

Abstract
Influenza virus defective interfering (DI) particles are naturally occurring noninfectious virions typically generated during in vitro serial passages in cell culture of the virus at a high multiplicity of infection. DI particles are recognized for the role they play in inhibiting viral replication and for the impact they have on the production of infectious virions. To date, influenza virus DI particles have been reported primarily as a phenomenon of cell culture and in experimentally infected embryonated chicken eggs. They have also been isolated from a respiratory infection of chickens. Using a sequencing approach, we characterize several subgenomic viral RNAs from human nasopharyngeal specimens infected with the influenza A(H1N1)pdm09 virus. The distribution of these in vivo-derived DI-like RNAs was similar to that of in vitro DIs, with the majority of the defective RNAs generated from the PB2 (segment 1) of the polymerase complex, followed by PB1 and PA. The lengths of the in vivo-derived DI-like segments also are similar to those of known in vitro DIs, and the in vivo-derived DI-like segments share internal deletions of the same segments. The presence of identical DI-like RNAs in patients linked by direct contact is compatible with transmission between them. The functional role of DI-like RNAs in natural infections remains to be established.
INTRODUCTION
Defective interfering (DI) particles are best described as noninfectious virions that carry an incomplete copy of their genome (1). They are defective for in vitro replication and compete with infectious viruses during replication (1, 2). Influenza virus DI particles are generated during multiple passages of infectious viruses in the host cell where the defective RNA is replicated in the presence of a helper (i.e., functionally competent) virus (3, 4). Influenza virus defective interfering particles have been observed during in vitro serial passages in cell culture of the virus at a high multiplicity of infection (5, 6). Defective RNAs have also been observed in experimentally infected embryonated chicken eggs (7). The only instance DI particles were found linked to a natural infection was during an H5N2 respiratory outbreak in chickens where defective interfering particles were seen in clonal isolates generated in embryonated eggs (8, 9). To date, there are no reports of their occurrence in human infections. Importantly, the influenza virus DI particles characterized thus far are of similar size and density as the infectious or standard virus, which differentiates them from DI particles of other viruses. In addition, structural and biological properties of the major proteins, like hemagglutinin and neuraminidase, are conserved (3).
The genome of influenza virus consists of eight negative-sense RNA segments (viral RNA [vRNA]) that encode 10 to 12 proteins, depending on the subtype. During virion assembly, one copy of each of the eight segments is required to produce a fully infectious virus particle (10). DI particles are identified as viruses containing at least one defective RNA segment with a large internal deletion. Although DI RNAs are believed to be the products of an aberrant replication event and are replicated during high-multiplicity passage, the generation of the subgenomic length vRNAs is sequence specific. DI RNA segments have a deletion of internal sequences of the parental vRNA while retaining certain 5′ and 3′ end-specific sequences of the progenitor vRNA. All influenza A virus DI RNAs retain identical terminal sequences derived from the first 13 nucleotides (nt) of the 5′ end (AGUAGAAACAAGG) and the last 12 nucleotides of the 3′ end (-CCUGCUUUCGCU-OH) of the segments and typically contain the critical packaging signals, which are within the terminal coding sequences of each gene segment (5, 11).
Those naturally occurring influenza virus DI RNAs documented thus far originate mostly from the polymerase genes (PB2, PB1, and PA), with a few cases of viral RNA deletions seen in other segments (7, 11, 12). DI vRNAs of almost all the major genes of influenza virus have been identified (10). In the first study identifying DI RNAs, the flanking sequences of internal deletions of the polymerase gene did not resemble a splicing sequence, an indication that this was the result of a replication event, rather than a splicing event (13). A coinoculation study with virus A/WSN (H1N1) (12) identified DI RNAs in mouse lungs, ranging in size between 350 and 450 nt and derived from PB1, PB2, PA, and matrix (M) segments with similar internal deletion, junction, and substitution sites as seen in earlier studies (7). In comparison to the full-length vRNAs, DI RNAs appear to be preferentially incorporated into the virions at the assembly step (14).
In the present study, we identified and characterized defective influenza virus RNAs by deep sequencing of primary clinical specimens collected from the upper respiratory tracts of symptomatic infected individuals. Critically, therefore, we consider DIs in nature rather than those in cell culture. The majority of these DI-like RNAs were derived from the polymerase subunit genes, with the highest number of these coming from PB2, followed by PB1 and PA, as observed in previous studies for culture-isolated viral particles (6, 7). We confirmed that these DI-like RNAs are present in the original upper respiratory tract specimens.
MATERIALS AND METHODS
Sample collection.
INSIGHT (International Network for Strategic Initiatives in Global HIV Trials) is a large, multinational clinical trial network funded by the National Institute of Allergy and Infectious Diseases (NIAID) of NIH. Following the appearance of the influenza A(H1N1)pdm09 virus in 2009 (15), INSIGHT is conducting two large, international observational cohort studies of patients with influenza-like illness (ILI): FLU 002, conducted in an outpatient setting, and FLU 003, enrolling patients who are hospitalized with ILI. Baseline specimens, including deep nasal and oropharyngeal specimens, are collected and transported to a central laboratory for analysis, and patients are monitored for clinical outcomes over 14 days (FLU 002) or 60 days (FLU 003). Deep nasal and oropharyngeal swabs were collected from INSIGHT FLU 002 and 003 study participants (after giving informed consent) with influenza-like illness and placed into a single 3-ml vial of universal transport medium (Diagnostic Hybrids or Copan Diagnostics, depending on the collection site). One-milliliter aliquots were prepared and stored at −70°C. Aliquoted samples were shipped on dry ice to the central laboratory repository (Advanced BioMedical Laboratories, Cinnaminson, NJ, USA). One aliquot was used for nucleic acid testing for influenza A virus subtypes: if influenza A(H1N1)pdm09 was detected, another 1-ml aliquot (unopened) was sent on dry ice to the J. Craig Venter Institute laboratory (JCVI) (Rockville, MD, USA) for influenza virus sequence analysis.
Sequencing and analysis.
Viral RNA was extracted from nasopharyngeal specimens and amplified by multisegment reverse transcription-PCR (RT-PCR) (16). The amplicons were processed for Sanger sequencing, as previously described (17). Problem samples, i.e., samples for which full virus segments could not be assembled, were sent for deep-sequence analysis on next-generation sequencing platforms. To do so, amplicons from the multisegment RT-PCR were bar coded using sequence-independent single-primer amplification (SISPA) as previously described (18). SISPA products were gel eluted, pooled, and sequenced on the 454/GS-FLX Roche platform. Twenty-six samples from the initial set of 150 received by JCVI were selected because they were problem samples and were processed in this manner. The 454 sequence reads were sorted by bar code, trimmed, and mapped to a reference influenza A virus genome using AMOScomp (19).
The alignments for the coverage plots demonstrating the mapping of all the deep-sequence reads on the reference segments were generated using the hmmalign program from the HMMER2 suite of programs (20) (http://hmmer.janelia.org/). The profile hidden Markov models (profile HMM) required by hmmalign to produce multiple-sequence alignments from 454 reads were built incrementally by the following procedure. (i) After the identifying bar code from each read was trimmed, an additional 26 bases were removed from the beginning and end of each read to ensure that no contamination by either bar code sequence or random hexamer primer remained. (ii) The reads were mapped using nucmer (MUMmer program [21]) onto the consensus sequence for each segment as determined by Sanger sequencing; only those reads that showed a full-length match were retained, and reads were reverse complemented when necessary. (iii) The entire set of trimmed reads was clustered by similarity using the program CD-HIT-EST (22) to create a nonredundant set of representative reads. The sequence identity threshold used varied from segment to segment. The purpose of this step was to produce a set of representative reads, manageable in number yet capable of capturing as much of the diversity present in the sample as possible. (iv) The set of representative reads was searched against the NCBI nonredundant nucleotide database, using BLAST. The full-length sequences from the top 100 hits for each representative read were pooled, and duplicate entries were removed. From these full-length sequences, 200 were selected at random and aligned using the muscle program. (v) After visual inspection and correction when necessary, an HMM was built and then used to align the representative reads obtained earlier by similarity clustering. This alignment of reads was inspected, corrected when necessary, and then used to build a profile HMM. (vi) The improved HMM was used to align a larger set of representative sequences, obtained by CD-HIT-EST clustering at an increased sequence identity threshold. This process was repeated several times until a satisfactory alignment was obtained.
Cloning of DI-like segments.
Extracted viral RNA was reamplified by multisegment RT-PCR (M-RT-PCR) using Uni12/13 primers and the One Step RT PCR kit (Invitrogen Corp., USA) (16). To amplify specific genomic segments, M-RT-PCR products were amplified with PB2-, PB1-, PA-, and nucleoprotein (NP)-specific primers.
The cDNAs were cloned as follows: PCR products ranging from 200 to 600 bp were gel purified, A tailed using Taq polymerase (Promega, USA) and dATP. The A-tailed PCR products were cloned in TOPO pCR4.1 according to the manufacturer's protocol (Invitrogen Corp., USA). The TOPO ligated PCR products were transformed into New England BioLabs (NEB) competent cells (New England BioLabs, USA). Colonies were grown in Luria-Bertani medium, and plasmids were extracted using the alkaline lysis method. Clones were screened by PCR for PB2, PB1, and PA segments using segment-specific primers. The inserts were then sequenced by the Sanger method. The primers used for amplification of the internal region of PB2 were as follows: PB2-F (F stands for forward) (5′GTGGACCATATGGCCATAATC3′) and PB2-R (R stands for reverse) (5′GACTCTTATCCCTCTCAGCGAC3′).
Alignment of DI reads.
Trimmed reads were mapped to reference segment sequences (PB2, PB1, and PA) using nucmer with a minimum cluster size of 20 and the maximum match option set to true. The middle of the reference sequences was removed based on where the truncated sequences mapped: from the most 3′ position of the 5′ DI-like sequence to the most 5′ position of the 3′ segment. Next, reads that mapped at both termini in the 5′-to-3′ orientation were aligned using Fast Statistical Alignment (FSA) program (23) with the fast option invoked. The alignment was degapped using a custom biopython script and then mapped again to obtain the coordinates of these subgenomic segments, using nucmer with the same settings as previously specified. Using the Bioperl graphics 2.21 module, a graphical map was generated using the tab-delimited output from the show-coords function of the mummer package. The sequences were aligned manually in BioEdit.
RESULTS
Sequence analysis of influenza A(H1N1)pdm09 virus strains.
Twenty-six specimens collected as part of the INSIGHT study (15) were analyzed by deep sequencing. Of the 26 specimens sequenced, only 13 had overrepresentation of the 5′ and 3′ ends of segments 1 (PB2), 2 (PB1), and 3 (PA), indicative of internal deletions in copies of the segments (the other 13 samples did not have any evidence of internally deleted segments). For the remaining segments (hemagglutinin [HA], nucleoprotein [NP], neurinaminidase [NA], matrix [M], and nonstructural [NS]), sequencing reads mapped across the complete segment. These samples with evidence of internal deletions were also sequenced by the Sanger method; full consensus segments could be assembled for the samples, except for INS077 and INS079 (GenBank accession numbers listed in Table 1).
Table 1.
GenBank accession numbers of INSIGHT clinical specimens used in our study and lengths of segments
| Clinical specimen | Accession no. and length of the following segmenta: |
|||||||
|---|---|---|---|---|---|---|---|---|
| 1 (PB2) | 2 (PB1) | 3 (PA) | 4 (HA) | 5 (NP) | 6 (NA) | 7 (MP) | 8 (NS) | |
| INS002 | CY055963 | CY055962 | CY055961 | CY055956 | CY055959 | CY055958 | CY055957 | CY055960 |
| 2,293 | 2,286 | 2,175 | 1,734 | 1,523 | 1,420 | 987 | 850 | |
| INS006 | CY055995 | CY055994 | CY055993 | CY055988 | CY055991 | CY055990 | CY055989 | CY055992 |
| 2,293 | 2,303 | 2,172 | 1,721 | 1,523 | 1,420 | 987 | 850 | |
| INS007 | CY083837 | CY083836 | CY083835 | CY083830 | CY083833 | CY083832 | CY083831 | CY083834 |
| 2,316 | 2,316 | 2,182 | 1,752 | 1,540 | 1,433 | 1,002 | 865 | |
| INS014 | CY083845 | CY083844 | CY083843 | CY083838 | CY083841 | CY083840 | CY083839 | CY083842 |
| 2,280 | 2,316 | 2,182 | 1,752 | 1,536 | 1,433 | 1,002 | 865 | |
| INS023 | CY083853 | CY083852 | CY083851 | CY083846 | CY083849 | CY083848 | CY083847 | CY083850 |
| 2,280 | 2,316 | 2,151 | 1,752 | 1,540 | 1,433 | 1,002 | 865 | |
| INS039 | CY083861 | CY083860 | CY083859 | CY083854 | CY083857 | CY083856 | CY083855 | CY083858 |
| 2,280 | 2,283 | 2,182 | 1,732 | 1,537 | 1,433 | 1,002 | 865 | |
| INS047 | CY083869 | CY083868 | CY083867 | CY083862 | CY083865 | CY083864 | CY083863 | CY083866 |
| 2,280 | 2,283 | 2,208 | 1,751 | 1,537 | 1,433 | 1,002 | 865 | |
| INS049 | CY083877 | CY083876 | CY083875 | CY083870 | CY083873 | CY083872 | CY083871 | CY083874 |
| 2,280 | 2,283 | 2,224 | 1,701 | 1,540 | 1,433 | 1,002 | 865 | |
| INS050 | CY083885 | CY083884 | CY083883 | CY083878 | CY083881 | CY083880 | CY083879 | CY083882 |
| 2,313 | 2,317 | 2,192 | 1,751 | 1,538 | 1,432 | 999 | 863 | |
| INS077*b | CY116603 | CY083682 | CY116601 | CY083677 | na | CY083679 | CY083678 | CY083680 |
| 312 | 539 | 221 | 241 | 146 | 1,002 | 865 | ||
| INS077* | CY116604 | na | CY116602 | CY083677 | na | na | na | na |
| 444 | 454 | 241 | ||||||
| INS079 | CY083689 | CY083688 | CY083687 | na | na | CY083685 | CY083684 | CY083686 |
| 2,301 | 2,166 | 2,127 | 536 | 1,002 | 865 | |||
| INS147 | CY083925 | CY083924 | CY083923 | CY083918 | CY083921 | CY083920 | CY083919 | CY083922 |
| 2,316 | 2,316 | 2,224 | 1,750 | 1,540 | 1,433 | 1,002 | 865 | |
| INS148 | CY083933 | CY083932 | CY083931 | CY083926 | CY083929 | CY083928 | CY083927 | CY083930 |
| 2,313 | 2,317 | 2,192 | 1,741 | 1,541 | 1,421 | 999 | 865 | |
| INS086 | CY083909 | CY083908 | CY083907 | CY083902 | CY083905 | CY083904 | CY083903 | CY083906 |
| 2,313 | 2,317 | 2,177 | 1,752 | 1,541 | 1,421 | 999 | 865 | |
| INS132 | CY083917 | CY083916 | CY083915 | CY083910 | CY083913 | CY083912 | CY083911 | CY083914 |
| 2,316 | 2,316 | 2,182 | 1,752 | 1,540 | 1,433 | 1,002 | 865 | |
GenBank accession numbers of INSIGHT clinical specimens used in our study and sequenced by the Sanger method. The lengths of the respective segments in nucleotides are shown in italic type.
INS77* contains multiple sequences for segments 1, 3, and 4.
The length and coverage of the sequence reads suggest an internal deletion, previously found to be associated with DI RNA in replicating influenza viruses (6). Figure 1 shows the alignment of the sequence reads on the PB2 segment for specimens A/San Diego/INS007/2009 (H1N1) and A/District of Columbia/INS047/2009 (H1N1), indicative of the internal deletion in the majority of the molecules sequenced. All 13 specimens contained PB2-derived DI-like RNAs with 5′ and 3′ ends; 7 samples had PB1, and only 3 samples had PA DI-like RNAs. From the length of the internal deletions observed in the sequence reads that mapped to the polymerase segments, the subgenomic segments appeared to range in size from 250 to 750 nt. While most of the reads mapping to these segments had a single internal deletion, a few reads had double deletions. In addition to the single internal deletion, we observed 3′ DI RNAs (6) from PB1 and PA segments for a few of the specimens. The remaining five segments of the specimens were also analyzed for DI-like sequences, but no short segments were observed; moreover, the segments had full coverage by deep sequencing.
Fig 1.

Example of mapping of deep-sequence reads on influenza virus segments. The sequencing reads from the GS-FLX (454) were mapped to the respective influenza A (H1N1) genomic segments. The plots depict the number of sequence reads that map to each nucleotide position across the genomic segment. The x axis represents the length of each segment, and each plot represents sequencing data for the indicated influenza virus segment from clinical isolate A/San Diego/INS007/2009 (H1N1).
PB2 and PB1 DI-like RNAs from the clinical specimens had internal deletion junctions very similar to the ones observed in previous studies for cultured virus isolates (7). In general, the junction site positions for the 5′ regions for all subgenomic RNAs (sgRNAs) found in our specimens occurred predominantly around nucleotide positions 200 to 300 from the ends of the parental segments. These positions are similar to previously observed internal deletion junction sites (7), suggesting that these coordinates are important to maintain packaging signals found on the 5′ and 3′ ends of the segments (10).
Cloning of subgenomic RNAs from clinical specimens.
To confirm the presence of DI-like RNAs in the primary specimens and map the junction sites of the deletions more clearly, we reextracted RNA from the primary clinical specimens that were shown to potentially have subgenomic segments. After M-RT-PCR, amplified fragments between 200 to 800 nt in size were gel extracted, cloned, and sequenced. We were not able to detect segment-specific fragments in all the specimens tested, possibly due to very low template concentrations for amplification. However, the products that were successfully amplified and sequenced support our deep sequencing data in identifying these DI-like RNAs (Fig. 2). The average length of the PB2, PB1, and PA subgenomic RNAs characterized by cloning (Table 2) was around 410 nt, within the range of the DI-like RNAs of the respective segments observed by deep sequencing.
Fig 2.

Alignment of DI-like sequences from deep-sequence data. (A and B) GS-FLX sequence reads of the PB2 segment of the clinical isolates A/San Diego/INS007/2009 (H1N1) (A) and A/District of Columbia/INS047/2009 (H1N1) (B) mapped to their respective reference sequences (CY083837 for INS007 and CY083869 for INS047). Positions are shown in thousands from 100 (0.1k) to 2,300 (2.3k).
Table 2.
Overlap sequences at the 5′ and 3′ junctions of cloned polymerase DI-like segmentsa
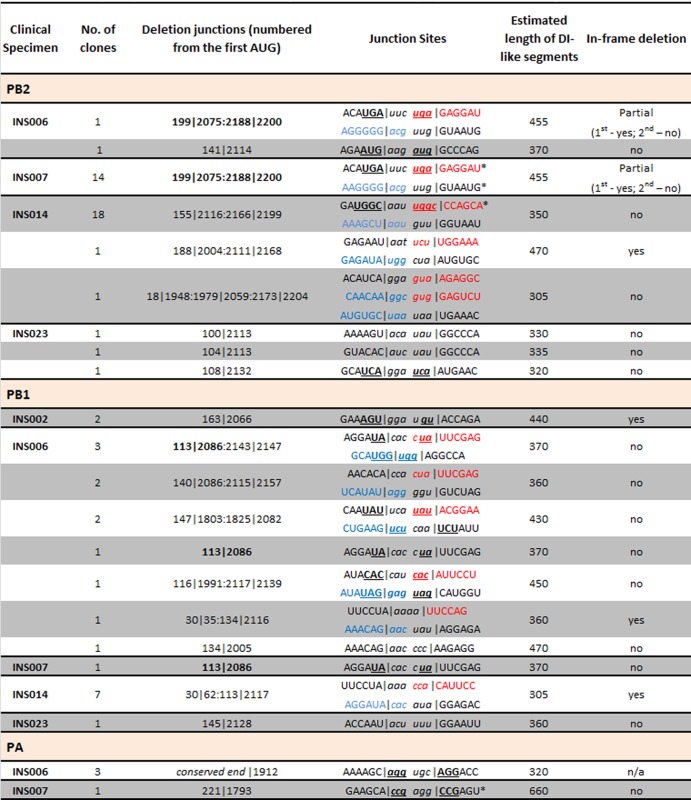
Uppercase text indicates the junction sites of the putative DI-like segments; italic text corresponds to the genomic sequence that is deleted just before the junction site. Underlined and bold text indicates junction sites where the deleted nucleotides are identical to the nucleotides on the opposite side of the junction. Colored text represents the junction sites of clones with 2 internal deletions, highlighting the 5′ (red) and 3′ (blue) ends of the central region of the subgenomic RNAs. Black pipes represent the deletion sites at both 5′ and 3′ junctions. Colons within the pipes separate fragments generated by multiple deletions in the same segment. Values in bold in the deletion junction column represent the same deletion junction in the PB1 segment of clones INS006 and INS007. Stars mark sites that were detected in both the deep sequencing data and cloned DI-like RNAs (shown in Fig. 4 for INS007 and INS014).
Table 2 shows the deletion junction sites of the cloned DI-like RNAs. Some of the specimens had multiple junction sites but often within a 30- to 50-nt region. In half of the junction sites in the sequenced clones, the junction occurs at a site where a few of the deleted nucleotides on one side of the junction are repeated in the conserved sequence on the other side of the junction (e.g., INS007 PB2 clones have a UGA before the junction that is repeated in the deleted portion of the segment on the other side of the junction); the length of the overlapping nucleotides of the 5′ and 3′ junction sites varied from 2 to 3 nt. In a few cases, the internal deletions led to in-frame deletions, indicating that truncated proteins could be translated from potentially expressed truncated genes.
Comparison of the internal deletion junctions in the cloned PB2 and PB1 cDNAs and the deep-sequence reads for the same segments demonstrates that the junction position is in some cases identical in both sets of sequences and across different specimens. Figure 3 provides an example for INS006, INS007, and INS0014 for PB2.
Fig 3.

Alignment of cloned DI-like sequences. (A and B) Alignment of clone sequences generated from the INSIGHT influenza virus-positive clinical samples mapped to the reference segment 1 (PB2) (A) and segment 2 (PB1) (B).
Notably, INS006 and INS007 have PB2 and PB1 DI-like RNAs that are identical (Table 2 and Fig. 3 and 4). These two specimens—along with INS002 for which cloning of the subgenomic RNAs was not as successful—belong to the same influenza virus contact network among students attending the University of California, San Diego (24). Such an evolutionary pattern is compatible with the transmission (as a minor population) of these DI-like RNAs among individuals. Although there is evidence for the transmission of defective (i.e., stop codon defined) viruses among individuals (25), including in influenza virus (26), this would represent the first evidence for the interperson transmission of DIs in influenza virus.
Fig 4.
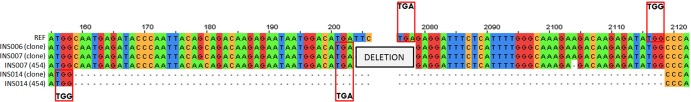
Graphical representation of the junction sites of cloned DI-like sequences compared to GS-FLX (454) sequences. Nucleotide sequences at the putative junctions between 5′ and 3′ regions of DI-like RNAs are shown. The top sequence is the reference (REF) sequence, whereas the remaining sequences are the cloned PB2 subgenomic RNAs of clinical specimens INS006, INS007, and INS014. The red boxes represent the overlapping sequences at the 5′ end and beginning of the 3′ region of the DI-like sequence. Table 2 shows detailed analysis of various junction sites observed with the INSIGHT samples.
Confirmation of DI-like RNAs.
To exclude the possibility that the subgenomic fragments identified were the result of PCR-specific errors, we repeated PB2 segment-specific RT-PCRs but comparing 2 different enzymes for the RT step: murine leukemia virus reverse transcriptase (MLV RT) and avian myeloblastosis virus reverse transcriptase (AMV-RT). The goal was to ensure that the detection of the fragments was not an effect of consistent reverse transcriptase cis or trans strand transfer. Figure 5 shows clear bands for a number of the specimens for which we characterized DI-like RNAs in the deep-sequence and cloned data. Banding patterns were similar for both enzymes. Furthermore, the sizes of the internally deleted RNAs were consistent with those identified in the deep-sequence and cloning experiments. The M-RT-PCR reactions that generated the short segment-specific fragments from the RNA of the putative DI-containing samples failed to do so from the control clinical sample, INS036 (specimen for which no subgenomic RNA was observed in the deep-sequence data), even though the PB2 sequence for this virus is identical in regions corresponding to the junction sites observed for subgenomic RNA in other clinical specimens. This supports our hypothesis that DI-like RNAs are real and the observed short segments in the sequence data are not the result of reverse transcriptase movement or Taq errors during M-RT-PCR.
Fig 5.
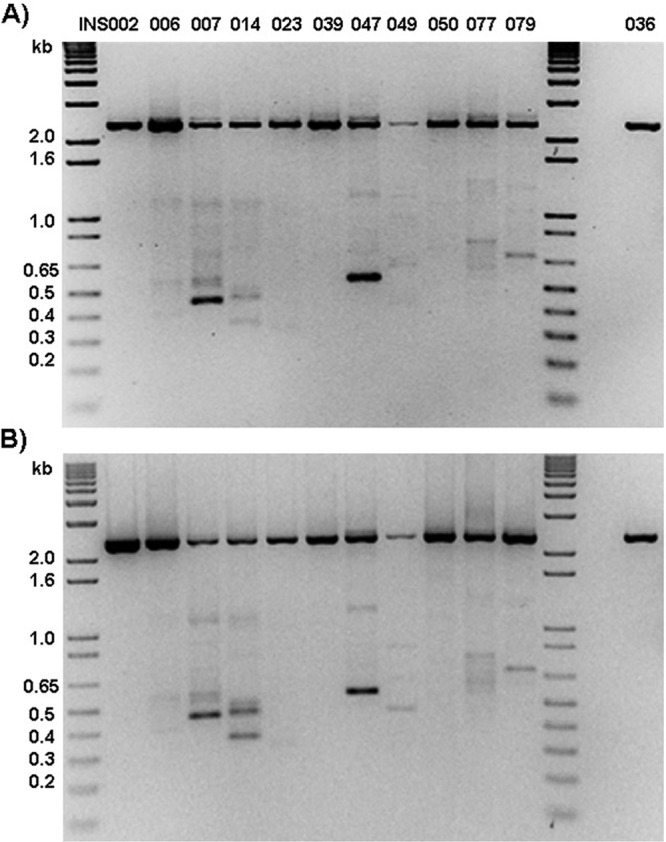
Detection of DI-like RNAs in clinical specimens. (A and B) RNA extracted from selected INSIGHT clinical specimens (INS002 to INS036) was subjected to PB2 segment-specific RT-PCR using SuperScript III (SSIII) first-strand synthesis kit MLV RT (Invitrogen) (A) or AMV RT (New England BioLabs) (B). INS036 is a negative control. The positions of molecular size standards (in kilobases) are shown to the left of the gels.
To further confirm the presence of subgenomic fragments directly from the original RNA extracted from the clinical specimens and before PCR, viral RNA was labeled and fractionated by polyacrylamide gel electrophoresis as described previously (27). We detected several short RNA species ranging from 200 to 600 nt (Fig. 6) in a few of the samples, but not in the control sample (INS086; sample for which the M-RT-PCR did not indicate the presence of DI-like RNAs), pointing to the existence of subgenomic RNAs with sizes that are similar to the DI-like segments we characterized.
Fig 6.

Gel electrophoresis of genomic RNA from INSIGHT clinical samples. Total RNA (∼30 ng) from the clinical samples was end labeled with 32P followed by separation of the genomic segments on a 6% native polyacrylamide gel. The autoradiograph was obtained using the Bio-Rad PhosphorImager. INS086 is a control sample for which no subgenomic RNAs were detected in the deep-sequence data. The positions of molecular size standards (M) (from 0.3 to 2.0 kilobases) are shown to the left of the gel.
DISCUSSION
The generation of defective particles of influenza viruses has been well documented in vitro and is suspected of competing with the replication of the wild-type virus. Here we provide a report of influenza virus defective genomic segments in vivo from clinical specimens from influenza A virus-infected individuals and observe identical DI-like RNA sequences in two individuals linked by contact, indicating possible transmission of defective viruses between them. Specifically, by sequencing, we identified several DI-like RNAs in nasopharyngeal swabs.
A number of studies describe the types and features of in vitro DI RNAs (6, 7, 11, 27), and the DI-like RNAs identified in our study are consistent with the characteristic features of the in vitro-generated DI RNAs. The similarity between the in vitro and in vivo subgenomic RNAs points to a mechanism that is conserved for the generation of defective particles. The DI-like RNAs we observed contained internal deletions, overlap sequences at the 5′ and 3′ junction sites, and in some cases, in-frame deletions. The lengths of the PB2, PB1, and PA subgenomic RNAs were similar to those reported previously for in vitro influenza virus DI RNAs (7, 11, 12) and were derived primarily from the polymerase segments, which is in agreement with previous in vitro reports. In addition, the DI-like RNAs have conserved 5′ and 3′ ends, essential for genome packaging (10), and a major internal deletion, as observed in the in vitro defective interfering RNAs. All the clones generated from each specimen were influenza virus specific, with 90% originating from PB2. We did not detect DI-like RNAs of nonpolymerase segments either by deep sequencing or cloning, but a few of the clones contained very short sequences (oligonucleotides) of M, HA, NP, and NA that did not contain 5′- or 3′-segment ends.
Comparison of the complete nucleotide sequence of the DI-like RNAs with the corresponding parental segment showed the presence of extensive internal deletions, reducing the length to 15 to 20% of the original segment length. PB2- and PB1-derived DI-like RNAs had equal contributions of the 5′ and 3′ region, whereas the PA subgenomic RNAs had disproportionate distribution with more 3′ region content. Analysis of the junctions revealed that the junction sites are flanked by overlap sequences, or identical sequences, indicative of a polymerase “skipping.” The sequences of the junction sites were similar to those of in vitro-derived DI RNAs. The length of the overlap sequences varied from as small as 2 nucleotides to up to 5 nucleotides. The overlap sequences were segment specific and conserved in different specimens analyzed in this study. The presence of the overlap sequences in the subgenomic RNAs indicates that a polymerase slippage event could be involved in the formation of the DI RNAs during natural infections. Some of the subgenomic RNAs from PB2, PB1, and PA had an in-frame deletion, indicating the presence of an open reading frame, which could eventually be expressed. In the past, it has been shown that influenza virus DI RNAs are transcribed into cRNAs and that some of the DI RNAs could be in vitro translated into polypeptides (27). It can be speculated that these clinically detected DI-like RNAs are expressed during the infection and could interfere with replication of the standard virus.
It remains unclear whether DI particle replication is favored because the polymerase protein prefers the smaller size of their vRNA segments over the longer intact segments or whether it is due to competition between large vRNA segments and more numerous small segments. Active transcription and translation of DI RNAs generates two transcripts and novel polypeptides of PB2. One hypothesis is that the translational products might have a role in virus multiplication, but to date, no experimental evidence has been found to support the influence of DI polypeptides on replication or to show they have an interfering advantage over the standard virus or other DI viruses (27). Recently, the presence of defective interfering particles was shown in sera of patients with acute dengue infections (28). The dengue virus (DENV) DI RNAs also had internal genomic deletions retaining the critical 5′ and 3′ ends of the genome. The DENV DI particles reduce the yield of the wild-type dengue virus in vitro. DENV and influenza virus DI RNAs share some features with respect to the length of the RNAs, contribution of the 5′ and 3′ regions, and nucleotide sequences of the junctions (data not shown).
The presence of DI-like RNAs in nasopharyngeal swabs of some of the patients raises the question of their origin; specifically whether the DIs are transmitted from person to person or arise after infection. Influenza virus infection stimulates both innate and adaptive immune responses through their genomic and antigenomic single-strand RNAs (29). Recent reports show that DI RNAs act as potent immune stimulators. Immunoprecipitation studies with RIG-I (retinoic acid-inducible gene 1) show that the pathogen receptors prefer influenza virus DI RNAs as ligands over the full-length genomic segments (30). It is possible that DI RNAs in the primary clinical specimens might play a significant role in interferon (IFN) induction during the natural infection of the host. Future studies could therefore be focused on determining the functional role of the identified DIs using reverse genetics and IFN induction reporter assays.
It has recently been shown that a molecularly cloned DI influenza A virus generated in vitro could be used as an intranasal treatment to protect mice and ferrets from subsequent influenza A virus infection (31, 32). In the case of mice, this defective virus was also able to protect against other unrelated heterologous viruses like influenza B virus (33). These studies have successfully demonstrated the potential of DI RNAs as an additional and novel approach to antiviral therapy. Similarly, the DI-like RNAs identified in the present study have the potential to be used to generate antiviral preparations closer to clinical or natural infections. For example, a DI influenza virus could be generated by reverse genetics using these DI clones and assessed for its interferon stimulation capability and for how well it protects against infection. Since the DI-like RNAs identified here have only 20% of the original PB2 and PB1 sequences, resulting in a DI virus with a nonfunctional polymerase complex, a helper virus would be necessary to produce negative-sense influenza virus genomic RNAs. All these issues were addressed in a recent study that demonstrated that defective interfering influenza viruses act as a natural antiviral agent and are a more effective treatment than commonly used antivirals like oseltamivir (34). In this study, pretreatment of ferrets with a DI virus called 244/PR8 generated from cloned DI RNA derived from segment 1 and incorporated into A/PR/8/34 significantly reduced fever, respiratory symptoms, cellular infiltration, and virus load in response to pandemic influenza virus A/California/04/09 infection. There was also a simultaneous increase in the population of DI RNA by more than 25,000-fold, suggesting a possible transmission of the DI virus to other cells in the respiratory tract, thereby increasing the population of virus-resistant cells containing DI RNA.
There is an ongoing debate about which segments undergo deletion during the generation of DIs from the in vitro infections. A few studies report only the subgenomic RNAs of the polymerase segments, whereas some argue for the presence of subgenomic RNAs of all the segments (5, 7, 11, 12, 35–38). Our observation of polymerase segment-derived DI-like RNAs as the major population in the clinical specimens supports the theory that polymerase segments may play an important role during interference.
Overall, our data point to the existence of defective interfering subgenomic RNAs of influenza A(H1N1)pdm09 virus in clinical specimens that share or resemble the well-characterized in vitro DI RNAs of influenza viruses. It has yet to be established whether the DI-like segments identified from the clinical samples interfere with the replication of the standard virus in vitro and the role, if any, of DIs in natural influenza infections has yet to be addressed. For example, do these particles interfere with replication of the influenza virus or progression from the upper respiratory tract to the lower respiratory tract or do they aid in induction of the innate immune pathways to restrict the virus? Future studies focusing on the detection of DIs in both the early and late phases of infection would provide important information about their role in the induction of host immune responses and replication interference.
ACKNOWLEDGMENTS
INSIGHT is funded by NIH grant UOI-AI068641, and the FLU 002 and FLU 003 studies are funded by SAIC Prime Contract HHSN261200800001E, NCI/NIAID. This research was supported with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, U.S. Department of Health and Human Services, under contract number HHSN272200900007C (D.E.W., E.G., E.C.H., R.H., and X.L.).
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
We thank the following study participants. The community representatives were D. Munroe, C. Rappoport, and S. Schwarze. The following individuals worked at coordinating centers: in Copenhagen, Denmark, B. Aagaard, D. Adzic, J. Grarup, P. Herrero, P. Jansson, J. Kjær, M. L. Jakobsen, B. Jensen, K. B. Jensen, H. Juncher, P. Lopez, A. Mocroft, M. Pearson, B. Portas, C. Sabin, and K. Tillmann; in London, England, A. Babiker, N. Braimah, Y. Collaco-Moraes, F. Hudson, I. Kummeling, F. Pacciarini, and N. Paton; at the Statistical and Data Management Center, Minneapolis, Minnesota, A. DuChene, M. George, M. Harrison, E. Krum, G. Larson, R. Nelson, K. Quan, S. Quan, C. Reilly, T. Schultz, G. Thompson, and N. Wyman; in Sydney, Australia, A. Avihingsanon, L. Cassar, K. Charoentonpuban, S. Emery, K. Laohajinda, T. Jupimai, I. Lanusse, A. Moricz, I. Otegui, K. Ruxrungtham, and R. Robson; in Washington, DC, E. Finley, F. Gordin, A. Sanchez, and M. L. Vjecha. The following individuals worked at specimen repositories and laboratories: J. Baxter, S. Brown (SAIC Frederick, Inc.), and M. Hoover (Advanced BioMedical Laboratories, Inc. [ABML]). B. Baseler (SAIC Frederick Inc.) and J. A. Metcalf worked at the National Institute of Allergy and Infectious Diseases. Other experts who participated in this study were N. Cox, L. Gubareva, K. Hancock, J. Katz, A. Klimov, and M. Shaw at the Centers for Disease Control and Prevention and L. Rubinson at the U.S. Department of Health and Human Services.
The INSIGHT FLU 002 clinical site investigators were as follows: in Argentina, L. Barcan, J. A. L. Corral, D. O. David, H. E. Laplume, M. B. Lasala, G. D. Lopardo, M. H. Losso, S. Lupo, and E. Warley; in Australia, M. Bloch, D. E. Dwyer, R. Moore, S. L. Pett, N. Roth, T. M. Soo, and E. Vlahakis; in Austria, H. Burgmann; in Belgium, N. Clumeck, S. De Wit, E. Florence, K. Kabeya, and J. Weckx; in Chile, C. Perez and M. J. Wolff; in Denmark, J. Gerstoft, J. D. Lundgren, and L. Ostergaard; in Estonia, K. Zilmer; in Germany, J. R. Bogner, N. H. Brockmeyer, G. Faetkenheuer, H. Klinker, A. Plettenberg, J. Rockstroh, and C. Stephan; in Greece, A. Antoniadou, G. Koratzanis, N. Koulouris, V. Polixronopoulos, H. Sambatakou, and N. Vasilopoulos; in Lithuania, S. Caplinskas; in Peru, A. La Rosa, F. Mendo, R. Salazar, and J. Valencia; in Poland, E. Bakowska, A. Horban, and B. Knysz; in Portugal, F. Antunes and M. Doroana; in South Africa, N. Padayatchi; in Spain, D. Dalmau, E. Fernandez-Cruz, J. M. Gatell, J. S. Sanz, and V. Soriano; in Thailand, P. Chetchotisakd, K. Ruxrungtham, and G. Suwanpimolkul; in the United Kingdom, C. L. S. Leen; in the United States, C. Cohen, D. L. Cohn, J. A. DeHovitz, W. El-Sadr, M. Glesby, F. M. Gordin, S. Hodder, N. Markowitz, R. M. Novak, R. Schooley, G. L. Simon, E. M. Tedaldi, Z. Temesgen, J. Timpone, D. Z. Uslan, and B. H. Wade.
The INSIGHT FLU 003 clinical site investigators were as follows: in Argentina, L. Barcan, J. A. L. Corral, D. O. David, H. E. Laplume, M. B. Lasala, G. D. Lopardo, M. H. Losso, and E. Warley; in Australia, D. E. Dwyer, J. Elliott, P. Konecny, J. McBride, and S. L. Pett; in Austria, H. Burgmann; in Belgium, N. Clumeck, S. De Wit, P. Jorens, and K. Kabeya; in Chile, M. J. Wolff; in China, T. C. Wu; in Denmark, J. Gerstoft, L. Mathiesen, H. Nielsen, L. Ostergaard, and S. S. Pedersen; in Germany, F. Bergmann, J. R. Bogner, N. H. Brockmeyer, G. Faetkenheuer, H. Klinker, J. Rockstroh, and C. Stephan; in Greece, A. Antoniadou, G. Koratzanis, N. Koulouris, V. Polixronopoulos, H. Sambatakou, and N. Vasilopoulos; in Norway, A. Maagaard; in Peru, F. Mendo and R. Salazar; in Poland, E. Bakowska and A. Horban; in South Africa, N. Padayatchi; in Spain, D. Dalmau, V. Estrada, E. Fernandez-Cruz, H. K. Freud, R. M. B. Garrido, J. M. Gatell, J. S. Moreno, J. R. Pano-Pardo, J. S. Sanz, and V. Soriano; in Thailand, P. Chetchotisakd, K. Ruxrungtham, and G. Suwanpimolkul; in the United Kingdom, B. J. Angus, D. R. Chadwick, D. Dockrell, C. L. S. Leen, M. Newport, and E. Wilkins; in the United States, H. Anderson III, J. V. Baker, D. L. Cohn, J. A. DeHovitz, W.A. El-Sadr, M. S. Freiberg, F. M. Gordin, R. Gulick, D. Gurka, S. Hodder, N. Markowitz, R. M. Novak, A. Paez, N. Patil, A. Reboli, M. Sands, R. Schooley, G. L. Simon, Z. Temesgen, J. Timpone, D. Z. Uslan, and B. H. Wade.
Footnotes
Published ahead of print 15 May 2013
REFERENCES
- 1. Huang AS, Baltimore D. 1970. Defective viral particles and viral disease processes. Nature 226:325–327 [DOI] [PubMed] [Google Scholar]
- 2. Janda JM, Davis AR, Nayak DP, De BK. 1979. Diversity and generation of defective interfering influenza virus particles. Virology 95:48–58 [DOI] [PubMed] [Google Scholar]
- 3. Nayak DP. 1980. Defective interfering influenza viruses. Annu. Rev. Microbiol. 34:619–644 [DOI] [PubMed] [Google Scholar]
- 4. Von Magnus P. 1954. Incomplete forms of influenza virus. Adv. Virus Res. 2:59–79 [DOI] [PubMed] [Google Scholar]
- 5. Davis AR, Nayak DP. 1979. Sequence relationships among defective interfering influenza viral RNAs. Proc. Natl. Acad. Sci. U. S. A. 76:3092–3096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nayak DP, Chambers TM, Akkina RK. 1985. Defective-interfering (DI) RNAs of influenza viruses: origin, structure, expression, and interference. Curr. Top. Microbiol. Immunol. 114:103–151 [DOI] [PubMed] [Google Scholar]
- 7. Jennings PA, Finch JT, Winter G, Robertson JS. 1983. Does the higher order structure of the influenza virus ribonucleoprotein guide sequence rearrangements in influenza viral RNA? Cell 34:619–627 [DOI] [PubMed] [Google Scholar]
- 8. Bean WJ, Kawaoka Y, Wood JM, Pearson JE, Webster RG. 1985. Characterization of virulent and avirulent A/chicken/Pennsylvania/83 influenza A viruses: potential role of defective interfering RNAs in nature. J. Virol. 54:151–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chambers TM, Webster RG. 1987. Defective interfering virus associated with A/Chicken/Pennsylvania/83 influenza virus. J. Virol. 61:1517–1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hutchinson EC, von Kirchbach JC, Gog JR, Digard P. 2010. Genome packaging in influenza A virus. J. Gen. Virol. 91:313–328 [DOI] [PubMed] [Google Scholar]
- 11. Davis AR, Hiti AL, Nayak DP. 1980. Influenza defective interfering viral RNA is formed by internal deletion of genomic RNA. Proc. Natl. Acad. Sci. U. S. A. 77:215–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Noble S, Dimmock NJ. 1995. Characterization of putative defective interfering (DI) A/WSN RNAs isolated from the lungs of mice protected from an otherwise lethal respiratory infection with influenza virus A/WSN (H1N1): a subset of the inoculum DI RNAs. Virology 210:9–19 [DOI] [PubMed] [Google Scholar]
- 13. Nayak DP, Sivasubramanian N, Davis AR, Cortini R, Sung J. 1982. Complete sequence analyses show that two defective interfering influenza viral RNAs contain a single internal deletion of a polymerase gene. Proc. Natl. Acad. Sci. U. S. A. 79:2216–2220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Odagiri T, Tashiro M. 1997. Segment-specific noncoding sequences of the influenza virus genome RNA are involved in the specific competition between defective interfering RNA and its progenitor RNA segment at the virion assembly step. J. Virol. 71:2138–2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dwyer DE. 2011. Surveillance of illness associated with pandemic (H1N1) 2009 virus infection among adults using a global clinical site network approach: the INSIGHT FLU 002 and FLU 003 studies. Vaccine 29(Suppl 2):B56–B62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhou B, Donnelly ME, Scholes DT, St. George K, Hatta M, Kawaoka Y, Wentworth DE. 2009. Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and swine origin human influenza A viruses. J. Virol. 83:10309–10313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ghedin E, Sengamalay NA, Shumway M, Zaborsky J, Feldblyum T, Subbu V, Spiro DJ, Sitz J, Koo H, Bolotov P, Dernovoy D, Tatusova T, Bao Y, St. George K, Taylor J, Lipman DJ, Fraser CM, Taubenberger JK, Salzberg SL. 2005. Large-scale sequencing of human influenza reveals the dynamic nature of viral genome evolution. Nature 437:1162–1166 [DOI] [PubMed] [Google Scholar]
- 18. Djikeng A, Halpin R, Kuzmickas R, Depasse J, Feldblyum J, Sengamalay N, Afonso C, Zhang X, Anderson NG, Ghedin E, Spiro DJ. 2008. Viral genome sequencing by random priming methods. BMC Genomics 9:5. 10.1186/1471-2164-9-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pop M, Phillippy A, Delcher AL, Salzberg SL. 2004. Comparative genome assembly. Brief. Bioinform. 5:237–248 [DOI] [PubMed] [Google Scholar]
- 20. Eddy SR. 1998. Profile hidden Markov models. Bioinformatics 14:755–763 [DOI] [PubMed] [Google Scholar]
- 21. Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. 2004. Versatile and open software for comparing large genomes. Genome Biol. 5:R12. 10.1186/gb-2004-5-2-r12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li W, Godzik A. 2006. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22:1658–1659 [DOI] [PubMed] [Google Scholar]
- 23. Bradley RK, Roberts A, Smoot M, Juvekar S, Do J, Dewey C, Holmes I, Pachter L. 2009. Fast statistical alignment. PLoS Comput. Biol. 5:e1000392. 10.1371/journal.pcbi.1000392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Holmes EC, Ghedin E, Halpin RA, Stockwell TB, Zhang XQ, Fleming R, Davey R, Benson CA, Mehta S, Taplitz R, Liu YT, Brouwer KC, Wentworth DE, Lin X, Schooley RT. 2011. Extensive geographical mixing of 2009 human H1N1 influenza A virus in a single university community. J. Virol. 85:6923–6929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aaskov J, Buzacott K, Thu HM, Lowry K, Holmes EC. 2006. Long-term transmission of defective RNA viruses in humans and Aedes mosquitoes. Science 311:236–238 [DOI] [PubMed] [Google Scholar]
- 26. Stack JC, Murcia PR, Grenfell BT, Wood JLN, Holmes EC. 2013. Inferring the inter-host transmission of influenza A virus using patterns of intra-host evolution. Proc. Roy Soc. B 280:20122173. 10.1098/rspb.2012.2173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Akkina RK, Chambers TM, Nayak DP. 1984. Expression of defective-interfering influenza virus-specific transcripts and polypeptides in infected cells. J. Virol. 51:395–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li D, Lott WB, Lowry K, Jones A, Thu HM, Aaskov J. 2011. Defective interfering viral particles in acute dengue infections. PLoS One 6:e19447. 10.1371/journal.pone.0019447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wolff T, Ludwig S. 2009. Influenza viruses control the vertebrate type I interferon system: factors, mechanisms, and consequences. J. Interferon Cytokine Res. 29:549–557 [DOI] [PubMed] [Google Scholar]
- 30. Baum A, Garcia-Sastre A. 2011. Differential recognition of viral RNA by RIG-I. Virulence 2:166–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dimmock NJ, Dove BK, Scott PD, Meng B, Taylor I, Cheung L, Hallis B, Marriott AC, Carroll MW, Easton AJ. 2012. Cloned defective interfering influenza virus protects ferrets from pandemic 2009 influenza A virus and allows protective immunity to be established. PLoS One 7:e49394. 10.1371/journal.pone.0049394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Easton AJ, Scott PD, Edworthy NL, Meng B, Marriott AC, Dimmock NJ. 2011. A novel broad-spectrum treatment for respiratory virus infections: influenza-based defective interfering virus provides protection against pneumovirus infection in vivo. Vaccine 29:2777–2784 [DOI] [PubMed] [Google Scholar]
- 33. Scott PD, Meng B, Marriott AC, Easton AJ, Dimmock NJ. 2011. Defective interfering influenza A virus protects in vivo against disease caused by a heterologous influenza B virus. J. Gen. Virol. 92:2122–2132 [DOI] [PubMed] [Google Scholar]
- 34. Dimmock NJ, Dove BK, Meng B, Scott PD, Taylor I, Cheung L, Hallis B, Marriott AC, Carroll MW, Easton AJ. 2012. Comparison of the protection of ferrets against pandemic 2009 influenza A virus (H1N1) by 244 DI influenza virus and oseltamivir. Antiviral Res. 96:376–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Duhaut SD, Dimmock NJ. 1998. Heterologous protection of mice from a lethal human H1N1 influenza A virus infection by H3N8 equine defective interfering virus: comparison of defective RNA sequences isolated from the DI inoculum and mouse lung. Virology 248:241–253 [DOI] [PubMed] [Google Scholar]
- 36. Liu C, Air GM. 1993. Selection and characterization of a neuraminidase-minus mutant of influenza virus and its rescue by cloned neuraminidase genes. Virology 194:403–407 [DOI] [PubMed] [Google Scholar]
- 37. Duhaut S, Dimmock NJ. 2000. Approximately 150 nucleotides from the 5′ end of an influenza A segment 1 defective virion RNA are needed for genome stability during passage of defective virus in infected cells. Virology 275:278–285 [DOI] [PubMed] [Google Scholar]
- 38. Sivasubramanian N, Nayak DP. 1983. Defective interfering influenza RNAs of polymerase 3 gene contain single as well as multiple internal deletions. Virology 124:232–237 [DOI] [PubMed] [Google Scholar]