

Abstract
CD55 limits excessive complement activation on the host cell surface by accelerating the decay of C3 convertases. In this study, we observed that hepatitis C virus (HCV) infection of hepatocytes or HCV core protein expression in transfected hepatocytes upregulated CD55 expression at the mRNA and protein levels. Further analysis suggested that the HCV core protein or full-length (FL) genome enhanced CD55 promoter activity in a luciferase-based assay, which was further augmented in the presence of interleukin-6. Mutation of the CREB or SP-1 binding site on the CD55 promoter impaired HCV core protein-mediated upregulation of CD55. HCV-infected or core protein-transfected Huh7.5 cells displayed greater viability in the presence of CD81 and CD55 antibodies and complement. Biochemical analysis revealed that CD55 was associated with cell culture-grown HCV after purification by sucrose density gradient ultracentrifugation. Consistent with this, a polyclonal antibody to CD55 captured cell culture-grown HCV. Blocking antibodies against CD55 or virus envelope glycoproteins in the presence of normal human serum as a source of complement inhibited HCV infection. The inhibition was enhanced in the presence of both the antibodies and serum complement. Collectively, these results suggest that HCV induces and associates with a negative regulator of the complement pathway, a likely mechanism for immune evasion.
INTRODUCTION
The complement system performs a vital effector function in the innate immune system by providing an efficient means for targeting and eliminating infected cells and invading microorganisms, including free viral particles (1–3). Activation of the complement cascade occurs primarily via the classical, alternative, or lectin pathway (2, 4). These three pathways activate C3 via cleavage to C3a and C3b by the C3 convertases. C5 convertases are generated by the association of C3b with the C3 convertases, which in turn cleaves C5 into C5a and C5b. The release of C5b initiates the nonenzymatic process of membrane-attack complex (MAC) formation that then sequentially recruits C6, C7, C8, and C9 proteins (1, 3, 5). The MAC forms a pore-like structure within the lipid envelope of the pathogen or the membrane of the infected cells that ultimately leads to lysis. In order to avert damage from excessive complement activation and MAC formation, host cells express membrane-bound regulatory proteins to limit these processes (6). Regulators of complement activation (RCA) are expressed on the surfaces of host cells and include CD46, CD55, and CD59 (7–9).
Hepatocytes are the primary sites for synthesis of complement components in vivo. During an acute-phase (AP) response, the biosynthesis of these components is increased by the action of AP response-associated cytokines interleukin-1 (IL-1), IL-6, and tumor necrosis factor alpha (TNF-α) (10, 11). Hepatocytes may be exposed to high local concentrations of complement components under these conditions and could promote complement-mediated damage (12–14). Decay-accelerating factor (DAF) or CD55 is one of the key RCA proteins and is expressed on all serum-exposed cells. CD55 limits excessive complement activation by inhibiting C3 convertase formation. It accelerates decay of C3 convertases, which slows cleavage of C3.
Previous reports suggested that complement attack on antibody-sensitized hepatoma cells following a 3-day incubation with a combination of AP cytokines resulted in an increased resistance to complement-mediated lysis and decreased C3b deposition (15). An increased hepatic synthesis of CD55 that could protect hepatocytes from high local concentrations of complement generated during the AP response was observed (15). More recently, primary human hepatocytes have been shown to be protected from complement-mediated cell lysis by an enhanced synthesis of complement-regulatory proteins, including CD55, induced by proinflammatory cytokines like gamma interferon, IL-1β, and TNF-α (16).
Recent studies have suggested that incorporation of the complement-regulatory protein CD59 in HCV in plasma as well as proteins derived from cell lines (17) protected HCV from antibody-dependent complement-mediated lysis. Analogously, HBV X protein has been implicated in increasing expression of CD59 in hepatoma cell lines. This resulted in a decrease in deposition of the C5b-C9 complex on the hepatocyte cell surface, which contributed to resistance of the cells to complement-dependent lysis (18).
In this study, we have shown that HCV core protein enhanced transcription and cell surface protein expression of CD55 in hepatocytes. Moreover, CD55 was associated with purified HCV that was grown in cells, and blocking antibodies against CD55 inhibited infection of HCV in cell culture. Our results suggest that induction of CD55 expression and its incorporation in HCV particles limit complement-mediated damage to infected cells and released virus.
MATERIALS AND METHODS
Patient materials.
Paired serum samples and liver biopsy specimens from chronically HCV-infected patients (n = 12) were used, as previously described (19, 20). Commercially available control liver RNA was purchased (Clonetics, CA; CloneTech, CA; and Lonza, NJ) and used in this study. Serum samples from healthy volunteers were used as controls. Serum and/or liver samples were collected from subjects with their written consent, and the human study protocol (protocol 10592) was approved by St. Louis University internal review.
Cells and transfections.
Immortalized human hepatocytes (IHH) were generated and maintained as previously described (21, 22). IHH were used for infection with HCV genotype 1a (clone H77) as previously described (23). Huh7.5 cells were transfected using Lipofectamine 2000 (Life Technologies, Inc., MD) with plasmid DNA from a mammalian expression vector (pcDNA3) containing the HCV genotype 1a full-length (FL) genome or protein-specific genomic region under the control of a cytomegalovirus (CMV) promoter. Stable colonies of transfectants were selected using neomycin and pooled for subsequent studies to avoid potential artifacts associated with clone-to-clone variation. Parental cells transfected with empty vector DNA were used in parallel as a negative control.
Flow cytometry.
Parental HCV-infected and HCV genotype 2a (clone JFH)-infected Huh7.5 cells were grown for 4 days, washed with phosphate-buffed saline (PBS), fixed with formaldehyde (final concentration, 2%), and incubated at 37°C for 10 min. Cells were centrifuged for 5 min (200 × g at 4°C) and washed with PBS containing 0.5% bovine serum albumin (BSA) and 0.1% NaN3. Cells were sequentially incubated for 60 min with a 1:100 dilution of anti-CD55 antibody (clone BRIC216; EMD Millipore, MA) in PBS containing 0.5% BSA and 0.1% NaN3. Cells were washed, a fluorescein isothiocyanate-conjugated anti-mouse IgG antibody (Santa Cruz, CA) was added at a 1:400 dilution, and the mixture was incubated for 30 min. Washed cells were resuspended in 90% ice-cold methanol for permeabilization, mixed gently, and placed at −20°C for 10 min. Next, cells were centrifuged and washed twice in PBS containing 0.5% BSA and 0.1% NaN3. A mouse anti-core proteinmonoclonal antibody (MAb; clone C7-50; Thermo Scientific) was added at a 1:100 dilution, and the cells were incubated for 60 min. Washed cells were incubated with an Alexa 647-conjugated rabbit anti-mouse IgG (Santa Cruz) at 1:400 dilutions for 30 min. Finally, cells were washed, resuspended in 500 μl cold PBS, and subjected to flow cytometric analysis (Becton, Dickinson) using software for processing data (Cell Quest software; BD Immunocytometry Systems).
Luciferase reporter assay.
The human CD55 promoter region was generated from genomic DNA of Huh7.5 cells. The CD55 promoter region was amplified by PCR from known sequences (24) using restriction site-containing synthetic oligonucleotide primers and cloned into a PGL3-Luc vector cassette as previously described (19). Huh7.5 cells were transfected with the CD55 promoter-driven luciferase reporter construct (200 ng/well) alone or together with HCV core or nonstructural proteins (NS2, NS3/4A, or NS5A) or a full-length infectious cDNA (HCV-FL) construct (500 ng/well) in a 24-well plate. In experiments assessing the effects of IL-1β and/or IL-6, cells were treated with the cytokine (10 ng/ml for IL-1β and 50 ng/ml for IL-6) at 24 h posttransfection. Cells were solubilized with reporter lysis buffer (Promega, WI) at 48 h after transfection, and the clarified lysates were subjected to the luciferase reporter assay using a luminometer (Opticomp II; MGM Instruments, CT).
Quantitative real-time PCR.
CD55 mRNA quantitation was performed by real-time PCR analysis using specific TaqMan primers and probes (Applied Biosystems assay identification [ID], Hs00892618_m1), after normalizing with 18S rRNA (Hs03928992-g1). RNA was isolated with the TRIzol reagent (Life Technologies) from liver biopsy specimens, HCV-infected Huh7.5 cells, or Huh7.5 cells transiently transfected with plasmids expressing specific HCV genomic regions. cDNA synthesis was performed using random hexamers and a SuperScript III first-strand synthesis kit (Invitrogen), and the levels of CD55 mRNA were evaluated.
Generation of site-specific mutations in CD55 promoter element.
Three separate mutations in the transcription factor binding sites of the CD55 promoter were generated. Briefly, the original pGL3-CD55-Luc plasmid construct was used to mutate the binding sites of three transcription factors (CREB, SP-1, and AP-1) in the CD55 promoter using a QuikChange II-E site-directed mutagenesis kit (Agilent Technologies) following the manufacturer's instructions. The primers used (5′-3′; with the mutation denoted by the underlined sequence) were CREB-(forward) (CCCACCCTTGGTGTGGCAGAGCCCCAGCCC), CREB-(reverse) (GGGCTGGGGCTCTGCCACACCAAGGGTGGG), SP-1-(forward) (CCCCAGCCCAGACCAAGCCCAAAGCACTCATTT), SP-1-(reverse) (AAATGAGTGCTTTGGGCTTGGTCTGGGCTGGGG), AP-1-(forward) (ACCCTTGGTGACGTGGAGCCCCAGCCC), and AP-1-(reverse) (GGGCTGGGGCTCCACGTCACCAAGGGT).
Cloned mutated DNAs with luciferase tags were used in the promoter assay.
Western blotting.
Proteins in cell lysates were resolved by 10% SDS-PAGE under reducing conditions, transferred onto a nitrocellulose membrane, and blotted with the appropriate primary antibody. Signal was detected using a peroxidase-conjugated secondary antibody (Bio-Rad, CA). Protein bands were visualized using an enhanced chemiluminescence detection kit (Super Signal West Pico; Thermo Chemical Company, IL). Cellular actin was detected for comparison of the protein load in each lane. The difference in intensities of the bands was obtained by densitometry scanning of Western blots and ImageJ software analyses.
Complement-mediated cytolysis assay.
Huh7.5 cells mock treated, infected with HCV genotype 2a, or constitutively expressing HCV core protein were maintained for 3 days. Cells were treated with serial dilutions of CD81 monoclonal antibody (isotype IgG2b; stock concentration, 200 μg/ml; Santa Cruz, CA) with or without human serum (5% dilution) as a source of complement (Quidel) in the presence or absence of function-blocking CD55 monoclonal antibody (isotype IgG1; clone BRIC216; EMD Millipore, MA).
Alternatively, Huh7.5 cells constitutively expressing HCV core protein were infected with vesicular stomatitis virus (VSV; Indiana) at a multiplicity of infection of 0.5. Cells were incubated for 16 h at 37°C before addition of serial dilutions of VSV G glycoprotein (VSV-G)-specific ascites (ATCC) and a 5% dilution of human serum as a source of complement (Quidel) in the presence or absence of function-blocking CD55 monoclonal antibody, as mentioned above. VSV-infected cells treated either with human serum alone or with VSV-specific antisera acted as negative controls. Cells were incubated for 4 h at 37°C before measurement of cell viability using a CellTiter 96 AQueous One cell viability assay (Promega Corporation, WI).
Density gradient-purified HCV preparation.
Cell culture-adapted HCV of genotype 2a (JFH1) was grown to an infectious titer ∼1,000-fold higher than that of the parent virus in Huh7.5 cells (25). Sucrose gradient-purified cell culture-grown virus was kindly provided to us by George Luo (University of Alabama at Birmingham). Biochemical analysis of gradient-purified HCV suggested the presence of viral proteins. Adenovirus was grown in 293 cells and purified by CsCl density gradient centrifugation as previously described (26). Gradient-purified virus preparations were estimated for protein content using a NanoOrange protein quantitation kit (Invitrogen, OR) following the supplier's procedure.
Suppression of HCV infection by CD55-specific antibody and human serum complement.
Cell culture-grown HCV genotype 1a or 2a (23) was used at a set virus number (∼100 focus-forming units [FFU]) for infection in Huh7.5 cells. Briefly, virus was incubated at 37°C in the presence or absence of CD55 antibody for 1 h. Cells were incubated with the virus-antibody mixture at 37°C for 5 h, rinsed, and incubated in medium for 96 h. Cells were rinsed with PBS and fixed with methanol for 20 min, followed by 0.2% Triton X-100 in PBS for 5 min. Cells were blocked with 3% BSA–PBS, followed by an overnight incubation at 4°C with an antibody specific to HCV NS5A protein (9E10; kindly provided by Charlie Rice, Rockefeller University, New York, NY). Secondary antibody (anti-mouse Alexa 488) was incubated over cells for 1 h at room temperature, and DAPI (4′,6-diamidino-2-phenylindole) was used for nuclear staining and contrast imaging. Infected cells were visualized under a Leica microscope, and fields were selected at random.
To quantify antibody- and/or complement-dependent suppression of HCV infection, real-time PCR was performed (27, 28). A predetermined number of FFU of cell culture-grown HCV was incubated with HCV envelope glycoprotein vaccine serum (1:100) in the presence or absence of a function-blocking mouse monoclonal antibody (1:50) to CD55. Untreated HCV and virus treated only with CD55 antibody were used as controls. After antibody addition, virus was incubated for 30 min at 37°C, followed by an additional 30 min of incubation in the presence of a normal human serum pool (1:20) as a source of complement. Virus-containing supernatants were incubated with Huh7.5 cells for 5 h at 37°C. Cells were rinsed twice and incubated at 37°C for 96 h. Cells were rinsed and lysed, and RNA was isolated using an RNeasy minikit (Qiagen). cDNA was generated using a SuperScript III kit (Invitrogen), followed by quantitative real-time PCR to measure levels of viral RNA (28). A standard curve generated from virus dilutions of known titer was used as a comparative control, and 18S rRNA was used as an internal control.
Biotinylation of human Ig to HCV E1/E2.
Sera (samples 19, 20, 21, and 34) that were from individuals vaccinated with the HCV envelope glycoproteins and that reacted against three E2-derived linear epitopes and one E1 epitope by enzyme-linked immunosorbent assay (ELISA) (29) were used to prepare Ig using protein A/G chromatography cartridges (Pierce). Antibodies to E1/E2 glycoproteins were biotinylated using an EZ-link sulfo-NHS biotinylation kit (Thermo Scientific, Rockford, IL) following the manufacturer's protocol.
HCV-capture ELISA using anti-CD55 antibody.
Rabbit anti-CD55 (Santa Cruz, CA) or a control rabbit antibody to an internal cellular protein, caspase 8 (BD Pharmingen), was coated on an ELISA plate (Corning Costar flat-bottom high-binding EIA/RIA 3690 plate) in PBS (pH 7.4; 0.4 μg/well) at 37°C for 2 h. The plate was blocked with 2% BSA in PBS. Cell culture-grown stock HCV genotype 1a or 2a (culture supernatant) of known titer (international units and numbers of FFU/ml) was added in triplicate to experimental or control wells. After incubation at 4°C overnight, wells were washed and bound virus or virus envelope glycoproteins were detected by serial addition of a biotin-labeled pooled anti-HCV envelope glycoprotein-specific human serum sample generated in a vaccine development study (29), avidin-horseradish peroxidase (HRP) conjugate (Pierce Immunopure streptavidin-HRP conjugate), and peroxidase substrate (Pierce Chemicals, IL). The absorbance at 450 nm was read and presented after subtraction of reagent-control values reacting against BSA-coated negative controls (<0.25).
Statistical analysis.
Results were expressed as the mean ± standard deviation (SD) from four independent experiments, and statistical analyses were performed using a two-tailed unpaired Student t test or one-way analysis of variance (ANOVA) in GraphPad Prism, version 5, software (GraphPad, La Jolla, CA). A P value of <0.05 was considered statistically significant.
RESULTS
HCV infection enhances CD55 expression.
CD55 mRNA expression in HCV-infected liver biopsy specimens in a cohort of patients studied previously (19, 20) was evaluated by real-time PCR analysis. CD55 expression was enhanced (∼3- to 4-fold) (P < 0.0001) in all 12 HCV-infected liver biopsy specimens compared with that in three healthy liver RNA samples (Fig. 1). The changes in CD55 level did not correlate with the stage of liver fibrosis or the results of laboratory tests (tests for rheumatoid factor, serum albumin, or alkaline phosphatase levels) in paired blood specimens from the same limited cohort of patients, indicating that liver fibrosis does not modulate CD55. The purpose of using the same cohort from previous studies for this analysis was to be able to work on a broad range of distinct but mechanistically linked complement components in the same cohort of chronically HCV-infected patients.
Fig 1.
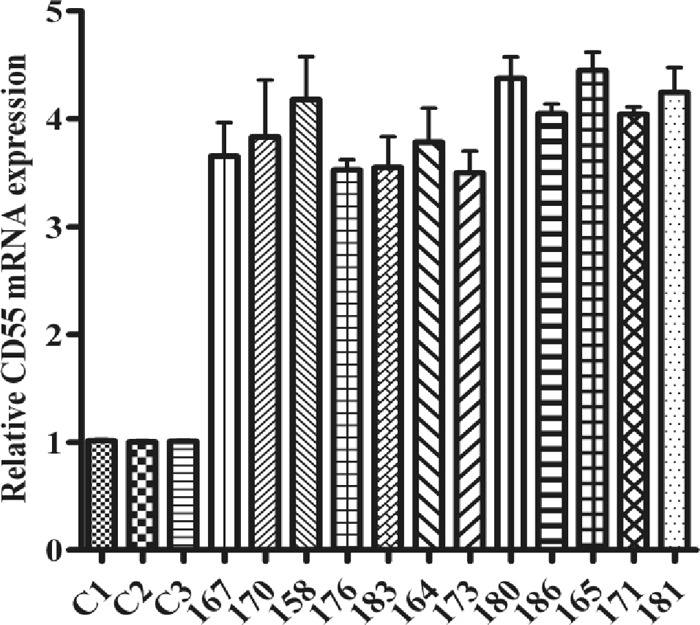
HCV infection upregulates CD55 mRNA expression. Upregulation of CD55 mRNA expression in HCV genotype 1a-infected paired liver biopsy specimens (marked with 3-digit numbers) compared with the expression of CD55 mRNA in non-HCV-infected control liver tissues (C1, C2, and C3) was determined by qRT-PCR. Results were normalized to those for 18S rRNA and shown as individual samples organized according to the stage of fibrosis of the patient liver (P < 0.0001, as a group of experimental samples compared to controls using one-way ANOVA).
CD55 expression increases upon HCV infection of hepatocytes.
We also compared CD55 mRNA expression levels in IHH infected with HCV genotype 1a and primary human hepatocytes (PHH) infected with cell culture-grown HCV genotype 2a with those in the corresponding uninfected parental cells by quantitative reverse transcription-PCR (qRT-PCR). An ∼2.5-fold or greater induction in CD55 mRNA was observed in IHH infected with HCV genotype 1a or PHH than in parental hepatocytes (Fig. 2A). We also observed a 6.3-fold increase in CD55 expression in HCV genotype 2a-infected Huh7.5 cell lysates compared to that in the mock-infected control by Western blotting using densitometry scanning analysis (Fig. 2B). The induction of CD55 expression in HCV-infected hepatocytes was reflected at the cell surface in both HCV genotype 1a-infected IHH (Fig. 2C) and HCV genotype 2a-infected Huh7.5 cells (data not shown), as determined by flow cytometry.
Fig 2.

CD55 expression increases upon HCV infection of hepatocytes. (A) Enhanced CD55 mRNA expression in IHH infected with HCV genotype 1a and PHH infected with HCV genotype 2a detected by qRT-PCR. Parental hepatocytes were used as a control for comparison. CD55 mRNA expression in parental hepatocytes was arbitrarily set at 1, and the results for the infected hepatocytes were statistically significantly different (P < 0.0001) using one-way ANOVA. (B) Detection of HCV core protein expression in HCV genotype 2a-infected Huh7.5 cells by Western blotting. Cellular actin expression was used as a loading control in each lane. (C) Staining with anti-CD55 antibody from HCV genotype 1a-infected IHH is shown by flow cytometry analysis.
CD55 expression is increased in the presence of HCV core protein.
Since we identified a transcription-regulatory role of core protein with a pleiotropic effect on multiple cellular genes (30), we investigated the role of HCV core protein on CD55 mRNA expression. Huh7.5 cells transfected with the HCV core protein gene exhibited an ∼5-fold increase (P < 0.001) in CD55 mRNA expression compared to empty vector-transfected cells (Fig. 3A). Similarly, we observed an ∼6-fold enhancement (P < 0.001) of CD55 transcript levels in cells transfected with an infectious HCV full-length cDNA. In contrast, transfection of HCV NS3/4A or NS5A failed to change CD55 levels compared to those in the vector control. We also observed 4.8-fold higher levels of CD55 protein expression in Huh7.5 cells stably expressing HCV core protein than in the mock-transfected control by Western blotting using densitometry scanning analysis (Fig. 3B). Flow cytometry analysis corroborated the increased cell surface expression of CD55 on Huh7.5 cells transfected with core protein (Fig. 3C). However, a similar treatment of IHH (Fig. 3D) resulted in no enhancement of CD55 expression by flow cytometry analysis, likely due to the presence of already enhanced CD55 levels in IHH due to immortalization by HCV core protein. Thus, upregulation of CD55 expression in HCV-infected hepatocytes may occur due to a transcriptional regulatory role of the virus core protein.
Fig 3.

Enhancement of CD55 expression in hepatocytes is mediated by HCV core protein. (A) Upregulation of CD55 mRNA expression in Huh7.5 cells transfected with HCV core protein or FL polyprotein compared with the level of expression in untreated control cells detected by real-time PCR. NS3/4A and NS5A did not affect CD55 expression. CD55 mRNA expression in untreated controls was arbitrarily set at 1, and the results were statistically significantly different (P < 0.0001) using one-way ANOVA. The experiments were done in triplicate and repeated three times. (B) Western blot analysis of lysates from Huh7.5 cells stably transfected with HCV core protein reveals upregulation of CD55. (C and D) Cell surface expression of membrane-bound CD55 on HCV core protein-transfected or vector-transfected Huh7.5 cells and IHH, as detected by flow cytometry.
CD55 promoter activity is enhanced by HCV core protein.
Our observation of enhanced CD55 mRNA levels in HCV infection prompted us to investigate whether this is caused by an upregulation in CD55 promoter activity resulting in an enhanced transcription rate. We used a CD55 promoter construct with a luciferase reporter gene tag and transfected it into Huh7.5 cells along with plasmids encoding individual HCV proteins (core protein, NS3/4A, or NS5A) as well as the full-length infectious HCV cDNA. Cells were lysed 48 h after transfection, and promoter activity was measured by luciferase assay. Our results suggested that CD55 promoter activity was enhanced in the presence of HCV polyprotein (Fig. 4A). Among the HCV proteins, this effect was apparent only in the presence of core protein, with an ∼8-fold enhancement in CD55 promoter activity (Fig. 4A). In comparison, CD55 promoter activity was unaffected in HCV NS3/4A- and NS5A-transfected cells.
Fig 4.
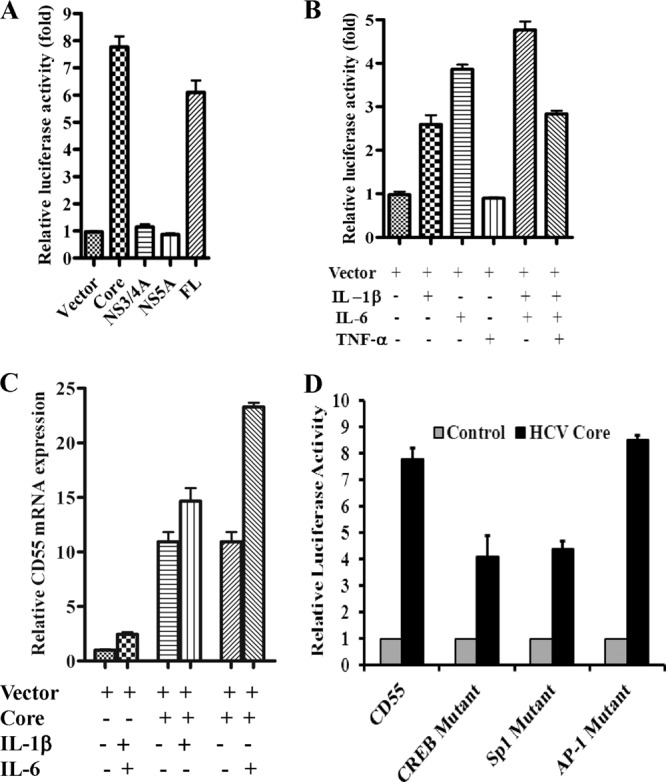
HCV core protein enhances CD55 promoter activity regulated by IL-1β and/or IL-6. (A) Enhancement of CD55 promoter activity by HCV core protein and FL polyprotein in a luciferase reporter assay. NS3/4A and NS5A did not display a major effect on CD55 promoter activity. The results obtained were statistically significantly different (P < 0.0001 for vector versus core protein and P < 0.0003 for vector versus FL polyprotein; the results for NS3/4A and NS5A were not significant with respect to the results for the vector) from the values for the vector control by the unpaired two-tailed t test. (B) Cytokine-mediated induction of CD55 promoter activity. IL-1β, IL-6, and TNF-α were used individually or in combination. Results obtained were statistically significantly different (P < 0.0001) from those for the uninduced control using one-way ANOVA. (C) IL-1β- and IL-6-mediated induction of CD55 mRNA expression in the presence of HCV core protein. Results obtained were statistically significantly different (P < 0.0001) from those for the uninduced control using one-way ANOVA. (D) Reduction in HCV core protein-mediated induction of luciferase expression from a CD55 promoter engineered with point mutations in transcription factor binding sites. The upregulation of CD55 promoter activity in Huh7.5 cells transfected with HCV core protein compared to the level of regulation in the vector control determined by a luciferase reporter assay is shown. A reduction in the levels of core protein-mediated induction was observed in the CD55 promoter harboring point mutations of both CREB as well as SP-1 binding sites, whereas an alteration in the AP-1 binding site did not affect core protein-mediated induction of CD55 promoter activity.
The expression of CD55 in hepatoma cells is reportedly regulated by IL-1β/IL-6 cytokines as well as TNF-α (15). We next examined the effect of these cytokines on CD55 promoter activity in Huh7.5 cells. We observed maximal levels of CD55 induction in the presence of IL-6 (∼4-fold) followed by IL-1β (∼2.7-fold) (Fig. 4B). TNF-α did not affect CD55 promoter activity either alone or when combined with IL-1β and IL-6. Previous reports had indicated that HCV core protein induces IL-6 expression in IHH (31), which can potentially act in an autocrine manner to stimulate CD55 expression. As such, we determined the impact of IL-1β and IL-6 on CD55 promoter activity in the presence of HCV core protein. While IL-1β caused negligible enhancement of CD55 promoter activity in core protein-transfected hepatocytes, addition of IL-6 resulted in an ∼2.2-fold induction of promoter activity (Fig. 4C).
Next, we investigated the mechanism of HCV core protein-mediated upregulation of CD55 expression. We generated luciferase-tagged CD55 promoter constructs containing loss-of-function point mutations in the binding sites of the transcription factors CREB, AP-1, and SP-1 (32). We observed a reduction in HCV core protein-mediated induction of CD55 promoter activity if the promoter encoded point mutations in CREB or SP-1 binding sites; this indicates that interaction of these transcription factors with their cognate binding sites is important for HCV core protein-mediated upregulation of CD55 (Fig. 4D). However, a mutation affecting the AP-1 binding site did not influence HCV core protein-mediated CD55 upregulation. Thus, our results suggest that induction of CD55 promoter activity by HCV core protein is mediated by CREB and SP-1 transcription factors.
Cells infected with HCV or expressing core protein inhibit antibody-dependent, complement-mediated cytolysis.
Previous reports indicated that enhanced CD55 expression in association with the acute-phase response protects hepatoma cells from complement-mediated cell damage (15). Here, we examined whether enhanced CD55 expression in hepatoma cells infected with HCV can inhibit complement-mediated cytolysis and thereby promote cell survival. Initially, HCV genotype 2a-infected Huh7.5 cells and uninfected control cells were treated with CD81-specific antibody in the presence or absence of a function-inhibiting CD55-neutralizing antibody (BRIC216). Antibody to CD81 was observed to induce a weak level of cell killing in control Huh7.5 cells upon the addition of complement. Incubation of CD81 antibody-treated cells revealed a consistent increase in cell viability in HCV-infected cells (∼35% inhibition of total cell death) compared to uninfected control cells (Fig. 5A). The addition of antibody to CD55 further decreased cell viability in HCV-infected cells, approaching levels similar to those apparent in control cells. In contrast, the addition of antibody to CD55 on control cells exhibited a negligible decrease in cell viability, suggesting that enhanced CD55 availability on the Huh7.5 cell surface is related to enhanced survival of HCV-infected cells. Experiments using HCV core protein-expressing stable transfectants in the place of HCV infection of Huh7.5 cells exhibited similar results (Fig. 5B).
Fig 5.

Inhibition of antibody-dependent complement-mediated cell death by HCV. (A and B) Huh7.5 cells were infected with HCV genotype 2a or left mock infected. Alternatively, Huh7.5 cells were stably transfected with the HCV core protein gene or vector. Cells were treated with anti-CD81 MAb (isotype 2a) at the indicated reciprocal dilutions and complement (Quidel) at a 1:5 dilution. The experiments were done in triplicate, were repeated three times, and gave consistent results. The difference in cell viability between control and HCV-infected cells (*, P < 0.0083 to 0.001) compared to that for core protein-expressing cells was statistically significant (**, P < 0.0557 to 0.238). (C) Huh7.5 cells, with or without constitutive expression of HCV core protein, were infected with VSV, followed by treatment with anti-VSV-G antibody and pooled human serum as a source of complement proteins with or without CD55-neutralizing antibody. Cell viability was measured by the CellTiter 96 AQueous One cell viability assay (Promega). The experiments were done in triplicate, were repeated three times, and gave consistent results. The difference in cell viability between control and HCV core protein-expressing cells was statistically significant (***, P = 0.0128 to P < 0.0001).
As HCV envelope glycoproteins do not assemble at the cell surface for budding, we used VSV as an efficient and sensitive viral model system for inhibition of complement-mediated lysis. Here, the enhanced availability of VSV G glycoprotein on the infected cell surface compared to that of cellular CD81 protein is believed to be able to convey a stronger indication of cell viability in response to HCV core protein-mediated CD55 overexpression in a complement-mediated cell lysis assay. For this experiment, Huh7.5 cells ectopically expressing HCV core protein were infected with VSV and treated with a VSV-G-specific antibody in the presence or absence of a CD55 antibody. As described above, an additional incubation with pooled human serum as a source of complement was used to trigger antibody-dependent complement-mediated cytolysis. The presence of HCV core protein induced cell viability that was significantly greater than the viability of vector-transfected control cells when cell death was triggered by antibody-dependent (anti-VSV) complement-mediated cytolysis after VSV infection (Fig. 5C). Similar to the experiments described above, ∼50% of all complement-mediated cell killing in this experimental system was inhibited by the presence of HCV core protein expression (∼80% cell killing versus 40% for HCV core protein-expressing cells). Also, the degree of cell viability conferred by HCV core protein expression was reduced upon addition of a neutralizing anti-CD55 antibody, suggesting that CD55 induction by HCV core protein is necessary for evasion of complement-mediated cell death in virus-infected cells. Together, our results suggested that the presence of HCV core protein alone or in HCV-infected cells confers protection via increased CD55 expression that would be beneficial to virus survival.
CD55 associates with HCV produced in cell culture.
Cell culture-adapted HCV of genotype 2a (JFH1) grows to an infectious titer ∼1,000-fold greater than that of its parent virus (33). We used sucrose gradient-purified cell culture-adapted HCV genotype 2a to examine the CD55 association with the virus particles. We also used a CsCl gradient-purified unrelated adenovirus as a negative control in this experiment. The cloned HCV core protein genomic region was transfected into Huh7.5 cells and used for analysis of CD55 protein expression by Western blotting. The presence of a CD55-specific band was detected in sucrose density gradient-purified cell culture-grown HCV genotype 2a but was absent from purified adenovirus even after loading a 15-fold excess (Fig. 6A). These results suggested that CD55 associates with or is incorporated into HCV.
Fig 6.

Association of CD55 proteins with HCV. (A) Sucrose density gradient-purified HCV was tested by Western blotting for detection of CD55. Gradient-purified adenovirus type 5 (Ad5) was used as a negative control. The amount of protein estimated from gradient-purified adenovirus type 5 was 15-fold higher than the amount of purified HCV loaded in Western blot analysis. (B) Cell culture-grown HCV was captured by antibody to CD55 coated on an ELISA plate. Captured HCV E1/E2 was detected by envelope glycoprotein-specific avidin-conjugated human antibodies. The results from 4 independent experiments are presented with standard errors. (C) Inhibition of cell culture-grown HCV genotype 1a or 2a infectivity by CD55-specific rabbit antiserum. (D) qRT-PCR analysis for inhibition of HCV genotype 2a infectivity in Huh7.5 cells by a function-blocking murine MAb (IgG1) to CD55, a suboptimal dilution of HCV-neutralizing anti-E1/E2 human serum (1/100), or both in the presence of pooled normal human serum (NHS) as a source of complement. Results from three independent experiments using samples in triplicate are presented. The difference in viral infectivity between the control and antibody treatments was statistically significant (*, P = 0.0139; **, P = 0.0003; ***, P < 0.0001).
In separate experiments, we examined for a direct association of CD55 with intact or disintegrated HCV envelope components using a capture ELISA. Infectious HCV from cell culture medium was captured with a rabbit anti-CD55 or a negative-control rabbit antiserum to an internal cellular protein (caspase 8), and then binding was detected with an antibody preparation from HCV envelope glycoprotein-vaccinated subjects. The results revealed that anti-CD55 antibody could efficiently capture both HCV genotype 1a and HCV genotype 2a from cell culture-grown stock virus (Fig. 6B), and an increased optical density correlated with a higher virus titer. To further verify the localization of CD55 on the HCV surface, we used a rabbit polyclonal antibody to CD55 (amino acids 35 to 353) for inhibition of a predetermined ∼100 fluorescent focus-forming units of virus. Use of anti-CD55 antibody (2 μg/ml) inhibited ≥50% fluorescent focus formation from HCV of both genotypes compared to that from untreated control virus (Fig. 6C). Together these results indicated that CD55 associates with the HCV surface and virus infectivity can be prevented by CD55-specific antibody.
To observe the effect of CD55 incorporation on virus fitness, we incubated HCV with a function-blocking CD55 monoclonal antibody (BRIC216; Millipore) alone or in the presence of a previously identified nonneutralizing concentration (1%) of heat-inactivated human serum from a recent HCV vaccine trial (29). Subsequently, pooled human serum from healthy volunteers was added at levels (10%) previously observed to have no negative effects upon virus fitness and further incubated at 37°C for 30 min prior to incubation with naive Huh7.5 cells. Incubation of HCV genotype 2a with antibody to CD55 in the presence of complement prior to addition on Huh7.5 cells modestly inhibited (<2-fold) HCV infection, as suggested from the viral RNA level within cells by qRT-PCR (Fig. 6D). A similar effect was also observed using HCV anti-E1/E2 human serum alone in the presence of complement (29). Addition of both antisera to E1/E2 and function-blocking CD55 resulted in an enhanced reduction (>5-fold) of HCV infectivity in Huh7.5 cells due to increased virus lysis under CD55-inhibiting conditions. These results further suggested that CD55 is physically associated with the HCV surface and may act to inhibit the innate immune function in HCV-infected individuals.
DISCUSSION
In this study, we observed an enhancement of CD55 expression at the mRNA and protein levels in hepatoma cells infected with HCV or ectopically expressing the core protein. Correspondingly, CD55 mRNA expression was also elevated in liver biopsy specimens from a small cohort of patients with chronic HCV infection. It is quite difficult to prepare cells for evaluation of multiple parameters (mRNA expression for quantitative real-time PCR and protein expression for flow cytometry or immunohistochemistry) with material from a single needle biopsy specimen from an individual patient. Our rationale for using human clinical materials was to provide a more physiological linkage to our cell culture studies, as a facile and tractable small-animal model for HCV infection is currently lacking. HCV core protein is a multifunctional regulator of gene expression and assumes different roles during the course of infection (30). Our results suggest that expression of HCV core protein augments CD55 promoter activity in a CREB or SP-1 transcription factor-dependent manner. The promoter and related transcription factors for CD55 expression in some cell lines have been identified (32). The CD55 promoter has different transcription factor binding sites for CREB, AP-1, and SP-1. In our study with Huh7.5 cells, we utilized the known information from the literature and mutated binding sites of the previously identified transcription factors. At this point, we cannot rule out the possibility that additional transcription factors and their cognate binding sites exist in the Huh7.5 CD55 promoter. We observed that point mutations at known CREB or SP-1 binding sites inhibited HCV core protein-mediated activation of the CD55 promoter. Double mutations in the already identified binding sites are likely to cause greater inhibition of CD55 promoter activity by HCV core protein. A detailed mechanism of HCV-mediated CD55 promoter regulation in Huh7.5 cells would require a more in-depth analysis. HCV appeared to upregulate CD55 on target cells to prevent complement-mediated opsonization or lysis, with a benefit of protecting the infected cells themselves from complement attack. We also detected the presence of CD55 in association with highly purified HCV through biochemical and immunological analysis.
A functional analysis for the outcome of enhanced CD55 expression was performed. HCV-infected or core protein-expressing hepatoma cells were treated with an antibody to cell surface tetraspanin protein CD81 in the presence or absence of a CD55-neutralizing antibody. Antibody-treated cells were exposed to human serum as a source of complement. An increase in cell viability was apparent in HCV-infected cells and HCV core protein-expressing cells compared to the viability of untreated negative-control cells. HCV core protein expression in hepatocytes also inhibited antibody-dependent complement-mediated cytolysis triggered by VSV infection as a model system. However, HCV core protein-mediated viability was reduced in the presence of blocking antibodies to CD55, further highlighting its role in the inhibition of complement-mediated lysis. We found CD55 to be associated with density gradient-purified cell culture-propagated HCV. In support of this, function-blocking antibody to CD55 or human anti-E1/E2 modestly neutralized HCV infectivity, and antibody to E1/E2 in the presence of anti-CD55 enhanced HCV neutralization in the presence of normal human serum as a source of complement.
Establishment of chronic HCV infection in most patients highlights that this virus is able to effectively evade innate and adaptive immune responses, including complement. We have reported previously that HCV core and NS5A proteins can transcriptionally repress induction of the complement components C3 and C4 by reducing expression of key transcription factors (19, 20). Complement-regulatory proteins CD46, CD55, and CD59 limit excessive complement-mediated damage to host cells. Each regulator attenuates complement activation at different stages in the enzymatic cascade. Whereas CD46 is a cofactor protein for the cleavage of C3b to iC3b (34), CD55 accelerates the decay of the C3 convertases (reviewed in reference 35). CD59, in contrast, attenuates MAC formation (36). Increased expression of a complement regulator(s) on the surface of host cells is associated with significant protection against complement-mediated lysis (8, 37–39). Viruses inhibit complement activation through a panoply of mechanisms. For instance, the genomes of herpesviruses and poxviruses encode homologs of complement regulators (40). As a different strategy, human immunodeficiency virus, cytomegalovirus, herpes simplex virus, Ebola virus, vaccinia virus, and influenza virus all escape antibody-dependent complement-mediated lysis by incorporating (hijacking) host RCA proteins onto the surface of the virion (reviewed in reference 17). They often produce complement binding proteins that attach the host's inhibitor to their membrane. Incorporation of CD55 into lentiviral particles rendered them more resistant to complement-mediated inactivation (41, 42). Analogously, parainfluenza virus type 5 (PIV5) and mumps virus (MuV) incorporate CD46 into progeny virions to gain partial resistance to neutralization by normal human serum. Complement regulators are often species specific, and their relative contribution to viral evasion from complement attack may vary. Future studies are planned to determine how the relative level of RCA upregulation in HCV-infected cells or the stoichiometry of incorporation into the virion modulates virus infectivity.
Upregulation of CD55 and CD59 expression serves to protect hepatocytes from high local concentrations of complement generated during the acute-phase response (15). This enhancement of CD55 is induced by acute-phase cytokines in an HCV-independent mechanism. A reduction in complement-mediated tumor lysis by higher CD55 levels could facilitate tumorigenesis and contribute to the pathogenesis of hepatocellular carcinoma that is observed during chronic HCV infection. Indeed, in both breast and colorectal cancer patients, expression of CD55 in tumors has been associated with a poor prognosis (43, 44). Also, earlier reports indicated that expression of CD46, another complement-regulatory protein, was significantly enhanced in HCC, apparently in an effort to escape from tumor-specific complement-mediated cytotoxicity (45). It will be interesting to investigate in detail the relationship between HCV infection, CD55 expression, and induction of HCC as a follow-up of the current study.
HCV incorporates CD59 into the viral lipid bilayer at biologically functional levels (17) and protects against antibody-dependent complement-mediated lysis. Thus, it is difficult to examine the role of CD55 in maintaining cell viability related to complement attack by depleting it alone in cells supporting HCV growth. A recent report (46) demonstrated that, among the three complement-regulatory proteins, HCV associates with CD59. To test if infected cells are putative sources for the acquisition of CD59 by HCV-Jc1, the authors analyzed Huh7.5 cells by flow cytometry. CD46 and CD59 were expressed efficiently and comparably on the cell surface, whereas less CD55 was detected. Expression of CD59 in Huh7.5 cells infected with HCV-Jc1, however, was not up- or downregulated, indicating that viral replication or proteins did not affect CD59 expression. Why CD55 was not detected in these virions remains uncertain, although it could be due to differences in the reactivity of antibodies or purification strategies.
In summary, upregulation of CD55 expression on HCV-infected hepatocytes can lead to increased resistance of the cells to cytokine-stimulated complement activation and subsequent complement-dependent lysis. This would enable HCV to evade complement attack and multiply to higher titers within infected cells. The CD55 association with intact HCV particles potentially renders them resistant to antibody-dependent or -independent complement-mediated lysis. Future in vivo studies will be designed to elucidate the contribution of RCAs to viral fitness during HCV infection and the survival of unrelated pathogens in HCV-infected individuals.
ACKNOWLEDGMENTS
We are grateful to Robert Belshe for helpful suggestions and Katia Los for technical assistance. We thank Lin Cowick for preparation of the manuscript.
This work was supported by research grant U54-AI057160 from the NIAID to the Midwest Regional Center of Excellence (MRCE) for Biodefense and Emerging Infectious Diseases Research and the Internal Blue Ribbon Fund of Saint Louis University.
Footnotes
Published ahead of print 8 May 2013
REFERENCES
- 1. Mollnes TE, Song WC, Lambris JD. 2002. Complement in inflammatory tissue damage and disease. Trends Immunol. 23:61–64 [DOI] [PubMed] [Google Scholar]
- 2. Gasque P. 2004. Complement: a unique innate immune sensor for danger signals. Mol. Immunol. 41:1089–1098 [DOI] [PubMed] [Google Scholar]
- 3. Kim DD, Song WC. 2006. Membrane complement regulatory proteins. Clin. Immunol. 118:127–136 [DOI] [PubMed] [Google Scholar]
- 4. Blue CE, Spiller OB, Blackbourn DJ. 2004. The relevance of complement to virus biology. Virology 319:176–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Walport MJ. 2001. Complement. First of two parts. N. Engl. J. Med. 344:1058–1066 [DOI] [PubMed] [Google Scholar]
- 6. Pangburn M, Ferreira VP, Cortes C. 2008. Discrimination between host and pathogens by the complement system. Vaccine 26(Suppl 8):I15–I21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Seya T, Atkinson JP. 1989. Functional properties of membrane cofactor protein of complement. Biochem. J. 64:581–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hourcade D, Liszewski MK, Krych-Goldberg M, Atkinson JP. 2000. Functional domains, structural variations and pathogen interactions of MCP, DAF and CR1. Immunopharmacology 49:103–116 [DOI] [PubMed] [Google Scholar]
- 9. Williams P, Chaudhry Y, Goodfellow IG, Billington J, Powell R, Spiller OB, Evans DJ, Lea S. 2003. Mapping CD55 function. The structure of two pathogen-binding domains at 1.7 Å. J. Biol. Chem. 278:10691–10696 [DOI] [PubMed] [Google Scholar]
- 10. Baumann H, Gauldie J. 1994. The acute phase response. Immunol. Today 15:74–80 [DOI] [PubMed] [Google Scholar]
- 11. Ramadori G, Christ B. 1999. Cytokines and the hepatic acute-phase response. Semin. Liver Dis. 19:141–155 [DOI] [PubMed] [Google Scholar]
- 12. Falus A, Rokita H, Walcz E, Brozik M, Hidvegi T, Meretey K. 1990. Hormonal regulation of complement biosynthesis in human cell lines. II. Upregulation of the biosynthesis of complement components C3, factor B and C1 inhibitor by IL-6 and IL-1 in human hepatoma cell line. Mol. Immunol. 27:197–201 [DOI] [PubMed] [Google Scholar]
- 13. Perissutti S, Tedesco F. 1994. Effect of cytokines on the secretion of the fifth and eighth complement components by HepG2 cells. Int. J. Clin. Lab. Res. 24:45–48 [DOI] [PubMed] [Google Scholar]
- 14. Scheurer B, Rittner C, Schneider PM. 1997. Expression of the human complement C8 subunits is independently regulated by interleukin 1 beta, interleukin 6, and interferon gamma. Immunopharmacology 38:167–175 [DOI] [PubMed] [Google Scholar]
- 15. Spiller OB, Criado-Garcia O, Rodriguez De Cordoba S, Morgan BP. 2000. Cytokine-mediated up-regulation of CD55 and CD59 protects human hepatoma cells from complement attack. Clin. Exp. Immunol. 121:234–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Halme J, Sachse M, Vogel H, Giese T, Klar E, Kirschfink M. 2009. Primary human hepatocytes are protected against complement by multiple regulators. Mol. Immunol. 46:2284–2289 [DOI] [PubMed] [Google Scholar]
- 17. Amet T, Ghabril M, Chalasani N, Byrd D, Hu N, Grantham A, Liu Z, Qin X, He JJ, Yu Q. 2012. CD59 incorporation protects hepatitis C virus against complement-mediated destruction. Hepatology 55:354–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shan C, Zhang S, Cui W, You X, Kong G, Du Y, Qiu L, Ye L, Zhang X. 2011. Hepatitis B virus X protein activates CD59 involving DNA binding and let-7i in protection of hepatoma and hepatic cells from complement attack. Carcinogenesis 32:1190–1197 [DOI] [PubMed] [Google Scholar]
- 19. Banerjee A, Mazumdar B, Meyer K, Di Bisceglie AM, Ray RB, Ray R. 2011. Transcriptional repression of C4 complement by hepatitis C virus proteins. J. Virol. 85:4157–4166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mazumdar B, Kim H, Meyer K, Bose SK, Di Bisceglie AM, Ray RB, Ray R. 2012. Hepatitis C virus proteins inhibit C3 complement production. J. Virol. 86:2221–2228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Basu A, Meyer K, Ray RB, Ray R. 2002. Hepatitis C virus core protein is necessary for the maintenance of immortalized human hepatocytes. Virology 298:53–62 [DOI] [PubMed] [Google Scholar]
- 22. Ray RB, Meyer K, Ray R. 2000. Hepatitis C virus core protein promotes immortalization of primary human hepatocytes. Virology 271:197–204 [DOI] [PubMed] [Google Scholar]
- 23. Kanda T, Basu A, Steele R, Wakita T, Ryerse JS, Ray R, Ray RB. 2006. Generation of infectious hepatitis C virus in immortalized human hepatocytes. J. Virol. 80:4633–4639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ewulonu UK, Ravi L, Medof ME. 1991. Characterization of the decay-accelerating factor gene promoter region. Proc. Natl. Acad. Sci. U. S. A. 88:4675–4679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jiang J, Luo G. 2009. Apolipoprotein E but not B is required for the formation of infectious hepatitis C virus particles. J. Virol. 83:12680–12691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ghosh AK, Steele R, Ray RB. 2005. Carboxyl-terminal repressor domain of MBP-1 is sufficient for regression of prostate tumor growth in nude mice. Cancer Res. 65:718–721 [PubMed] [Google Scholar]
- 27. Engle RE, Russell RS, Purcell RH, Bukh J. 2008. Development of a TaqMan assay for the six major genotypes of hepatitis C virus: comparison with commercial assays. J. Med. Virol. 80:72–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Meyer K, Banerjee A, Frey SE, Belshe RB, Ray R. 2011. A weak neutralizing antibody response to hepatitis C virus envelope glycoprotein enhances virus infection. PLoS One 6:e23699. 10.1371/journal.pone.0023699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ray R, Meyer K, Banerjee A, Basu A, Coates S, Abrignani S, Houghton M, Frey SE, Belshe RB. 2010. Characterization of antibodies induced by vaccination with hepatitis C virus envelope glycoproteins. J. Infect. Dis. 202:862–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Banerjee A, Ray RB, Ray R. 2010. Oncogenic potential of hepatitis C virus proteins. Viruses 2:2108–2133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Basu A, Meyer K, Lai KK, Saito K, Di Bisceglie AM, Grosso AM, Ray RB, Ray R. 2006. Microarray analyses and molecular profiling of Stat3 signaling pathway induced by hepatitis C virus core protein in human hepatocytes. Virology 349:347–358 [DOI] [PubMed] [Google Scholar]
- 32. Thomas DJ, Lublin DM. 1993. Identification of 5′-flanking regions affecting the expression of the human decay accelerating factor gene and their role in tissue-specific expression. J. Immunol. 150:151–160 [PubMed] [Google Scholar]
- 33. Jiang J, Luo G. 2012. Cell culture-adaptive mutations promote viral protein-protein interactions and morphogenesis of infectious hepatitis C virus. J. Virol. 86:8987–8997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liszewski MK, Post TW, Atkinson JP. 1991. Membrane cofactor protein (MCP or CD46): newest member of the regulators of complement activation gene cluster. Annu. Rev. Immunol. 9:431–455 [DOI] [PubMed] [Google Scholar]
- 35. Lublin DM, Atkinson JP. 1989. Decay-accelerating factor: biochemistry, molecular biology, and function. Annu. Rev. Immunol. 7:35–58 [DOI] [PubMed] [Google Scholar]
- 36. Kimberley FC, Sivasankar B, Paul Morgan B. 2007. Alternative roles for CD59. Mol. Immunol. 44:73–81 [DOI] [PubMed] [Google Scholar]
- 37. Liszewski MK, Farries TC, Lublin DM, Rooney IA, Atkinson JP. 1996. Control of the complement system. Adv. Immunol. 61:201–283 [DOI] [PubMed] [Google Scholar]
- 38. Richards A, Kavanagh D, Atkinson JP. 2007. Inherited complement regulatory protein deficiency predisposes to human disease in acute injury and chronic inflammatory states. Adv. Immunol. 96:141–177 [DOI] [PubMed] [Google Scholar]
- 39. Kavanagh D, Richards A, Atkinson JP. 2008. Complement regulatory genes and hemolytic uremic syndrome. Annu. Rev. Med. 59:293–309 [DOI] [PubMed] [Google Scholar]
- 40. Favoreel HW, Van de Walle GR, Nauwynck HJ, Pensaert MB. 2003. Virus complement evasion strategies. J. Gen. Virol. 84:1–15 [DOI] [PubMed] [Google Scholar]
- 41. Huser A, Rudolph M, Hofmann C. 2001. Incorporation of decay-accelerating factor into the baculovirus envelope generates complement-resistant gene transfer vectors. Nat. Biotechnol. 19:451–455 [DOI] [PubMed] [Google Scholar]
- 42. Kaname Y, Tani H, Kataoka C, Shiokawa M, Taguwa S, Abe T, Moriishi K, Kinoshita T, Matsuura Y. 2010. Acquisition of complement resistance through incorporation of CD55/decay-accelerating factor into viral particles bearing baculovirus GP64. J. Virol. 84:3210–3219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Durrant LG, Chapman MA, Buckley DJ, Spendlove I, Robins RA, Armitage NC. 2003. Enhanced expression of the complement regulatory protein CD55 predicts a poor prognosis in colorectal cancer patients. Cancer Immunol. Immunother. 52:638–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ikeda J, Morii E, Liu Y, Qiu Y, Nakamichi N, Jokoji R, Miyoshi Y, Noguchi S, Aozasa K. 2008. Prognostic significance of CD55 expression in breast cancer. Clin. Cancer Res. 14:4780–4786 [DOI] [PubMed] [Google Scholar]
- 45. Kinugasa N, Higashi T, Nouso K, Nakatsukasa H, Kobayashi Y, Ishizaki M, Toshikuni N, Yoshida K, Uematsu S, Tsuji T. 1999. Expression of membrane cofactor protein (MCP, CD46) in human liver diseases. Br. J. Cancer 80:1820–1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ejaz A, Steinmann E, Banki Z, Anggakusuma, Khalid S, Lengauer S, Wilhelm C, Zoller H, Schloegl A, Steinmann J, Grabski E, Kleines M, Pietschmann T, Stoiber H. 2012. Specific acquisition of functional CD59 but not CD46 or CD55 by hepatitis C virus. PLoS One 7:e45770. 10.1371/journal.pone.0045770 [DOI] [PMC free article] [PubMed] [Google Scholar]