

Abstract
Almost half of the human genome is composed of transposable elements. The genomic structures and life cycles of some of these elements suggest they are a result of waves of retroviral infection and transposition over millions of years. The reduction of retrotransposition activity in primates compared to that in nonprimates, such as mice, has been attributed to the positive selection of several antiretroviral factors, such as apolipoprotein B mRNA editing enzymes. Among these, APOBEC3G is known to mutate G to A within the context of GG in the genome of endogenous as well as several exogenous retroelements (the underlining marks the G that is mutated). On the other hand, APOBEC3F and to a lesser extent other APOBEC3 members induce G-to-A changes within the nucleotide GA. It is known that these enzymes can induce deleterious mutations in the genome of retroviral sequences, but the evolution and/or inactivation of retroelements as a result of mutation by these proteins is not clear. Here, we analyze the mutation signatures of these proteins on large populations of long interspersed nuclear element (LINE), short interspersed nuclear element (SINE), and endogenous retrovirus (ERV) families in the human genome to infer possible evolutionary pressure and/or hypermutation events. Sequence context dependency of mutation by APOBEC3 allows investigation of the changes in the genome of retroelements by inspecting the depletion of G and enrichment of A within the APOBEC3 target and product motifs, respectively. Analysis of approximately 22,000 LINE-1 (L1), 24,000 SINE Alu, and 3,000 ERV sequences showed a footprint of GG→AG mutation by APOBEC3G and GA→AA mutation by other members of the APOBEC3 family (e.g., APOBEC3F) on the genome of ERV-K and ERV-1 elements but not on those of ERV-L, LINE, or SINE.
INTRODUCTION
Nearly half of the mammalian genome is derived from mobile or transposable elements (1–3). The retroelements, which make up ∼42% of the human genome, are divided into two classes based on the existence of long terminal repeat (LTR) sequences in their genome. The LTR retroelements include endogenous retroviruses (ERVs; ∼8% of the human genome), and the non-LTR retroelements include long and short interspersed nuclear elements (LINE and SINE; ∼34% of the human genome) (2).
The human endogenous retroviruses (HERVs) are believed to be the remnants of the ancient exogenous retroviruses that infected germ line cells (4). There are more than 98,000 copies of HERV sequences in the modern human genome (5, 6); however, despite recent amplification events, no active HERV has been reported so far (7). The majority of HERV sequences show multiple deleterious mutations and/or major deletions, sometimes leading to the creation of solo LTRs (8, 9). It is worth noting that the majority of HERV sequences are common between humans and other primates. Therefore, HERV sequences are not necessarily human specific. However, the ERV sequences obtained from the human genome are also referred to as HERVs. We use these two terms interchangeably in this paper.
LINE-1 (L1), with over 500,000 sequence copies, is the only active autonomous non-LTR retroelement in the modern human genome (10). However, only ∼80 to 100 copies are active at this time (11). In addition to self-copying, the L1 elements assist nonautonomous SINE to retrotranspose (12–14). With more than 1 million copies, the Alu elements are the most prominent SINE in humans and thus are considered the most successful transposons (14).
Retroelements have played major roles in genome evolution (1, 15, 16). Endogenized retroviruses may provide a selective advantage to a host fighting against exogenous counterparts (2). Also, there is evidence to support vital roles for retroelements in developing placenta (17–19). Nevertheless uncontrolled retrotransposition can lead to genetic diseases (12, 20, 21). To avoid the potentially adverse impact of uncontrolled transposition and infection, human cells use a wide range of mechanisms, including CpG methylation, histone modification, small RNA silencing, and inhibition by restricting enzymes (22–29). It is known that restriction enzymes inhibit the activity of endogenous retroelements in a way similar to that of their exogenous counterparts, such as human immunodeficiency virus (HIV) (15, 30).
Compared to that in nonprimates, such as mice, the activity of retroelements has decreased dramatically in primates (3, 31, 32). This reduction has been attributed partly to the evolution and positive selection of several viral restriction factor genes, such as the APOBEC (apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like) genes in primates (26, 32).
Humans and primates have more APOBEC gene members than other mammalian species, such as mice, cats, and cows (33). The mouse genome has a single APOBEC3 gene (mA3) whose product has left a mutational footprint (GAA→AAA) on the genome of endogenous murine leukemia viruses (34). The human APOBEC gene family includes seven members, APOBEC3A, APOBEC3B, APOBEC3C, APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H (24). Several members of this family have been under positive selection and rapid expansion (26, 35, 36). Among the human APOBEC3 family, APOBEC3G (hA3G) and APOBEC3F (hA3F) are known for their potent mutagenicity. These two enzymes inhibit the activity of a wide range of endogenous and exogenous retroelements (37–40). They target the negative-strand DNA intermediate during reverse transcription and deaminate cytosine (C) residues to uracil (U). This process results in a guanine (G)-to-adenine (A) replacement on the positive strand (24, 26). The affected sequences, referred to as “hypermutated,” usually contain signatures of mutations at multiple sites of the genome. These footprints can be found on a proportion of sequences obtained from individuals infected with exogenous retroviruses, such as HIV and simian immunodeficiency virus (SIV). They are also easily detectable in the in vitro studies of cells transfected with hA3G and/or hA3F genes. Most importantly, a few reports have identified signatures of mutation by hA3G on the genome of a number of endogenous retroviruses from the HERV-K family (41–43). This observation, as well as the coinciding evolution and expansion of the APOBEC3 family with the emergence of HERVs, may imply that hA3G and hA3F have been in action against retroviruses for millions of years (24). The majority of endogenous retroelements are thought to have accumulated mutations that have rendered them inactive (8, 24). This might suggest a role for APOBEC3 enzymes in the inactivation and/or evolution of retroelements.
We argue that if endogenous retroelements have been inactivated or evolved by a G-to-A mutation induced by hA3G and/or hA3F, this should be evidenced in their genome in the form of underrepresentation of hA3G target motifs and overrepresentation of product motifs. Importantly, G-to-A mutation by hA3G and hA3F is highly sequence motif dependent (41–43). It is known that hA3G preferably targets the underlined G within GG, but hA3F prefers G within the GA motif (note that in this report, we refer to the hA3G and hA3F target and product motifs with respect to the positive strand). Therefore, analysis of the representation of these target motifs and their corresponding product motifs resulting from G-to-A mutation would reveal possible footprints of hA3G and/or hA3F. The footprints may be found in two forms: (i) highly underrepresented target motifs and overrepresented product motifs resulting from many G-to-A mutations by hA3G and/or hA3F and (ii) a less severe but significant decrease in the representation of target motifs and an increase in the representation of product motifs resulting from stepwise and evolutionary accumulation of G-to-A changes induced by hA3G and/or hA3F. The first type of footprint has been found for hA3G on two individual HERV-K sequences in studies of a limited number of human-specific HERV-K elements (41–43). This could be described as the effect of APOBEC3 on individual sequences. These sequences would be seen as outliers from the population. The second type could be described as the evolution (and perhaps gradual inactivation) of the retroelement population by APOBEC3. These types of changes, which would be expected to be observed in the entire population, have not previously been investigated.
In this paper, we use a recently developed method (44, 45) to identify individual sequences affected by hA3G, hA3F, or both enzymes. Additionally, we look for footprints of evolutionary pressure by these enzymes on retroelements at a population level. The genomes of tens of thousands of retroelements from families L1, SINE Alu, ERV-1, ERV-K, and ERV-L were interrogated for signatures of context-dependent G-to-A mutations.
It is worth noting that all members of the human APOBEC3 family except APOBEC3G preferably target G within GA (46). Within those proteins with a GA→AA mutation preference (APOBEC3A, APOBEC3B, APOBEC3C, APOBEC3D, APOBEC3F, and APOBEC3H) (46), APOBEC3F is the most mutagenic, and hence for simplicity this is the enzyme we refer to as being responsible for the GA→AA changes discussed in this study. However, any reported GA→AA changes could well be due to the other members of this family.
MATERIALS AND METHODS
Sequence data.
The sequences of all 23 human chromosomes were obtained from the University of California, Santa Cruz (UCSC), genome browser, build 37, February 2009 version (hg19, GRCh37 Genome Reference Consortium Human Reference 37 [GCA_000001405.1]) (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/chromosomes/). The genomic coordinates of the location of retroelements on the human chromosome sequences were collected from the RepeatMasker human genome build 37 annotation database described in the rmsk.txt.gz file (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/). These chromosomal coordinates were then used to extract, from the human genome, the sequences of L1, the LTR ERV family, and SINE Alu using MATLAB scripts. It is worth noting that we did not use BLAT search using reference sequences of retroelements. A total of 951,864 L1, 21,361 LTR ERV family, and 1,194,734 SINE Alu sequences were initially extracted. The complete genomes of L1 and LTR are ∼6 kb and ∼5 kb in size, respectively. But many of these elements are truncated (1) and appear as shorter fragments in the human genome. In order to have a robust measurement of motif representation, the sequences needed to be large enough to contain an adequate number of hA3G and hA3F target and product motifs. We therefore applied a length threshold of 3 kb and excluded from the analysis L1 and ERV sequences shorter than this threshold. The SINE sequences are much shorter DNA elements, usually with a length of <500 bp. We included in the analysis SINE Alu sequences of >300 bp.
As a positive control, we merged two populations of in vivo and in vitro hypermutated HIV-1 sequences. The former contained 83 hypermutated full-length HIV-1 sequences from the Los Alamos National Laboratory (LANL) database. The latter had 19 in vitro hypermutated HIV-1 sequences described by Armitage et al. (41). We have recently uniquely identified the sources (hA3G or hA3F) of hypermutation for these sequences (45).
As negative controls, we used sequences of normal (nonhypermutated) HIV-1 and human reference genes. We have recently shown that the nonhypermutated HIV-1 population does not contain a footprint of G-to-A mutation by hA3G and/or hA3F (44). The human genes are also not expected to have been targeted by these enzymes. A total of 2,378 full-length (>7,000-bp) nonhypermutated HIV-1 sequences were downloaded from the LANL database (http://www.hiv.lanl.gov), and 4,322 human reference genes (>3,000 bp) were downloaded from the UCSC annotation database. Table 1 summarizes the types and compositions of the sequences used in this paper.
Table 1.
A summary of the sequences used in this study
| Retroelement type | No. of sequences | Avg length (bp) | % A | % C | % G | % T |
|---|---|---|---|---|---|---|
| LTR ERV | ||||||
| ERV-1 | 1,956 | 4,379 | 28.8 | 24.6 | 19.0 | 27.6 |
| ERV-K | 321 | 4,723 | 31.6 | 21.0 | 19.4 | 28.0 |
| ERV-L | 665 | 4,450 | 28.7 | 21.0 | 23.4 | 26.9 |
| L1 | 21,930 | 4,788 | 40.1 | 20.3 | 18.4 | 21.1 |
| SINE Alu | 24,000 | 309 | 30.7 | 23.1 | 28.1 | 18.0 |
| Human reference genes | 4,322 | 5,231 | 26.5 | 23.8 | 24.2 | 25.5 |
| Nonhypermutated HIV-1 | 2,378 | 9,030 | 36.2 | 17.6 | 24.0 | 22.1 |
| Hypermutated HIV-1 | ||||||
| In vivo | 83 | 8,700 | 39.1 | 17.5 | 21.3 | 22.2 |
| In vitro | 19 | 8,030 | 37.5 | 17.7 | 22.2 | 22.5 |
Preprocessing of sequences.
The mRNA from the human genes as well as L1 and SINE Alu sequences contain 3′ poly(A) tails. If not removed before analyzing the representation of motifs, these large stretches of adenosines result in biased motif representations and therefore elevated hA3G diagnostic ratio (DRhA3G) and DRhA3F values that are irrelevant to the G-to-A mutations by APOBEC3 enzymes. The poly(A) tails were removed from the sequences using the trimest program of the European Molecular Biology Open Software Suite (EMBOSS; version 6.4.0) (47). The Alu elements also contain a second poly(A) sequence in the middle of their genome that is believed to have resulted from the integration of Alu elements into the genomes of one another (14, 48, 49). Similar to the poly(A) tails, these internal poly(A) sequences need to be removed to obtain an unbiased representation of APOBEC3 target and product motifs. We employed an in-house MATLAB code by using the algorithm used in the trimest program of EMBOSS to remove, from the Alu genomes, the internal poly(A) sequences.
Analysis of motif representation using Markov models.
To quantify the representation of hA3G and hA3F target and product motifs, the observed probability of the motif, pobs(k-mer), was compared with its expected probability, pexp(k-mer), using D = pobs/pexp (44, 50). The overrepresented and underrepresented motifs are characterized by D values of ≫1 and D values of ≪1, respectively. The pobs(k-mer) is defined as the ratio of the number of times the k-mer appears in the sequence to the total number of all k-mers of the same length. The pexp(k-mer) was calculated using the observed probabilities of sub-k-mers (see the following equation and references 44 and 45 for details): D(AG) = pobs(AG)/pexp(AG) = pobs(AG)/[pobs(A) × pobs(G)].
We defined two diagnostic ratios (DRs), one for each APOBEC3 enzyme (45). These DRs are the normalized ratios of the representation of the product over the target motif for each k-mer pair. Examples are given in the following equations: DRhA3G(AG,GG) = D(AG)/[D(AG) + D(GG)] and DRhA3F(AA,GA) = D(AA)/[D(AA) + D(GA)].
DRs of a random sequence with D values of ∼1 are expected to be around 0.5. Mutation by hA3G increases D(product) and decreases D(target), resulting in an increased DR. The footprints of hA3G or hA3F on the endogenous retroelements are characterized by an elevated DRhA3G or DRhA3F, respectively.
RESULTS
ERV-K.
A plot of DRhA3G versus DRhA3F for ERV-K sequences is shown in Fig. 1. For comparison, the data of a nonhypermutated HIV-1 population (green) and hypermutated sequences by hA3G (purple) and hA3F (blue), adapted from reference 45, are also shown in the same figure. The nonhypermutated HIV-1 population that has not been affected by hA3G or hA3F appears as a cluster in the intersection of 0.5 DRhA3G and 0.5 DRhA3F. On the other hand, sequences affected by hA3G and hA3F show elevated DRhA3G and DRhA3F and thus appear extended along the axes of DRhA3G and DRhA3F, respectively. The following three observations are made for the ERV-K sequences.
Fig 1.
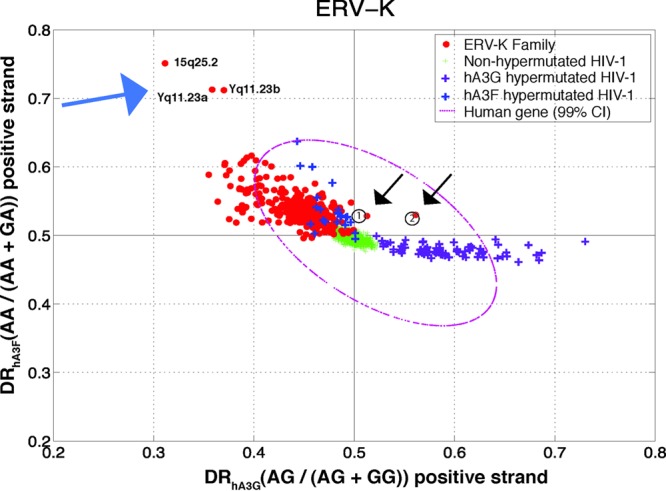
Diagnostic ratio of hA3G versus that of hA3F for the positive strand of the ERV-K family. Each point in this plot is a sequence belonging to ERV-K (red circles), nonhypermutated HIV-1 (green plus signs), HIV-1 hypermutated by hA3G (purple plus signs), HIV-1 hypermutated by hA3F (blue plus signs). The 99% confidence interval (CI) of human genes is shown by a magenta dotted oval. The calculated DR of hA3G and that of hA3F for the previously reported hypermutated HERV-K sequences are indicated by circled numbers 1 (HERV-K-158c3) and 2 (HERV-K-11c21). Genomic locations of three sequences with signatures of hA3F (indicated by blue arrow) are as follows: chr15.+0.84829019.84832364, chrY.-0.26397836.26401035, and chrY.+0.27561401.27564601. The chromosomal locations were extracted using a RepeatMasker tag. Each sequence description has a chromosome number, strand, starting position, and end position, each separated by a period.
(i) Contrary to the human genes and the nonhypermutated HIV-1 sequences that locate as clusters around 0.5 DR, almost the entire ERV-K population exhibits an increased DRhA3F, thus appearing in the top left-hand quarter of Fig. 1. The ERV-K population has extended in the same direction as those of hypermutated HIV-1 by hA3F.
(ii) Contrary to the homogenous and tight oval-like nonhypermutated HIV-1 population, the ERV-K sequences show a wide dispersion, possibly implying their older evolutionary mutation and inactivation history.
(iii) There are three individual ERV-K sequences with highly elevated DRhA3F and two with increased DRhA3G. These sequences, shown by arrows in Fig. 1, contain footprints of a high number of G-to-A mutations within GA and GG, respectively.
Subramanian et al. have recently reported a comprehensive catalogue of the human ERV-K sequences (51). Analysis of the sequences reported in that study and comparison to the sequences analyzed here revealed that the three sequences with the mutational signature of hA3F are the HERV-K elements at loci Yq11.23a, Yq11.23b, and 15q25.2 that belong to the LTR5B family. Compared to the other members of the HERV-K family, these three elements show a large number of context-dependent G-to-A mutations. To investigate whether the observed hypermutation pattern is a result of one or more hypermutation events, we analyzed 1,000-bp sequences flanking the hypermutated HERV-K elements from the 5′ and 3′ ends. The alignment of the flanking regions (Fig. 2) revealed that these three elements share the same flanking sequences, suggesting that they are a result of a single hypermutation event followed by duplication of genomic segments containing the hypermutated HERV-K element.
Fig 2.

Alignment of portions of the flanking sequences of three hypermutated HERV-K elements at 15q25.2, Yq11.23a, and Yq11.23b. There is a 129-bp deletion in the sequence flanking the 3′ end of the element 15q25.2. The element Yq11.23a is on the negative DNA strand. The darker the color is at each position, the higher the similarity is between sequences. Black color indicates 100% identity. This image was generated using the Geneious software.
Additionally, we investigated the DRhA3G and DRhA3F values of the flanking sequences to rule out the possibility that the elevated DR values of hypermutated HERV-K elements are due to the high DR values of their integration sites in the human chromosome, which is known to be highly polymorphic. The results presented in Fig. 3 show that the flanking sequences of nonhypermutated and hypermutated HERV-K sequences form a single cluster. This is in contrast to the results presented in Fig. 1 in which hypermutated HERV-K sequences appeared as outliers. This analysis confirmed that the observed hypermutation signature on these elements is not an artifact, due to the human chromosome being polymorphic.
Fig 3.
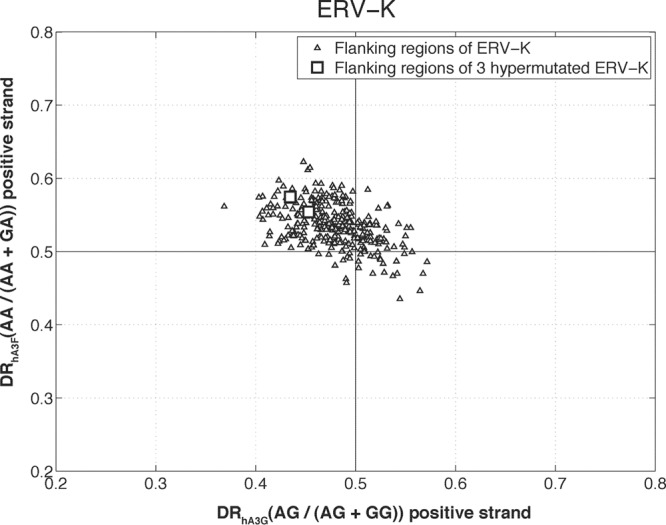
The plot of DRhA3G versus DRhA3F for the sequences flanking the HERV-K elements. The flanking sequences of the three hypermutated HERV-K elements (two of which are overlapping) are shown by squares. As displayed, the flanking sequences form a single cluster without an outlier in the hA3F direction.
The present study reports, for the first time, the footprint of hA3F on the HERV-K genomes. But a few studies have previously identified signatures of hypermutation by hA3G on two HERV-K elements, 11c21 and 158c3 (41). The reference genomes of these two elements are known as HERV-K60 and HERV-KI, respectively (43). In the report by Subramanian et al. (51), these are shown by 21q21.1 and 3q21.2, respectively. These genomes have also been referred to, in a different study, as KCHR21/ch21-0189 and KI/ch3-1271, respectively (42). To investigate whether the two sequences identified by our method as hypermutated by hA3G are identical to the previously known hypermutated HERV-K sequences, we took the following steps.
We downloaded the previously reported hypermutated HERV-K sequences 11c21 and 158c3 and performed a BLAT search to find the best-match sequences in the human build 37 that was employed in our study. The BLAT search returned sequences that existed in our HERV-K data set and were located at loci almost identical to those given by RepeatMasker but that had some deviation of coverage span, perhaps due to a different build version. As shown in Fig. 1 by circled numbers, the previously reported hypermutated sequences have an elevated DRhA3G value, by which they are identifiable from the rest of the HERV-K population.
The results presented so far were related to a G-to-A mutation footprint on the positive strand (i.e., a C-to-T mutation on the negative strand). To further confirm that the observed pattern was attributable to the mutational footprint of APOBEC3 and not merely the result of random G-to-A or other changes, we investigated the same effect on the negative-strand of ERV sequences. Both hA3G and hA3F are known to change G to A almost exclusively on the positive strand (26). This means the viral negative strand is not expected to contain a context-dependent G-to-A footprint. Therefore, the DRhA3G and DRhA3F of the negative strands of the above-mentioned hypermutated ERV sequences are expected to be within the range of those of the rest of the ERV family. However, if the pattern were due to random mutation or other factors, then it would be expected to be present on both strands. As displayed in Fig. 4, the negative strands of nonhypermutated and hypermutated HIV-1 sequences show the same distributions in terms of DRhA3G and DRhA3F. The ERV-K sequences also form a cluster without any apparent outlier. This further confirms the hypermutation status of these five sequences. In contrast to the positive strand, where the nonhypermutated HIV sequences cluster around the DR values of 0.5 (see Fig. 1), the negative strands of the HIV population show an elevated DRhA3F (see Fig. 4). This might be due to sequence context-dependent C-to-T changes on the positive strand or G-to-A changes on the negative strand. Given that nonhypermutated and hypermutated sequences cluster together in Fig. 4, this pattern is unlikely to be a result of hypermutation by APOBEC3 enzymes.
Fig 4.
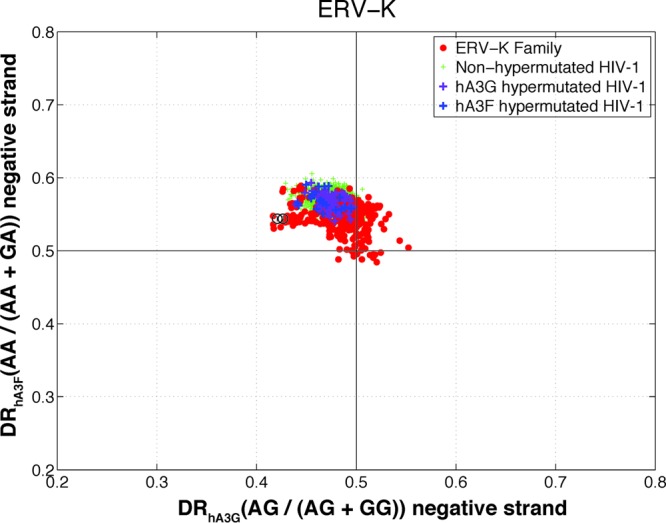
Diagnostic ratio of hA3G versus that of hA3F for the negative strand of the ERV-K family. Each point in this plot is a sequence belonging to ERV-K (red circles), nonhypermutated HIV-1 (green plus signs), HIV-1 hypermutated by hA3G (purple plus signs), and HIV-1 hypermutated by hA3F (blue plus signs). The calculated DR of hA3G and that of hA3F for the negative strand of the previously reported hypermutated HERV-K sequences are indicated by circled numbers 1 (HERV-K-158c3) and 2 (HERV-K-11c21).
ERV-1, ERV-L, LINE, and SINE.
The diagnostic ratio plots for ERV-1, ERV-L, L1, and Alu are shown in Fig. 5. The ERV-1 sequences are more diverse and show a wider distribution of DRs than the ERV-K and ERV-L classes. In contrast to the ERV-K family, the ERV-1 and ERV-L sequences form more clustered populations, although with some outliers. The ERV-1 population exhibits an elevated DRhA3F but to a lesser extent than that of ERV-K. On the other hand, the ERV-L population does not show an increased DRhA3F (however, there are some individual ERV-L elements with an elevated DRhA3F). Therefore, ERV sequences in the human genome can be ordered ERV-K > ERV-1 > ERV-L in terms of the magnitude of GA→AA changes on their genome at the population level. However, it is worth noting that the absolute value of DRs cannot be used to infer evolutionary impacts.
Fig 5.

Diagnostic ratio of hA3G versus that of hA3F in ERV-1 (A), ERV-L (B), L1 (C), and SINE Alu (D) families. Each point in this plot is a sequence belonging to ERV-1 (red circles) in panel A, ERV-L (red circles) in panel B, L1 (red circles) in panel C, SINE-Alu (red circles) in panel D, nonhypermutated HIV-1 (green plus signs), HIV-1 hypermutated by hA3G (purple plus signs), HIV-1 hypermutated by hA3F (blue plus signs). The 99% confidence interval (CI) of human genes is shown by a magenta dotted oval.
Within the ERV-1 and ERV-L families, there are a number of sequences (shown by arrows in Fig. 5A and B) that locate outside the general population clusters. In the case of the ERV-1 family, 13 out of 38 elements of an ERV-1 subfamily known as ERV-S71 (also known as HERV-S71 or S71) appear as a separate cluster (shown by a black arrow in Fig. 5A). These sequences extend in a direction that is characteristic of mutation by both hA3G and hA3F. The ERV-S71 family is a distinct subclass compared to the other ERV-1 subclasses. A deletion in the pol region and an absence of the 5′ LTR have been reported for this ERV-1 subclass (52) that resembles the ecotropic murine leukemia virus AKV and primate proviruses such as baboon endogenous virus (BaEV) and simian sarcoma virus (SSV) (53). Previous studies have found signatures of mutation by murine APOBEC3 on the genomes of murine leukemia viruses (34, 54, 55). To confirm the signatures of hA3G and hA3F on the genomes of these 13 members of the ERVS71 subfamily, we generated a consensus sequence from the remaining 25 ERV-S71 members that clustered with the general ERV-1 population. Then we aligned the 13 suspected sequences with this consensus sequence and used the program Hypermut2 from the Los Alamos National Laboratory website (http://www.hiv.lanl.gov/content/sequence/HYPERMUT/hypermut.html) to investigate the mutations by hA3G and hA3F. The hypermutation footprint of hA3G (GG→AG) and hA3F (GA→AA) was found, with a high confidence level (P < 2.6 × 10−15), on all 13 sequences. The results are presented in Fig. 6 and Table 2. As displayed in Fig. 6, these sequences show very similar hypermutation patterns. This prompted us to speculate that these signatures are a result of a single hypermutation event followed by genomic segmental duplications. To test this hypothesis, we analyzed 1,500 bp of the 5′ and 3′ sequences flanking the hypermutated HERV-S71 elements. Using alignment (Fig. 7), it was found that the hypermutated HERV-S71 elements share the same flanking sequences, thus pointing to a single hypermutation event followed by several segmental duplication events.
Fig 6.

Analysis of the hypermutation footprint of hA3G and hA3F on 13 HERV-S71 sequences from the ERV-1 family. Sequences were aligned using MAFT, and a consensus sequence was generated from 25 HERV-S71 sequences that had normal DR values. Thirteen suspected hypermutated HERV-S71 sequences were aligned with the consensus sequence, and their hypermutation status was investigated using the Hypermut2 program from the Los Alamos National Laboratory. The vertical axis shows the number of mutations in each sequence against the consensus sequence. The horizontal axis shows a brief description of each of the HERV-S71 sequences (chromosomal location described by the RepeatMasker tag). Each sequence description has chromosome number, strand, starting position, and end position, each separated by a period. The bars represent total numbers of G-to-A mutations as well as those within different sequence contexts.
Table 2.
Result of hypermutation analysis by Hypermut2 for 13 HERVS71 sequences from the ERV-1 family
| Sequence | Fisher exact P valuea |
|---|---|
| chr11.−.67653255.67656426 | 1.22E−17 |
| chr11.+.71417587.71420776 | 9.99E−19 |
| chr4.−.4035668.4038849 | 5.77E−19 |
| chr7.−.7031700.7034884 | 6.97E−19 |
| chr4.+.9602012.9605198 | 3.12E−18 |
| chr3.−.129899217.129902403 | 5.89E−16 |
| chr8.−.12381112.12384285 | 4.20E−17 |
| chr8.+.7996414.7999582 | 4.20E−17 |
| chr3.−.75552888.75556064 | 4.37E−19 |
| chr11.−.3534266.3537462 | 6.22E−17 |
| chr3.+.125551334.125554491 | 1.52E−18 |
| chr12.−.8473773.8476964 | 2.61E−15 |
| chr4.+.9070675.9073870 | 3.98E−18 |
P values of ≪0.05 show significant hypermutation by APOBEC3 enzymes.
Fig 7.

Alignment of a portion of the flanking sequences of 13 hypermutated HERV-S71 elements. The darker the color is at each position, the higher the similarity is between sequences. Black color indicates 100% identity. This image was generated using the Geneious software.
The ERV-1 sequence that is shown by a blue arrow in Fig. 5A belongs to the subfamily of LTR25 and appears to have a signature of mutation by hA3F. This sequence was compared, using Hypermut2, to the consensus sequence made from the available 22 LTR25 sequences. The results, presented in Fig. 8, indicated a significant GA→AA footprint on this genome.
Fig 8.
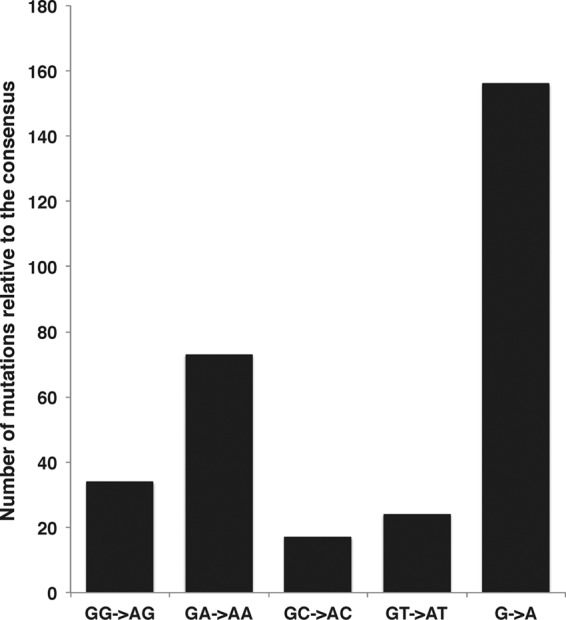
Analysis of the hypermutation footprint of hA3G and hA3F on a suspected LTR25 sequence from the ERV-1 family. Twenty-two LTR25 sequences were aligned using MAFT, and a consensus sequence was generated to compare against the potentially hypermutated LTR25 sequence, using the Hypermut2 program from the Los Alamos National Laboratory. The vertical axis shows the number of mutations against the consensus sequence. The bars represent the total number of G-to-A changes as well as changes within different sequence contexts.
In the case of ERV-L, the outlier sequences (shown by black arrows) belong to the subfamilies of HERV-L66 (n = 2), HERV-L74 (n = 2), LTR52 (n = 1), and LTR57 (n = 2). For these subfamilies, there was not a sequence that clustered with the normal (nonhypermutated) ERV-L family; therefore, it was not possible to confirm, using alignment and Hypermut2, the signatures of hA3F.
We also investigated the footprint of APOBEC3 on the negative strand of the ERV-1 and ERV-L sequences. As stated earlier, the negative strand is not expected to contain a footprint. The results of the analysis of negative strands are shown in Fig. 9. Noticeably, the 13 HERV-S71 sequences that showed signature of hA3G and hA3F and the single LTR25 sequence with the hA3F footprint on their positive strand do not exhibit such patterns on their negative strand. This further confirms that these sequences have been targeted by APOBEC3 enzymes. Interestingly, a separate cluster containing all LTR12B and LTR12D sequences emerges from the analysis of negative strands (shown by the black arrow in Fig. 9A). These sequences show a very high DRhA3G on their negative strand but not on their positive strand. This pattern is the opposite of the general signature of hA3G in terms of strand specificity. Currently, this pattern is under investigation in our laboratory.
Fig 9.

Diagnostic ratio plots of the negative strand for ERV-1 (A) and ERV-L (B) families. Each point in this plot is a sequence belonging to ERV-1 (red circles in panel A), ERV-L (red circles in panel B), normal HIV-1 (green plus signs), HIV-1 hypermutated by hA3G (purple plus signs), HIV-1 hypermutated by hA3F (blue plus signs), and HERV-S71 of ERV-1 family (yellow circles in panel A). The black arrow in panel A points to LTR12B and LTR12D sequence cluster.
L1 and SINE Alu.
As indicated in Fig. 5, neither L1 nor SINE Alu sequences show an elevated DR for hA3G and/or hA3F enzymes. This implies, contrary to the ERV sequences, that the LINE and SINE elements do not show a mutational footprint of hA3G and/or hA3F. However, the SINE elements show a decreased DRhA3F that suggests a mutational footprint in the opposite direction of the hA3F signature. It implies that the genomes of the SINE Alu population contain a footprint of A-to-G mutation in the context of AA. This might be a signature of mutation by ADAR adenosine deaminase enzymes that are known to induce an A-to-G mutation in Alu elements (56–58). The L1 population, on the other hand, seems to have a decreased DRhA3G that might point to an A-to-G footprint in the context of AG dinucleotides.
DISCUSSION
Although there is strong evidence that some retroelements have vital cellular roles, retrotransposition is potentially harmful, in particular when it disturbs genes (59, 60). One of the mechanisms by which cells control retrotransposition is editing by restriction enzymes, such as APOBEC. Several members of the APOBEC family have been shown to inhibit retrotransposition in yeasts, mice, and humans (24, 27, 34, 61, 62).
The ability of APOBEC3 enzymes to hypermutate exogenous as well as endogenous retroviruses (27, 32, 63) and more importantly the discovery of G-to-A mutation footprints by the mouse APOBEC3 (mA3) on the genome of endogenous murine leukemia viruses (34) as well as those of human APOBEC3G (hA3G) on individual HERV-K genomes (41–43) prompted us to speculate that hA3 proteins have played a role in the evolution and/or inactivation of endogenous retroelements. This hypothesis is further supported by the general consensus that the majority of retroelements are inactive and their genome contains multiple deleterious mutations (8).
Lentiviruses such as HIV encode a virion infectivity factor (Vif) gene that can prevent hypermutation by hA3G and hA3F (64, 65). This gene does not exist in the genome of retroelements, and no other gene has been shown to have a similar function in retroelements (66). This further supports our hypothesis that retroelements are likely to have been affected by hA3G and/or hA3F.
Given that LINE and SINE are nuclear elements, they are not expected to contain a footprint of mutation by hA3G and hA3F, which are cytoplasmic proteins. Studies have shown that APOBEC3 enzymes do not mutate L1 elements (61, 67, 68), which reverse transcribe in the nuclei (67, 69). For some members of the APOBEC3 family, such as APOBEC3A and APOBEC3B, that have been detected in the nuclei, deamination-independent mechanisms are thought to be the dominant method in inhibiting retrotransposition (67, 70, 71). It has been suggested that the single-stranded nucleic acid substrate may not be available for editing by APOBEC3 members during the target-primed reverse transcription of non-LTR elements (27).
Similarly, studies have shown that inhibition of SINE Alu elements is also by deamination-independent mechanisms (72). It is known that hA3G inhibits SINE Alu by sequestering its RNA into high-molecular-mass complexes and keeping it away from the nuclear machinery, including L1 targeting (72), that is required by these nonautonomous elements for replication. Moreover, association of hA3F with the L1 ORF2 protein can hinder the mobilization of Alu elements (71). The inhibition of L1 machinery (thus indirect restriction of SINE Alu retrotransposition) by other members of the APOBEC3 family has also been suggested (71, 73).
ERVs, on the other hand, are structurally similar to the modern exogenous retroviruses and even have a common gene order. These genomic fossils are regarded as ancestral sequences of exogenous retroviruses (74, 75). ERVs reverse transcribe in the cytoplasm in a way that is similar to that of their exogenous counterparts. Therefore, it is reasonable to postulate that they might have been evolved at a population level and/or have been inactivated at a single-genome level by APOBEC3s.
It is believed that no human ERV element is active or infectious (8), and the most recent replication events in human date back millions of years (43), although we note that a recent in vitro study suggests that infectious HERV-K elements can potentially emerge via recombination (76). Experiments have shown that the APOBEC3 family can actively inhibit retrotransposition of LTR retroelements in yeasts, mice, and humans (42, 61–63, 77, 78), but whether the LTR elements have evolved or become inactivated from mutations by APOBEC3s is unclear. What is known so far is that a few HERV-K sequences contain the footprint of the G-to-A mutation induced by hA3G only (79). Interestingly, in a recent communication by Carmi et al. (79), pairwise comparison of retroelement genomes has identified sequences with clusters of G-to-A mutations that have been attributed by the authors to the APOBEC3 enzymes. However, in that study, the motif dependency of the G-to-A mutation, which is a hallmark of hA3G and hA3F, was not investigated. It is worth noting that cytosine methylation within CG (i.e., CpG) followed by deamination in one of the DNA strands can result in a CG→CA mutation on the other strand. Therefore, a cluster of G-to-A changes that are used in the study by Carmi et al. (79) as an evidence of APOPBEC mutation could well be attributed to CpG methylation and spontaneous deamination. Given that we consider the sequence context dependency of G-to-A mutations, G-to-A mutations (i.e., random or in other sequence contexts) by other mechanisms do not affect our analysis.
In this paper, we used a recently developed method based on motif representation (45) to investigate the footprint of editing by hA3 enzymes on the genome of endogenous retroelements. We defined variables using the representation data to include, in the analysis, context dependency of mutation by individual enzymes. We identified both several individual ERV sequences with footprints of mutations by hA3F or hA3G and some ERV classes (e.g., ERV-K) in which the populations of ERV elements seem to be affected by hA3F. This suggests an ongoing role for these enzymes in controlling retrotransposition.
Further studies are needed to identify the molecular source of GA→AA footprints from among the members of the APOBEC3 family which preferentially mutate G within GA motifs. The use of hA3F in this work was only to represent the six members of the APOBEC3 family with a GA→AA mutation preference.
The method used in the present work is a conservative approach. This is because we mainly attempted to identify hypermutated sequences (outliers of the population in the APOBEC3 directions) by analyzing subfamilies of the same retroelement family (e.g., all ERV-K subfamilies of HERV-K3, HERV-K9, HERV-K11, HERV-K22) at the same time. An alternative and less conservative approach would be to analyze the sequences of each individual subfamily separately. However, in order to infer hypermutation signatures (with high confidence levels) from such analyses, there needs to be an adequately large number of member sequences with sufficient length in each subfamily.
ACKNOWLEDGMENTS
F.A. is supported by a postgraduate scholarship from the Australian Government and the University of New South Wales. M.P.D. is a senior research fellow funded by the Australian National Health and Medical Research Council (NHMRC). D.E. is funded by an Early Career Fellowship from NHMRC.
We declare no conflicts of interests.
Footnotes
Published ahead of print 22 May 2013
REFERENCES
- 1. Kazazian HH., Jr 2004. Mobile elements: drivers of genome evolution. Science 303:1626–1632 [DOI] [PubMed] [Google Scholar]
- 2. Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409:860–921 [DOI] [PubMed] [Google Scholar]
- 3. Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, Antonarakis SE, Attwood J, Baertsch R, Bailey J, Barlow K, Beck S, Berry E, Birren B, Bloom T, Bork P, Botcherby M, Bray N, Brent MR, Brown DG, Brown SD, Bult C, Burton J, Butler J, Campbell RD, Carninci P, Cawley S, Chiaromonte F, Chinwalla AT, Church DM, Clamp M, Clee C, Collins FS, Cook LL, Copley RR, Coulson A, Couronne O, Cuff J, Curwen V, Cutts T, Daly M, David R, Davies J, Delehaunty KD, Deri J, Dermitzakis ET, et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420:520–562 [DOI] [PubMed] [Google Scholar]
- 4. Cullen BR. 2006. Role and mechanism of action of the APOBEC3 family of antiretroviral resistance factors. J. Virol. 80:1067–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Belshaw R, Pereira V, Katzourakis A, Talbot G, Paces J, Burt A, Tristem M. 2004. Long-term reinfection of the human genome by endogenous retroviruses. Proc. Natl. Acad. Sci. U. S. A. 101:4894–4899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Belshaw R, Dawson ALA, Woolven-Allen J, Redding J, Burt A, Tristem M. 2005. Genomewide screening reveals high levels of insertional polymorphism in the human endogenous retrovirus family HERV-K(HML2): implications for present-day activity. J Virol. 79:12507–12514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bannert N, Kurth R. 2004. Retroelements and the human genome: new perspectives on an old relation. Proc. Natl. Acad. Sci. U. S. A. 101:14572–14579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hughes JF, Coffin JM. 2004. Human endogenous retrovirus K solo-LTR formation and insertional polymorphisms: implications for human and viral evolution. Proc. Natl. Acad. Sci. U. S. A. 101:1668–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kijak GH, Janini LM, Tovanabutra S, Sanders-Buell E, Arroyo MA, Robb ML, Michael NL, Birx DL, McCutchan FE. 2008. Variable contexts and levels of hypermutation in HIV-1 proviral genomes recovered from primary peripheral blood mononuclear cells. Virology 376:101–111 [DOI] [PubMed] [Google Scholar]
- 10. Khatua AK, Taylor HE, Hildreth JE, Popik W. 2010. Inhibition of LINE-1 and Alu retrotransposition by exosomes encapsidating APOBEC3G and APOBEC3F. Virology 400:68–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Beck CR, Collier P, Macfarlane C, Malig M, Kidd JM, Eichler EE, Badge RM, Moran JV. 2010. LINE-1 retrotransposition activity in human genomes. Cell 141:1159–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deininger PL, Batzer MA. 1999. Alu repeats and human disease. Mol. Genet. Metab. 67:183–193 [DOI] [PubMed] [Google Scholar]
- 13. Dewannieux M, Esnault C, Heidmann T. 2003. LINE-mediated retrotransposition of marked Alu sequences. Nat. Genet. 35:41–48 [DOI] [PubMed] [Google Scholar]
- 14. Rowold DJ, Herrera RJ. 2000. Alu elements and the human genome. Genetica 108:57–72 [DOI] [PubMed] [Google Scholar]
- 15. Deininger PL, Batzer MA. 2002. Mammalian retroelements. Genome Res. 12:1455–1465 [DOI] [PubMed] [Google Scholar]
- 16. Deininger PL, Moran JV, Batzer MA, Kazazian HH. 2003. Mobile elements and mammalian genome evolution. Curr. Opin. Genet. Dev. 13:651–658 [DOI] [PubMed] [Google Scholar]
- 17. Mi S, Lee X, Li X, Veldman GM, Finnerty H, Racie L, LaVallie E, Tang XY, Edouard P, Howes S, Keith JC, McCoy JM. 2000. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 403:785–789 [DOI] [PubMed] [Google Scholar]
- 18. Rote NS, Chakrabarti S, Stetzer BP. 2004. The role of human endogenous retroviruses in trophoblast differentiation and placental development. Placenta 25:673–683 [DOI] [PubMed] [Google Scholar]
- 19. Sekita Y, Wagatsuma H, Nakamura K, Ono R, Kagami M, Wakisaka N, Hino T, Suzuki-Migishima R, Kohda T, Ogura A, Ogata T, Yokoyama M, Kaneko-Ishino T, Ishino F. 2008. Role of retrotransposon-derived imprinted gene, Rtl1, in the feto-maternal interface of mouse placenta. Nat. Genet. 40:243–248 [DOI] [PubMed] [Google Scholar]
- 20. Kazazian HH. 1999. An estimated frequency of endogenous insertional mutations in humans. Nat. Genet. 22:130. [DOI] [PubMed] [Google Scholar]
- 21. Symer DE, Connelly C, Szak ST, Caputo EM, Cost GJ, Parmigiani G, Boeke JD. 2002. Human l1 retrotransposition is associated with genetic instability in vivo. Cell 110:327–338 [DOI] [PubMed] [Google Scholar]
- 22. Arjan-Odedra S, Swanson CM, Sherer NM, Wolinsky SM, Malim MH. 2012. Endogenous MOV10 inhibits the retrotransposition of endogenous retroelements but not the replication of exogenous retroviruses. Retrovirology 9:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barlow DP. 1993. Methylation and imprinting: from host defense to gene regulation? Science 260:309–310 [DOI] [PubMed] [Google Scholar]
- 24. Chiu YL, Greene WC. 2008. The APOBEC3 cytidine deaminases: an innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu. Rev. Immunol. 26:317–353 [DOI] [PubMed] [Google Scholar]
- 25. Goodier JL, Cheung LE, Kazazian HH. 2012. MOV10 RNA helicase is a potent inhibitor of retrotransposition in cells. PLoS Genet. 8:e1002941. 10.1371/journal.pgen.1002941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Harris RS, Liddament MT. 2004. Retroviral restriction by APOBEC proteins. Nat. Rev. Immunol. 4:868–877 [DOI] [PubMed] [Google Scholar]
- 27. Holmes RK, Malim MH, Bishop KN. 2007. APOBEC-mediated viral restriction: not simply editing? Trends Biochem. Sci. 32:118–128 [DOI] [PubMed] [Google Scholar]
- 28. Kazazian HH. 2011. Host factors involved in L1 retrotransposition, p 203–205 Mobile DNA: finding treasure in junk. FT Press, Upper Saddle River, NJ [Google Scholar]
- 29. Yoder JA, Walsh CP, Bestor TH. 1997. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 13:335–340 [DOI] [PubMed] [Google Scholar]
- 30. Löwer R, Löwer J, Kurth R. 1996. The viruses in all of us—characteristics and biological significance of human endogenous retrovirus sequences. Proc. Natl. Acad. Sci. U. S. A. 93:5177–5184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Maksakova IA, Romanish MT, Gagnier L, Dunn CA, van de Lagemaat LN, Mager DL. 2006. Retroviral elements and their hosts: insertional mutagenesis in the mouse germ line. PLoS Genet. 2:e2. 10.1371/journal.pgen.0020002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Turelli P, Trono D. 2005. Editing at the crossroad of innate and adaptive immunity. Science 307:1061–1065 [DOI] [PubMed] [Google Scholar]
- 33. LaRue RS, Jónsson SR, Silverstein KA, Lajoie M, Bertrand D, El-Mabrouk N, Hötzel I, Andrésdóttir V, Smith TP, Harris RS. 2008. The artiodactyl APOBEC3 innate immune repertoire shows evidence for a multi-functional domain organization that existed in the ancestor of placental mammals. BMC Mol. Biol. 9:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jern P, Stoye JP, Coffin JM. 2007. Role of APOBEC3 in genetic diversity among endogenous murine leukemia viruses. PLoS Genet. 3:2014–2022. 10.1371/journal.pgen.0030183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sawyer SL, Emerman M, Malik HS. 2004. Ancient adaptive evolution of the primate antiviral DNA-editing enzyme APOBEC3G. PLoS Biol. 2:E275. 10.1371/journal.pbio.0020275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang J, Webb DM. 2004. Rapid evolution of primate antiviral enzyme APOBEC3G. Hum. Mol. Genet. 13:1785–1791 [DOI] [PubMed] [Google Scholar]
- 37. Holmes RK, Koning FA, Bishop KN, Malim MH. 2007. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J. Biol. Chem. 282:2587–2595 [DOI] [PubMed] [Google Scholar]
- 38. Liddament MT, Brown WL, Schumacher AJ, Harris RS. 2004. APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr. Biol. 14:1385–1391 [DOI] [PubMed] [Google Scholar]
- 39. Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. 2003. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 424:99–103 [DOI] [PubMed] [Google Scholar]
- 40. Pathak VK, Temin HM. 1990. Broad spectrum of in vivo forward mutations, hypermutations, and mutational hotspots in a retroviral shuttle vector after a single replication cycle: deletions and deletions with insertions. Proc. Natl. Acad. Sci. U. S. A. 87:6024–6028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Armitage AE, Katzourakis A, de Oliveira T, Welch JJ, Belshaw R, Bishop KN, Kramer B, McMichael AJ, Rambaut A, Iversen AK. 2008. Conserved footprints of APOBEC3G on hypermutated human immunodeficiency virus type 1 and human endogenous retrovirus HERV-K(HML2) sequences. J. Virol. 82:8743–8761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Esnault C, Priet S, Ribet D, Heidmann O, Heidmann T. 2008. Restriction by APOBEC3 proteins of endogenous retroviruses with an extracellular life cycle: ex vivo effects and in vivo “traces” on the murine IAPE and human HERV-K elements. Retrovirology 5:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lee YN, Malim MH, Bieniasz PD. 2008. Hypermutation of an ancient human retrovirus by APOBEC3G. J. Virol. 82:8762–8770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ebrahimi D, Anwar F, Davenport MP. 2011. APOBEC3 has not left an evolutionary footprint on the HIV-1 genome. J. Virol. 85:9139–9146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ebrahimi D, Anwar F, Davenport MP. 2012. APOBEC3G and APOBEC3F rarely co-mutate the same HIV genome. Retrovirology 9:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hultquist JF, Lengyel JA, Refsland EW, LaRue RS, Lackey L, Brown WL, Harris RS. 2011. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J. Virol. 85:11220–11234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rice P, Longden I, Bleasby A. 2000. EMBOSS: the European molecular biology open software suite. Trends Genet. 16:276–277 [DOI] [PubMed] [Google Scholar]
- 48. Dridi S. 2012. Alu mobile elements: from junk DNA to genomic gems. Scientifica 2012:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Renwick PJ, Birley AJ, Hultén MA. 1997. Study of Alu sequences at the hypoxanthine phosphoribosyltransferase (HPRT) encoding region of man. Gene 184:155–162 [DOI] [PubMed] [Google Scholar]
- 50. Philips GJ, Arnold J, Ivarie R. 1987. Mono- through hexanucleotide composition of the Escherichia coli genome—a Markov chain analysis. Nucleic Acids Res. 15:2611–2626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Subramanian RP, Wildschutte JH, Russo C, Coffin JM. 2011. Identification, characterization, and comparative genomic distribution of the HERV-K (HML-2) group of human endogenous retroviruses. Retrovirology 8:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yi J-M, Kim H-S. 2007. Expression and phylogenetic analyses of human endogenous retrovirus HC2 belonging to the HERV-T family in human tissues and cancer cells. J. Hum. Genet. 52:285–296 [DOI] [PubMed] [Google Scholar]
- 53. Werner T, Brack-Werner R, Leib-Mösch C, Backhaus H, Erfle V, Hehlmann R. 1990. S71 is a phylogenetically distinct human endogenous retroviral element with structural and sequence homology to simian sarcoma virus SSV. Virology 174:225–238 [DOI] [PubMed] [Google Scholar]
- 54. Langlois M-A, Kemmerich K, Rada C, Neuberger MS. 2009. The AKV murine leukemia virus is restricted and hypermutated by mouse APOBEC3. J. Virol. 83:11550–11559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Perez-Caballero D, Soll SJ, Bieniasz PD. 2008. Evidence for restriction of ancient primate gammaretroviruses by APOBEC3 but not TRIM5alpha proteins. PLoS Pathog. 4:e1000181. 10.1371/journal.ppat.1000181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kim DDY, Kim TTY, Walsh T, Kobayashi Y, Matise TC, Buyske S, Gabriel A. 2004. Widespread RNA editing of embedded Alu elements in the human transcriptome. Genome Res. 14:1719–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Levanon EY, Eisenberg E, Yelin R, Nemzer S, Hallegger M, Shemesh R, Fligelman ZY, Shoshan A, Pollock SR, Sztybel D, Olshansky M, Rechavi G, Jantsch MF. 2004. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat. Biotechnol. 22:1001–1005 [DOI] [PubMed] [Google Scholar]
- 58. Paz-Yaacov N, Levanon EY, Nevo E, Kinar Y, Harmelin A, Jacob-Hirsch J, Amariglio N, Eisenberg E, Rechavi G. 2010. Adenosine-to-inosine RNA editing shapes transcriptome diversity in primates. Proc. Natl. Acad. Sci. U. S. A. 107:12174–12179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Callinan A, Batzer MA. 2006. Retrotransposable elements and human disease. Genome 1:104–115 [DOI] [PubMed] [Google Scholar]
- 60. Chen J-M, Férec C, Cooper DN. 2006. LINE-1 endonuclease-dependent retrotranspositional events causing human genetic disease: mutation detection bias and multiple mechanisms of target gene disruption. J. Biomed. Biotech. 2006:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Esnault C, Heidmann O, Delebecque F, Dewannieux M, Ribet D, Hance AJ, Heidmann T, Schwartz O. 2005. APOBEC3G cytidine deaminase inhibits retrotransposition of endogenous retroviruses. Nature 433:430–433 [DOI] [PubMed] [Google Scholar]
- 62. Schumacher AJ, Nissley DV, Harris RS. 2005. APOBEC3G hypermutates genomic DNA and inhibits Ty1 retrotransposition in yeast. Proc. Natl. Acad. Sci. U. S. A. 102:9854–9859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Esnault C, Millet J, Schwartz O, Heidmann T. 2006. Dual inhibitory effects of APOBEC family proteins on retrotransposition of mammalian endogenous retroviruses. Nucleic Acids Res. 34:1522–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Stopak K, de Noronha C, Yonemoto W, Greene WC. 2003. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol. Cell 12:591–601 [DOI] [PubMed] [Google Scholar]
- 65. Zheng YH, Irwin D, Kurosu T, Tokunaga K, Sata T, Peterlin BM. 2004. Human APOBEC3F is another host factor that blocks human immunodeficiency virus type 1 replication. J. Virol. 78:6073–6076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Oberste MS, Gonda MA. 1992. Conservation of amino-acid sequence motifs in lentivirus Vif proteins. Virus Genes 6:95–102 [DOI] [PubMed] [Google Scholar]
- 67. Bogerd HP, Wiegand HL, Hulme AE, Garcia-Perez JL, O'Shea KS, Moran JV, Cullen BR. 2006. Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc. Natl. Acad. Sci. U. S. A. 103:8780–8785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Turelli P, Vianin S, Trono D. 2004. The innate antiretroviral factor APOBEC3G does not affect human LINE-1 retrotransposition in a cell culture assay. J. Biol. Chem. 279:43371–43373 [DOI] [PubMed] [Google Scholar]
- 69. Cost GJ, Feng Q, Jacquier A, Boeke JD. 2002. Human L1 element target-primed reverse transcription in vitro. EMBO J. 21:5899–5910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ikeda T, Abd El Galil KH, Tokunaga K, Maeda K, Sata T, Sakaguchi N, Heidmann T, Koito A. 2011. Intrinsic restriction activity by apolipoprotein B mRNA editing enzyme APOBEC1 against the mobility of autonomous retrotransposons. Nucleic Acids Res. 39:5538–5554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Stenglein MD, Harris RS. 2006. APOBEC3B and APOBEC3F inhibit L1 retrotransposition by a DNA deamination-independent mechanism. J. Biol. Chem. 281:16837–16841 [DOI] [PubMed] [Google Scholar]
- 72. Chiu YL, Witkowska HE, Hall SC, Santiago M, Soros VB, Esnault C, Heidmann T, Greene WC. 2006. High-molecular-mass APOBEC3G complexes restrict Alu retrotransposition. Proc. Natl. Acad. Sci. U. S. A. 103:15588–15593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hulme AE, Bogerd HP, Cullen BR, Moran JV. 2007. Selective inhibition of Alu retrotransposition by APOBEC3G. Gene 390:199–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Boeke JD, Stoye JP. 1997. Retrotransposons, endogenous retroviruses, and the evolution of retroelements. In Coffin JM, Hughes SH, Varmus HE. (ed), Retroviruses. CSHL Press, New York, NY [Google Scholar]
- 75. Temin HM. 1992. Origin and general nature of retroviruses, p 1–18 In Levy JA. (ed), The Retroviridae. Plenum Press, New York, NY [Google Scholar]
- 76. Dewannieux M, Harper F, Richaud A, Letzelter C, Ribet D, Pierron G, Heidmann T. 2006. Identification of an infectious progenitor for the multiple-copy HERV-K human endogenous retroelements. Genome Res. 16:1548–1556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Chen H, Lilley CE, Yu Q, Lee DV, Chou J, Narvaiza I, Landau NR, Weitzman MD. 2006. APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr. Biol. 16:480–485 [DOI] [PubMed] [Google Scholar]
- 78. Lee YN, Bieniasz PD. 2007. Reconstitution of an infectious human endogenous retrovirus. PLoS Pathog. 3:e10. 10.1371/journal.ppat.0030010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Carmi S, Church GM, Levanon EY. 2011. Large-scale DNA editing of retrotransposons accelerates mammalian genome evolution. Nat. Commun. 2:519. [DOI] [PubMed] [Google Scholar]