The mean scores ± standard error of the mean are shown. ***, P < 0.0001. (d) Flow cytometric analysis of Alexa Fluor 647-labeled OVA (OVA-647) uptake in differentiated Kb/A THP-1 cells with or without TBPM expression, as described for panel a. The PLAP-positive population is shown for transduced cells.
In contrast, we again observed that inhibition of AP-1 activity by expression of TBPM dramatically reduced cross-presentation of the SIINFEKL epitope following incubation of the cells with soluble OVA (P < 0.0001; Fig. 4b, top, with quantification shown in Fig. 4c). TBPM did not alter the ability of Kb/A expressed by APCs to bind synthetic SIINFEKL peptide (Fig. 4b, top). Thus, these experiments indicate a highly specific role for AP-1 in cross-presentation of exogenous antigens by MHC-I in APCs.
To rule out the possibility that inhibition of AP-1 affected the internalization of exogenous antigen, we measured the effect of TBPM on the uptake of a fluorescently tagged OVA protein (OVA-647). We found that all cell populations uniformly took up OVA-647 to similar levels and that this was not affected by TBPM expression (Fig. 4d). Thus, AP-1 activity is not necessary for antigen uptake.
AP-1 activity and the MHC-I cytoplasmic tail tyrosine are both needed to direct MHC to AP-1+ organelles in macrophages.
In HIV-infected T lymphocytes, Nef-dependent AP-1 binding to the MHC-I cytoplasmic tail does not affect MHC-I transport through the secretory pathway until MHC-I reaches the TGN (11). However, transport from the TGN to the plasma membrane is dramatically reduced by this interaction and MHC-I is instead targeted from the TGN to AP-1-positive (AP-1+) organelles and, ultimately, to lysosomes (11). In uninfected macrophages, we hypothesized that AP-1 similarly affected post-TGN trafficking of MHC-I molecules but that in macrophages, MHC-I would be targeted to specialized antigen presentation compartments. Therefore, we examined MHC-I localization in human macrophages with or without TBPM expression. In these studies, TBPM was expressed using retroviral vectors that also expressed a marker gene (PLAP; Fig. 5a). In control differentiated macrophages, we observed that MHC-I was localized to both intracellular compartments and the cell surface (Fig. 5b, top). Notably, the intracellular MHC-I colocalized strikingly with AP-1 (Fig. 5b). In contrast, inhibition of AP-1 with TBPM resulted in a dramatic relocalization of MHC-I from intracellular compartments and reduced MHC-I colocalization with AP-1 (Fig. 5b, bottom, with quantification shown in Fig. 5c; P < 0.02). Inhibition of AP-1 with TBPM expression resulted in a corresponding 2-fold increase in MHC-I surface expression over that of the vector control, as measured by flow cytometry (Fig. 5d, with quantification shown in Fig. 5e; P < 0.001). The exact same pattern was observed when the AP-1 signal was mutated to eliminate the required tyrosine residue (AXXXAXXD; Fig. 5f to i). These data support a model in which the role of AP-1 is to bind the tyrosine-based signal in the MHC-I cytoplasmic tail and redirect MHC-I from the TGN to a novel intracellular antigen presentation compartment that is best defined by AP-1 staining.
Fig 5.

AP-1 targets MHC-I molecules containing a cytoplasmic tail tyrosine to intracellular AP-1+ compartments prior to the cell surface. (a) Schematic of retroviral vectors used to express TBPM and PLAP. (b) Immunofluorescent, confocal microscopic analysis of THP-1 HA-A2 cells transduced with the indicated PLAP-expressing lentiviral vector, differentiated, and stained with antibodies against the indicated protein. MHC-I HA-HLA-A2 and AP-1 colocalization appear yellow in merge panels. PLAP (expressed on the cell surface) and HA-HLA-A2 colocalization appear purple in merge panels. (c) Quantification of AP-1 and MHC-I colocalization in PLAP-positive cells. Eleven images per condition were assessed by 5 blinded reviewers scoring the percentage of AP-1 colocalizing with MHC-I in each cell. The mean ± the standard error of the mean is shown. **, P < 0.02. (d) Flow cytometric analysis of MHC-I HLA-A2 surface expression in THP-1 cells transduced with the indicated retroviral vector. For vector and TBPM samples, PLAP-positive cells are shown. (e) Summary plot demonstrating the effect of TBPM on MHC-I surface expression relative to that of the vector control measured by flow cytometry. ***, P < 0.002 (n = 13). (f) Immunofluorescent, confocal microscopic analysis of differentiated THP-1 cells expressing the designated HA-tagged MHC-I HLA-A2 protein and stained with antibodies against the indicated protein. HLA-A2 and AP-1 colocalization appears yellow in merge panels. (g) Quantification of colocalization of AP-1 and MHC-I was scored by a panel of 4 blinded reviewers. The mean scores ± standard error of the mean are shown. ***, P < 0.001 (n = 4 cells per condition). (h) Flow cytometric analysis of HA-tagged MHC-I surface expression in THP-1 cells. The control was parental THP-1 cells treated in parallel. (i) Quantification of the surface expression of the indicated HA-tagged MHC-I measured by flow cytometry. *, P < 0.05 (n = 8). (j and k) Flow cytometric analysis of MHC-I surface stability in differentiated (j) or undifferentiated (k) THP-1 cells expressing the indicated HA-tagged HLA-A2 molecule. The mean percent remaining ± SD (n = 3) is shown. (l) Flow cytometric analysis of surface transport of newly synthesized and stored MHC-I HLA-A2 in THP-1 cells transduced with the indicated lentiviral vector, differentiated, and stripped of stainable surface MHC-I in a low-pH buffer at time zero. Results for GFP-positive cells are shown. (m) Flow cytometric analysis of surface transport of stored MHC-I HLA-A2 in THP-1 HA-A2 cells transduced with the indicated lentiviral vector, differentiated, treated with cycloheximide to inhibit new protein synthesis, and stripped of stainable surface MHC-I as described for panel l. The zero time point is the point at which surface-stainable MHC-I has been stripped from the cells. Data are representative of two independent experiments.
AP-1 activity is required for forward transport of stored MHC-I.
While our studies with Nef provide precedent for AP-1 acting primarily at the TGN and sorting newly synthesized MHC-I to intracellular compartments (11), it is also possible that AP-1 is acting at the cell surface and promoting internalization of surface MHC-I to intracellular compartments. To test this, we examined the internalization of wild-type MHC-I HLA-A2 compared to that of MHC-I HLA-A2 lacking the cytoplasmic tyrosine signal (Y320A). We found that wild-type HLA-A2 is stably maintained on the cell surface in differentiated macrophages (Fig. 5j) and that mutation of the tyrosine signal did not inhibit internalization. Indeed, we noticed somewhat more internalization of HLA-A2 in the absence of the tyrosine signal. Thus, the cytoplasmic tail tyrosine signal does not serve as an endocytosis signal in our system either in differentiated macrophages or in undifferentiated monocytes (Fig. 5j and k).
To better understand the role of AP-1 on intracellular transport and recycling, we examined MHC-I trafficking of both newly synthesized and recycled MHC-I molecules in the presence of TBPM using a flow cytometric assay (24). We found that inhibition of AP-1 activity by TBPM increased the rate of transport of total intracellular MHC-I (newly synthesized plus stored) compared to that in the vector controls (Fig. 5l). This result is consistent with our model that AP-1 redirects newly synthesized MHC-I molecules at the trans-Golgi network to intracellular compartments rather than directly to the cell surface.
In contrast, when new protein synthesis was inhibited, we observed that loss of AP-1 activity resulted in a reduction in the amount of MHC-I transported from stored compartments to the cell surface (Fig. 5m). Taken together, these data support a model in which AP-1 sorts MHC-I into intracellular compartments and the MHC-I stored in these compartments is eventually expressed at the cell surface. These data are consistent with a role for AP-1 in targeting MHC-I to intracellular antigen presentation compartments prior to presentation at the cell surface.
Nef disrupts cross-presentation by MHC-I molecules containing HLA-A and HLA-B cytoplasmic tails.
In T lymphocytes, AP-1 does not naturally interact with the MHC-I cytoplasmic tail, but this interaction can be induced by expression of the HIV-1 Nef protein. In T cells, AP-1 binding to MHC-I HLA-A and HLA-B molecules disrupts endogenous antigen presentation and targets MHC-I into the endolysosomal pathway for accelerated degradation. As another way of confirming the physiological AP-1 interaction with MHC-I in APCs, we directly compared the ability of Nef to disrupt surface MHC-I expression in APCs versus T cells. We found that Nef was substantially more effective at downmodulating total MHC-I from the cell surface in T cells than primary human macrophages and human macrophage cell lines (Fig. 6a, with quantification shown in Fig. 6b) and that this was not explained by differences in Nef expression in these cell types (Fig. 6c). This is most likely because MHC-I is already directed into an AP-1-dependent pathway in APCs and Nef only minimally alters it.
Fig 6.

HIV Nef inhibits cross-priming and cross-presentation by cells containing a cytoplasmic tail tyrosine. (a) Flow cytometric analysis of MHC-I surface levels in the indicated cell type transduced with an adenoviral vector expressing Nef or the empty-vector control (vector). The fold reduction in surface MHC-I HLA-A2 by Nef expression is indicated in each plot. Dashed histograms represent negative controls (isotype control for macrophage experiments or the CEM parental line for CEM-A2 samples). CEM-A2, CD4+ T cells stably expressing HLA-A2. (b) Quantification of MHC-I downmodulation by Nef in THP-1 or primary macrophages versus CEM-A2 cells. ***, P < 0.0001 (n = 14); **, P < 0.02 (n = 3). (c) Western blot analysis of Nef levels in transduced cells from the experiment whose results are presented in panel a. Western blots were also probed for tubulin as a loading control. (d) IL-2 production by primary CD8+ OT-1 mouse T lymphocytes cocultured in triplicate with differentiated Kb/A THP-1 cells that had been treated with Nef, as indicated, and pulsed with OVA. Where indicated, cells were pulsed with SIINFEKL peptide instead of OVA as a positive control. ***, P < 0.002. (e) Flow cytometric analysis of Kb/A or Kb/C expression levels in THP-1 cells treated with an adenoviral vector expressing Nef or a control adenoviral vector (d). The fold downmodulation of MHC-I by Nef is indicated. The control was differentiated THP-1 parental cells treated in parallel. (f) Immunofluorescent, confocal microscopy of differentiated Kb/A THP-1 cells transduced with Nef or adenoviral vector control, as indicated, pulsed with OVA, and stained with 25.D1.16. The control was differentiated THP-1 parental cells treated in parallel. (g) Quantification of cross-presentation signal by a panel of 5 blinded reviewers scoring the presence (score, 10) or absence (score, 0) of the 25.D1.16 signal in each cell. The mean scores for each condition ± standard error of the mean are shown (n = 19, 24, and 19 for untransduced, adenoviral vector-expressing, and Nef-expressing cells, respectively). ***, P < 0.0001. (h) Flow cytometric analysis of Alexa Fluor 647-labeled OVA (OVA-647) uptake in differentiated Kb/A THP-1 cells with or without the indicated adenoviral vector. The control was differentiated Kb/A THP-1 cells that were not incubated with OVA. (i) Immunofluorescent, confocal microscopy of differentiated Kb/C THP-1 cells transduced with Nef or adenoviral vector control, as indicated, pulsed with OVA, and stained with 25.D1.16. The control was differentiated THP-1 parental cells treated in parallel. (j) Quantification of cross-presentation signal by a panel of 5 blinded reviewers scoring the presence (score, 10) or absence (score, 0) of the 25.D1.16 signal in each cell. n = 19, 21, and 17 for untransduced, adenoviral vector-expressing, and Nef-expressing cells, respectively.
While Nef-dependent AP-1 binding of MHC-I aims to disrupt normal antigen presentation and send MHC-I to lysosomes, we propose that the natural interaction of AP-1 in APCs ultimately sends MHC-I molecules loaded with exogenous antigen back to the cell surface. Thus, we expected that Nef might disrupt cross-presentation by dysregulating the AP-1 interaction and altering normal trafficking. Indeed, we found that expression of the HIV-1 Nef protein in human macrophages reduced cross-priming of primary naive OT-1 mouse CD8+ T cells by 10-fold (Fig. 6d; P < 0.002). This effect of Nef was not due to total downmodulation of surface MHC-I, because Nef did not significantly alter the ability of cells to bind synthetic SIINFEKL peptide and prime OT-1 CD8+ T cells (Fig. 6d). Moreover, Nef exerted only a 2-fold reduction of surface Kb/A in APCs (Fig. 6e, left). We confirmed this result using microscopy, where we observed a cross-presentation signal in untransduced and control vector-expressing cells; however, this signal was significantly reduced in the presence of Nef (P < 0.0001; Fig. 6f, with quantification shown in Fig. 6g). The effect of Nef was not due to defects in antigen uptake, based on the fact that we did not observe reductions in uptake of OVA by differentiated THP-1 cells expressing Nef (Fig. 6h).
Nef does not affect cross-presentation by MHC-I molecules containing the HLA-C cytoplasmic tail.
MHC-I HLA-C proteins lack two amino acids within the AP-1 binding sequence (Fig. 3f and g), and because of this, Nef does not downmodulate HLA-C in CD4+ T cells (6). We confirmed that Nef had no significant effect on Kb/C surface expression in THP-1 cells (Fig. 6e, right), and we demonstrated that Nef expression did not significantly affect cross-presentation of soluble OVA epitopes by Kb/C (Fig. 6i, with quantification shown in Fig. 6j). Thus, the effect of Nef on cross-presentation by MHC-I molecules containing HLA-A and HLA-B tails is highly specific and related to cytoplasmic tail sequences previously implicated in AP-1 binding.
Nef dysregulates an interaction between MHC-I and AP-1 in macrophages.
To test whether the effect of Nef on MHC-I expression and cross-presentation was due to inefficient or dysregulated recruitment of AP-1 to the MHC-I cytoplasmic tail, we performed immunoprecipitation experiments with MHC-I HLA-A2 in differentiated THP-1 macrophages. While we again observed an AP-1 association with MHC-I in the absence of Nef in the untransduced and vector controls (Fig. 7, lanes 2 and 3), we also observed a dramatic enhancement of AP-1 coimmunoprecipitation with HLA-A2 when Nef was expressed in macrophages (Fig. 7, lane 4). This is in contrast to the results for T cells, where AP-1 was observed to coimmunoprecipitate with MHC-I only in the presence of Nef (11). These data confirm that AP-1 plays a previously unidentified role in normal MHC-I trafficking in APCs and, moreover, indicate that Nef disrupts MHC-I trafficking in macrophages by overly stabilizing and dysregulating this interaction.
Fig 7.
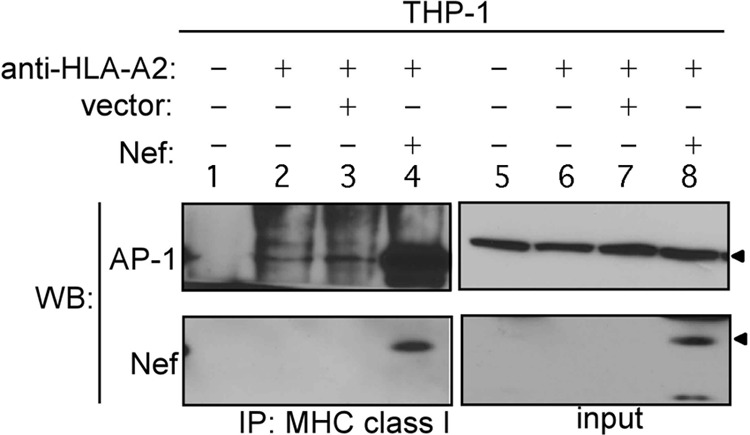
Immunoprecipitation and Western blot analysis of MHC-I HLA-A2 and AP-1 or Nef in untransduced THP-1 cells or THP-1 cells transduced with Nef or control vector. Lane 1, lysates from untransduced THP-1 cells immunoprecipitated with an isotype control antibody; input, whole-cell lysates of the indicated sample. Arrowheads inicate the positions of AP-1 and Nef on the blot.
DISCUSSION
The MHC-I cross-presentation pathway is essential for the development of CTLs against pathogens that do not productively infect APCs. Despite its importance, the molecular mechanism by which MHC-I is directed into the cross-presentation pathway is not known. Here we provide insights uncovered by studying the pathways targeted by HIV. We report that AP-1, which disrupts MHC-I transport to the cell surface in Nef-expressing T cells, naturally interacts with MHC-I in uninfected APCs. Similar to the Nef-dependent interaction, the natural interaction between AP-1 and MHC-I required a conserved tyrosine (Y320) in the MHC-I cytoplasmic tail. In addition, natural AP-1 activity reduced the transport of newly synthesized MHC-I to the cell surface. However, unlike Nef-dependent AP-1 trafficking, the natural AP-1-dependent pathway ultimately led to the transport of stored MHC-I molecules to the cell surface for antigen presentation.
The data supporting a role for AP-1 in cross-presentation that we have generated are robust and highly specific. We observed a requirement for AP-1 in both primary APCs and APC lines using both a dominant negative AP-1 mutant and RNA silencing. We found that AP-1 activity was required for cross-presentation by endogenous murine MHC-I molecules and by chimeric MHC-I molecules containing HLA-A/B cytoplasmic tails. In contrast, AP-1 activity was not needed for cross-presentation by MHC-I molecules containing HLA-C cytoplasmic tails, which lack the required tyrosine. Additionally, AP-1 activity was not required for endogenous antigen presentation via the classical MHC-I pathway.
It is not yet known what signal stabilizes the natural MHC-I–AP-1 interaction in APCs. A cell-type-specific Nef-like factor or posttranslational modification may activate the tyrosine-binding motif, allowing AP-1 recognition (Fig. 8). In addition to AP-1, we expect that cross-presentation requires some of the same cellular factors as endogenous antigen presentation, such as the transporter associated with antigen processing (TAP) and the proteasome, as has previously been shown (29–31). This expectation is due to the fact that MHC-I proteins must be appropriately loaded with endogenous peptides to efficiently exit the endoplasmic reticulum (ER) (32) and traffic to the TGN, where they would be accessible to AP-1 for subsequent transport to antigen-exchange compartments. TAP and the proteasome may additionally be required for transport of antigenic peptides into the phagosomal compartment (29–31). Moreover, CD74 may play a role in preventing premature peptide binding, which would facilitate endosomal uptake of peptides (33).
Fig 8.

Model of cross-presentation. (a) A subset of MHC-I loaded with endogenous peptide in the ER undergoes a posttranslational modification that allows regulated binding to AP-1 in APCs. Association with AP-1 redirects MHC-I into AP-1+ cross-presentation compartments. AP-1 is then released, antigen exchange occurs, and MHC-I exits to the plasma membrane for cross-presentation. Expression of the AP-1 dominant negative mutant TBPM, AP-1 silencing, or mutation of the cytoplasmic tail tyrosine disrupts the cross-presentation pathway and MHC-I traffics directly to the cell surface. (b) In HIV-infected cells, Nef (red rectangle) binds to the MHC-I cytoplasmic tail early in the secretory pathway. Subsequent binding to AP-1 leads to an overly stable complex. AP-1 is not efficiently released, and effective cross-presentation does not occur. Recycling of this overly stable complex to the TGN results in a buildup of unbound MHC-I that traffics to the plasma membrane to present endogenous peptides.
In agreement with our results, another group has shown that the MHC-I cytoplasmic tail tyrosine is required for the generation of CTL responses to viruses in vivo in a murine model system. However, the mechanism by which this tyrosine promoted cross-presentation was not known (10). Our studies provide a crucial missing link in this pathway by showing that Y320 is necessary for AP-1 binding and that AP-1 is needed to direct MHC-I to cellular compartments for acquisition and presentation of exogenous soluble antigen (Fig. 8). We furthermore show that in humans, the more recently evolved MHC-I HLA-C allotype lacking the cytoplasmic tail tyrosine utilizes an AP-1-independent pathway for cross-presentation. The HLA-C cytoplasmic tail contains a dihydrophobic signal that regulates trafficking and expression at the cell surface (13). Thus, cross-presentation by HLA-C may be regulated by a unique trafficking factor that binds to this signal.
Nef selectively downmodulates MHC-I molecules containing the AP-1 tyrosine signal (HLA-A and HLA-B) but not MHC-I molecules that lack the tyrosine (HLA-C) in HIV-infected T cells (4, 34). It has been proposed that this is beneficial to the virus because HLA-C expression is needed to inhibit natural killer (NK) cell lysis (4, 34). The requirement for HLA-C expression to inhibit NK cells may be an Achilles heel for the virus, with the consequence that HLA-C-restricted CTLs are uniquely efficacious against HIV infection. However, HLA-C is normally expressed at levels as many as 10 times lower than the levels of HLA-A and HLA-B expression on primary T lymphocytes (13), and thus, the role of HLA-C in CTL recognition may be limited by expression level. Consistent with this, genetic variations leading to higher HLA-C surface expression are protective in HIV disease (35; reviewed in reference 5). While HLA-C levels are comparatively low on primary T lymphocytes, HLA-C is upregulated in differentiated APCs to levels comparable to those of the HLA-A and HLA-B allotypes (13). Thus, the role of HLA-C may primarily relate to a unique function in APCs (reviewed in reference 5).
In addition to reducing MHC-I HLA-A and HLA-B surface expression in T lymphocytes, our new data provide direct evidence that HIV Nef disrupts cross-presentation by the HLA-A and HLA-B allotypes in APCs. Consistent with an effect of Nef on cross-presentation, wild-type simian immunodeficiency virus (SIV) elicits a more limited anti-SIV CD8+ T cell response in vivo than mutant SIVs harboring nef alleles that are defective at MHC-I downmodulation (3). Thus, therapeutic approaches that disrupt the activity of Nef may lead to the development of a more effective anti-HIV immune response in vivo.
Despite the dramatic effect of Nef on cross-presentation that we observed, Nef was less effective at reducing total surface MHC-I in macrophages than T lymphocytes. This may result from dysregulation of the Nef–MHC-I–AP-1 complex in cell types that naturally support an AP-1–MHC-I complex (Fig. 8). This hypothesis is consistent with other studies demonstrating that dominant active ARF-1 mutants that hyperstabilize the Nef–MHC-I–AP-1 complex in T cells also inhibit MHC-I downmodulation by Nef (36). From these observations, we propose that efficient MHC-I downmodulation by HIV requires that Nef and/or AP-1 have the capacity to dynamically cycle off the affected molecules to recruit downstream proteins in the degradation pathway and to target the newly synthesized cargo molecules (Fig. 8). Similarly, the inhibitory effect of Nef on cross-presentation of soluble antigen indicates that though AP-1 is necessary for this pathway, dysregulation of the AP-1–MHC-I complex by Nef binding is inhibitory to the overall process.
In sum, here we report a previously unidentified role for AP-1 in directing the MHC-I HLA-A and HLA-B allotypes to a novel cross-presentation pathway. These results have potential implications for vaccine development because novel adjuvants that stimulate cross-presentation may induce more effective CD8+ T cell responses (37–39). Our results also highlight that AP-1 is a key host factor for HIV pathogenesis, which may aid the development of better treatments for HIV-infected people. Finally, our results reveal the existence of a unique cross-presentation pathway utilized by HLA-C which is resistant to the effects of HIV Nef and thus may be particularly important in HIV disease.
ACKNOWLEDGMENTS
This work was funded by U.S. National Institutes of Health grants RO1 AI051192, RO1 AI046998, and RO1 AIO66131. D.A.K. was supported by the Irvington Institute Fellowship Program of the Cancer Research Institute. Services were provided by the University of Michigan Flow Cytometry Core, Sequencing Core (supported by National Institutes of Health grant P30CAO46592), Vector Core, and Hybridoma Core (supported by National Institutes of Health grant P30AR048310).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
J. A. Ganesh, J. Leonard, K. Leopold, L. McNamara, J. Norman, A. Onafuwa-Nuga, F. Taschuk, and S. Leonard assisted in scoring microscopy images. We thank P. Cresswell, E. Gagnon, and J. Moran for critical reading of the manuscript.
Footnotes
Published ahead of print 15 May 2013
REFERENCES
- 1. Collins K, Chen B, Kalams S, Walker B, Baltimore D. 1998. HIV-1 Nef protein protects infected primary human cells from killing by cytotoxic T lymphocytes. Nature 391:397–401 [DOI] [PubMed] [Google Scholar]
- 2. Schwartz O, Marechal V, Le Gall S, Lemonnier F, Heard J. 1996. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat. Med. 2:338–342 [DOI] [PubMed] [Google Scholar]
- 3. Swigut T, Alexander L, Morgan J, Lifson J, Mansfield KG, Lang S, Johnson RP, Skowronski J, Desrosiers R. 2004. Impact of Nef-mediated downregulation of major histocompatibility complex class I on immune response to simian immunodeficiency virus. J. Virol. 78:13335–13344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Le Gall S, Erdtmann L, Benichou S, Berlloz-Torrent C, Liu L, Benarous R, Heard J, Schwartz O. 1998. Nef interacts with mu subunit of clathrin adaptor complexes and reveals a cryptic sorting signal in MHC I molecules. Immunity 8:483–495 [DOI] [PubMed] [Google Scholar]
- 5. Kulpa DA, Collins KL. 2011. The emerging role of HLA-C in HIV-1 infection. Immunology 134:116–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wonderlich ER, Williams M, Collins KL. 2008. The tyrosine-binding pocket in the adaptor protein 1 (AP-1) mu1 subunit is necessary for Nef to recruit AP-1 to the major histocompatibility complex class I cytoplasmic tail. J. Biol. Chem. 283:3011–3022 [DOI] [PubMed] [Google Scholar]
- 7. Robinson MS. 2004. Adaptable adaptors for coated vesicles. Trends Cell Biol. 14:167–174 [DOI] [PubMed] [Google Scholar]
- 8. Jia X, Singh R, Homann S, Yang H, Guatelli J, Xiong Y. 2012. Structural basis of evasion of cellular adaptive immunity by HIV-1 Nef. Nat. Struct. Mol. Biol. 19:701–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Amigorena S, Savina A. 2010. Intracellular mechanisms of antigen cross presentation in dendritic cells. Curr. Opin. Immunol. 22:109–117 [DOI] [PubMed] [Google Scholar]
- 10. Lizee G, Basha G, Tiong J, Julien JP, Tian M, Biron KE, Jefferies WA. 2003. Control of dendritic cell cross-presentation by the major histocompatibility complex class I cytoplasmic domain. Nat. Immunol. 4:1065–1073 [DOI] [PubMed] [Google Scholar]
- 11. Roeth JF, Williams M, Kasper MR, Filzen TM, Collins KL. 2004. HIV-1 Nef disrupts MHC-I trafficking by recruiting AP-1 to the MHC-I cytoplasmic tail. J. Cell Biol. 167:903–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schaefer MR, Wonderlich ER, Roeth JF, Leonard JA, Collins KL. 2008. HIV-1 Nef targets MHC-I and CD4 for degradation via a final common beta-COP-dependent pathway in T cells. PLoS Pathog. 4:e1000131. 10.1371/journal.ppat.1000131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schaefer MR, Williams M, Kulpa DA, Blakely PK, Yaffee AQ, Collins KL. 2008. A novel trafficking signal within the HLA-C cytoplasmic tail allows regulated expression upon differentiation of macrophages. J. Immunol. 180:7804–7817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Carter CC, Onafuwa-Nuga A, McNamara LA, Riddell J, IV, Bixby D, Savona MR, Collins KL. 2010. HIV-1 infects multipotent progenitor cells causing cell death and establishing latent cellular reservoirs. Nat. Med. 16:446–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Van Parijs L, Refaeli Y, Lord JD, Nelson BH, Abbas AK, Baltimore D. 1999. Uncoupling IL-2 signals that regulate T cell proliferation, survival, and Fas-mediated activation-induced cell death. Immunity 11:281–288 [DOI] [PubMed] [Google Scholar]
- 16. Robinson A, Meredith C, Austen BM. 1986. Isolation and properties of the signal region from ovalbumin. FEBS Lett. 203:243–246 [DOI] [PubMed] [Google Scholar]
- 17. Williams M, Roeth JF, Kasper MR, Filzen T, Collins KL. 2005. Human immunodeficiency virus type 1 Nef domains required for disruption of major histocompatibility complex class I trafficking are also necessary for coprecipitation of Nef with HLA-A2. J. Virol. 79:632–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pear WS, Nolan GP, Scott ML, Baltimore D. 1993. Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. U. S. A. 90:8392–8396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Naviaux RK, Costanzi E, Haas M, Verma IM. 1996. The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses. J. Virol. 70:5701–5705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Burns JC, Friedmann T, Driever W, Burrascano M, Yee JK. 1993. Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc. Natl. Acad. Sci. U. S. A. 90:8033–8037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. 1998. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 72:8463–8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hopkins N. 1993. High titers of retrovirus (vesicular stomatitis virus) pseudotypes, at last. Proc. Natl. Acad. Sci. U. S. A. 90:8759–8760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Parham P, Brodsky FM. 1981. Partial purification and some properties of BB7.2. A cytotoxic monoclonal antibody with specificity for HLA-A2 and a variant of HLA-A28. Hum. Immunol. 3:277–299 [DOI] [PubMed] [Google Scholar]
- 24. Kasper MR, Collins KL. 2003. Nef-mediated disruption of HLA-A2 transport to the cell surface in T cells. J. Virol. 77:3041–3049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Traub LM, Kornfeld S, Ungewickell E. 1995. Different domains of the AP-1 adaptor complex are required for Golgi membrane binding and clathrin recruitment. J. Biol. Chem. 270:4933–4942 [DOI] [PubMed] [Google Scholar]
- 26. National Institutes of Health 2002. Public Health Service policy on humane care and use of laboratory animals. Office of Laboratory Animal Welfare, National Institutes of Health, Bethesda, MD: http://grants.nih.gov/grants/olaw/references/phspol.htm [Google Scholar]
- 27. Porgador A, Yewdell JW, Deng Y, Bennink JR, Germain RN. 1997. Localization, quantitation, and in situ detection of specific peptide-MHC class I complexes using a monoclonal antibody. Immunity 6:715–726 [DOI] [PubMed] [Google Scholar]
- 28. Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. 1994. T cell receptor antagonist peptides induce positive selection. Cell 76:17–27 [DOI] [PubMed] [Google Scholar]
- 29. Ackerman AL, Kyritsis C, Tampe R, Cresswell P. 2003. Early phagosomes in dendritic cells form a cellular compartment sufficient for cross presentation of exogenous antigens. Proc. Natl. Acad. Sci. U. S. A. 100:12889–12894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guermonprez P, Saveanu L, Kleijmeer M, Davoust J, Van Endert P, Amigorena S. 2003. ER-phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature 425:397–402 [DOI] [PubMed] [Google Scholar]
- 31. Houde M, Bertholet S, Gagnon E, Brunet S, Goyette G, Laplante A, Princiotta MF, Thibault P, Sacks D, Desjardins M. 2003. Phagosomes are competent organelles for antigen cross-presentation. Nature 425:402–406 [DOI] [PubMed] [Google Scholar]
- 32. Donaldson JG, Williams DB. 2009. Intracellular assembly and trafficking of MHC class I molecules. Traffic 10:1745–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Basha G, Omilusik K, Chavez-Steenbock A, Reinicke AT, Lack N, Choi KB, Jefferies WA. 2012. A CD74-dependent MHC class I endolysosomal cross-presentation pathway. Nat. Immunol. 13:237–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cohen GB, Gandhi RT, Davis DM, Mandelboim O, Chen BK, Strominger JL, Baltimore D. 1999. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity 10:661–671 [DOI] [PubMed] [Google Scholar]
- 35. Thomas R, Apps R, Qi Y, Gao X, Male V, O'hUigin C, O'Connor G, Ge D, Fellay J, Martin JN, Margolick J, Goedert JJ, Buchbinder S, Kirk GD, Martin MP, Telenti A, Deeks SG, Walker BD, Goldstein D, McVicar DW, Moffett A, Carrington M. 2009. HLA-C cell surface expression and control of HIV/AIDS correlate with a variant upstream of HLA-C. Nat. Genet. 41:1290–1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wonderlich ER, Leonard JA, Kulpa DA, Leopold KE, Norman JM, Collins KL. 2011. ADP ribosylation factor 1 activity is required to recruit AP-1 to the major histocompatibility complex class I (MHC-I) cytoplasmic tail and disrupt MHC-I trafficking in HIV-1-infected primary T cells. J. Virol. 85:12216–12226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Najar HM, Dutz JP. 2007. Topical TLR9 agonists induce more efficient cross-presentation of injected protein antigen than parenteral TLR9 agonists do. Eur. J. Immunol. 37:2242–2256 [DOI] [PubMed] [Google Scholar]
- 38. Robson NC, McAlpine T, Knights AJ, Schnurr M, Shin A, Chen W, Maraskovsky E, Cebon J. 2010. Processing and cross-presentation of individual HLA-A, -B, or -C epitopes from NY-ESO-1 or an HLA-A epitope for Melan-A differ according to the mode of antigen delivery. Blood 116:218–225 [DOI] [PubMed] [Google Scholar]
- 39. Schnurr M, Orban M, Robson NC, Shin A, Braley H, Airey D, Cebon J, Maraskovsky E, Endres S. 2009. ISCOMATRIX adjuvant induces efficient cross-presentation of tumor antigen by dendritic cells via rapid cytosolic antigen delivery and processing via tripeptidyl peptidase II. J. Immunol. 182:1253–1259 [DOI] [PubMed] [Google Scholar]