

Abstract
Inflammatory cytokines and chemokines play important roles in inflammation during viral infection. Hepatitis C virus (HCV) is a hepatotropic RNA virus that is closely associated with chronic liver inflammation, fibrosis, and hepatocellular carcinoma. During the progression of HCV-related diseases, hepatic stellate cells (HSCs) contribute to the inflammatory response triggered by HCV infection. However, the underlying molecular mechanisms that mediate HSC-induced chronic inflammation during HCV infection are not fully understood. By coculturing HSCs with HCV-infected hepatocytes in vitro, we found that HSCs stimulated HCV-infected hepatocytes, leading to the expression of proinflammatory cytokines and chemokines such as interleukin-6 (IL-6), IL-8, macrophage inflammatory protein 1α (MIP-1α), and MIP-1β. Moreover, we found that this effect was mediated by IL-1α, which was secreted by HSCs. HCV infection enhanced production of CCAAT/enhancer binding protein (C/EBP) β mRNA, and HSC-dependent IL-1α production contributed to the stimulation of C/EBPβ target cytokines and chemokines in HCV-infected hepatocytes. Consistent with this result, knockdown of mRNA for C/EBPβ in HCV-infected hepatocytes resulted in decreased production of cytokines and chemokines after the addition of HSC conditioned medium. Induction of cytokines and chemokines in hepatocytes by the HSC conditioned medium required a yet to be identified postentry event during productive HCV infection. The cross talk between HSCs and HCV-infected hepatocytes is a key feature of inflammation-mediated, HCV-related diseases.
INTRODUCTION
Hepatitis C virus (HCV) can cause chronic liver disease, which can progress to fibrosis, cirrhosis, and hepatocellular carcinoma (HCC) (1). Clearance of HCV during the acute phase of infection is associated with a robust CD4 and CD8 T-cell response to multiple viral epitopes (2). However, clearance of HCV infection often fails because of an intermediate cytotoxic T-cell response that is unable to eliminate the infection but causes hepatocyte destruction. T-cell-mediated hepatocytotoxicity poses a high risk for progression to chronic liver inflammation and damage (3). During chronic HCV infection, chemokine-chemokine receptor interactions are particularly important for the recruitment of T cells to sites of inflammation in the liver. Liver-infiltrating lymphocytes in HCV patients exhibit increased expression of CXCR3 and CCR5 (4). Moreover, intrahepatic chemokines, such as RANTES, macrophage inflammatory protein 1α (MIP-1α), MIP-1β, and IP-10, are elevated in HCV patients (5), and intrahepatic proinflammatory cytokine levels are correlated with the severity of inflammation and liver fibrosis (6).
The induction of proinflammatory cytokines and chemokines is triggered by viral proteins and double-stranded RNA (dsRNA) from HCV. The HCV core protein induces inflammatory cytokines through the STAT3 signaling pathway (7). Retinoic acid-inducible gene I (RIG-I) and Toll-like receptor 3 (TLR-3) are cellular sensors that recognize HCV dsRNA, resulting in production of chemokines such as interleukin-8 (IL-8), RANTES, MIP-1α, and MIP-1β (8, 9). Recently, an alternative mechanism for HCV-induced inflammation was reported. It was demonstrated that NS5B, the viral RNA-dependent RNA polymerase (RdRp), catalyzes production of small RNA species that trigger an innate immune response, leading to the production of both interferon (IFN) and inflammatory cytokines (10).
Hepatic stellate cells (HSCs) represent 5 to 8% of the total human liver cells and reside in the Disse space (11). Activation or transdifferentiation of HSCs is regulated by growth factors, including transforming growth factor β (TGF-β), which are associated with pathological conditions such as liver injury, cirrhosis, and cancer (11, 12). During liver injury, quiescent HSCs become activated and convert into highly proliferative, myofibroblast-like cells, which produce inflammatory and fibrogenic mediators (13). In a human hepatoma model, the cross talk between tumor hepatocytes and activated HSCs induced an inflammatory response, and the amounts of cytokines and chemokines associated with hepatocyte-HSC cross talk correlated to HCC progression (14).
Although direct induction of liver inflammation by HCV infection through cellular sensors or HCV proteins is well documented, little is known about the mechanisms governing the proinflammatory cytokines and chemokines that are produced during the interactions between HCV-infected hepatocytes and HSCs. Here, we show that HSCs can act as an inflammatory mediator to HCV-infected cells. Infection of hepatocytes with HCV resulted in increased CCAAT/enhancer binding protein (C/EBPβ) production. Conditioned medium (CM) from HSCs induced hepatocyte production of inflammatory cytokines and chemokines, such as IL-6, IL-8, and MIP-1β, which are potential targets of C/EBPβ. Stimulation of these cytokines and chemokines in HCV-infected cells by HSC CM was suppressed by knockdown of mRNA for C/EBPβ. From the chemokines secreted by HSCs, IL-1α was identified as the inducer of MIP-1β. These results suggest HSCs may contribute to virus infection-associated liver inflammation through cross talk with HCV-infected hepatocytes.
MATERIALS AND METHODS
Cells.
LX2 cells (kindly provided by S. Friedman), NP-2-CCR5 cells (kindly provided by T. Hoshino), and Huh7.5 cells (kindly provided by C. Rice) were cultured in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) supplemented with 10% fetal bovine serum (FBS), 100 μg/ml penicillin and streptomycin, and 100 U/ml nonessential amino acids (Invitrogen). To maintain the quality of the cells, stored frozen stocks were thawed every 3 months and used in the experiments.
Plasmids.
DNA fragments encoding each of the HCV nonstructural proteins were generated from a full-length cDNA clone of JFH1 by PCR. The fragments were cloned into pCAG-GS/N-Flag, in which the sequence encoding a Flag tag is inserted at the 5′ terminus of the cloning site of pCAG-GS.
Virus.
Infectious HCV in cell culture (HCVcc) was produced by transfection of Huh7.5 cells with in vitro-transcribed RNA derived from JFH1 (kindly provided by T. Wakita) or TNS2J1 (the chimeric HCV genome containing HCV-1b in the structure region and JFH1 non-structural-protein-coding regions). UV-irradiated JFH1 was prepared by irradiation with a UV lamp of 254-nm wavelength at a distance of 6 cm for 1 min.
HCV infection.
Huh7.5 cells were infected with JFH1 at a multiplicity of infection (MOI) of 5. Under this condition, 80 to 90% of the cells became positive for HCV core protein after 3 days.
Preparation of conditioned medium from Huh7.5 or LX2 cells.
Huh7.5 or LX2 cells (1 × 106) were seeded in 10 ml of medium in a 100-mm dish for 3 days. Supernatants were collected and filtered through 0.45-μm-pore-size filters.
Chemotaxis of NP-2-CCR5 cells.
A 60-fold concentration of Huh7.5 or LX2 CM was generated by tapping in a filter membrane that cut off the 100-kDa-molecular-mass marker protein (100,000-molecular-weight-cutoff filter) and was used for experimental stimulations. Huh7.5 or JFH1-infected Huh7.5 cells (Huh7.5/JFH1 cells) were treated with each concentrated CM. After 24 h of treatment, the medium was changed to serum-free DMEM for 24 h and then used for chemotaxis assays. Chemotaxis of NP-2-CCR5 cells was measured in a 48-well chemotaxis chamber (Neuro Probe). The chamber consisted of a 48-well upper chamber and a 48-well lower chamber separated by a polycarbonate filter (pore size, 8 μm) coated with rat tail collagen. The lower wells were filled with each conditioned medium. The NP-2-CCR5 cells were washed and suspended in serum-free DMEM in the absence or presence of 0.1 nM maraviroc and then divided in the upper wells (5,000 cells per well). After incubation at 37°C for 180 min, the cells that had migrated into the lower well of the 48-well chemotaxis chamber were counted by Diff-Quik staining.
Quantitative RT-PCR.
Total RNA was extracted from cells using RNeasy minikits (Qiagen), and cDNA was prepared with SuperscriptIII (Invitrogen) using oligo(dT) primers. Quantitative real-time PCR (qRT-PCR) was performed with Fast SYBR green master mix (Applied Biosystems), and fluorescent signals were analyzed with the Fast RT-PCR system (Applied Biosystems). The PCR primer pairs are described in Table 1.
Table 1.
Real-time PCR primers
| Primer | Sequence (5′–3′) |
|---|---|
| CXCL1-F | GCAGGGAATTCACCCCAAGAAC |
| CXCL1-R | CTATGGGGGATGCAGGATTGAG |
| CXCL2-F | CCAACTGACCAGAAGGAAGGAG |
| CXCL2-R | ATGGCCTCCAGGTCATCATCAG |
| CXCL5-F | TGAGAGAGCTGCGTTGCGTTTG |
| CXCL5-R | TTCCTTCCCGTTCTTCAGGGAG |
| CXCL6-F | CTGCGTTGCACTTGTTTACGCG |
| CXCL6-R | GGGTCCAGACAAACTTGCTTCC |
| IL-1alpha-F | AGCTATGGCCCACTCCATGAAG |
| IL-1alpha-R | ACATTAGGCGCAATCCAGGTGG |
| IL-6-F | CCCCCAGGAGAAGATTCCAAAG |
| IL-6-R | TTCTGCCAGTGCCTCTTTGCTG |
| IL-7-F | ATTCCGTGCTGCTCGCAAGTTG |
| IL-7-R | AACCTGGCCAGTGCAGTTCAAC |
| IL-8-F | CTGTTAAATCTGGCAACCCTAGTCT |
| IL-8-R | CAAGGCACAGTGGAACAAGGA |
| MIP-1alpha-F | GCTGACTACTTTGAGACGAGC |
| MIP-1alpha-R | CCAGTCCATAGAAGAGGTAGC |
| MIP-1beta-F | CAGCGCTCTCAGCACCAATGG |
| MIP-1beta-R | GATCAGCACAGACTTGCTTGCTTC |
| C/EBP-beta-F | CTCGCAGGTCAAGAGCAAG |
| C/EBP-beta-R | GACAGCTGCTCCACCTTCTT |
| Collagen-F | AACATGACCAAAAACCAAAAGTG |
| Collagen-R | CATTGTTTCCTGTGTCTTCTGG |
| IL-1R-F | CCTGTCTTATGGCGTTGCAGGC |
| IL-1R-R | AGTGCCCTGGGCTGCTATTGAC |
siRNA transfection.
Small interfering RNA (siRNA) was transfected using Lipofectamine RNAiMAX reagent (Invitrogen) according to the manufacturer's protocol. The duplex nucleotides of siRNA specific to the mRNA for C/EBPβ (5′-GAAGAAACGUCUAUGUGUA-3′) and the Mission siRNA universal negative control were purchased from Sigma. Synthetic siRNA specific to mRNA for IL-1 receptor-associated kinase 1 (IRAK1) (5′-CCCGGGCAAUUCAGUUUCUACAUCA-3′) and the Stealth RNA interference (RNAi) negative control duplex were purchased from Invitrogen.
Cytokine antibody array.
LX2 cells (1 × 106) were seeded in 10 ml of medium in a 100-mm dish for 2 days. The supernatant was then changed to 0.2% FBS-DMEM. Two days after incubation, the supernatants were collected and then concentrated by using a 100,000-molecular-weight-cutoff filter. The trapped and flowthrough fractions were dialyzed with phosphate-buffered saline (PBS) for 18 h. The amount of protein in each fraction was determined using a bicinchoninate protein assay kit (Nacalai Tesque). Three milligrams of each fraction was subjected to the cytokine antibody array.
The expression levels of 507 human proteins in the trap and flowthrough fractions from the LX2 cells were determined using biotin label-based human antibody array I (Raybiotech) according to the manufacturer's protocol.
RESULTS
LX2 cells induce MIP-1β expression in JFH1-infected Huh7.5 cells.
Our preliminary results indicated that coculturing human hepatic stellate cells (HSCs) with HCV-infected cells stimulated the expression of MIP-1β, which was found to be one of most upregulated chemokines. Here, we focused on its role as a marker of inflammation.
To investigate whether human HSCs play a role in the proinflammatory response of HCV-infected cells, JFH1-infected Huh7.5 cells were cocultured with LX2 cells, which are an HSC line generated by spontaneous immortalization in low-serum conditions (15). The expression of MIP-1β mRNA was then determined by qRT-PCR. Compared to the level in uninfected Huh7.5 cells, HCV infection induced a low level of MIP-1β expression (Fig. 1A, Huh7.5/JFH1). Moreover, MIP-1β expression was significantly enhanced in JFH1-infected Huh7.5 cells after they were cocultured with LX2 cells (Fig. 1A, Huh7.5/JFH1+LX2). Importantly, MIP-1β expression was undetectable or very low in LX2 cells alone or in uninfected Huh7.5 cells cocultured with LX2 (Fig. 1A, LX2 and Huh7.5+LX2). Interestingly, increased MIP-1β expression in JFH1-infected Huh7.5 cells was specifically induced by cocultivation with LX2 cells. Cocultivation with other cell lines, such as 293T, HeLa, and THP-1, had no effect on MIP-1β expression in JFH1-infected Huh7.5 cells (Fig. 1B). These results suggest that Huh7.5 cells produce MIP-1β in response to HCV infection and that LX2 cells increase MIP-1β expression in HCV-infected Huh7.5 cells.
Fig 1.

Conditioned medium from LX2 induces MIP-1β expression in JFH1-infected Huh7.5 cells. (A) Huh7.5 cells (1 × 105 cells) and JFH1-infected Huh7.5 (Huh7.5/JFH1) cells (1 × 105 cells) were cultured alone or in the presence of LX2 cells (1 × 105 cells) for 24 h. The level of MIP-1β was measured by qRT-PCR. Quantitative analysis of the PCR data was performed using the 2−ΔΔCT method, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) CT values were used for normalization. The fold changes are relative to values for Huh7.5 cells. (B) Huh7.5 cells and JFH1-infected Huh7.5 cells (Huh7.5/JFH1) were cultured alone or in the presence of LX2, 293T, HeLa, or THP-1 cells for 24 h. The expression level of MIP-1β was measured by qRT-PCR as described for panel A. (C) Huh7.5 cells and JFH1-infected Huh7.5 cells were treated with conditioned medium from Huh7.5 (Huh7.5 CM) or LX2 (LX2 CM) cells for 24 h. The expression level of MIP-1β was measured by qRT-PCR as described for panel A. (D) Huh7.5, JFH1-infected Huh7.5 (Huh7.5/JFH1), Huh7/NNC, Huh7.5/TNS2J1, and Huh7.5/SGR-JFH1 cells were treated with CM from Huh7.5 or LX2 cells for 24 h. The expression level of MIP-1β was measured by qRT-PCR as described for panel A. (E) Huh7.5 and JFH1-infected Huh7.5 (Huh7.5/JFH1) cells were treated with Huh7.5 CM, LX2 CM, or LI90 CM for 24 h. The expression level of MIP-1β was measured by qRT-PCR as described for panel A. (F) LX2 cells were treated with 2.5 ng/ml TGF-β1 for 24 h, and then the level of collagen mRNA in these cells was determined by qRT-PCR (left). Huh7.5 or JFH1-infected Huh7.5 cells were treated with LX2 CM or TGF-β1-stimulated LX2 CM for 24 h. The expression level of MIP-1β was measured by qRT-PCR as described for panel A (right). The results are representative of three independent experiments, and the error bars represent the standard deviation of the means.
We next determined whether MIP-1β induction by LX2 cells in HCV-infected cells was mediated by a secreted soluble factor(s). Huh7.5 and JFH1-infected Huh7.5 cells were treated with conditioned medium (CM) from Huh7.5 or LX2 cells (Fig. 1C). As expected, Huh7.5 cells had no response to Huh7.5 CM or LX2 CM. However, we observed that LX2 CM, but not Huh7.5 CM, stimulated MIP-1β expression in JFH1-infected Huh7.5 cells. These results indicated that LX2 cells secrete a factor that stimulates MIP-1β expression.
To address whether MIP-1β stimulation after culturing with LX2 CM is dependent on HCV genotype or on the maintenance of the HCV replicon in cells, we used Huh7.5 cells that carry different types of the HCV genome. Huh7/NNC cells are Huh7 cells that contain the noninfectious full HCV genotype 1b, Huh7.5/SGR-JFH1 cells contain the subgenome replicon of JFH1, and Huh7.5/TNS2J1 cells have the infectious chimeric HCV genome, which consists of an HCV-1b-derived sequence in the structural-protein-coding region and a JFH-derived sequence in nonstructural-protein-coding region (Fig. 1D). In Huh7.5/TNS2J1 cells, MIP-1β expression was significantly induced by LX2 CM (47-fold), though there was a lower level of expression than what was observed in JFH1-infected cells (272-fold). By contrast, NNC and SGR-JFH1 had no effect on MIP-1β expression. These results demonstrate that LX2 CM-induced stimulation of MIP-1β expression may require a productive HCV infection (see also Fig. 6).
Fig 6.

HCV entry is required for induction of MIP-1β expression by LX2 CM. (A) Huh7.5, JFH1-infected Huh7.5 (Huh7.5/JFH1), and JFH1-CL3B-infected Huh7.5 (Huh7.5/JFH1-CL3B) cells were treated with Huh7.5 CM or LX2 CM for 24 h. The level of MIP-1β expression was analyzed by qRT-PCR as described for Fig. 1A. (B) Huh7.5 cells were treated with Huh7.5 CM or LX2 CM. After 24 h of treatment, cells were infected with JFH1 for 2, 4, or 6 h. The level of MIP-1β expression was analyzed by qRT-PCR as described for Fig. 1A. (C) Huh7.5 cells were treated with Huh7.5 CM or LX2 CM in the presence of an isotype control IgG or anti-CD81 antibody (1 μg/ml). After 24 h of treatment, the cells were infected with JFH1 in the presence of an isotype control IgG or anti-CD81 antibody (1 μg/ml) for 2, 4, or 6 h. The level of MIP-1β expression was analyzed by qRT-PCR as described for Fig. 1A. (D) HCV JFH1 was UV irradiated and used to infect Huh7.5 cells. At 24 h after infection, cells were treated with Huh7.5 CM or LX2 CM. The level of MIP-1β expression was analyzed by qRT-PCR as described for Fig. 1A. (E) Huh7.5 cells expressing the indicated HCV proteins were treated with Huh7.5 CM or LX2 CM for 24 h. The levels of MIP-1β expression were analyzed by qRT-PCR as described for Fig. 1A. The results are representative of three independent experiments, and the error bars represent the standard deviations of the means.
Because LX2 cells are a human hepatic stellate cell line that was established by immortalization, we confirmed our findings by using another human hepatic stellate cell line, LI90, which was derived from a human mesenchymal liver tumor (16). When uninfected or JFH1-infected Huh7.5 cells were treated with LI90 CM, MIP-1β expression was increased only in the JFH1-infected Huh7.5 cells. These results are similar to those found after addition of LX2 CM to JFH1-infected Huh7.5 cells (Fig. 1E).
Activated hepatic stellate cells play a critical role in inflammation, yet the functional impact they have on hepatocytes has not yet been determined. Therefore, we evaluated whether the activation of LX2 cells affects the expression of MIP-1β in JFH1-infected Huh7.5 cells. LX2 cells were treated with TGF-β1, and the mRNA expression of the collagen gene, a marker of HSC activation, was measured (Fig. 1F, left). LX2 CM from activated cells significantly enhanced MIP-1β expression in JFH1-infected Huh7.5 cells but not in uninfected Huh7.5 cells, compared to the increase with nonactivated LX2 CM (Fig. 1F, right). In parallel experiments, TGF-β1-treated Huh7.5 CM did not affect MIP-1β expression in Huh7.5 or JFH1-infected Huh7.5 cells.
The supernatant from JFH1-infected Huh7.5 cells cultured with LX2 CM induces migration of NP-2-CCR5 cells.
MIP-1β is a physiological ligand for the CCR5 receptor. To test whether MIP-1β produced by HCV-infected hepatocytes that have been cultured with LX2 CM has this activity, we performed a chemotactic assay using NP-2-CCR5 cells, a human glioma-derived cell line expressing CCR5 on its cell surface (Fig. 2). The treatment of NP-2-CCR5 cells with supernatant from LX2 CM-stimulated JFH1-infected Huh7.5 cells increased their migration by 2-fold compared to treatment with supernatant from uninfected Huh7.5 cells treated with LX2 CM. This increase in NP-2-CCR5 cell migration was blocked with maraviroc (a CCR5 antagonist) treatment. These results indicated that LX2 CM induces secretion of a physiologically functional MIP-1β by JFH1-infected Huh7.5 cells.
Fig 2.
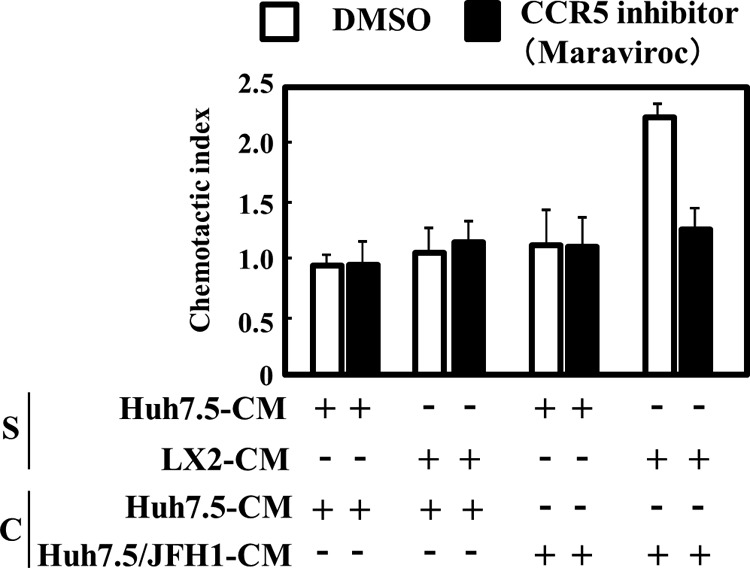
Conditioned medium from JFH1-infected Huh7.5 cells treated with LX2-conditioned medium induces chemotaxis of NP-2-CCR5 cells. Huh7.5 CM or LX2 CM was concentrated 60-fold using a 100,000-molecular-weight-cutoff membrane filter. Huh7.5 and Huh7.5/JFH1 cells were treated with each concentrated medium (S). After 24 h of treatment, the medium was changed to serum free DMEM for 24 h. The chemotactic activity of each conditioned medium (C) was determined by using 5 nM maraviroc (CCR5 inhibitor) and NP-2-CCR5 cells, as described in Materials and Methods. The results are representative of three independent experiments, and the error bars represent the standard deviations of the means. DMSO, dimethyl sulfoxide.
Identification of the factor in LX2 CM responsible for MIP-1β stimulation.
We first fractionated culture medium of Huh7.5 or LX2 cells using a membrane filter which cut off the 100-kDa-molecular-mass marker protein and then collected the trapped and flowthrough fractions. As expected, MIP-1β expression in uninfected Huh7.5 or JFH1-infected Huh7.5 cells did not increase after treatment with the trap or the flowthrough fraction of the Huh7.5 CM (Fig. 3A). By contrast, the trap fraction of LX2 CM enhanced MIP-1β expression in JFH1-infected Huh7.5 cells, suggesting that the stimulator in the LX2 CM was enriched in the 100-kDa-molecular-mass-cutoff filter. To further analyze the trap fraction of LX2 CM, we created a cytokine antibody array (Fig. 3B). By analyzing 507 cytokines and chemokines in the array, we found four candidates (TSG-14, monocyte chemoattractant protein 2 [MCP-2], MCP-3, and IL-1α), which were more concentrated by the 100-kDa-molecular-mass-cutoff filter than by the flowthrough fraction of LX2 CM (Fig. 3B, compare 100K-Flow through to 100K-Trap). We tested the effects of these candidates on stimulation of MIP-1β expression (Fig. 4A). Although recombinant TSG-14, MCP-2, or MCP-3 did not stimulate MIP-1β expression in Huh7.5 cells or JFH1-infected cells, recombinant IL-1α induced MIP-1β expression in only JFH1-infected Huh7.5 cells. This effect was dose dependent (Fig. 4B). To evaluate whether IL-1α is required for MIP-1β stimulation in JFH1-infected Huh7.5 cells by LX2 CM, we used a neutralizing antibody against IL-1α and the IL-1 receptor antagonist. LX2 CM stimulated MIP-1β expression in JFH1-infected Huh7.5 cells, whereas anti-IL-1α and the IL-1 receptor antagonist (IL-1RA) blocked MIP-1β stimulation (Fig. 4C). Additionally, the neutralizing antibody against IL-1β had no effect (data not shown). It is not clear why IL-1α of about 30 kDa was concentrated into the trap fraction. However, it is likely that IL-1α is formed at a large mass with other proteins in the culture medium to be contained. Moreover, knockdown of IRAK1, which is essential for the downstream signal of IL-1R, impaired the response to LX2 CM (Fig. 4D). These results suggest that IL-1α contributes to stimulation of MIP-1β expression by LX2 CM and that JFH1-infected Huh7.5 cells are highly sensitive to IL-1α. Furthermore, considering the molecular weight of IL-1α, it is possible that an unknown amount of IL-1α, undetectable by the cytokine antibody array, passed through the filter, which caused activation of MIP-1b by the 100,000-molecular-weight-cutoff flowthrough fraction. Alternatively, a factor(s) other than IL-1α in the 100,000-molecular-weight-cutoff flowthrough fraction might have been responsible for the activation.
Fig 3.

Identification of a factor(s) in the LX2 conditioned medium that is responsible for induction of MIP-1β expression. (A) Huh7.5 CM or LX2 CM was concentrated using a 100,000-molecular-weight-cutoff membrane filter. Huh7.5 and JFH1-infected Huh7.5 (Huh7.5/JFH1) cells were treated with each unconcentrated conditioned medium, with the flowthrough fraction (100K-FT), or with the trap fraction (100K-T). The MIP-1β expression level was analyzed by qRT-PCR as described for Fig. 1A. (B) LX2 CM was concentrated using a 100,000-molecular-weight-cutoff membrane filter. A cytokine antibody array (RayBiotech) was used for the simultaneous detection of 507 inflammatory factors. The antibody-coated membrane was incubated with the trap fraction (100K-Trap) or with the flowthrough fraction (100K-Flow through). Representative spots (TSG-14, MCP-1, MCP-3, and IL-1α) are shown.
Fig 4.

IL-1α induces MIP-1β expression in JFH1-infected Huh7.5 cells. (A) Huh7.5 and JFH1-infected Huh7.5 (Huh7.5/JFH1) cells were treated with recombinant MCP-2 (10 ng/ml), recombinant MCP-3 (rMCB-3; 10 ng/ml), recombinant TSG14 (10 ng/ml), or recombinant IL-1α (10 pg/ml) for 24 h. The level of MIP-1β expression was analyzed by qRT-PCR as described for Fig. 1A. (B) Huh7.5 and JFH1-infected Huh7.5 (Huh7.5/JFH1) cells were treated with various amounts of recombinant IL-1α (0, 0.1, 1.0, 10, or 100 pg/ml) for 24 h. The level of MIP-1β expression was analyzed by qRT-PCR as described for Fig. 1A. (C) JFH1-infected Huh7.5 cells were treated with Huh7.5 CM or LX2 CM along with an isotype control (0.2 μg/ml), anti-IL-1α (0.2 μg/ml), or IL-1RA (100 μg/ml) for 24 h. The level of MIP-1β expression was analyzed by qRT-PCR as described for Fig. 1A. (D) Huh7.5 and Huh7.5/JFH1 cells were transfected with 50 nM control siRNA (siControl) or 50 nM IRAK1 siRNA (siIRAK1). At 48 h after transfection, cells were treated with Huh7.5 CM or LX2 CM. The level of MIP-1β expression was analyzed by qRT-PCR as described for Fig. 1A. The results are representative of three independent experiments, and the error bars represent the standard deviations of the means.
The transcription factor C/EBPβ mediates LX2 CM-stimulated MIP-1β production.
IL-1 is one of the most important proinflammatory cytokines and binds to the cell surface IL-1 type I receptor to activate downstream signaling pathways such as IKK–NF-κB, extracellular signal-regulated kinase (ERK), Jun N-terminal protein kinase (JNK), p38, and C/EBPβ. Moreover, the MIP-1β promoter contains a C/EBPβ motif located between bp −222 and −100, and the C/EBPβ promoter is required for a functional response to IL-1β in human chondrocytes (17). To evaluate whether LX2 CM-stimulated MIP-1β expression involves C/EBPβ stimulation, the level of C/EBPβ mRNA was measured in uninfected and HCV-infected cells. C/EBPβ expression was higher in Huh7.5/JFH1 and Huh7.5/TNS2J1 cells than in Huh7.5, Huh7/NNC, and Huh7.5/SGR-JFH1 cells (Fig. 5A). This result correlates with MIP-1β stimulation shown in Fig. 1D. Additionally, LX2 CM induced low levels of C/EBPβ expression in Huh7.5/JFH1 and Huh7.5/TNS2J1 cells; induction was likely caused by IL-1α (Fig. 5A). To further confirm that the enhancement of MIP-1β expression is mediated by C/EBPβ, we performed experiments where uninfected or JFH1-infected Huh7.5 cells were transduced with either a control or a C/EBPβ-specific siRNA and then treated with Huh7.5 CM or LX2 CM. Quantitative RT-PCR analysis demonstrated that the siRNAs targeting C/EBPβ significantly suppressed endogenous C/EBPβ expression (Fig. 5B). Depletion of C/EBPβ significantly reduced the MIP-1β expression stimulated by LX2 CM in JFH1-infected Huh7.5 cells. Residual stimulation of MIP-1β seems to be attributable to imperfect knockdown of C/EBPβ (70 to 80%). However, currently we cannot rule out the possibility of the involvement of other transcription factor(s) in the activation of MIP-1β expression. Furthermore, cytokines (IL-6, IL-8, CXCL2, CXCL1, MIP-1β, and MIP-1α) that were enhanced by C/EBPβ expression were upregulated by LX2 CM only in JFH1-infected Huh7.5 cells (Fig. 5D). There was no induction of C/EBPβ-independent cytokines (IL-7, CXCL6, CXCL5, IL-1α, and IL-1R) after the addition of LX2 CM. None of these cytokines were induced by LX2 CM in uninfected Huh7.5 cells. These data indicate that HCV stimulates C/EBPβ expression, which confers upon HCV-infected Huh7.5 cells the ability to produce proinflammatory cytokines in response to LX2 CM.
Fig 5.

Conditioned medium from LX2 induces C/EBPβ-mediated proinflammatory gene expression in JFH1-infected Huh7.5 cells. (A) The indicated cells were treated with Huh7.5 CM or LX2 CM for 24 h. The level of C/EBPβ expression was analyzed by qRT-PCR as described for Fig. 1A. (B and C) Huh7.5 cells and JFH1-infected Huh7.5 cells were transfected with 50 nM control siRNA or siC/EBPβ. After 24 h of transfection, the cells were treated with Huh7.5 CM or LX2 CM for 24 h. The levels of C/EBPβ (B) and MIP-1β (C) expression were analyzed by qRT-PCR as described for Fig. 1A. (D) Huh7.5 cells and JFH1-infected Huh7.5 cells were treated with Huh7.5 CM or LX2 CM for 24 h. The levels of the indicated cytokines and chemokines were measured by qRT-PCR, as described for Fig. 1A. The results are representative of three independent experiments.
Early steps of the HCV life cycle trigger MIP-1β stimulation by LX2 CM.
We observed that Huh7.5/TNS2J1 and Huh7.5/JFH1 cells, but not Huh7/NNC or Huh7.5/SGR-JFH1 cells, could respond to LX2 CM (Fig. 1D), suggesting that the production of infectious HCV is required for MIP-1β induction. However, it remains to be determined whether infectious HCV particles actually induce MIP-1β expression in the presence of LX2 CM. To address this question, we used JFH1-CL3B, which is a virus that is defective in the production of virus particles because of mutations in domain III of NS5A; however, the self-replication ability of its genome is normal (18). As shown in Fig. 6A, JFH1-CL3B genome-bearing Huh7.5 cells responded to neither Huh7.5 CM nor LX2 CM. These data suggest that productive infection of HCV is an essential event for MIP-1β induction. To characterize the mechanism that mediates LX2 CM-stimulated MIP-1β induction in HCV-infected cells, we studied the temporal kinetics of MIP-1β expression. In JFH1-infected Huh7.5 cells, MIP-1β expression did not increase in the first 2 h after infection but it started to increase after 4 h in the presence of LX2 CM (Fig. 6B). Furthermore, MIP-1β induction in Huh7.5/JFH1 cells that were cultured in the presence of LX2 CM was suppressed by pretreatment of the cells with a neutralizing antibody against anti-CD81. To further address whether MIP-1β induction by LX2 CM in Huh7.5/JFH1 cells is due to the binding of HCV E1/E2 to the receptor, JFH1-infected cells were UV irradiated and used to infect Huh7.5 cells. Huh7.5 cells infected with UV-irradiated JFH1 did not show MIP-1β induction stimulated by LX2 CM (Fig. 6D). In addition, ectopic expression of HCV proteins in Huh7.5 cells did not respond to the LX2 CM (Fig. 6E). These results suggest that MIP-1β expression is required for HCV infection at postentry.
DISCUSSION
HCV-specific CTLs are concentrated within the liver during chronic infection, and they may control viral replication and contribute to progressive liver disease (19). Recruitment of T cells to the liver is required for the expression of CCR5 on activated T cells (20). Intrahepatic expression of the ligands for CCR5 (RANTES, MIP-1β, and MIP-1α) is elevated in HCV patients, and these chemokines have been linked to a high degree of liver inflammation (21). However, the mechanism mediating the stimulation of these cytokines is unclear. In this study, we demonstrated that cross talk between HCV-infected hepatocytes and human stellate cells (HSCs) induced inflammatory cytokines and chemokines. Importantly, uninfected hepatocytes are tolerant of HSC stimulation. Of note, HCV has no direct effect on LX2 cells with regard to MIP-1β stimulation because MIP-1β expression in Huh7.5/JFH1 cells was induced by treatment with CM from JFH1-treated LX2 cells as well as LX2 CM.
We identified IL-1α as an inducer of cytokines and chemokines in HCV-infected hepatocytes. These cells are highly sensitive to IL-1α (Fig. 4). Previous reports have indicated that the HCV core and NS3 proteins, which induced IL-1 receptor-associated kinase (IRAK) activity, triggered inflammatory cell activation through TLR-2 (22). IRAK activation by HCV infection may contribute to the induction of cytokines and chemokines by HSCs. We addressed this point and found that knockdown of IRAK1 in HCV-infected hepatocytes indicated that the response to HSC CM was impaired (Fig. 4D). By contrast, another group reported that HCV NS5A inhibits recruitment of IRAK to MyD88, resulting in suppression of the TLR signaling pathways in macrophage cell lines that stably expressed HCV NS5A (23). Currently, there is no evidence to suggest that HCV infects macrophages or that macrophages uptake functional NS5A to modulate TLR signaling. These conflicting results may be explained by the differences in cell types.
Our results suggest that IL-1α produced from hepatic stellate cells contributes to the inflammation by HCV infection in vivo. Another report shows augmented expression of IL-1α, tumor necrosis factor alpha (TNF-α), and IL-2 in HCV-infected patients compared to controls (24). Further, IL-1α plays roles in the liver to induce the acute-phase response of inflammation and autoactivation of Kupffer cells (25). Brain tissue from HCV patients, which is positive for HCV RNA and virus proteins, showed a significantly higher level of IL-1α than HCV-negative controls. Higher levels of proinflammatory cytokines such as IL-1α, IL-1β, TNF-α, IL-12, and IL-18 correlate with neurocognitive dysfunction among HCV patients. These results suggest that IL-1α is an important factor of HCV-related inflammation (26).
The relative importance of C/EBPβ in the stimulation of IL-1-inducible genes, such as the MIP-1α, MIP-1β, and IL-6 genes, is still not fully understood. Our results indicate that HCV-infected cells, which exhibited a 3- to 5-fold enhancement of C/EBPβ mRNA expression compared to uninfected cells (Fig. 5A), responded to IL-1α for the induction of MIP-1β mRNA synthesis. MIP-1β expression required the presence of C/EBPβ because knockdown of C/EBPβ significantly decreased LX2 CM-induced MIP-1β expression (Fig. 5C). To further address the relationship between C/EBPβ and MIP-1β, we analyzed the levels of C/EBPβ and MIP-1β mRNAs in uninfected Huh7.5 and Huh7.5/JFH1 cells in the presence of IL-1α (1 to 100 pg/ml) (data not shown). Compared with Huh7.5 cells, C/EBPβ expression was modestly induced in Huh7.5/JFH1 cells (about 4-fold) in the presence of 100 pg/ml IL-1α. By contrast, MIP-1β expression in Huh7.5/JFH1 cells at this dose of IL-1α was strongly induced (300-fold). These results indicate that IL-1α-induced MIP-1β mRNA induction in Huh7.5/JFH1 cells cannot be fully explained by the slight increase of C/EBPβ mRNA expression. It is known that C/EBPβ is modified posttranslationally (27, 28). C/EBPβ is phosphorylated by the Ras/mitogen-activated protein kinase (Ras/MAPK) pathway (29, 30), and a Ca2+/calmodulin-dependent protein kinase (31) or ERK1/ERK2 stimulates the transactivation potential of C/EBPβ (32). C/EBPβ in HCV-infected cells may also be activated by a posttranslational modification in response to IL-1 treatment. It is possible that activation of the MAPK (33) and ERK pathways (34) by HCV infection induces phosphorylation of C/EBPβ, leading to a highly sensitive IL-1 response. Further studies will be required to clarify the details of how HCV modulates increased MIP1-β production through both the C/EBPβ and IL-1 receptor signaling pathways.
This study revealed that induction of inflammatory cytokines and chemokines by cross talk between HSCs and HCV-infected hepatocytes primarily involves the activation of C/EBPβ-dependent gene transcription, such as transcription of the IL-6, IL-8, CXCL-1, and MIP-1β genes (Fig. 5). A recent study demonstrated that the HCV NS5A protein induces C/EBPβ expression (35). However, treatment of Huh7.5 cells that expressed NS5A with HCS CM did not induce MIP-1β expression significantly (Fig. 6E). We also observed only a marginal increase in MIP-1β mRNA level by solitary expression of each HCV protein in Huh7.5 cells by addition of LX2 CM (Fig. 6E).
Moreover, previous reports have suggested that ER stress, which is induced by HCV infection, leads to the generation of mature sterol regulatory element-binding protein 1 (SREBP-1) (36, 37) and that the mature SREBP-1c can bind to the C/EBPβ promoter (36, 38), leading to induction of C/EBPβ. Additionally, SREBP-1c promoter activity was upregulated in core-transgenic mice in a PA28γ-dependent manner (39). Indeed, we observed that endoplasmic reticulum (ER) stress-related genes and SREBP-1c expression were enhanced by HCV infection. However, induction of SREBP-1c is not involved in this process because HCV-infected cells that had SREBP-1c knocked down could still respond to HSC CM (data not shown).
Chronic inflammation is triggered by many events that also can increase the risk of developing cancer. For example, Helicobacter pylori is associated with gastric cancer, and inflammatory bowel disease is associated with colon cancer. In particular, IL-6 is one of the clinical targets for cancer-related inflammation (2). The level of IL-6 expression correlates with a rapid progression from hepatitis to hepatocellular carcinomas (40). The Ras and Jak/Stat pathway, which is downstream of the IL-6 receptor, is activated in hepatocellular carcinomas that have a poor prognosis (41). Additionally, IL-8 is a proinflammatory cytokine that specifically attracts and activates human neutrophils. In HCV patients, pegylated IFN-α-2a (PEG-IFN-α-2a) and ribavirin therapy decreased the neutrophil count in virologic responders compared to nonresponders (42). Enhancement of expression of these cytokines as well as MIP-1β, described here, suggests that the cross talk between HCV-infected hepatocytes and HSCs polarizes the cytokine profile toward a Th2-type immune response.
In conclusion, we have demonstrated that the cross talk between HSCs and HCV-infected hepatocytes results in the induction of inflammatory cytokines and chemokines, which promote the migration of CCR5-expressing cells in in vitro experiments. These results suggest that the induction of inflammatory cytokines and chemokines by HCV infection may recruit inflammatory cells such as cytotoxic T lymphocytes (CTL) and neutrophils to the liver, which induces liver cell injury leading to chronic hepatitis.
ACKNOWLEDGMENTS
We are grateful to H. Hoshino at Gunma University, C. Rice at The Rockefeller University, S. Friedman at Mount Sinai School of Medicine, and T. Wakita at the National Institute of Infectious Diseases for their contributions of research materials and to H. Yamamoto and R. Shiina for their technical assistance.
This work was partly supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), by the MEXT-Supported Program for the Strategic Research Foundation at Private Universities, and by grants-in-aid for research on hepatitis from the Ministry of Health, Labor, and Welfare of Japan.
Footnotes
Published ahead of print 15 May 2013
REFERENCES
- 1. Bartosch B, Thimme R, Blum HE, Zoulim F. 2009. Hepatitis C virus-induced hepatocarcinogenesis. J. Hepatol. 51:810–820 [DOI] [PubMed] [Google Scholar]
- 2. Bowen DG, Walker CM. 2005. Adaptive immune responses in acute and chronic hepatitis C virus infection. Nature 436:946–952 [DOI] [PubMed] [Google Scholar]
- 3. Liaw YF, Lee CS, Tsai SL, Liaw BW, Chen TC, Sheen IS, Chu CM. 1995. T-cell-mediated autologous hepatocytotoxicity in patients with chronic hepatitis C virus infection. Hepatology 22:1368–1373 [PubMed] [Google Scholar]
- 4. Shields PL, Morland CM, Salmon M, Qin S, Hubscher SG, Adams DH. 1999. Chemokine and chemokine receptor interactions provide a mechanism for selective T cell recruitment to specific liver compartments within hepatitis C-infected liver. J. Immunol. 163:6236–6243 [PubMed] [Google Scholar]
- 5. Harvey CE, Post JJ, Palladinetti P, Freeman AJ, Ffrench RA, Kumar RK, Marinos G, Lloyd AR. 2003. Expression of the chemokine IP-10 (CXCL10) by hepatocytes in chronic hepatitis C virus infection correlates with histological severity and lobular inflammation. J. Leukoc. Biol. 74:360–369 [DOI] [PubMed] [Google Scholar]
- 6. Napoli J, Bishop GA, McGuinness PH, Painter DM, McCaughan GW. 1996. Progressive liver injury in chronic hepatitis C infection correlates with increased intrahepatic expression of Th1-associated cytokines. Hepatology 24:759–765 [DOI] [PubMed] [Google Scholar]
- 7. Basu A, Meyer K, Lai KK, Saito K, Di Bisceglie AM, Grosso LE, Ray RB, Ray R. 2006. Microarray analyses and molecular profiling of Stat3 signaling pathway induced by hepatitis C virus core protein in human hepatocytes. Virology 349:347–358 [DOI] [PubMed] [Google Scholar]
- 8. Li K, Li NL, Wei D, Pfeffer SR, Fan M, Pfeffer LM. 2012. Activation of chemokine and inflammatory cytokine response in hepatitis C virus-infected hepatocytes depends on Toll-like receptor 3 sensing of hepatitis C virus double-stranded RNA intermediates. Hepatology 55:666–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wagoner J, Austin M, Green J, Imaizumi T, Casola A, Brasier A, Khabar KS, Wakita T, Gale M, Jr, Polyak SJ. 2007. Regulation of CXCL-8 (interleukin-8) induction by double-stranded RNA signaling pathways during hepatitis C virus infection. J. Virol. 81:309–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yu GY, He G, Li CY, Tang M, Grivennikov S, Tsai WT, Wu MS, Hsu CW, Tsai Y, Wang LH, Karin M. 2012. Hepatic expression of HCV RNA-dependent RNA polymerase triggers innate immune signaling and cytokine production. Mol. Cell 48:313–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Friedman SL. 2008. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 88:125–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Friedman SL. 2008. Mechanisms of hepatic fibrogenesis. Gastroenterology 134:1655–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hernandez-Gea V, Friedman SL. 2011. Pathogenesis of liver fibrosis. Annu. Rev. Pathol. 6:425–456 [DOI] [PubMed] [Google Scholar]
- 14. Coulouarn C, Corlu A, Glaise D, Guénon I, Thorgeirsson SS, Clément B. 2012. Hepatocyte-stellate cell cross-talk in the liver engenders a permissive inflammatory microenvironment that drives progression in hepatocellular carcinoma. Cancer Res. 72:2533–2542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xu L, Hui AY, Albanis E, Arthur MJ, O'Byrne SM, Blaner WS, Mukherjee P, Friedman SL, Eng FJ. 2005. Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut 54:142–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Murakami K, Abe T, Miyazawa M, Yamaguchi M, Masuda T, Matsuura T, Nagamori S, Takeuchi K, Abe K, Kyogoku M. 1995. Establishment of a new human cell line, LI90, exhibiting characteristics of hepatic Ito (fat-storing) cells. Lab. Invest. 72:731–739 [PubMed] [Google Scholar]
- 17. Zhang Z, Bryan JL, DeLassus E, Chang LW, Liao W, Sandell LJ. 2010. CCAAT/enhancer-binding protein β and NF-κB mediate high level expression of chemokine genes CCL3 and CCL4 by human chondrocytes in response to IL-1β. J. Biol. Chem. 285:33092–33103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Masaki T, Suzuki R, Murakami K, Aizaki H, Ishii K, Murayama A, Date T, Matsuura Y, Miyamura T, Wakita T, Suzuki T. 2008. Interaction of hepatitis C virus nonstructural protein 5A with core protein is critical for the production of infectious virus particles. J. Virol. 82:7964–7976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Freeman AJ, Pan Y, Harvey CE, Post JJ, Law MG, White PA, Rawlinson WD, Lloyd AR, Marinos G, Ffrench RA. 2003. The presence of an intrahepatic cytotoxic T lymphocyte response is associated with low viral load in patients with chronic hepatitis C virus infection. J. Hepatol. 38:349–356 [DOI] [PubMed] [Google Scholar]
- 20. Zeremski M, Petrovic LM, Talal AH. 2007. The role of chemokines as inflammatory mediators in chronic hepatitis C virus infection. J. Viral Hepat. 14:675–687 [DOI] [PubMed] [Google Scholar]
- 21. Wald O, Weiss ID, Galun E, Peled A. 2007. Chemokines in hepatitis C virus infection: pathogenesis, prognosis and therapeutics. Cytokine 39:50–62 [DOI] [PubMed] [Google Scholar]
- 22. Dolganiuc A, Oak S, Kodys K, Golenbock DT, Finberg RW, Kurt-Jones E, Szabo G. 2004. Hepatitis C core and nonstructural 3 proteins trigger toll-like receptor 2-mediated pathways and inflammatory activation. Gastroenterology 127:1513–1524 [DOI] [PubMed] [Google Scholar]
- 23. Abe T, Kaname Y, Hamamoto I, Tsuda Y, Wen X, Taguwa S, Moriishi K, Takeuchi O, Kawai T, Kanto T, Hayashi N, Akira S, Matsuura Y. 2007. Hepatitis C virus nonstructural protein 5A modulates the toll-like receptor-MyD88-dependent signaling pathway in macrophage cell lines. J. Virol. 81:8953–8966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kasprzak A, Zabel M, Biczysko W, Wysocki J, Adamek A, Spachacz R, Surdyk-Zasada J. 2004. Expression of cytokines (TNF-alpha, IL-1alpha, and IL-2) in chronic hepatitis C: comparative hybridocytochemical and immunocytochemical study in children and adult patients. J. Histochem. Cytochem. 52:29–38 [DOI] [PubMed] [Google Scholar]
- 25. Winwood PJ, Arthur MJ. 1993. Kupffer cells: their activation and role in animal models of liver injury and human liver disease. Semin. Liver Dis. 13:50–59 [DOI] [PubMed] [Google Scholar]
- 26. Wilkinson J, Radkowski M, Eschbacher JM, Laskus T. 2010. Activation of brain macrophages/microglia cells in hepatitis C infection. Gut 59:1394–1400 [DOI] [PubMed] [Google Scholar]
- 27. Harrison JR, Kelly PL, Pilbeam CC. 2000. Involvement of CCAAT enhancer binding protein transcription factors in the regulation of prostaglandin G/H synthase 2 expression by interleukin-1 in osteoblastic MC3T3-E1 cells. J. Bone Miner. Res. 15:1138–1146 [DOI] [PubMed] [Google Scholar]
- 28. Akira S, Isshiki H, Sugita T, Tanabe O, Kinoshita S, Nishio Y, Nakajima T, Hirano T, Kishimoto T. 1990. A nuclear factor for IL-6 expression (NF-IL6) is a member of a C/EBP family. EMBO. J. 9:1897–1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nakajima T, Kinoshita S, Sasagawa T, Sasaki K, Naruto M, Kishimoto T, Akira S. 1993. Phosphorylation at threonine-235 by a ras-dependent mitogen-activated protein kinase cascade is essential for transcription factor NF-IL6. Proc. Natl. Acad. Sci. U. S. A. 90:2207–2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhu S, Yoon K, Sterneck E, Johnson PF, Smart RC. 2002. CCAAT/enhancer binding protein-beta is a mediator of keratinocyte survival and skin tumorigenesis involving oncogenic Ras signaling. Proc. Natl. Acad. Sci. U. S. A. 99:207–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wegner M, Cao Z, Rosenfeld MG. 1992. Calcium-regulated phosphorylation within the leucine zipper of C/EBP beta. Science 256:370–373 [DOI] [PubMed] [Google Scholar]
- 32. Giltiay NV, Karakashian AA, Alimov AP, Ligthle S, Nikolova-Karakashian MN. 2005. Ceramide- and ERK-dependent pathway for the activation of CCAAT/enhancer binding protein by interleukin-1beta in hepatocytes. J. Lipid Res. 46:2497–2505 [DOI] [PubMed] [Google Scholar]
- 33. George A, Panda S, Kudmulwar D, Chhatbar SP, Nayak SC, Krishnan HH. 2012. Hepatitis C virus NS5A binds to the mRNA cap-binding eukaryotic translation initiation 4F (eIF4F) complex and up-regulates host translation initiation machinery through eIF4E-binding protein 1 inactivation. J. Biol. Chem. 287:5042–5058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhao LJ, Wang L, Ren H, Cao J, Li L, Ke JS, Qi ZT. 2005. Hepatitis C virus E2 protein promotes human hepatoma cell proliferation through the MAPK/ERK signaling pathway via cellular receptors. Exp. Cell Res. 305:23–32 [DOI] [PubMed] [Google Scholar]
- 35. Qadri I, Choudhury M, Rahman SM, Knotts TA, Janssen RC, Schaack J, Iwahashi M, Puljak L, Simon FR, Kilic G, Fitz JG, Friedman JE. 2012. Increased phosphoenolpyruvate carboxykinase gene expression and steatosis during hepatitis C virus subgenome replication: role of nonstructural component 5A and CCAAT/enhancer-binding protein β. J. Biol. Chem. 287:37340–37351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lei X, Zhang S, Barbour SE, Bohrer A, Ford EL, Koizumi A, Papa FR, Ramanadham S. 2010. Spontaneous development of endoplasmic reticulum stress that can lead to diabetes mellitus is associated with higher calcium-independent phospholipase A2 expression: a role for regulation by SREBP-1. J. Biol. Chem. 285:6693–6705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Joyce MA, Walters KA, Lamb SE, Yeh MM, Zhu LF, Kneteman N, Doyle JS, Katze MG, Tyrrell DL. 2009. HCV induces oxidative and ER stress, and sensitizes infected cells to apoptosis in SCID/Alb-uPA mice. PLoS Pathog. 5:e1000291. 10.1371/journal.ppat.1000291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Le Lay S, Lefrère I, Trautwein C, Dugail I, Krief S. 2002. Insulin and sterol-regulatory element-binding protein-1c (SREBP-1C) regulation of gene expression in 3T3-L1 adipocytes. Identification of CCAAT/enhancer-binding protein beta as an SREBP-1C target. J. Biol. Chem. 277:35625–35634 [DOI] [PubMed] [Google Scholar]
- 39. Moriishi K, Mochizuki R, Moriya K, Miyamoto H, Mori Y, Abe T, Murata S, Tanaka K, Miyamura T, Suzuki T, Koike K, Matsuura Y. 2007. Critical role of PA28gamma in hepatitis C virus-associated steatogenesis and hepatocarcinogenesis. Proc. Natl. Acad. Sci. U. S. A. 104:1661–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wong VW, Yu J, Cheng AS, Wong GL, Chan HY, Chu ES, Ng EK, Chan FK, Sung JJ, Chan HL. 2009. High serum interleukin-6 level predicts future hepatocellular carcinoma development in patients with chronic hepatitis B. Int. J. Cancer 124:2766–2770 [DOI] [PubMed] [Google Scholar]
- 41. Calvisi DF, Ladu S, Gorden A, Farina M, Conner EA, Lee JS, Factor VM, Thorgeirsson SS. 2006. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology 130:1117–1128 [DOI] [PubMed] [Google Scholar]
- 42. Chung RT, Poordad FF, Hassanein T, Zhou X, Lentz E, Prabhakar A, Di Bisceglie AM. 2010. Association of host pharmacodynamic effects with virologic response to pegylated interferon alfa-2a/ribavirin in chronic hepatitis C. Hepatology 52:1906–1914 [DOI] [PMC free article] [PubMed] [Google Scholar]