

Abstract
Paramyxovirus V proteins block Toll-like receptor 7 (TLR7)- and TLR9-dependent signaling leading to alpha interferon production. Our recent study has provided evidence that interaction of the V proteins with IRF7 is important for the blockade. However, the detailed mechanisms still remain unclear. Here we reexamined the interaction of the human parainfluenza virus type 2 (HPIV2) V protein with signaling molecules involved in TLR7/9-dependent signaling. Immunoprecipitation experiments in HEK293T cells transfected with V protein and one of the signaling molecules revealed that the V protein interacted with not only IRF7 but also TRAF6, IKKα, and MyD88. Whereas overexpression of TRAF6 markedly enhanced the level of V protein associating with IRF7, IKKα, and MyD88 in HEK293T cells, the level of V protein associating with TRAF6 was little affected by overexpression of IRF7, IKKα, and MyD88. Moreover, knockdown or knockout of endogenous TRAF6 in HEK293T or mouse embryonic fibroblast cells resulted in dissociation of the V protein from IRF7, IKKα, and MyD88. These results demonstrate that binding of the V protein to IRF7, IKKα, and MyD88 is largely indirect and mediated by endogenous TRAF6. It was found that the V protein inhibited TRAF6-mediated lysine 63 (K63)-linked polyubiquitination of IRF7, which is prerequisite for IRF7 activation. Disruption of the tryptophan-rich motif of the V protein significantly affected its TRAF6-binding efficiency, which correlated well with the magnitude of inhibition of K63-linked polyubiquitination and the resultant activation of IRF7. Taken together, these results suggest that the HPIV2 V protein prevents TLR7/9-dependent interferon induction by inhibiting TRAF6-mediated K63-linked polyubiquitination of IRF7.
INTRODUCTION
Interferons (IFNs) play pivotal roles in host innate immunity to protect against various virus infections. In respiratory virus infection, a major population of cells responsible for production of alpha IFN (IFN-α) includes alveolar macrophages, conventional dendritic cells (cDCs), and plasmacytoid DCs (pDCs) (1). Of these cell types, pDCs are unique in their capacity to secrete large amounts of IFN-α (2). However, pDCs are not recruited unless viruses overcome the first defense line to cause systemic infection or can effectively prevent cDCs and alveolar macrophages from producing IFN-α (1). Since the signaling pathway leading to IFN-α production in pDCs is different from that in cDCs and alveolar macrophages, pDCs can combat even viruses that have acquired the ability to successfully limit IFN-α production of alveolar macrophages and cDCs during evolution. Whereas cDCs and alveolar macrophages employ the signaling pathway dependent on retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), including RIG-I and melanoma differentiation-associated protein 5 (MDA5), pDCs utilize the Toll-like receptor 7 (TLR7)- and TLR9-dependent signaling pathway to produce IFN-α. It was found that most paramyxoviruses, including parainfluenza viruses, have evolved the V protein that blocks the TLR7/9-dependent pathway as well as the RLR-dependent pathway to counteract such a highly organized IFN production system (3–12). Studies on the blocking mechanisms for the RLR-dependent pathway uncovered that the paramyxovirus V protein physically associated with MDA5 and prevented double-stranded RNA from binding to MDA5, thereby inhibiting MDA5-dependent signaling (13, 14). In addition to this common mechanism targeting MDA5, some paramyxovirus V proteins have acquired an additional mechanism that targets a downstream step. The V protein of the human parainfluenza virus type 2 (HPIV2), mumps virus, and parainfluenza virus 5 in the genus Rubulavirus interacts with inducible I-kappa B (IκB) kinase (IKKi)/TANK-binding kinase 1 (TBK1) and inhibits activation of interferon regulatory factor 3 (IRF3) by acting as alternative substrates for IKKi/TBK1 (15). On the other hand, the V proteins of Sendai virus (SeV), measles virus (MeV), and Newcastle disease virus interact with IRF3 and suppress the transcriptional activity of IRF3 (16). In contrast to these extensive studies on the blockade of RLR-dependent signaling, the molecular basis for the blockade of TLR7/9-dependent signaling has not been well characterized. This study was thus intended to elucidate how the paramyxovirus V proteins block TLR7/9-dependent signaling leading to IFN-α.
pDCs, unlike other cell types, express a definite amount of cytosolic latent IRF7 in addition to endosomal TLR7 and TLR9 (17, 18). Engagement of TLR7 and TLR9 with guanosine-rich and uridine-rich single-stranded RNA and unmethylated DNA with CpG motifs, respectively, induces complex formation of IRF7 with myeloid differentiation factor 88 (MyD88), interleukin-1 (IL-1) receptor-associated kinase 4 (IRAK4), tumor necrosis factor receptor-associated factor 6 (TRAF6), TRAF3, IRAK1, IκB kinase alpha (IKKα), and osteopontin (19–25). IRF7 is phosphorylated by the serine/threonine kinases, IRAK1, and/or IKKα, forms a dimer, and translocates into the nucleus to activate the IFN-α genes (reviewed in references 26 to 29). Little is known about roles of each signaling molecule in the signaling pathway, but it is proposed that lysine 63 (K63)-linked polyubiquitination, which is mediated by K63 of ubiquitin (Ub), of IRF7 by TRAF6 and subsequent phosphorylation of IRF7 by IRAK1 and/or IKKα is prerequisite for activation of IRF7 (19, 21, 23, 30). K63-linked polyubiquitination is generally involved in activation of various signal transduction cascades and is different from K48-linked polyubiquitination leading to degradation of proteins (31). A recent study has uncovered a role of viperin, one of the IFN-stimulated genes, in the TLR7/9-dependent signaling pathway (32). Viperin recruits TRAF6 and IRAK1 to a lipid droplet, thereby activating IRAK1 that catalyzes phosphorylation of IRF7.
It is difficult to analyze the molecular mechanisms involved in the blockade of TLR7/9-dependent signaling using pDCs, because pDCs are less amenable to biochemical analysis, including plasmid transfection. However, a reconstitution system of TLR7/9-dependent signaling in HEK293T cells by transfection with MyD88, TRAF6, IKKα, and IRF7, together with IFN-α6 promoter-driven reporter plasmid, which is activated by IRF7 but not IRF3, made it possible to study molecular basis of the underlying mechanisms (8). Using this reconstitution system, it was found that the MeV V protein served as a decoy of substrate for IKKα and blocked TLR7/9-dependent IFN induction (8, 33). A subsequent study further revealed that the blockade of TLR7/9-dependent signaling was a common feature of the V protein of most paramyxoviruses, including SeV, HPIV2, bovine parainfluenza virus type 3, MeV, and the emerging highly pathogenic Nipah virus, suggesting its significant role in evading host innate immunity (12).
The V protein of most paramyxoviruses, including HPIV2, interacts with IRF7 (12). A tryptophan (Trp)-rich motif in the HPIV2 V protein is important for binding to IRF7. The IRF7-binding efficiency of the HPIV2 V mutants, in which amino acid residues in the Trp-rich motif or zinc-finger-like motif of the HPIV2 V-unique C-terminal domain were replaced with other amino acid residues, correlated well with the magnitude of inhibition of IRF7 activation, suggesting that the HPIV2 V protein targets IRF7 for the blockade of TLR7/9-dependent signaling. Nevertheless it was found that the V protein was incapable of inhibiting IPS-1-mediated activation of IRF7 in the RLR-dependent pathway (12). This raises the possibility that the V protein targets another or other signaling molecules for the blockade of TLR7/9-dependent signaling pathway.
In the present study, we sought to find out a real target of the HPIV2 V protein for the blockade of TLR7/9-dependent signaling. Coimmunoprecipitation experiments with HEK293T cells uncovered interaction of the HPIV2 V protein with not only IRF7 but also MyD88, IKKα, and TRAF6. However, the interactions with MyD88, IKKα, and IRF7 were found to be indirect and largely mediated by endogenous TRAF6. Analysis of the underlying mechanisms revealed that the HPIV2 V protein inhibited the TRAF6-mediated K63-linked polyubiquitination of IRF7 through the interaction with TRAF6, thereby blocking TLR7/9-dependent signaling leading to IFN-α production.
MATERIALS AND METHODS
Cells and viruses.
Human peripheral blood mononuclear cells (PBMCs) were prepared from whole blood of healthy donors by Lymphoprep density gradient centrifugation (Axis-Shield, Oslo, Norway). pDCs were isolated from human PBMCs by magnetically activated cell sorting using the Diamond plasmacytoid dendritic cell isolation kit II (Miltenyi Biotec, Auburn, CA) according to the manufacturer's instructions and were resuspended in RPMI (Nacalai Tesque) supplemented with 10% fetal bovine serum (FBS), human IL-3 (10 ng/ml; Peprotech), 2 mM l-glutamine, penicillin (100 IU/ml), and streptomycin (100 μg/ml). Mouse embryonic fibroblast (MEF) and TRAF6 knockout (TRAF6-KO) MEF cells were kindly provided by J. Inoue (Tokyo, Japan). In addition to these MEF cells, HEK293T and Vero cells were cultured in Dulbecco's modified Eagle's medium containing 2 mM l-glutamine, penicillin (100 IU/ml), streptomycin (100 μg/ml), and 10% fetal bovine serum (34). Sf9 insect cells were grown in TC-100 medium (Sigma, St. Louis, MO) supplemented with 0.26% tryptose phosphate broth and 10% fetal bovine serum (35). The HPIV2 wild type (Toshiba strain) and recombinant HPIV2 lacking expression of the V protein, HPIV2V(−), were propagated in Vero cells (36).
Measurement of IFN-α.
Levels of human IFN-α in culture media were determined by enzyme-linked immunosorbent assay (ELISA) with the human IFN alpha ELISA kit (PBL Interferon Source, Piscataway, NJ) according to the manufacturer's instructions.
Plasmids and siRNAs.
The construction of a mammalian expression plasmid, pCA7, encoding P, V, VC12, VC345, VC6, VW123, VW1, VW2, VW3 (derived from HPIV2 Toshiba strain), MyD88, TRAF6, IKKα, or IRF7 (of mouse origin) with an N-terminal 3× FLAG, V5, or myc tag was described previously (12). A mammalian expression plasmid encoding IRAK1, IRAK4, or viperin of human origin with N-terminal V5 tag was newly created by insertion of a cDNA fragment containing each open reading frame into the multicloning site of pCA7. For the purpose of expression in Sf9 insect cells, a cDNA fragment containing the open reading frame of FLAG-V, V5-MyD88, V5-TRAF6, V5-IKKα, or V5-IRF7 was cloned in the insect expression plasmid pAF (35). A cDNA fragment for mutants of IRF7 and Ub was generated by PCR-based overlap mutagenesis as described previously (12) and cloned into the multicloning site of pCA7. The mutants include FLAG-tagged IRF7(K3R), in which key lysine residues for K63-linked ubiquitination are mutated to arginine (K398R, K400R, and K406R) (30), FLAG-tagged IRF7(2A), in which key serine residues for phosphorylation are mutated to alanine (S425A and S426A) (37), and hemagglutinin (HA)-tagged Ub K63, in which all lysine residues but K63 were replaced with arginine (K6R, K11R, K27R, K29R, K33R, and K48R) (38). pEFneo-RIG-IN, which expresses the N-terminal portion (amino acids [aa] 1 to 229) of human RIG-I, a constitutively active form of RIG-I (39), and pEFneo-IPS-1 were a gift from T. Komatsu (Aichi, Japan). pCAGGs-puro/N-FLAG-NS3/4A, which expresses FLAG-tagged NS3/4A of hepatitis C virus (HCV), was a gift from T. Abe (Osaka, Japan) (40). The small interfering RNA (siRNA) targeting human TRAF6 (GenBank accession no. NM_145803) and the scrambled siRNA were purchased from TaKaRa Bio, Japan. The TRAF6 siRNA consists of 5′-CCACGAAGAGAUAAUGGAUTT-3′ (sense) and 5′-AUCCAUUAUCUCUUCGUGGTT-3′ (antisense) oligonucleotides (41). Lipofectamine RNAiMAX transfection reagent was used for siRNA transfection according to the manufacturer's instructions. The sequence fidelity of all plasmids newly created was confirmed by sequence analysis.
Immunoprecipitation.
HEK293T (∼5 × 105/well), wild-type MEF, TRAF6-KO MEF (∼2.5 × 104/well), or Sf9 (∼1.0 × 106/well) cells in a 6-well plate were transfected with various combinations of plasmids, using polyethyleneimine (PEI) (Polysciences) for mammalian cells or UniFector (Gentaur) for insect cells. In certain experiments, the transfected cells were further infected with HPIV2 at a multiplicity of infection (MOI) of 1.0. After incubation for an appropriate length of time, cells were lysed in 400 μl of a lysis buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% Triton X-100, and protease inhibitor cocktail). Cell lysates were then incubated with anti-FLAG mouse monoclonal antibody (MAb) (1E6; Wako) or anti-V5 mouse MAb (SV5-Pk1; Invitrogen) together with protein G-Sepharose 4 Fast Flow (GE Healthcare Bio-Science) at 4°C for 2 h. After being washed five times with the lysis buffer, proteins were eluted from the beads by boiling with Laemmli sample buffer (50 mM Tris-HCl [pH 6.8], 2% sodium dodecyl sulfate [SDS], 0.1% bromophenol blue, 10% glycerol, and 5% 2-mercaptoethanol), and then the samples were subjected to immunoblot analysis.
Immunoblot analysis.
Samples were resolved by SDS-polyacrylamide (10 to 15%) gel electrophoresis and then electroblotted onto a membrane filter (Immobilon-P; Millipore). The membrane was blocked in phosphate-buffered saline (PBS) containing 5% skim milk and 0.05% Tween 20, followed by incubation at room temperature for 2 h with anti-FLAG mouse MAb (1E6), anti-V5 mouse MAb (SV5-Pk1), anti-myc mouse MAb (9E10; Wako), anti-HA mouse MAb (4B2; Wako), anti-actin mouse MAb (AC-74; Sigma), anti-TRAF6 rabbit MAb (EP592Y; Epitomics), anti-phospho-IRF7 (anti-pIRF7) rabbit polyclonal antibody (Cell Signaling Technology), or anti-HPIV2 V/P mouse MAb (315-1) (42). The membrane was then incubated at room temperature for 1 h with horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG antibody (GE Healthcare Bio-Science). Immunoreactive bands were visualized using the enhanced chemiluminescence ECL Advance substrate (GE Healthcare Bio-Science).
In vivo ubiquitination assay.
The in vivo ubiquitination assay was performed as described previously, with slight modifications (30). HEK293T cells cultured in a 6-well plate were transfected with HA-Ub K63 (750 ng), myc-TRAF6 (50 ng), V5-IRF7 (150 ng), and viral protein (P, V, VW123, or VW2) (500 ng) by using PEI reagent. At 24 h posttransfection, cells were lysed in 40 μl of a denaturing buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% Triton X-100, 1% SDS, 1 mM dithiothreitol [DTT], 10 mM N-ethylmaleimide, 10 mM PR-619 [Lifesensors], and protease inhibitor cocktail) and incubated for 15 min on ice. The cell lysates were denatured at 95°C for 10 min to dissociate noncovalently bound proteins, diluted 10-fold with the lysis buffer, and then passed 10 times through 1-ml syringe with 26-gauge needle. After centrifugation, supernatants were subjected to immunoprecipitation with anti-V5 antibody.
Reporter assay.
HEK293T cells cultured in a 24-well plate were transfected in triplicate with various combinations of plasmids that express FLAG-V, FLAG-VC12, FLAG-VC345, FLAG-VC6, FLAG-VW123, FLAG-VW1, FLAG-VW2, FLAG-VW3 (50 ng/well), myc-MyD88 (5 ng/well), myc-TRAF6 (5 ng/well), myc-IKKα (5 ng/well), V5-IRF7 (15 ng/well), FLAG-IRF7 (15 ng/well), FLAG-IRF7(K3R) (15 ng/well), FLAG-IRF7(2A) (15 ng/well), RIG-IN (25 ng/well), IPS-1 (25 ng/well), and FLAG-NS3/4A (50 ng/well), together with the IFN-α6 promoter-driven reporter plasmid (80 ng/well) and pRL-TK (10 ng/well) (Promega) using PEI reagent. The total mass of transfected DNA was held constant in all experiments by adding an appropriate amount of pCA7 empty plasmid. Cells were lysed at 24 h posttransfection, and relative luciferase activity was determined by dual-luciferase reporter assay system (Promega). Activation of the IFN-α6 promoter was evaluated by calculating the mean value of relative luciferase activities from three independent experiments (12).
RESULTS
The HPIV2 V protein as a negative regulator of IFN-α production in pDCs.
The HPIV2 V protein blocks TLR7/9-dependent signaling, which is reconstituted in HEK293T cells, but there is no evidence that the V protein with this ability actually functions as a negative regulator of IFN-α production in pDCs. We thus checked if HPIV2 infection rendered pDCs unresponsive to a TLR7 ligand. pDCs were isolated from human PBMCs and infected with HPIV2 at an MOI of 10. At 18 h postinfection, a culture medium was replaced with a fresh medium containing R848, a ligand against TLR7 (Fig. 1A). Whereas R848 significantly stimulated IFN-α production of mock-infected pDCs, prior HPIV2 infection suppressed the response of pDCs to R848, indicating that the TLR7/9-dependent signaling is prevented in HPIV2-infected pDCs. We next examined whether the V protein was involved in the regulation of IFN-α production of pDCs. pDCs were infected with HPIV2 or HPIV2V(−), which lacks expression of the V protein, at an MOI of 0.1, and production of IFN-α was monitored (Fig. 1B). As shown in Fig. 1B, HPIV2V(−) induced IFN-α about 7 times more than the HPIV2 wild type. This confirms an important role of the V protein in negative regulation of IFN-α production in pDCs.
Fig 1.
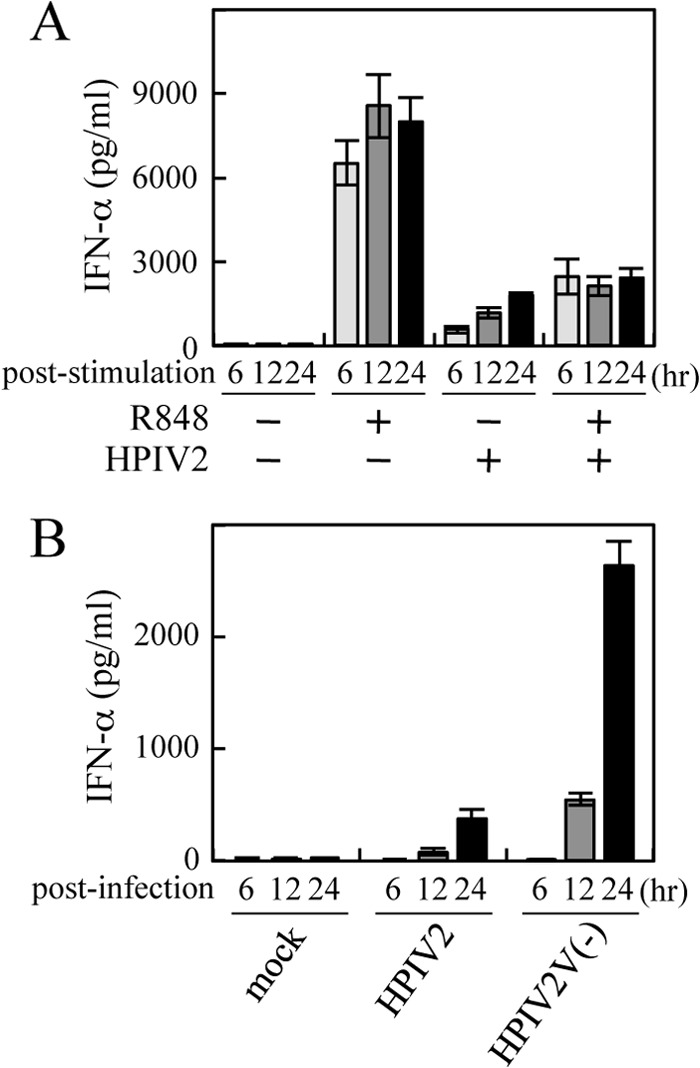
Production of IFN-α in human pDCs infected with HPIV2 or HPIV2V(−). (A) Human pDCs (∼104 cells/well) in a 96-well plate were mock infected or infected with HPIV2 at an MOI of 10. At 18 h postinfection, media were replaced with fresh media or media containing 10 mM R848. Levels of IFN-α in media were determined by ELISA at the indicated time points after stimulation with R848. (B) Human pDCs (∼104 cells/well) were infected with HPIV2 or HPIV2V(−) at an MOI of 0.1. Levels of IFN-α in media were determined by the ELISA at the indicated time points after infection.
Interaction of the HPIV2 V protein with MyD88, TRAF6, IKKα, and IRF7.
Our recent study showed that interaction of the paramyxovirus V proteins with IRF7 was important for the inhibition of IRF7 activation in the TLR7/9-dependent pathway (12). On the other hand, it was found that the HPIV2 V protein could not inhibit IRF7 activation stimulated by overexpression of IPS-1, a key mitochondrial kinase in the RLR-dependent pathway. This raises a fundamental question of whether the HPIV2 V protein really targets IRF7 for the blockade. We thus decided to reexamine the interaction between the HPIV2 V protein and signaling molecules of the TLR7/9-dependent pathway by coimmunoprecipitation experiments.
V5-tagged MyD88, TRAF6, IKKα, IRF7, IRAK1, IRAK4, or viperin was transfected into HEK293T cells together with FLAG-tagged HPIV2 V, and the transfected cells were subjected to immunoprecipitation. As shown in Fig. 2A and B, FLAG-V was coimmunoprecipitated with anti-V5 antibody in cells transfected with V5-tagged MyD88, TRAF6, IKKα, and IRF7, but not in cells transfected with V5-tagged IRAK1, IRAK4, and viperin. Conversely, V5-tagged IKKα, TRAF6, and IRF7, except for MyD88, were coimmunoprecipitated with anti-FLAG antibody (Fig. 2C). These results suggest that not only IRF7 but also MyD88, TRAF6, and IKKα are candidate targets of the V protein. The reason for a missing band of MyD88 to be coimmunoprecipitated in Fig. 2C was unclear, but it might be due to a lower expression level of V5-MyD88 in a cell lysate compared with that of other signaling molecules. The interaction of the V protein with MyD88, TRAF6, IKKα, and IRF7 was also observed when the V protein was supplied by HPIV2 infection instead of plasmid transfection (Fig. 2D).
Fig 2.

Interaction of the HPIV2 V protein with signaling molecules of the TLR7/9-dependent pathway. HEK293T cells (A to D) or Sf9 insect cells (E and F) in a 6-well plate were transfected with various combinations of the indicated plasmids (500 ng/well each). In panel D, transfected cells were infected with HPIV2 at an MOI of 1.0 at 12 h posttransfection. These cells then were lysed at 24 h posttransfection (A, B, C, E, and F) or postinfection (D), and the lysates were subjected to immunoprecipitation (IP) with anti-V5 (A, B, D, and E) or anti-FLAG antibody (C and F) followed by immunoblot (IB) analysis with anti-FLAG, anti-V5, or anti-PIV2 V/P antibody. The whole-cell lysates prepared for IP were also subjected to IB. Asterisks indicate positions of the antibody light chain (Ab LC) and heavy chain (Ab HC).
HEK293T cells stably express endogenous MyD88, IKKα, and TRAF6, which are reported to interact with IRF7 expressed by plasmid transfection (19, 23, 30, 43). This leads to a presumption that these endogenous molecules may mediate binding of FLAG-V to a V5-tagged signaling molecule. To diminish the effect of endogenous signaling molecules, we performed a similar experiment using Sf9 insect cells, which are genetically distant from mammalian HEK293T cells. As shown in Fig. 2E, the level of FLAG-V coimmunoprecipitated with anti-V5 antibody in insect cells transfected with V5-tagged MyD88 and IKKα was remarkably reduced, although no significant change was seen in the level of FLAG-V coimmunoprecipitated in insect cells transfected with V5-TRAF6 and V5-IRF7. The reduction was also confirmed in immunoprecipitation experiments with anti-FLAG antibody instead of anti-V5 antibody (Fig. 2F). These results suggest that binding of the V protein to MyD88 and IKKα in HEK293T cells is indirect and mediated by a certain endogenous molecule or molecules.
The HPIV2 V protein binds to IRF7 via TRAF6.
HEK293T cells do not express a detectable level of IRF7 without stimulation with cytokines, such as IFN. Therefore, it is very unlikely that binding of FLAG-V to V5-TRAF6 is mediated by IRF7. On the other hand, it is possible that the V-IRF7 binding is mediated by endogenous TRAF6, since a distinct level of endogenous TRAF6 is stably expressed in HEK293T cells (30). To assess this possibility, we examined the effect of changing the TRAF6 expression level on the V-IRF7 binding. FLAG-V and myc-IRF7 were transfected into HEK293T cells together with or without V5-TRAF6, and coimmunoprecipitation experiments were performed. The level of myc-IRF7 coimmunoprecipitated with FLAG-V was enhanced by expression of V5-TRAF6 (Fig. 3A). On the contrary, expression of myc-IRF7 did not affect the level of V5-TRAF6 coimmunoprecipitated with FLAG-V (Fig. 3B). Knockdown of endogenous human TRAF6 (hTRAF6) with siRNA specific to hTRAF6 but not mouse TRAF6 resulted in significant reduction of the level of V5-IRF7-associated FLAG-V, which was restored by expression of myc-tagged mouse TRAF6 but not MyD88 (Fig. 3C). A similar result was obtained in cells where the V protein was supplied by HPIV2 infection instead of transfection with the V expression plasmid (Fig. 3D). Eventually we used TRAF6-KO MEF cells to confirm dependency of the V-IRF7 binding on endogenous TRAF6. As shown in Fig. 3E, FLAG-V was coimmunoprecipitated with V5-IRF7 in wild-type MEF cells like HEK293T cells, while the level of FLAG-V that coimmunoprecipitated with V5-IRF7 was significantly reduced in TRAF6-KO MEF cells. Again, expression of myc-tagged mouse TRAF6 resulted in restoration of the level of FLAG-V coimmunoprecipitated with V5-IRF7, although the restoration was inefficient. The inefficient restoration appeared to be due to low expression of myc-tagged TRAF6 in TRAF6-KO MEF cells (Fig. 3E). It is empirically known that transfection efficiency in MEF cells is considerably lower than that in HEK293T cells. Taken together, these results demonstrate that binding of the V protein to IRF7 is largely mediated by endogenous TRAF6.
Fig 3.

Effect of the TRAF6 expression level on the V-IRF7 interaction. HEK293T (A to D), wild-type MEF (E), or TRAF6-KO MEF (E) cells in a 6-well plate were transfected with various combinations of indicated plasmids (500 ng/well each). In panel D, transfected cells were infected with HPIV2 at an MOI of 1.0 at 12 h posttransfection. In panels C and D, TRAF6-specific or scrambled siRNA (50 pmol/well) was mock transfected (−) or transfected 12 h prior to the plasmid transfection. The cells were lysed 24 h after the plasmid transfection (A, B, C, and E) or HPIV2 infection (D), and the lysates were subjected to immunoprecipitation (IP) with the anti-FLAG (A and B) or anti-V5 (C, D, and E) antibody, followed by immunoblot analysis (IB) with the anti-myc, anti-V5, anti-FLAG, or anti-HPIV2 V/P antibody. The whole-cell lysates prepared for IP were also subjected to IB with the anti-myc, anti-V5, anti-FLAG, anti-TRAF6, anti-PIV2 V/P, or anti-actin antibody. Asterisks indicate positions of the antibody light chain (Ab LC) and heavy chain (Ab HC). ehTRAF6, endogenous human TRAF6; eTRAF6, endogenous mouse TRAF6.
Binding of the HPIV2 V protein to MyD88 and IKKα is also mediated by TRAF6.
Binding of the V protein to MyD88 and IKKα in HEK293T cells appears to be indirect, because the MyD88- and IKKα-binding efficiency of the V protein was significantly reduced in insect cells, as described above (Fig. 2E and F). To determine whether binding of the V protein to MyD88 and IKKα was also mediated by endogenous TRAF6, similar coimmunoprecipitation experiments were performed. HEK293T cells were transfected with FLAG-V and V5-MyD88 (or V5-IKKα), together with or without myc-TRAF6. Expression of myc-TRAF6 resulted in a significant increase in the level of V5-MyD88 and V5-IKKα coimmunoprecipitated with FLAG-V (Fig. 4AD). On the other hand, expression of V5-MyD88 and V5-IKKα exerted only a marginal effect on the level of V5-TRAF6 coimmunoprecipitated with FLAG-V (Fig. 4BE). Knockdown of endogenous TRAF6 with the hTRAF6 siRNA resulted in obvious reduction of the level of V5-MyD88- and V5-IKKα-associated FLAG-V, which was restored by expression of mouse myc-TRAF6 (Fig. 4C and F). These results suggest that binding of the V protein to MyD88 and IKKα is also mediated by TRAF6.
Fig 4.

Effect of the TRAF6 expression level on the V-MyD88 and V-IKKα interactions. HEK293T cells in a 6-well plate were transfected with various combinations of the indicated plasmids (400 ng/well each). In panels C and F, HEK293T cells were mock transfected (−) or transfected with TRAF6-specific or scrambled siRNA (50 pmol/well) 12 h prior to the plasmid transfection. The cells were lysed 24 h after the plasmid transfection, and the lysates were subjected to immunoprecipitation (IP) with the anti-FLAG (A, B, D, and E) or anti-V5 (C and F) antibody, followed by immunoblot analysis (IB) with the anti-V5, anti-FLAG, or anti-myc antibody. The whole-cell lysates prepared for IP were also subjected to IB with the anti-V5, anti-FLAG, anti-myc, anti-TRAF6, or anti-actin antibody. ehTRAF6, endogenous human TRAF6.
Importance of the V-TRAF6 binding for the blockade of TLR7/9-dependent signaling.
The HPIV2 V protein is synthesized from a faithful copy of the P/V gene, whereas the P protein is from the edited mRNA in which an additional two G nucleotides are inserted at the editing site during de novo viral mRNA synthesis (44). Accordingly the N-terminal region of the V protein (V/P; aa 1 to 172) is shared by the P protein (Fig. 5A). On the other hand, the C-terminal unique region (aa 173 to 223) is highly conserved among paramyxoviruses, with invariantly spaced His and Cys residues forming the zinc-finger-like motif. Just upstream of the Cys cluster, there is a Trp-rich motif, Trp-(X)3-Trp-(X)9-Typ, presumably implicated in protein-protein interaction (45). To check if the V-TRAF6 binding is important for the blockade of TLR7/9-dependent signaling, V mutants with amino acid substitutions of Cys or Trp residues were examined for the TRAF6-binding capacity and the ability to block TLR7/9-dependent signaling. Coimmunoprecipitation experiments showed that VC12 (C193A C197A), VC345 (C209A C211A C214A), and VC6 (C221A) were similar to wild-type V in the TRAF6-binding capacity (Fig. 5B), while VW123 (W178H W182E W192A) and VW2 (W182E) almost completely lost the binding capacity (Fig. 5BC). These results suggest that the Trp-rich motif consisting of three Trp residues, especially at position W2, is important for TRAF6 binding. The ability to block TLR7/9-dependent signaling was assessed by using the reconstitution system, in which HEK293T cells were transfected with IFN-α6 promoter-driven luciferase reporter plasmid and signaling molecules of the TLR7/9-dependent pathway together with a V mutant. As shown in Fig. 5D, VC12, VC345, and VC6 exhibited an inhibitory effect comparable to that of V, while VW123 and VW2 showed little inhibitory effect on activation of the IFN-α6 promoter in the reconstitution system. A similar inhibitory pattern was observed in ELISA for IFN-α in cell culture media (Fig. 5E). Overall, there is a good correlation between the TRAF6-binding capacity and the inhibitory effect on IFN-α induction (Fig. 5B to E), demonstrating that the TRAF6 binding is required for the blockade of TLR7/9-dependent signaling.
Fig 5.

Importance of the V-TRAF6 binding for the blockade of TLR7/9-dependent IFN induction. (A) Schematic diagram of the C-terminal unique region of the HPIV2 V protein. W1, W2, and W3 indicate positions of Trp residues in the Trp-rich motif. Conserved His and Cys residues forming the zinc finger-like motif are underlined. Positions (C1 to C6) of the Cys residues are also shown. (B and C) HEK293T cells in a 6-well plate were transfected with various combinations of the indicated plasmids (500 ng/well each). The transfected cells were lysed at 24 h posttransfection, and the lysates were subjected to immunoprecipitation (IP) with the anti-FLAG antibody, followed by immunoblot analysis (IB) with the anti-FLAG or anti-V5 antibody. The whole-cell lysates prepared for IP were also subjected to IB. (D) HEK293T cells in a 24-well plate were transfected with various combinations of myc-MyD88, myc-TRAF6, myc-IKKα, V5-IRF7, FLAG-V, FLAG-VC12, FLAG-VC345, FLAG-VC6, FLAG-VW123, FLAG-VW1, FLAG-VW2, and FLAG-VW3, together with an IFN-α6 promoter-driven reporter plasmid and pRL-TK. At 24 h posttransfection, relative luciferase activity was determined by a dual-luciferase assay system. (E) HEK293T cells in a 24-well plate were transfected with indicated plasmids without the reporter plasmid and pRL-TK. At 36 h posttransfection, levels of IFN-α in culture media were determined by ELISA. VC12, V(C193A C197A); VC345, V(C209A C211A C214A); VC6, V(C218A); VW123, V(W178H W182E W192A); VW1, V(W178H); VW2, V(W182E); VW3, V(W192A).
The HPIV2 V protein inhibits TRAF6-mediated polyubiquitination of IRF7.
TRAF6 serves as a ubiquitin E3 ligase and catalyzes synthesis of K63-linked polyubiquitination of IRF7 (19, 30). Since the K63-linked ubiquitin ligase activity of TRAF6 is a prerequisite for activation of IRF7 in the TLR7/9-dependent pathway, we examined effect of the V protein on TRAF6-mediated K63-linked polyubiquitination and subsequent phosphorylation of IRF7. V5-IRF7 was transfected into HEK293T cells together with or without myc-TRAF6. Coexpression of myc-TRAF6 resulted in the appearance of a high-molecular-weight ladder of bands above IRF7 and of a phosphorylated IRF7 band (Fig. 6A). The ladder, which likely represents polyubiquitinated IRF7, disappeared in the presence of V but not VW123 and VW2. To determine whether the ladder contained K63-linked polyubiquitinated IRF7, an in vivo ubiquitination assay was performed using HA-Ub K63, in which all lysine residues except K63 of Ub were replaced with arginine. HA-Ub K63, V5-IRF7, and myc-TRAF6 were transfected into HEK293T cells, and the cell lysate was subjected to immunoprecipitation with anti-V5 antibody followed by immunoblot analysis with anti-HA antibody to detect HA-Ub K63-linked IRF7. As shown in Fig. 6B, a smear of K63-linked polyubiquitinated IRF7 bands was seen in cells cotransfected with myc-TRAF6, while it disappeared in the presence of V but not P, VW123, and VW2. These results suggest that the V protein inhibits TRAF6-mediated K63-linked polyubiquitination of IRF7 through the interaction with TRAF6.
Fig 6.
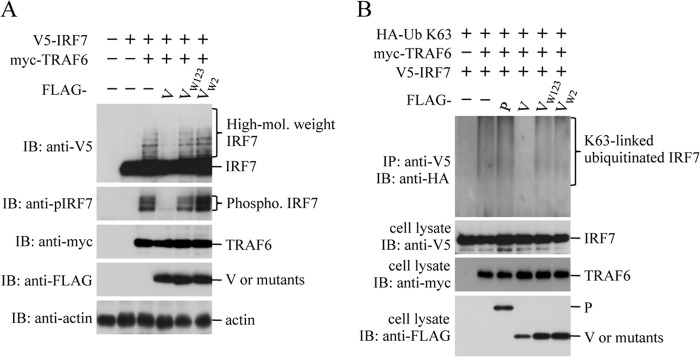
Effect of the HPIV2 V protein on TRAF6-mediated K63-linked polyubiquitination of IRF7. (A) HEK293T cells in a 6-well plate were transfected with various combinations of myc-TRAF6 (50 ng), V5-IRF7 (150 ng), FLAG-V (500 ng), FLAG-VW123 (500 ng), and FLAG-VW2 (500 ng). At 24 h posttransfection, the transfected cells were lysed in the RIPA buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 0.1% SDS, 1% NP-40, 0.5% sodium deoxycholate, protease inhibitor [Roche], and phosphatase inhibitor [Nacalai]), and the lysates were subjected to immunoblot analysis (IB) with the anti-V5, anti-pIRF7, anti-myc, anti-FLAG, or anti-actin antibody. (B) For the in vivo ubiquitination assay, HEK293T cells in a 6-well plate were transfected with various combinations of the indicated plasmids. At 24 h posttransfection, cells were lysed in the denaturing buffer and boiled at 95°C for 10 min as described in Materials and Methods. The cell lysates were then subjected to immunoprecipitation (IP) with the anti-V5 antibody, followed by IB with anti-HA antibody. The whole-cell lysates prepared for IP were also subjected to IB with anit-V5, anti-myc, or anti-FLAG antibody.
The HPIV2 V protein does not inhibit IPS-1-mediated activation of IRF7.
TRAF6 is involved in activation of IRF7 not only in the TLR7/9-dependent pathway but also in the RLR-dependent pathway (46). In the RLR-dependent pathway, IPS-1 localized at the mitochondrial outer membrane associates with RIG-I and MDA5 via the CARD-CARD interaction essential for triggering downstream signaling (47). However, our previous study uncovered that the V protein could not inhibit IPS-1-mediated activation of IRF7. Indeed, a reporter assay in the reconstitution system showed that neither V nor the V mutants (VW123, VW1, VW2, and VW3) inhibited IPS1-mediated activation of IRF7, while it was significantly inhibited by HCV NS3/4A, which leads to degradation of IPS-1 (Fig. 7A). This suggests that the ability of the V protein to inhibit K63-linked polyubiquitination of IRF7 does not affect IPS-1-mediated activation of IRF7. To determine whether the K63-linked polyubiquitination is required for activation of IRF7 in the RLR-dependent pathway, we examined activation of a ubiquitination-defective mutant, IRF7(K3R), in which three key lysine residues for K63-linked polyubiquitination are mutated to arginine (30). Whereas expression of the set of MyD88, TRAF6, and IKKα could not activate IRF7(K3R), it was activated by both IPS-1 and RIG-IN, a constitutively active form of RIG-I (Fig. 7B). On the other hand, neither IPS-1 nor RIG-IN could activate a phosphorylation-defective mutant, IRF7(2A), in which two key serine residues for phosphorylation are mutated to alanine (37). These results demonstrate that K63-linked polyubiquitination of IRF7 is required for activation of IRF7 in the TLR7/9-dependent pathway but not in the RLR-dependent pathway.
Fig 7.
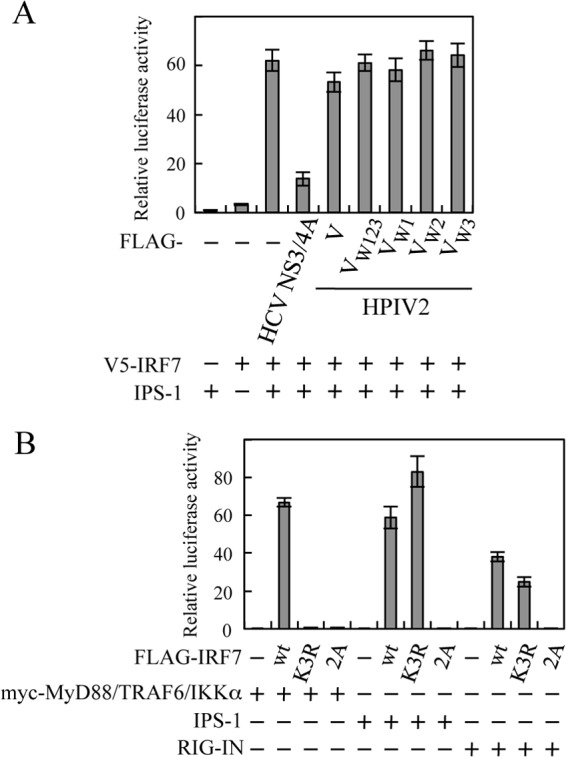
Effect of the HPIV2 V protein on IPS1-mediated activation of IRF7. (A) HEK293T cells were transfected with IPS-1, V5-IRF7, IFN-α6 promoter-driven reporter plasmid, and pRL-TK, together with FLAG-NS3/4A, FLAG-V, FLAG-VW123, FLAG-VW1, FLAG-VW2, or FLAG-VW3. (B) HEK293T cells in a 24-well plate were transfected with various combinations of myc-MyD88, myc-TRAF6, myc-IKKα, IPS-1, RIG-IN, FLAG-IRF7, FLAG-IRF7(K3R), and FLAG-IRF7(2A), together with the IFN-α6 promoter-driven reporter plasmid and pRL-TK. At 24 h posttransfection, relative luciferase activity was determined by a dual-luciferase assay system.
DISCUSSION
The present study provides several lines of evidence that the HPIV2 V protein targets TRAF6 for the blockade of TLR7-dependent signaling leading to IFN-α production. First, the HPIV2 V protein binds to a number of signaling molecules, including IRF7, IKKα, MyD88, and TRAF6 in HEK293T cells (Fig. 2), but binding of the V protein to IRF7, IKKα, and MyD88, except for TRAF6, was proved to be indirect and mediated by TRAF6 (Fig. 3 and Fig. 4). It is difficult to provide strict evidence for direct binding of the V protein to TRAF6, but at least none of the signaling molecules examined in the TLR7/9-dependent signaling pathway appears to mediate the V-TRAF6 binding (Fig. 3 and 4). Second, the HPIV2 V protein inhibits TRAF6-mediated K63-linked polyubiquitination of IRF7 (Fig. 6), which is a prerequisite for activation of IRF7 (19, 30). In accordance with this inhibition, subsequent phosphorylation of IRF7 was also inhibited (Fig. 6). Third, the V-TRAF6 binding is critical for the inhibition of TRAF6-mediated K63-linked polyubiquitination of IRF7 and for the blockade of TLR7/9-dependent signaling, because the Trp mutants VW123 and VW2, which have a defect in the TRAF6-binding capacity, have lost the ability to inhibit TRAF6-mediated K63-linked polyubiquitination of IRF7 as well as to block TLR7/9-dependent IFN induction (Fig. 5 and 6). Furthermore, there is a good correlation between the TRAF6-binding capacity and the ability to block TLR7/9-dependent signaling in the V mutants (Fig. 5). From these findings, we conclude that the HPIV2 V protein inhibits TRAF6-mediated K63-linked polyubiquitination of IRF7 through the interaction with TRAF6 to block TLR7/9-dependent signaling leading to IFN-α production.
The above conclusion is consistent with the results obtained previously (12). Our previous study showed that the level of V and the same V mutants (VC12, VC345, VC6, VW123, VW1, VW2, and VW3) that coimmunoprecipitated with IRF7 in HEK293T cells correlated well with magnitude of the inhibitory effect on TLR7/9-dependent IFN induction. Since the V protein binds to IRF7 via endogenous TRAF6 in HEK293T cells, the IRF7-binding efficiency of the V and V mutants must represent the efficiency of binding to TRAF6 that associates with IRF7. Indeed, we could confirm that the IRF7-binding efficiency of the V mutants in HEK293T cells (12) correlated well with their TRAF6-binding efficiency (Fig. 5BC). The previous study further demonstrated that the inhibitory domain of IRF7 was responsible for the V-IRF7 interaction (12). This result is also in good agreement with the acknowledged conclusion that TRAF6 binds to the inhibitory domain of IRF7 (19).
TRAF6 participates in activation of IRF7 via not only TLR7/9-dependent signaling but also RLR-dependent signaling. However, the V protein could not inhibit IPS1-mediated activation of IRF7 in the RLR-dependent pathway (Fig. 7A). The IRF7(K3R) mutant, in which K63-linked polyubiquitination sites are disrupted, could be activated by IPS-1 stimulation (Fig. 7B), indicating that K63-linked polyubiquitination of IRF7 is not required for activation of IRF7 in the RLR-dependent pathway. Indeed, IRF7 can be activated by even a TRAF6 mutant that has a defect in E3 ubiquitin ligase activity (46). Accordingly, TRAF6 plays a distinct role in the RLR-dependent pathway and does not serve as an E3 ubiquitin ligase for IRF7. This is one reason why the V protein is incapable of inhibiting the IPS-1-mediated IRF7 activation.
TRAF6 plays important roles not only in activation of IRF7 but also in activation of transcription factor nuclear factor kappaB (NF-κB), which controls induction of proinflammatory cytokines, in signaling pathways dependent on any of the TLRs, including TLR7/9 (48). This raises the possibility that the V protein inhibits activation of NF-κB by various stimuli via TLRs. Indeed, it was reported that expression of the MeV V protein suppressed NF-κB activation induced by overexpression of RIG-IN, IPS-1, MyD88, and TRIF (49). Similar inhibition of MyD88-induced activation of NF-κB was observed for the HPIV2 V protein (unpublished result). Engagement of TLRs with their respective ligands promotes TRAF6-mediated K63-linked polyubiquitination of TRAF6 itself and NF-κB essential modifier (NEMO)/IKKγ (50, 51). Ubiquitinated NEMO and TRAF6 subsequently recruit a protein kinase complex involving transforming growth factor-β-activated kinase 1 (TAK1) and TAK1 binding proteins (TAB1, TAB2, and TAB3) (51), which activates the IKK complex consisting of IKKα, IKKβ, and NEMO/IKKγ. The IKK complex phosphorylates IκB proteins, resulting in polyubiquitination and degradation of the IκB proteins, allowing NF-κB to translocate into the nucleus. It was found that the MeV V protein interacted with the Rel homology domain of the NF-κB subunit p65 (RelA) and inhibited nuclear translocation of p65 (49). However, the present study offers an additional hypothesis that the V protein inhibits activation of NF-κB by inhibiting TRAF6-mediated K63-linked polyubiquitination of target proteins. Therefore, it is of significance to examine effect of the V protein on K63-linked ubiquitination of substrates of TRAF6 in all of the TLR-dependent pathways.
In addition to the detailed analysis of the blocking mechanism using the artificial system reconstituted in HEK293T cells, we examined IFN-α production of pDCs infected with HPIV2 ex vivo (Fig. 1). Infection with HPIV2 rendered pDCs unresponsive to R848, as already reported for measles virus (strain Schwarz) and human respiratory syncytial virus (HRSV) (A2 strain) (33). Comparison of IFN-α production between HPIV2-infected and HPIV2V(−)-infected pDCs elucidated a crucial role of the V protein in restricting IFN-α production of infected pDCs. Therefore, it is likely that the V protein also functions in vivo as a negative regulator of IFN-α production in infected pDCs. Since pDCs do not participate in production of IFN-α when virus infection is localized to a small region or cDCs and alveolar macrophages can produce IFN-α in sufficient amounts (1), it should be determined whether HPIV2 infection really leads to recruitment of pDCs in vivo. It was reported that infection with HRSV, human metapneumovirus, and mouse pneumovirus, as well as SeV, leads to recruitment of pDCs in vivo, probably due to their high ability to suppress RLR-dependent IFN-α induction in cDCs and alveolar macrophages (1, 52–54). Thus, it is likely that HPIV2 infection also leads to recruitment of pDCs, because HPIV2 has a similar ability to inhibit RLR-dependent IFN induction, comparable to those of SeV and HRSV (36, 55, 56). In SeV infection, at least a portion of pDCs were found to produce IFN-α in vivo (1). This result raises an open question as to whether the ability to block TLR7/9-dependent signaling is operative in vivo (57). To settle this question, it should be determined by in vivo experiments whether there is a pDC population infected with HPIV2 and whether infected pDCs are inferior to uninfected pDCs in IFN-α production.
In summary, we demonstrate that the HPIV2 V protein binds to TRAF6 and inhibits TRAF6-mediated K63-linked polyubiquitination of IRF7 for the blockade of TLR7/9-dependent signaling leading to IFN-α. In addition to HPIV2, the V proteins of other paramyxoviruses, such as MeV and Nipah virus, interact with IRF7 and block TLR7/9-dependent signaling. Accordingly, it is of great interest to check whether the V-IRF7 interactions seen in these paramyxoviruses are also mediated by TRAF6 and whether the inhibition of K63-linked ubiquitination of IRF7 is a common feature of the V proteins of all paramyxoviruses. A study of this issue is now in progress.
ACKNOWLEDGMENTS
We thank J. Inoue (Tokyo) for providing wild-type and TRAF6-KO MEF cells, Y. Yanagi (Fukuoka) for providing pCA7, and J. Miyazaki (Osaka) for permission to use the CAG promoter of pCA7. Sequence analysis was performed using the ABI PRISM 3130xl Genetic Analyzer in the Central Research Laboratory, Shiga University of Medical Science.
This work was supported by JSPS KAKENHI grant no. 11020436, 22590414, and 25460563, and by grants from the Shiga University of Medical Science, Wajinkai, and the Yakult Foundation, Japan.
Footnotes
Published ahead of print 15 May 2013
REFERENCES
- 1. Kumagai Y, Takeuchi O, Kato H, Kumar H, Matsui K, Morii E, Aozasa K, Kawai T, Akira S. 2007. Alveolar macrophages are the primary interferon-alpha producer in pulmonary infection with RNA viruses. Immunity 27:240–252 [DOI] [PubMed] [Google Scholar]
- 2. Cao W, Liu YJ. 2007. Innate immune functions of plasmacytoid dendritic cells. Curr. Opin. Immunol. 19:24–30 [DOI] [PubMed] [Google Scholar]
- 3. Abe T, Sato M, Saigo Y, Tamai M. 2003. Interferon gamma expression and clinical features in patients with acute retinal necrosis syndrome. Graefes Arch. Clin. Exp. Ophthalmol. 241:982–987 [DOI] [PubMed] [Google Scholar]
- 4. Poole E, He B, Lamb RA, Randall RE, Goodbourn S. 2002. The V proteins of simian virus 5 and other paramyxoviruses inhibit induction of interferon-beta. Virology 303:33–46 [DOI] [PubMed] [Google Scholar]
- 5. Andrejeva J, Childs KS, Young DF, Carlos TS, Stock N, Goodbourn S, Randall RE. 2004. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc. Natl. Acad. Sci. U. S. A. 101:17264–17269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Komatsu T, Takeuchi K, Yokoo J, Gotoh B. 2004. C and V proteins of Sendai virus target signaling pathways leading to IRF-3 activation for the negative regulation of interferon-beta production. Virology 325:137–148 [DOI] [PubMed] [Google Scholar]
- 7. Komatsu T, Takeuchi K, Gotoh B. 2007. Bovine parainfluenza virus type 3 accessory proteins that suppress beta interferon production. Microbes Infect. 9:954–962 [DOI] [PubMed] [Google Scholar]
- 8. Pfaller CK, Conzelmann KK. 2008. Measles virus V protein is a decoy substrate for IkappaB kinase alpha and prevents Toll-like receptor 7/9-mediated interferon induction. J. Virol. 82:12365–12373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nakatsu Y, Takeda M, Ohno S, Shirogane Y, Iwasaki M, Yanagi Y. 2008. Measles virus circumvents the host interferon response by different actions of the C and V proteins. J. Virol. 82:8296–8306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ikegame S, Takeda M, Ohno S, Nakatsu Y, Nakanishi Y, Yanagi Y. 2010. Both RIG-I and MDA5 RNA helicases contribute to the induction of alpha/beta interferon in measles virus-infected human cells. J. Virol. 84:372–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ramachandran A, Horvath CM. 2010. Dissociation of paramyxovirus interferon evasion activities: universal and virus-specific requirements for conserved V protein amino acids in MDA5 interference. J. Virol. 84:11152–11163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kitagawa Y, Yamaguchi M, Zhou M, Komatsu T, Nishio M, Sugiyama T, Takeuchi K, Itoh M, Gotoh B. 2011. A tryptophan-rich motif in the human parainfluenza virus type 2 V protein is critical for the blockade of Toll-like receptor 7 (TLR7)- and TLR9-dependent signaling. J. Virol. 85:4606–4611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Childs K, Stock N, Ross C, Andrejeva J, Hilton L, Skinner M, Randall R, Goodbourn S. 2007. mda-5, but not RIG-I, is a common target for paramyxovirus V proteins. Virology 359:190–200 [DOI] [PubMed] [Google Scholar]
- 14. Childs KS, Andrejeva J, Randall RE, Goodbourn S. 2009. Mechanism of mda-5 inhibition by paramyxovirus V proteins. J. Virol. 83:1465–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lu LL, Puri M, Horvath CM, Sen GC. 2008. Select paramyxoviral V proteins inhibit IRF3 activation by acting as alternative substrates for inhibitor of kappaB kinase epsilon (IKKe)/TBK1. J. Biol. Chem. 283:14269–14276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Irie T, Kiyotani K, Igarashi T, Yoshida A, Sakaguchi T. 2012. Inhibition of interferon regulatory factor 3 activation by paramyxovirus V protein. J. Virol. 86:7136–7145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdorfer B, Giese T, Endres S, Hartmann G. 2002. Quantitative expression of Toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J. Immunol. 168:4531–4537 [DOI] [PubMed] [Google Scholar]
- 18. Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. 2005. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434:772–777 [DOI] [PubMed] [Google Scholar]
- 19. Kawai T, Sato S, Ishii KJ, Coban C, Hemmi H, Yamamoto M, Terai K, Matsuda M, Inoue J, Uematsu S, Takeuchi O, Akira S. 2004. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat. Immunol. 5:1061–1068 [DOI] [PubMed] [Google Scholar]
- 20. Honda K, Yanai H, Mizutani T, Negishi H, Shimada N, Suzuki N, Ohba Y, Takaoka A, Yeh WC, Taniguchi T. 2004. Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. Proc. Natl. Acad. Sci. U. S. A. 101:15416–15421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Uematsu S, Sato S, Yamamoto M, Hirotani T, Kato H, Takeshita F, Matsuda M, Coban C, Ishii KJ, Kawai T, Takeuchi O, Akira S. 2005. Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR)7- and TLR9-mediated interferon-α induction. J. Exp. Med. 201:915–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Oganesyan G, Saha SK, Guo B, He JQ, Shahangian A, Zarnegar B, Perry A, Cheng G. 2006. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature 439:208–211 [DOI] [PubMed] [Google Scholar]
- 23. Hoshino K, Sugiyama T, Matsumoto M, Tanaka T, Saito M, Hemmi H, Ohara O, Akira S, Kaisho T. 2006. IkappaB kinase-alpha is critical for interferon-alpha production induced by Toll-like receptors 7 and 9. Nature 440:949–953 [DOI] [PubMed] [Google Scholar]
- 24. Hacker H, Redecke V, Blagoev B, Kratchmarova I, Hsu LC, Wang GG, Kamps MP, Raz E, Wagner H, Hacker G, Mann M, Karin M. 2006. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 439:204–207 [DOI] [PubMed] [Google Scholar]
- 25. Shinohara ML, Lu L, Bu J, Werneck MB, Kobayashi KS, Glimcher LH, Cantor H. 2006. Osteopontin expression is essential for interferon-alpha production by plasmacytoid dendritic cells. Nat. Immunol. 7:498–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kawai T, Akira S. 2009. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 21:317–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kumar H, Kawai T, Akira S. 2009. Toll-like receptors and innate immunity. Biochem. Biophys. Res. Commun. 388:621–625 [DOI] [PubMed] [Google Scholar]
- 28. Blasius AL, Beutler B. 2010. Intracellular Toll-like receptors. Immunity 32:305–315 [DOI] [PubMed] [Google Scholar]
- 29. Kawai T, Akira S. 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11:373–384 [DOI] [PubMed] [Google Scholar]
- 30. Ning S, Campos AD, Darnay BG, Bentz GL, Pagano JS. 2008. TRAF6 and the three C-terminal lysine sites on IRF7 are required for its ubiquitination-mediated activation by the tumor necrosis factor receptor family member latent membrane protein 1. Mol. Cell. Biol. 28:6536–6546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ikeda F, Crosetto N, Dikic I. 2010. What determines the specificity and outcomes of ubiquitin signaling? Cell 143:677–681 [DOI] [PubMed] [Google Scholar]
- 32. Saitoh T, Satoh T, Yamamoto N, Uematsu S, Takeuchi O, Kawai T, Akira S. 2011. Antiviral protein Viperin promotes Toll-like receptor 7- and Toll-like receptor 9-mediated type I interferon production in plasmacytoid dendritic cells. Immunity 34:352–363 [DOI] [PubMed] [Google Scholar]
- 33. Schlender J, Hornung V, Finke S, Gunthner-Biller M, Marozin S, Brzozka K, Moghim S, Endres S, Hartmann G, Conzelmann KK. 2005. Inhibition of Toll-like receptor 7- and 9-mediated alpha/beta interferon production in human plasmacytoid dendritic cells by respiratory syncytial virus and measles virus. J. Virol. 79:5507–5515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gotoh B, Takeuchi K, Komatsu T, Yokoo J. 2003. The STAT2 activation process is a crucial target of Sendai virus C protein for the blockade of alpha interferon signaling. J. Virol. 77:3360–3370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kitagawa Y, Tani H, Limn CK, Matsunaga TM, Moriishi K, Matsuura Y. 2005. Ligand-directed gene targeting to mammalian cells by pseudotype baculoviruses. J. Virol. 79:3639–3652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nishio M, Tsurudome M, Ito M, Garcin D, Kolakofsky D, Ito Y. 2005. Identification of paramyxovirus V protein residues essential for STAT protein degradation and promotion of virus replication. J. Virol. 79:8591–8601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lin R, Mamane Y, Hiscott J. 2000. Multiple regulatory domains control IRF-7 activity in response to virus infection. J. Biol. Chem. 275:34320–34327 [DOI] [PubMed] [Google Scholar]
- 38. Jura N, Scotto-Lavino E, Sobczyk A, Bar-Sagi D. 2006. Differential modification of Ras proteins by ubiquitination. Mol. Cell 21:679–687 [DOI] [PubMed] [Google Scholar]
- 39. Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, Foy E, Loo YM, Gale M, Jr, Akira S, Yonehara S, Kato A, Fujita T. 2005. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 175:2851–2858 [DOI] [PubMed] [Google Scholar]
- 40. Abe T, Kaname Y, Hamamoto I, Tsuda Y, Wen X, Taguwa S, Moriishi K, Takeuchi O, Kawai T, Kanto T, Hayashi N, Akira S, Matsuura Y. 2007. Hepatitis C virus nonstructural protein 5A modulates the Toll-like receptor-MyD88-dependent signaling pathway in macrophage cell lines. J. Virol. 81:8953–8966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Muroi M, Tanamoto K. 2008. TRAF6 distinctively mediates MyD88- and IRAK-1-induced activation of NF-kappaB. J. Leukoc. Biol. 83:702–707 [DOI] [PubMed] [Google Scholar]
- 42. Nishio M, Tsurudome M, Ito M, Watanabe N, Kawano M, Komada H, Ito Y. 1997. Human parainfluenza virus type 2 phosphoprotein: mapping of monoclonal antibody epitopes and location of the multimerization domain. J. Gen. Virol. 78:1303–1308 [DOI] [PubMed] [Google Scholar]
- 43. Balkhi MY, Fitzgerald KA, Pitha PM. 2010. IKKalpha negatively regulates IRF-5 function in a MyD88-TRAF6 pathway. Cell. Signal. 22:117–127 [DOI] [PubMed] [Google Scholar]
- 44. Lamb RA, Parks GD. 2007. Paramyxoviridae: the viruses and their replication, p 1449–1496 Knipe DM, Howley PM. (ed), Fields virology, 5th ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 45. Nishio M, Garcin D, Simonet V, Kolakofsky D. 2002. The carboxyl segment of the mumps virus V protein associates with Stat proteins in vitro via a tryptophan-rich motif. Virology 300:92–99 [DOI] [PubMed] [Google Scholar]
- 46. Konno H, Yamamoto T, Yamazaki K, Gohda J, Akiyama T, Semba K, Goto H, Kato A, Yujiri T, Imai T, Kawaguchi Y, Su B, Takeuchi O, Akira S, Tsunetsugu-Yokota Y, Inoue J. 2009. TRAF6 establishes innate immune responses by activating NF-kappaB and IRF7 upon sensing cytosolic viral RNA and DNA. PLoS One 4:e5674. 10.1371/journal.pone.0005674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. 2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6:981–988 [DOI] [PubMed] [Google Scholar]
- 48. Kawai T, Akira S. 2007. Signaling to NF-kappaB by Toll-like receptors. Trends Mol. Med. 13:460–469 [DOI] [PubMed] [Google Scholar]
- 49. Schuhmann KM, Pfaller CK, Conzelmann KK. 2011. The measles virus V protein binds to p65 (RelA) to suppress NF-κB activity. J. Virol. 85:3162–3171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen F, Bhatia D, Chang Q, Castranova V. 2006. Finding NEMO by K63-linked polyubiquitin chain. Cell Death Differ. 13:1835–1838 [DOI] [PubMed] [Google Scholar]
- 51. Adhikari A, Xu M, Chen ZJ. 2007. Ubiquitin-mediated activation of TAK1 and IKK. Oncogene 26:3214–3226 [DOI] [PubMed] [Google Scholar]
- 52. Davidson S, Kaiko G, Loh Z, Lalwani A, Zhang V, Spann K, Foo SY, Hansbro N, Uematsu S, Akira S, Matthaei KI, Rosenberg HF, Foster PS, Phipps S. 2011. Plasmacytoid dendritic cells promote host defense against acute pneumovirus infection via the TLR7-MyD88-dependent signaling pathway. J. Immunol. 186:5938–5948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Guerrero-Plata A, Kolli D, Hong C, Casola A, Garofalo RP. 2009. Subversion of pulmonary dendritic cell function by paramyxovirus infections. J. Immunol. 182:3072–3083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Grayson MH, Ramos MS, Rohlfing MM, Kitchens R, Wang HD, Gould A, Agapov E, Holtzman MJ. 2007. Controls for lung dendritic cell maturation and migration during respiratory viral infection. J. Immunol. 179:1438–1448 [DOI] [PubMed] [Google Scholar]
- 55. Randall RE, Goodbourn S. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 89:1–47 [DOI] [PubMed] [Google Scholar]
- 56. Nishio M, Tsurudome M, Ito M, Kawano M, Komada H, Ito Y. 2001. High resistance of human parainfluenza type 2 virus protein-expressing cells to the antiviral and anti-cell proliferative activities of alpha/beta interferons: cysteine-rich V-specific domain is required for high resistance to the interferons. J. Virol. 75:9165–9176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gerlier D, Lyles DS. 2011. Interplay between innate immunity and negative-strand RNA viruses: towards a rational model. Microbiol. Mol. Biol. Rev. 75:468–490 [DOI] [PMC free article] [PubMed] [Google Scholar]