

Abstract
ORF59 of Kaposi's sarcoma-associated herpesvirus (KSHV) plays an essential role in viral lytic replication by providing DNA processivity activity to the viral DNA polymerase (ORF9). ORF59 forms a homodimer in the cytoplasm and binds and translocates ORF9 into the nucleus, where it secures ORF9 to the origin of lytic DNA replication (oriLyt) in order to synthesize long DNA fragments during replication. ORF59 binds to oriLyt through an immediate early protein, replication and transcription activator (RTA). Here, we show that viral kinase (ORF36) phosphorylates serines between amino acids 376 and 379 of ORF59 and replacement of the Ser378 residue with alanine significantly impairs phosphorylation. Although mutating these serine residues had no effect on binding between ORF59 and ORF9, viral polymerase, or ORF36, the viral kinase, it significantly reduced the ability of ORF59 to bind to RTA. The results for the mutant in which Ser376 to Ser379 were replaced by alanine showed that both Ser378 and Ser379 contribute to binding to RTA. Additionally, the Ser376, Ser378, and Ser379 residues were found to be critical for binding of ORF59 to oriLyt and its processivity function. Ablation of these phosphorylation sites reduced the production of virion particles, suggesting that phosphorylation is critical for ORF59 activity and viral DNA synthesis.
INTRODUCTION
Kaposi's sarcoma (KS)-associated herpesvirus (KSHV), also known as human herpesvirus 8 (HHV8), is a member of the Gammaherpesvirus family. KSHV is associated with KS as well as several lymphoproliferative diseases, including primary effusion lymphoma (PEL) and multicentric Castleman's disease (MCD) (1, 2). Similar to other herpesviruses, KSHV establishes a lifelong latency following a primary infection (3). During latency, the KSHV genome is maintained as a nonintegrated episome tethered to the host chromosome via viral latency-associated nuclear antigen (LANA) protein, encoded by ORF73 (4). Limited numbers of viral genes are expressed during latency, and replication of the virus is dependent on host cell replication machinery (5). KSHV latency can be disrupted by various stimuli, and disruption results in the expression of various lytic genes and the production of infectious virions. KSHV replication and transcription activator (RTA), encoded by ORF50, is necessary and sufficient for the activation of KSHV lytic replication and production of viral particles (6–8). Several chemical inducers, such as 12-O-tetradecanoyl-phorbol-13-acetate (TPA) and sodium butyrate (NaB), have successfully been used to trigger lytic replication (9).
In KSHV-induced malignancies, the majority of the tumor cells are latently infected with the virus; however, a small proportion (2 to 5%) of the latent tumor cells undergoes lytic replication (10, 11). This spontaneous reactivation is proposed to produce homologs of cellular cytokines, which act in a paracrine manner for tumor progression, as well as generation of virions for further spread of infection (10). These cytokines are capable of promoting tumorigenesis by activating pathways involved in inflammation, angiogenesis, and enhanced proliferation, invasion, and dissemination of tumor cells (12–23). Consequently, the study of lytic replication is essential not only for understanding the mechanism of lytic DNA replication but also for developing new treatment strategies for preventing KSHV-associated malignancies.
KSHV lytic replication requires eight viral proteins, including ORF9 (DNA polymerase), ORF6 (single-stranded DNA binding protein), ORF40/41 (primase-associated factor), ORF44 (helicase), ORF56 (primase), ORF59 (processivity factor), ORF50 (RTA), and ORF K8 (K-bZIP) (24). RTA is the most important protein required for the initiation of lytic replication, similar to the requirements of other herpesviruses. RTA binds to the C/EBPα and the RTA response element (RRE) of the origin of lytic replication (oriLyt) and thereby initiates lytic replication through transcription activation as well as recruitment of additional factors (20, 25–30).
One of the factors recruited by RTA is the viral processivity factor, ORF59. Rossetto et al. have shown that the binding of ORF59 to the C/EBPα binding motif within oriLyt is crucial for its function and is dependent on the presence of RTA (31). ORF59 forms a homodimer, which translocates viral polymerase (ORF9) into the nucleus for efficient synthesis (processivity function) of DNA fragments (32). The ORF59 homolog in another herpesvirus, human cytomegalovirus (hCMV), ppUL44, is phosphorylated by a viral Ser/Thr kinase, ppUL97, which modulates ppUL44's ability to localize to the nucleus (33). BMRF1, the Epstein-Barr virus (EBV) processivity factor, is phosphorylated by BGLF4 viral kinase within a hinge region-like domain known to be important for transmitting conformational changes (34). As a result, BGLF4 phosphorylation enhances the transactivation activity of Zta (an RTA homolog) and the synergistic activation of lytic replication at oriLyt (35). Similarly, ORF59 is a phosphoprotein which is phosphorylated by KSHV viral Ser/Thr kinase (ORF36), but the consequences of this modification were not determined previously (36). Here, we show that ORF36 phosphorylates ORF59 primarily at Ser378, which is critical for ORF59's ability to bind to RTA and oriLyt. Furthermore, replacing the phosphorylating serines of these sites with alanines significantly reduced viral production.
MATERIALS AND METHODS
DNA constructs.
ORF36-Flag/Myc, ORF59-GST full-length, and ORF59-GST segments were constructed by PCR amplification and cloning using a wild-type (wt) KSHV genome as the template. ORF59 deletion mutants were assembled from two PCRs: (i) one using sense ORF59 primer and antisense ORF59 mutant primer and (ii) a second using sense ORF59 mutant primer and antisense ORF59 primer. Mutants with specific deletions were screened and confirmed by sequencing. ORF59 point mutants were constructed using a QuikChange Lightning Multi site-directed mutagenesis kit as directed by the manufacturer (Agilent Technologies Inc., Santa Clara, CA). These ORF59 deletion and point mutants were cloned into glutathione S-transferase (GST), pxi-hemagglutinin (HA), and pLVX-Flag vectors by PCR amplification. An ORF36 kinase-dead (kd) mutant (ORF36 K108Q) was generated in a two-step approach similar to the approach used for the ORF59 deletion mutants. The resulting PCR product was ligated, transformed, and screened for the K108Q mutation. All primers used for cloning and targeted mutagenesis are listed in Table 1. Bacterial artificial chromosomes (BAC) containing the KSHV genome but lacking ORF59 (BAC36 ΔORF59) and plasmids ORF9-Flag, K8-Flag, and 8088sc were gifts from Gregory Pari (University of Nevada, Reno, NV).
Table 1.
Sequences of primers used in this study
| Clone or mutant | Sensea | Primer oligonucleotide sequence (5′–3′) |
|---|---|---|
| Clones | ||
| ORF36-pA3F/pA3 M | S | TTTGAATTCCGCCATGCGGTGGAAGAATGGAGAG |
| AS | TTTGCGGCCGCGAAAACAAGTCCGCGGGTGTGGGG | |
| pLVX-ORF59-Flag | S | TTTGAATTCTCCTGTGGATTTTCACTATGGGGTC |
| AS | TCAGCGGCCGCGAAAACAAGTCCGCGGGTGTGGGG | |
| ORF59-HA-pxi | S | TTTGAATTCCACCATGCCTGTGGATTTTCACTATGGG |
| AS | TTTGGATCCTTAGCCCTAAGCGTAATCTGGAACATCGTATGGGTACCAACCCGGGACTTTACACAGTCT | |
| ORF59-GST | S | TTTGGATCCATGCCTGTGGATTTTCACTAT |
| AS | TTTGAATTCGTAGGAAATGGTGGTCCTGA | |
| ORF59 1-132aa GST | S | TTTGAATTCCACCATGCCTGTGGATTTTCACTATGGG |
| AS | TTTGAATTCGTAGGAAATGGTGGTCCTGA | |
| ORF59 133-264aa GST | S | TTTGGATCCGGGGACAACCTCACCTCCACC |
| AS | TTTGAATTCCCAACCCGGGACTTTACACAGTCT | |
| ORF59 265-396aa GST | S | TTTGGATCCTTTACCCCCGGGCTGATTTGG |
| AS | TTTGAATTCGTAGGAAATGGTGGTCCTGA | |
| Mutants | ||
| ORF36-K108Q-pA3F | S | GTGTGCAGCAGTTTGATAGCCGCCGGG |
| AS | AACTGCTGCACACACAGATCCTCGGAGA | |
| ORF59-d304-318 (DM1) | S | CCGATCTGTGATACCGGACTCCAGGAGGCA |
| AS | AGTCCGGTATCACAGATCGGTTTACCTCTA | |
| ORF59-d333-349 (DM2) | S | GTCTCCGGATGAGGACGCTGCCTCTGTCAC |
| AS | CAGCGTCCTCATCCGGAGACTCCAATTCCG | |
| ORF59-d376-379 (DM3) | S | CAAGAGGCGCCAGTCCAGGGATCGTGGGAA |
| AS | CCCTGGACTGGCGCCTCTTGTGAGGCCTCT | |
| ORF59 S305A, S312A, S318A (PM1) | AACCGATCTGTGGCAGCCGAGGGAGGCGAGTCTGCACAAAAGGTTCCCGATGCAATACCGGACTCC | |
| ORF59 S333A, S349A (PM2) | TTGGAGTCTCCGGATGCACCCCCTCTCACACCA | |
| ORF59 S376A, S378A, S379A (PM3) | CCTCACAAGAGGCGCGCAGACGCGGCCCAGTCCAGGGATCGT | |
| ORF59 S376A | CCTCACAAGAGGCGCGCAGACTCGAGCCAGTCCAGGGATCGT | |
| ORF59 S378A | CCTCACAAGAGGCGCTCAGACGCGAGCCAGTCCAGGGATCGT | |
| ORF59 S379A | CCTCACAAGAGGCGCTCAGACTCGGCCCAGTCCAGGGATCGT |
S, sense; AS, antisense.
Cell culture, transfection, and transduction by lentivirus.
293T cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% bovine growth serum (HyClone, Logan, UT), 2 mM l-glutamine, 25 U/ml penicillin, and 25 μg/ml streptomycin. All cultures were incubated at 37°C in a humidified chamber supplemented with 5% CO2. Cells were transfected by the calcium phosphate method as previously described (37), with minor modification. Briefly, 500 μl 1× HBS (140 mM NaCl, 0.75 mM Na2HPO4·2H2O, 25 mM HEPES, 5 mM KCl, 6 mM dextrose, pH 7.1) was added to DNA and the components were mixed. Thirty microliters of 2.5 M CaCl2 was added to the solution and the components were mixed. After 20 min at room temperature, the solution was evenly added dropwise onto the 60 to 70% confluent 293T cells.
For generating lentivirus, 10 μg of lentiviral vector, 7.5 μg CMV-dR8.2 packaging plasmid, and 2.5 μg pCMV-VSVG envelope plasmids were transfected into a 100-mm dish of 293T cells using 1× HBS. At 24 h posttransfection, virus was induced with 1 mM NaB in DMEM (with 5% bovine growth serum) with 100 mM HEPES for 10 h. The supernatant was collected three times thereafter at 12- to 16-h intervals. The supernatant was filtered through a 0.45-μm-pore-size filter and ultracentrifuged at 25,000 rpm for 1.5 h using a Beckman Coulter Optima L-90K ultracentrifuge (Beckman Coulter, Inc., Brea, CA). The virus pellet was resuspended in DMEM and added to the target cells.
BACmid DNA was transfected with the Metafectene Pro reagent (Biontex Laboratories GmbH, San Diego, CA) due to the larger size of the plasmid and the fragility of the DNA. Ninety percent confluent cells on a 6-well plate were transfected with 5 to 10 μg of BACmid DNA in 200 μl serum and antibiotic-free medium, after mixing with 6 μl Metafectene Pro reagent. The mixture was added onto the cells dropwise after 15 to 20 min of incubation at room temperature. The BACmid contained green fluorescent protein (GFP), which allowed efficient evaluation of transfection efficiency.
Immunoprecipitation, Western blotting, and antibodies.
Transfected cells were harvested, washed with ice-cold phosphate-buffered saline (PBS), and lysed in 0.5 ml ice-cold radioimmunoprecipitation assay (RIPA) buffer (1% Nonidet P-40 [NP-40], 10 mM Tris [pH 7.5], 2 mM EDTA, 150 mM NaCl) supplemented with protease inhibitors (1 mM phenylmethylsulfonyl fluoride, 1 μg/ml aprotinin, 1 μg/ml pepstatin, 1 μg/ml leupeptin). Cell debris was removed by centrifugation at 13,000 × g (10 min and 4°C), and lysates were precleared by 1 h of rotation at 4°C with 30 μl of protein A-protein G-conjugated Sepharose beads. After approximately 5% of the lysate was saved for use as an input control, the protein of interest was captured by rotating the remainder of the lysate with 1 μg of the appropriate antibody overnight at 4°C. Immune complexes were captured with 30 μl of protein A-protein G-conjugated Sepharose beads by rotating for 2 h at 4°C. The beads were pelleted and washed three times with RIPA buffer. Proteins immunoprecipitated for kinase assay were washed with RIPA buffer containing 300 mM NaCl to reduce any contaminating proteins. For Western blot assay, input lysates and immunoprecipitated (IP) complexes were boiled for 5 to 10 min in Laemmli buffer, resolved by SDS-PAGE, and transferred as per the manufacturer's recommendation (Bio-Rad Laboratories). The nitrocellulose membrane was probed with appropriate antibodies, followed by incubation with infrared dye-tagged secondary antibody, and viewed on an Odyssey imager (LICOR Inc., Lincoln, NE).
The following antibodies were used: mouse anti-Flag (M2; Sigma-Aldrich, St. Louis, MO), rabbit anti-Flag (F7425; Sigma-Aldrich, St. Louis, MO), mouse anti-RTA (mouse hybridoma), mouse anti-LANA (mouse hybridoma), rabbit anti-HA (6908; Sigma-Aldrich, St. Louis, MO), mouse anti-GST (A00014; GenScript Corp.), mouse anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH; G8140; US Biologicals), and rabbit anti-Myc (SAB4300605; Sigma-Aldrich, St. Louis, MO).
Purification of GST fusion proteins.
Escherichia coli BL21(DE3) cells were transformed with the plasmid constructs for each GST fusion protein. Bacterial culture was incubated until the optical density at 600 nm (OD600) was approximately 0.6, at which time the cultures were induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 4 h at 37°C. The bacteria were pelleted, washed once with 5 ml STE buffer (100 mM NaCl, 10 mM Tris, 1 mM EDTA, pH 7.5), resuspended in 5 ml NETN buffer (0.5% NP-40, 100 mM NaCl, 20 mM Tris, 1 mM EDTA, pH 8.0), supplemented with protease inhibitors, and incubated on ice for 15 min. A volume of 75 μl of 1 M dithiothreitol (DTT) and 900 μl of a 10% solution of Sarkosyl in STE buffer were added, and the suspension was sonicated on ice (for 2 min at 40% amplitude with a 20-s-on and 20-s-off sonication cycle) to lyse the cells. The lysates were centrifuged (13,000 × g, 10 min, 4°C) to separate the unsolubilized fraction. Clear supernatant was transferred to a fresh tube, to which 1.5 ml of 10% Triton X-100 in STE buffer and 200 μl of glutathione-Sepharose beads were added. The tube was rotated overnight at 4°C, after which the purified protein bound to glutathione was collected by centrifugation (2 min, 600 × g, 4°C) and washed five times with NETN buffer supplemented with protease inhibitors. The level of purification was determined by SDS-PAGE, and the purified proteins were stored at 4°C.
In vitro translation and binding assay.
For in vitro-translated ORF36, we used a TNT T7 quick-coupled transcription-translation system (Promega, Madison, WI). Briefly, a 50-μl reaction mixture was set up with 2 μg ORF36-Flag plasmid, 40 μl TNT T7 quick master mix, 1 mM [35S]methionine, and 1 μl T7 TNT enhancer. The mixture was incubated at 30°C for 90 min, after which the radioactively labeled protein was used for the binding experiments. In vitro-translated ORF36 was incubated with GST, GST-precipitated ORF59, and its segments by rotating overnight at 4°C. As a control, the luciferase gene was also in vitro translated and rotated with GST and ORF59-GST. The beads were collected and washed with NETN buffer (0.5% NP-40, 100 mM NaCl, 20 mM Tris, 1 mM EDTA, pH 8.0) supplemented with protease inhibitors. The proteins were then visualized with a Coomassie stain, and the gel was dried using a Bio-Rad Gel Air dryer (Hercules, CA). The radioactive gel was exposed to a phosphorimager plate, and the phosphorylated proteins were imaged using a Storm 820 apparatus from Amersham Biosciences (GE Healthcare Inc., Waukesha, WI).
In vitro kinase assay.
Approximately 20 μg purified kinase protein and 20 μg of substrate protein per sample were washed with kinase wash buffer (20 mM HEPES, pH 7.5, 5 mM MnCl2, 10 mM β-mercaptoethanol) containing complete protease and phosphatase inhibitors and resuspended in 20 μl of kinase wash buffer for the reaction. Kinase and the control proteins were resuspended in 10 μl kinase wash buffer with 10 mM cold ATP (3.5 μl of a 100 mM stock) and 0.2 U Ci/μl [γ-32P]ATP (0.7 μl of the stock), and the mixture was added to the substrate. The mixture was incubated in a 37°C water bath for 30 min, after which the reaction was stopped with 15 μl Laemmli buffer. The samples were heated to 95°C for 5 to 10 min and loaded on a 10% SDS-polyacrylamide gel to resolve the proteins. The proteins were then visualized with a Coomassie stain and the gel was dried using a Bio-Rad Gel Air dryer (Hercules, CA). The radioactive gel was exposed to a phosphorimager plate, and the phosphorylated proteins were imaged using a Storm 820 apparatus from Amersham Biosciences (GE Healthcare Inc., Waukesha, WI).
ChIP analysis.
Chromatin immunoprecipitation (ChIP) was performed as described previously (38). Briefly, cells were cross-linked with 3% formaldehyde by rocking for 10 min at room temperature, followed by addition of 125 mM glycine to stop the cross-linking reaction. Cells were washed with cold PBS containing protease inhibitors (1 μg/ml leupeptin, 1 μg/ml aprotinin, 1 μg/ml pepstatin, 1 mM phenylmethylsulfonyl fluoride). Cells were resuspended in 1 ml cell lysis buffer [5 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES), KOH (pH 8.0), 85 mM KCl, 0.5% NP-40] containing protease inhibitors and were incubated on ice for 10 min. Cells were subjected to Dounce homogenization for efficient lysis, followed by centrifugation at 2,500 rpm for 5 min at 4°C. Nuclei were resuspended in nuclear lysis buffer (50 mM Tris [pH 8.0], 10 mM EDTA, and 1% SDS containing protease inhibitors), followed by incubation on ice for 10 min. The chromatin was sonicated to an average length of 700 bp, and cell debris was removed by centrifugation at high speed for 10 min at 4°C. The supernatant containing the sonicated chromatin was diluted 5-fold with ChIP dilution buffer (0.01% SDS, 1.0% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris [pH 8.1], 167 mM NaCl, including protease inhibitor). Samples were precleared with a salmon sperm DNA-protein A-protein G-Sepharose slurry for 1 h at 4°C with constant rotation. The supernatant was collected after a brief centrifugation (2,000 rpm at 4°C). Ten percent of the total supernatant was saved for use as the input in Western blotting, and the remaining 90% was divided into three fractions, to which was added 1 μg of (i) a control antibody, (ii) anti-Flag antibody (M2; Sigma-Aldrich), or (iii) anti-RTA antibody, respectively. The reaction complexes were rotated overnight at 4°C, followed by precipitation of the immune complex by using a salmon sperm DNA-protein A-protein G slurry. Beads were then washed sequentially with a low-salt buffer (0.1% SDS, 1.0% Triton X-100, 2 mM EDTA, 20 mM Tris [pH 8.1], 150 mM NaCl), a high-salt buffer (0.1% SDS, 1.0% Triton X-100, 2 mM EDTA, 20 mM Tris [pH 8.1], 500 mM NaCl), and an LiCl wash buffer (0.25 M LiCl, 1.0% NP-40, 1% deoxycholate, 1 mM EDTA, 10 mM Tris [pH 8.0]) and twice in Tris-EDTA. Ten percent of the immunoprecipitated chromatin was taken for Western blot assay. Chromatin was eluted using an elution buffer (1% SDS, 0.1 M NaHCO3) and reverse cross-linked by adding 0.3 M NaCl at 65°C overnight. Eluted DNA was precipitated, treated with proteinase K at 45°C for 2 h, and purified. Purified DNA was used as a template for PCR amplification of the RRE region of KSHV oriLyt.
Quantitative RT-PCR.
Quantitative real-time (RT) PCR was performed in a total volume of 20 μl including 10 μl of SYBR green PCR 2× master mix (Applied Biosystems). For viral production, we used 0.5 μM each KSHV LANA primer (forward, 5′-TTGCCTATACCAGGAAGTCCCACA-3′; reverse, 5′-GGAGGAAGACGTGGTTACGGG-3′). The relative numbers of viral particles were quantified using a standard curve and the ΔΔCT method (where CT is threshold cycle). To obtain the relative KSHV genome copy number, we used GAPDH (forward, 5′-CAGCAAGAGCACAAGAGGAAGA-3′; reverse, 5′-TTGATGGTACATGACAAGGTGCGG-3′) for the ΔΔCT method of quantitation. Chromatin immunoprecipitated DNA with ORF59, point mutant 3 (PM3), and RTA proteins was analyzed using oriLyt RRE primers (forward, 5′-CTCTGGGTGGTTTCGGTAGA-3′; reverse, 5′-CCTCGTTACGGGTAAATCCA-3′). Purified DNA samples, the ChIP fraction, and the input DNA samples were amplified by PCR for 3 min at 95°C and 40 cycles of 15 s at 95°C, 30 s at 51°C, and 30 s at 72°C on an ABI StepOne Plus real-time PCR machine (Applied Biosystems). A melting curve analysis was performed to verify the specificity of the amplified product, and each sample was tested in triplicate. The relative numbers of copies of RRE bound to ORF59, its mutant, and RTA were quantified using the ΔΔCT method and the vector-complemented sample as a reference.
Indirect immunofluorescence microscopy.
To check the colocalization of ORF59 and its point mutants with ORF36, ORF9, and RTA, we transfected 293L cells that were plated on glass coverslips using 1× HBS. At 24 h posttransfection, cells were fixed with 3% paraformaldehyde for 20 min at room temperature, washed twice with 1× PBS, and then permeabilized with 0.2% Triton X-100 for 10 min at room temperature. The cells were washed twice with 1× PBS and blocked in 0.4% fish skin gelatin (Sigma-Aldrich) and 0.05% Triton X-100 for 40 min at room temperature. The cells were washed twice with 1× PBS and incubated with primary antibody (0.2% fish skin gelatin, 0.05% Triton X-100, 0.5 μg antibody) for 1 h at room temperature. The cells were washed with PBS and treated with secondary antibody (0.2% fish skin gelatin, 0.05% Triton X-100, a 1:20,000 dilution of stock antibody). Alexa Fluor (AF)-conjugated antibodies (AF 594 and AF 647) were used for the detection of RTA and ORF59, as indicated in the immune localization panels. The cells were then washed with PBS, and the nuclei were stained with TO-PRO-3 (Molecular Probe) for 10 min. Images were obtained using a laser scanning confocal microscope (Carl Zeiss, Inc.) and processed with ZEN imaging software (Carl Zeiss, Inc.) to assign the appropriate color.
Generation of BAC36 ΔORF59.
Mutagenesis of BAC36 was performed using the Red recombination method as previously described (39, 40). Briefly, a kanamycin (Kan) resistance cassette (Kan cassette) flanked by the FLP recombination target (FRT) sequence was PCR amplified with primers containing sequence homologous to ORF59. The cassette was transformed into BAC36-containing E. coli EL350. These bacteria carry a bacteriophage lambda prophage with the genes exo, bet, and gam. These genes are under the control of a temperature-sensitive repressor, thus allowing their expression at 42°C. Gam inhibits RecBCD nuclease from degrading linear DNA, while Exo and Beta provide double-stranded break repair recombinase. EL350 bacteria were induced at 42°C for 15 min to activate Gam, Exo, Beta, and bacteriophage lambda λ recombinase, allowing the PCR product to be inserted at the site of ORF59 homologous to the ends of the product. Transformed bacteria were plated on kanamycin plates and grown at 32°C to prevent further recombination. Southern blotting was used for screening the BAC36 ΔORF59 intermediate. Successful integration of the Kan cassette gave a 5,883-bp fragment when the BACmid was digested with PstI. The Kan cassette was removed by FLP recombination, which resulted in an FRT site scar at the site of recombination. This region of ORF59 in the resulting BAC36 was PCR amplified and verified for interrupted ORF59 protein by sequencing.
trans Complementation of BAC36 ΔORF59 with ORF59 and a point mutant.
293L cells expressing the ORF59 wt, PM3, and a vector control were generated by transducing them with a lentiviral vector, pLVX-puro (Clontech Laboratories), containing ORF59-HA and PM3-HA. Cells were selected with puromycin to obtain a pure population of cells before transfecting them with BAC36 ΔORF59. Cells maintaining the KSHV bacterial artificial chromosome (BAC) genome were selected with hygromycin, and the pool-selected cells, with almost 100% green fluorescence, were used for the detection of the latently maintained KSHV genome. Latently maintained KSHV genome copy numbers were determined by extracting DNA from these cells using a modified Hirt procedure (41) and quantifying the KSHV genome in a semiquantitative real-time PCR assay. These cells were treated with sodium butyrate and TPA to induce lytic reactivation, which was confirmed by the detection of the RTA protein in a Western blot. Virions produced after lytic reactivations were quantified by collection of the supernatant at 4 days postinduction and extraction of viral DNA for real-time PCR after centrifugation to remove any cells. The relative numbers of virions produced were determined by the detection of the KSHV genome in the supernatant using the ΔΔCT method and BAC36 ΔORF59 with an empty vector as a reference. Induction of lytic reactivation and virion production was confirmed by comparing the virions produced from equal numbers of cells with and without induction of the lytic cycle. These experiments were performed three independent times, and the data presented are the averages of three experiments.
RESULTS
As part of the replication complex, ORF59, the KSHV processivity factor, plays a critical role in initiating KSHV lytic DNA replication. It forms a homodimer in the cytoplasm, binds to viral polymerase (ORF9) to transport it into the nucleus, and helps in synthesizing longer DNA fragments (32). It has a high affinity for double-stranded DNA (dsDNA) and is found at the origin of lytic replication (oriLyt) (36). ORF59 is recruited to the origin through the viral replication and transcription activator (RTA), as demonstrated previously using a chromatin immunoprecipitation assay (31). In other herpesviruses, phosphorylation of the processivity factor plays an important role for its function. A homolog of ORF59 in hCMV, ppUL44, is phosphorylated by viral kinase (ppUL97) to modulate its ability to localize to the nucleus. BMRF1, the EBV processivity factor, is phosphorylated by BGLF4, a viral kinase for optimal oriLyt-dependent viral DNA replication (35).
KSHV kinase (ORF36) binds to ORF59 between aa 1 and 264 and phosphorylates between aa 265 and 396.
Previous studies have indicated that ORF59 is a phosphoprotein, but the significance of phosphorylation was not determined (36). Although detectable binding between a kinase and its substrate may not be necessary for phosphorylation to occur, we determined that ORF36 did in fact bind to ORF59 (Fig. 1A, fourth lane). This binding was specific, as the vector control lane did not show any detectable levels of ORF59 (Fig. 1A, ORF59-Flag lane). In order to determine the domain of ORF59 interaction, an in vitro binding assay with ORF59-GST and three ORF59-GST-fused segments, segments from amino acids (aa) 1 to 132, aa 133 to 264, and aa 265 to 396 (Fig. 1B and C), was performed and identified that ORF36 binds between aa 133 and 264 with a higher affinity and between aa 1 and 264 at a slightly lower affinity (Fig. 1D; compare lanes 6 and 7). As a control, we used in vitro-translated luciferase protein, which showed no binding to ORF59-GST, and the GST without the fusion protein confirmed the specificity of binding between ORF36 and ORF59 (Fig. 1D, lane 3). Furthermore, in vitro analysis also confirmed that no other proteins were necessary for the binding between ORF36 and ORF59.
Fig 1.

KSHV kinase (ORF36) binds to ORF59 between aa 1 and 264. (A) Binding between full-length ORF59 and ORF36 was established by cotransfecting ORF36-Flag and ORF59-HA plasmids and immunoprecipitating with anti-Flag antibody. The Western blot shows specific binding between ORF36 and ORF59, as the vector did not precipitate any ORF59. (B) Schematic of ORF59 GST-tagged segments used for in vitro binding assay. NLS, nuclear localization signal. (C) GST-fused full-length ORF59 and ORF59 segments were expressed in E. coli as stated in Materials and Methods and visualized on a Coomassie gel. Red asterisks, each of the purified proteins. (D) In order to identify the site of interaction, full-length ORF59 as well as its GST-fused segments (aa 1 to 132, aa 133 to 264, aa 265 to 396) were used for an in vitro binding assay with in vitro-translated [35S]methionine-labeled ORF36. As a control, luciferase was translated in vitro and did not bind to either GST or ORF59-GST. Radioactively labeled ORF36 was detected with ORF59-GST as well as the segments from aa 1 to 132 and aa 133 to 264, binding with a higher affinity to the latter segment.
In order to determine the site of phosphorylation, we used ORF59-GST-tagged segments for a kinase assay with ORF36 (viral kinase)-immunoprecipitated ORF36-Flag expressed in 293T cells. ORF36-Flag and Flag vector immunoprecipitates were washed with RIPA buffer containing higher salt (300 mM NaCl) to ensure that no additional protein carried over for the kinase assay, as the cellular kinases may also be pulled down, which may convolute the results. Immunoprecipitated ORF36-Flag and Flag vector were used for kinase assay with ORF59-GST and GST-tagged ORF59 fragments (aa 1 to 132, aa 133 to 264, aa 265 to 396) (Fig. 2A). Our results show that ORF59 is heavily phosphorylated by ORF36 (Fig. 2B, lane 3). Phosphorylation of ORF59 fragments identified that the site of phosphorylation lies between aa 265 and 396 (Fig. 2B, lane 6). Since ORF36 requires autophosphorylation for its kinase activity, detection of ORF36 phosphorylation in all the lanes confirmed the presence of active kinase (Fig. 2B, lanes 2 to 6).
Fig 2.

ORF36 phosphorylates ORF59 between aa 265 and 396. (A) Coomassie-stained gel of GST-ORF59 and segments used in the kinase assay. Lanes 1 and 3, ORF59-GST; lanes 4 to 6, ORF59-GST segments from aa 1 to 132, aa 133 to 264, and aa 265 to 396, respectively; lane 2, GST protein used in the assay. (B) Autoradiography image of the in vitro kinase assay performed on ORF59 and its GST-fused segments using immunoprecipitated ORF36-Flag as the kinase and the Flag vector as a control for kinase. Lane 1, GST-ORF59 as a negative control showing no phosphorylation when ORF36 was not present; lane 3, ORF59-GST phosphorylation when ORF36 kinase was present (asterisk). Among the GST-ORF59 domains, the region from aa 265 to 396 (lane 6) was phosphorylated by ORF36 (red asterisk). Lanes 2 to 6, ORF36 autophosphorylation, which is essential for its kinase activity.
As a negative control, we performed a kinase assay on ORF59-GST with a Flag vector-immunoprecipitated complex as well as ORF36 with GST as a substrate. Consequently, we detected no phosphorylation of ORF59-GST when assayed with the Flag vector (Fig. 2B, lane 1), and although there was autophosphorylation of ORF36, the absence of GST phosphorylation confirmed the specificity of the assay (Fig. 2A and B, lane 2).
ORF36 phosphorylated ORF59 at Ser between aa 265 and 396.
In order to identify the probable phosphorylation sites on ORF59 between aa 265 and 396, we used a program, KinasePhos (http://kinasephos.mbc.nctu.edu.tw/; National Chiao Tang University, Taiwan), to determine the phosphorylation sites. The program predicted one threonine and several serine residues to be the potential sites of phosphorylation (Fig. 3A). We grouped these residues to make three ORF59 deletion mutants (DM1, DM2, DM3) (Fig. 3B). The sequences in Fig. 3B outlined with a red box denote the sequences deleted from full-length ORF59. ORF59 deletion mutants were constructed by sequential PCR, and the presence of deletions was confirmed by sequence analysis. These deletion mutants were cloned in frame with GST to produce GST fusion proteins.
Fig 3.

ORF36 phosphorylates ORF59 between aa 376 and 379. (A) Results of KinasePhos program analysis identifying possible phosphorylation sites on ORF59 between aa 364 and 396. The sites were grouped as indicated by the red boxes in panel B to generate three ORF59 deletion mutants (DMs). (B) Schematic of ORF59 deletion mutants used for mapping of the sites of phosphorylation, and the deleted sequences are marked with red boxes. Potential phosphorylation sites (S/T) are shown as red text. (C) In vitro kinase assay done with ORF59 GST-fused deletion mutants. (Top) Autoradiography image of ORF59 DM1, DM2, and DM3 with control Flag vector (lanes 1 to 3) and with immunoprecipitated ORF36-Flag (lanes 4 to 6). As seen in lane 6, ORF59 DM3 was not phosphorylated by ORF36, which thus identified aa 376 to 379 to be the specific site of phosphorylation by ORF36. Autophosphorylation of ORF36 (required for its kinase activity) is indicated in lanes 4 to 6. (Bottom) The proteins used in the kinase assay. Lanes 1 through 3, ORF59-GST DM1, DM2, and DM3 with Flag, respectively; lanes 4 to 6, DM1, DM2, and DM3 with ORF36, respectively. (D) A positive control was included with a known ORF36 substrate, K8. (Left) Coomassie-stained gel of K8 and GST; (right) kinase assay showing autophosphorylation of K8 (lane 3) but not GST (lane 4). Autophosphorylation of ORF36 (active kinase) is indicated by an arrowhead (lanes 3 and 4). (E) As a control, ORF59 was used for an in vitro kinase assay with ORF36-Flag and the kd mutant ORF36 (K108Q). The results showed specific phosphorylation of ORF59 with ORF36-Flag (lane 2) but not with the Flag vector (lane 1). Lane 3, the kd mutant of ORF36 was unable to phosphorylate ORF59 (ORF59) or itself (ORF36), suggesting the specificity of ORF59 phosphorylation by ORF36 kinase.
GST and GST-tagged ORF59, DM1, DM2, and DM3 proteins were purified and used for in vitro kinase assay. Immunoprecipitated ORF36-Flag and Flag vector were washed with RIPA buffer with 300 mM NaCl to remove any additional carryover proteins. Consequently, the results of the kinase assay showed that ORF36 failed to phosphorylate ORF59 DM3 (Fig. 3C, lane 6) but not DM1 or DM2 (Fig. 3C, lanes 4 and 5). Autophosphorylation of ORF36 in this assay confirms the presence of active kinase in all three lanes (Fig. 3C, lanes 4 to 6). The phosphorylation of these truncation mutants by ORF36 was specific, as the immunoprecipitate with the Flag vector control was unable to phosphorylate these mutants (Fig. 3C, lanes 1 to 3). This suggested that ORF36 phosphorylates ORF59 at residue Ser376, Ser378, or Ser379.
For a positive control, we used K8, a known substrate of ORF36, to test the activity of the kinase used in this assay, and as seen in Fig. 3D, lane 3, ORF36 was able to autophosphorylate as well as phosphorylate K8. As a negative control, GST was used as a substrate with ORF36, which showed autophosphorylation, indicating active kinase; however, no signal for GST phosphorylation was detected, suggesting the specificity of the assay (Fig. 3D, lane 4). We also tested the specificity of phosphorylation by ORF36 by using a kd mutant of ORF36 (K108Q). The kd mutant of ORF36 was generated by a two-step approach similar to the approach used to generate ORF59 deletion mutants. Kinase assay with ORF36 K108Q showed that ORF59 was specifically phosphorylated by the viral kinase, as the kd mutant was unable to phosphorylate ORF59 (Fig. 3E, lane 2). Abolition of kinase activity in the kd mutant was evidenced by the lack of autophosphorylation of ORF36 as well (Fig. 3E, lane 3).
Moreover, we were concerned that a large deletion in ORF59 deletion mutants may have an effect on the protein conformation and its tertiary structure; therefore, we generated ORF59 point mutants (PM1, PM2, PM3) to verify our findings and conduct further experiments. The point mutants, where each individual serine residue was mutated to an alanine, were generated using a QuikChange Lightning Multi site-directed mutagenesis kit (Fig. 4B). As with the ORF59 deletion mutants, ORF59 PM1, PM2, and PM3 were used for in vitro kinase assay with immunoprecipitated ORF36 or just the Flag vector as a control. As expected, ORF59 PM3 was not phosphorylated by ORF36, while ORF59 PM1 and PM2 were phosphorylated by ORF36 (Fig. 4B, bottom; compare lane 6 with lanes 4 and 5). ORF36 autophosphorylation is shown in Fig. 4B as a control of kinase activity. As a control, K8 was phosphorylated by ORF36, as demonstrated earlier, and GST lacked a signal, as expected (Fig. 4C, lanes 3 and 4).
Fig 4.
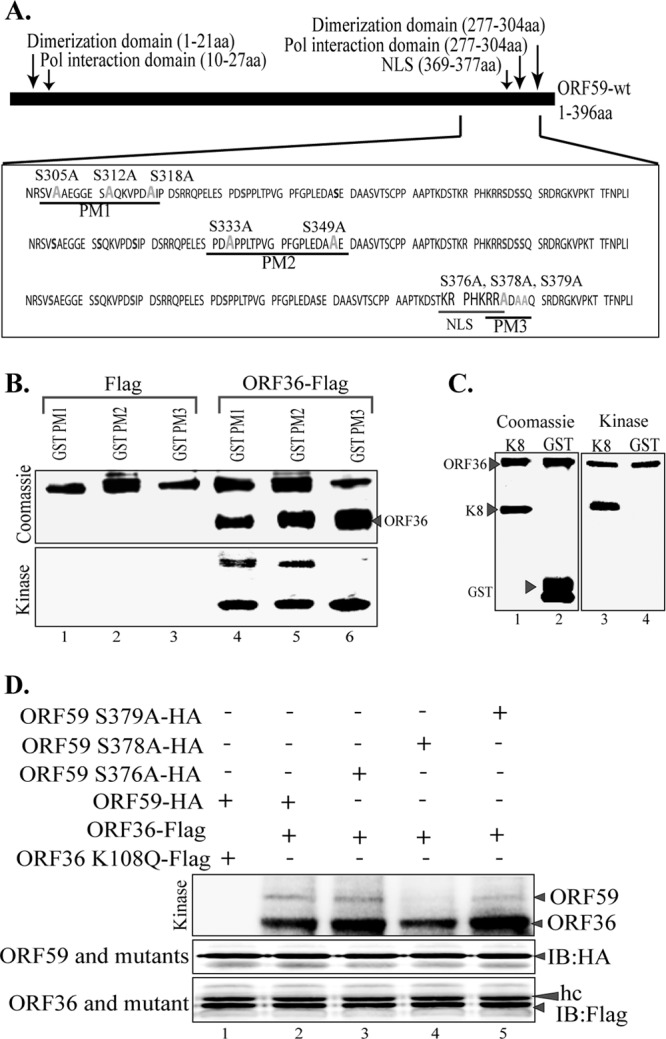
ORF36 phosphorylates ORF59 at Ser376, Ser378, and Ser379. (A) Schematic of ORF59 point mutants (PM1, PM2, PM3) used in the identification of specific phosphorylation targets by ORF36 kinase. The serine residues identified in the ProsKinase program as potential phosphorylation targets are marked in red. The serine residues mutated to alanines are marked in green. (B) In vitro kinase assay with ORF59 GST-fused point mutants. (Bottom) Autoradiography image of ORF59 PM1, PM2, and PM3 with the control Flag vector (lanes 1 to 3) and with immunoprecipitated ORF36-Flag (lanes 4 to 6). ORF59 PM3 was not phosphorylated by ORF36, which thus identified Ser376, Ser378, and Ser379 to be the specific sites of ORF36 phosphorylation. Lanes 4 to 6 show that autophosphorylation of ORF36 is essential for its kinase function. (Top) Proteins used in the kinase assay. Lanes 1 to 3, ORF59-GST PM1, PM2, and PM3 with Flag, respectively; lanes 4 to 6, PM1, PM2, and PM3 with ORF36 kinase, respectively. Lanes 4 to 6 of the Coomassie panel show the levels of ORF36 used for the in vitro kinase assay. (C) K8 was used as a positive control for ORF36 phosphorylation. (Left) Coomassie-stained gel of K8 (substrate) and GST, as a control; (right) phosphorylation of K8 as well as autophosphorylation of ORF36 but no phosphorylation of just GST. (D) ORF59 with serine 378 mutated to alanine lacks ORF36-mediated phosphorylation. Mutants with a single amino acid mutation at Ser376A (lane 3), Ser378A (lane 4), and Ser379A (lane 5) were immunoprecipitated and subjected to the kinase assay with ORF36. ORF59 with Ser378 mutated to A showed almost no phosphorylation (lane 4). The Ser379A mutant (lane 5) also had slightly reduced phosphorylation compared to that of Ser376A (lane 3) and wt ORF59 (lane 2). The kd mutant (ORF36 K108Q) was unable to phosphorylate ORF59 (lane 1). Immunoprecipitated ORF59 and its mutants were detected by anti-HA Western blotting (IB: HA). ORF36 and mutant were detected by anti-Flag Western blotting (IB: Flag). hc, heavy chain.
In an attempt to determine the exact residue of PM3 phosphorylated by ORF36, we generated single-amino-acid alanine substitution mutants of ORF59 at residues 376, 378, and 379. Wild-type ORF59 (Fig. 4D, lane 2) and its single-amino-acid alanine substitution mutants (Fig. 4D, lanes 3 to 5) were subjected to kinase assay with ORF36. Detection of phosphorylation showed that ORF59 containing Ser378 mutated to alanine (Fig. 4D, lane 4) had significantly reduced levels of phosphorylation, suggesting that residue 378 is the primary site of ORF36-mediated phosphorylation. The levels of ORF59 and its substitution mutants as well as the kinase, ORF36, were similar in all the lanes (Fig. 4D, immunoblot with HA [IB: HA] and IB: Flag, respectively), suggesting that the ablation of phosphorylation was due to the lack of a phosphorylation site in the Ser378-to-alanine mutant (Fig. 4D, lane 4). The specificity of ORF59 phosphorylation was once again confirmed, since the kd mutant of ORF36 did not phosphorylate ORF59 (Fig. 4D, lane 1).
The ORF59 point mutant lacks the ability to bind to RTA.
Considering the functions of ORF59 and how the absence of phosphorylation may affect the virus, we evaluated the binding between ORF59 and its point mutants with the already known binding partners (31, 32). First, we determined the binding of ORF59 PM1, PM2, and PM3 to ORF36 by coimmunoprecipitation (CoIP) assays (Fig. 5A). Our data determined that the binding of ORF36 to ORF59 containing substitution mutations was unaffected, as all three point mutants (PM1, PM2, PM3) precipitated ORF36, a result comparable to that for wt ORF59 (Fig. 5A, IB: Myc; compare lane 7 with lanes 8 to 10). The vector control did not precipitate ORF36, confirming the specificity of their interaction (Fig. 5A, lane 6). We further determined whether the kd mutant of ORF36 was capable of binding to ORF59 by performing coimmunoprecipitation. Our data showed significantly reduced levels of ORF59 coprecipitation for kd ORF36 compared to that for active ORF36 (Fig. 5B, IB: HA; compare lane 8 with lane 9). Additionally, we further analyzed the binding of ORF36 to single-alanine-substitution mutant PM3, and no effect on its binding to ORF36 was found, as expected (Fig. 5B, IB: HA, lanes 9 to 12).
Fig 5.
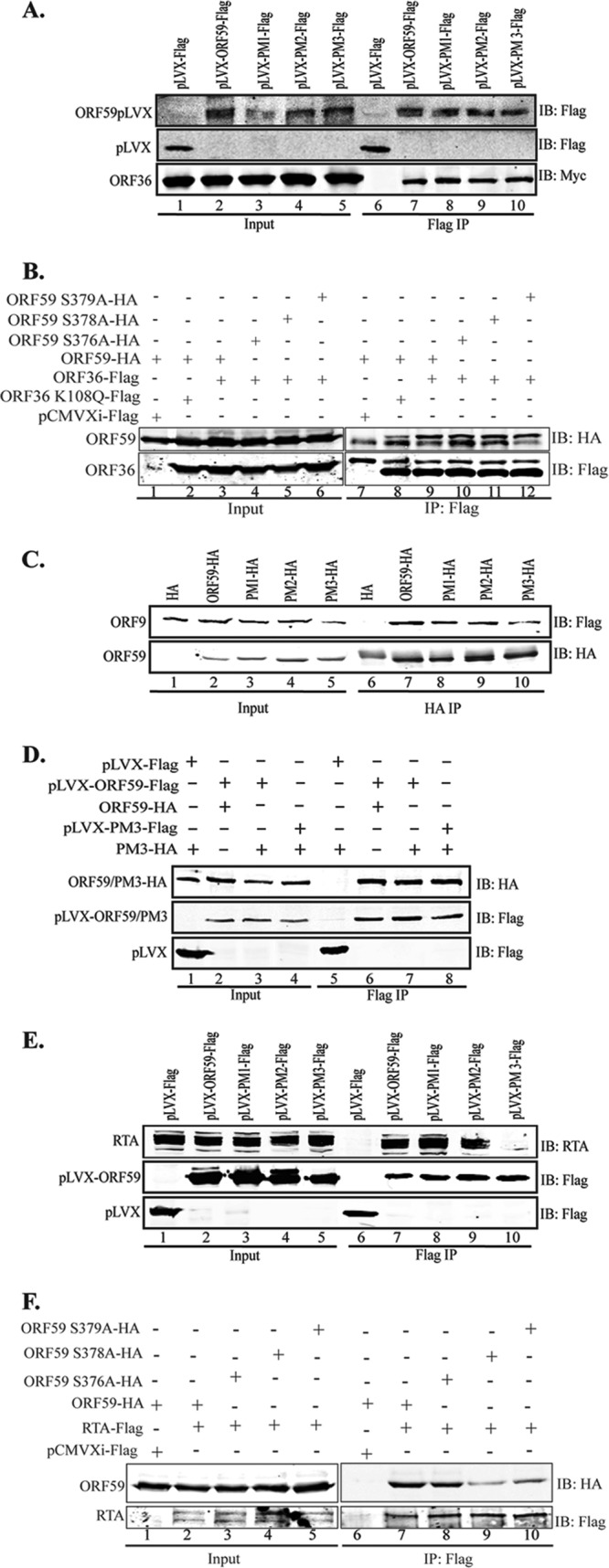
ORF59 S376A, S378A, and S379A kinase mutants do not bind to RTA or oriLyt. (A) Binding between ORF36 and ORF59 point mutants was determined by CoIP assay. The ORF36-Myc plasmid was cotransfected with either empty pLVX vector, pLVX-ORF59-Flag, pLVX-PM1-Flag, pLVX-PM2-Flag, or pLVX-PM3-Flag. Flag immunoprecipitation complexes were resolved on a Western blot and probed with anti-Myc and anti-Flag antibodies. Lanes 1 to 5, inputs; lanes 7 to 10, immunoprecipitated ORF59 and its point mutants precipitating with ORF36-Myc. (B) ORF36 binding with single-amino-acid-substitution mutants of ORF59. HA-tagged ORF59 was coexpressed with Flag vector (lane 1), kd ORF36 (lane 2), or ORF36 kinase (lane 3). Immunoprecipitation with anti-Flag antibody to precipitate ORF36 coprecipitated ORF59 (IB: HA, lane 9). The kd mutant of ORF36 showed significantly reduced coprecipitation of ORF59 (lane 8), suggesting that phosphorylation was important for the binding. Single-amino-acid substitution mutants of ORF59 with mutations at Ser376 (lane 10), Ser378 (lane 11), and Ser379 (lane 12) showed binding comparable to that of wt ORF59. (C) Binding between ORF9 and ORF59 point mutants was determined by cotransfecting ORF9-Flag with either the HA vector, ORF59-HA, PM1-HA, PM2-HA, or PM3-HA. Anti-HA immunoprecipitation complexes were resolved on an SDS-polyacrylamide gel and probed with anti-Flag and anti-HA antibodies. Lanes 1 to 5, inputs; lanes 7 to 10, immunoprecipitated ORF59. Coprecipitation of ORF9 showed binding comparable to that of wt ORF59 as well as that of its point mutants. (D) The serine residue mutated to alanine in PM3 does not affect ORF59 dimerization. A GFP-Flag-tagged lentivirus vector with PM3-HA, pLVX-ORF59-Flag with ORF59-HA, pLVX-ORF59-Flag with PM3-HA, or pLVX-PM3-Flag with PM3-HA was transfected into 293T cells and immunoprecipitated with anti-Flag antibody. The immunoprecipitated complex was resolved on an SDS-polyacrylamide gel and stained with anti-Flag and anti-HA antibodies. Lanes 1 to 4, inputs; lanes 6 to 8, dimerization was not affected by the S376A, S378A, or S379A mutation. (E) Binding between RTA and ORF59 point mutants determined by CoIP. The RTA-expressing plasmid was cotransfected with empty pLVX vector, pLVX-ORF59-Flag, pLVX-PM1-Flag, pLVX-PM2-Flag, or pLVX-PM3-Flag. The anti-Flag IP complex was resolved on an SDS-polyacrylamide gel and probed with anti-RTA and anti-Flag antibodies. Lanes 1 to 5, inputs; lanes 7 to 10, immunoprecipitated ORF59. The blot shows binding of RTA to PM1 (lane 3) and PM2 (lane 9) but not to PM3 (lane 10). (F) RTA binding to single-amino-acid-substitution mutants of ORF59. RTA-Flag was cotransfected with either wt ORF59 (lane 2) or its single-amino-acid alanine substitution mutant with a mutation at Ser376 (lane 3), Ser378 (lane 4), or Ser379 (lane 5). Immunoprecipitation of RTA with anti-Flag antibody efficiently coprecipitated wt ORF59 (lane 7) but showed reduced precipitation of the Ser378 (lane 9) and Ser379 (lane 10) mutants.
Due to the proximity of these point mutations to ORF9, the viral polymerase binding domain, we wanted to determine if these mutations in ORF59 affected its ability to bind to or colocalize with the viral polymerase. We cotransfected ORF9-Flag plasmid with HA vector or HA-tagged ORF59 and its point mutants. The CoIP was done with anti-HA antibody to precipitate ORF59 and its mutants. Figure 5C shows that ORF9 was efficiently coimmunoprecipitated with ORF59 and its point mutants (Fig. 5C, IB: Flag, lanes 7 to 10). A lack of ORF9 precipitation with the HA empty vector confirms the specificity of interactions of these proteins (Fig. 5C, IB: Flag, lane 6). These data indicate that phosphorylation does not affect the ability of ORF59 to bind to the viral DNA polymerase. Additionally, the introduction of single alanine substitutions at the serines of PM3 (Ser376, Ser378, and Ser379) did not affect its binding to ORF9, as expected (data not shown). Viral polymerase translocates into the nucleus along with ORF59; therefore, we determined the localization of ORF9 with alanine mutants of ORF59. Our colocalization data showed that ORF9 efficiently translocated to the nucleus with wt ORF59 as well as the points mutants (data not shown), confirming that ORF59 was still able to recruit viral polymerase and transport it into the nucleus.
Furthermore, we wanted to determine whether mutation of Ser376, Ser378, or Ser379 to alanine has any effect on the ability of ORF59 to dimerize. To this end, we cotransfected Flag-tagged ORF59 with HA-tagged ORF59 (Fig. 5D, lane 2) as a positive control, Flag-tagged ORF59 (ORF59-pLVX-Flag) with HA-tagged point mutant 3 (PM3-HA) (Fig. 5D, lane 3), or Flag-tagged PM3 with HA-tagged PM3 (Fig. 5D, lane 4) into 293T cells to immunoprecipitate with anti-Flag antibody. If dimerization occurred, the Flag-tagged protein would be able to bring down its HA-tagged counterpart. We demonstrated that ORF59 was able to dimerize with PM3 at a level similar to that with wt ORF59 (Fig. 5D, lanes 6 and 7). We also showed that PM3 dimerizes with itself at relatively the same level as it does with wt ORF59, which further verifies that dimerization was not affected by phosphorylation at serine residue 376, 378, or 379 (Fig. 5D, lane 8). The vector control did not precipitate ORF59 PM3, confirming the specificity of the association identified above (Fig. 5D, lane 5).
As previously stated, the interaction between ORF59 and RTA is critical for the function of ORF59 as a processivity factor (31). We determined the binding of ORF59 point mutants and RTA by cotransfecting an RTA expression plasmid with GFP-Flag-tagged vector, ORF59, or ORF59 PM1, PM2, or PM3 pLVX-Flag. Immunoprecipitation of ORF59 and its point mutants showed that PM1 and PM2 were still able to bind to RTA (Fig. 5E, lanes 8 and 9), while the PM3 mutants containing the S376A, S378A, and S379A mutations were unable to bind to RTA (Fig. 5E, IB: RTA; compare lane 10 with lanes 8 and 9). In an attempt to determine the exact residues of PM3 responsible for binding to RTA, we used ORF59 containing single-amino-acid alanine substitutions at serine residues 376, 378, and 379. Immunoprecipitation of RTA showed efficient coprecipitation of wt ORF59 as well as ORF59 with the Ser376A mutant (Fig. 5F, IB: HA, lanes 7 and 8). Interestingly, mutation of the Ser378 residue to alanine significantly reduced the binding of RTA to ORF59 (Fig. 5F, IB: HA, lane 9). Since Ser379A also showed slightly reduced binding to RTA (Fig. 5F, IB: HA; compare lane 10 with lanes 8 and 9), we suggest that both serines at residues 378 and 379 play critical roles in binding to RTA.
Point mutants of ORF59 do not affect its localization with ORF36 but do affect its localization with RTA.
Since the binding of ORF36 to ORF59 was unaffected even after the replacement of the serines between aa 376 and 379, we wanted to determine their localization in the cellular compartment by immunolocalization assay. As expected, ORF59 and its point mutants (PM1 to PM3) localized to the nucleus in a similar fashion as ORF36, with the majority of the spot colocalizing to the same nuclear compartments determined by yellow signals in the overlay panels (Fig. 6A). We further determined the localization of RTA with ORF59 and its point mutants by cotransfecting GFP-fused ORF59 along with RTA expression plasmids, which were detected by anti-RTA antibody. The yellow signals in the overlay panels show the localization of RTA and ORF59 in the same nuclear compartments (Fig. 6B). PM1 and PM2 showed colocalization almost similar to that of wt ORF59, but PM3, which had Ser376, Ser378, and Ser379 mutated to alanines, showed the least yellow signal in the overlay panel, suggesting that the association of PM1 and PM2 is dependent on these serines (Fig. 6B). These data corroborate the immunoprecipitation data and are significant, as they show that ORF36-mediated phosphorylation of ORF59 plays an important role in their interaction.
Fig 6.

The ORF59 point mutant can associate with ORF36 but not with RTA. (A) 293L cells were transfected with GFP-fused ORF59 and its point mutants (PM1 to PM3) along with Ds-Red-fused ORF36. The cells were fixed, permeabilized, stained with TO-PRO-3 and nuclear stain, and imaged with a laser confocal microscope. The colocalization of these two proteins, depicted as yellow signals (Overlay), in wt ORF59 as well as the point mutants is shown. (B) For determining the nuclear localization of ORF59 and RTA, GFP-fused ORF59 and its point mutants (PM1 to PM3) were cotransfected with RTA-expressing plasmid. Cells were fixed, permeabilized, and stained with mouse anti-RTA primary antibody. We used Alexa Fluor 594 (red) to visualize RTA localization. The overlay of red and green (yellow) indicates colocalization of RTA with ORF59, PM1, and PM2 but not with PM3. (C) Localization of RTA with ORF59 and its point mutants in BAC36 ΔORF59 cells. BAC36 ΔORF59 was transfected into 293L cells expressing either wtORF59 or point mutants, followed by selection for a pure population. Cells were induced to undergo lytic reactivation, followed by fixing and permeabilizing so that they could be stained with anti-ORF59 and anti-RTA antibodies. Alexa Fluor 594 was used for the detection of ORF59, and Alexa Fluor 647 was used for the detection of the RTA protein. The overlay panels show colocalization, detected by the yellow signal, in wt ORF59 and PM1 and PM2 but that the colocalization was significantly reduced in PM3. DIC, differential inference contrast.
Additionally, we confirmed the association of RTA with ORF59 and its point mutants in BAC36 ΔORF59-containing 293L cells complemented with either wt ORF59 or the point mutants. Induced cells were subjected to the detection of ORF59 and RTA using specific antibodies. Localization of ORF59 with Alexa Fluor 594 and RTA with Alexa Fluor 647 showed yellow colocalizing signals in wt ORF59 as well as in PM1 and PM2 (Fig. 6C). However, BAC36 ΔORF59 complemented with PM3 showed the fewest yellow signals, confirming the results presented above and the requirement of serine for its association with RTA.
ORF59 PM3 lacks the ability to bind to oriLyt.
Since the binding of ORF59 PM3 to RTA was severely affected, we wanted to determine whether this mutant was also affected in its ability to get recruited to the oriLyt chromatin, which is required for processivity function. We performed a chromatin immunoprecipitation assay in an overexpression system after transfection of 293L cells with RTA-GFP or oriLyt (8088sc) containing the plasmid pLVX-GFP-Flag vector, pLVX-GFP-ORF59-Flag, or pLVX-GFP-PM3-Flag. Sonicated chromatins were immunoprecipitated with anti-Flag, anti-RTA, and IgG-matched control antibodies. A fraction of the immunoprecipitated chromatin was analyzed in a Western blot to ensure that these antibodies were efficiently precipitating chromatins (Fig. 7A and B). DNA extracted from the immunoprecipitated chromatin was used for quantitative PCR (qPCR) with primers targeted to amplify the RTA response element (RRE) of the oriLyt sequence. The results, analyzed using the ΔΔCT method, showed a dramatic decrease in the ability of ORF59 to bind to oriLyt when Ser376, Ser378, and Ser379 (PM3) of ORF59 were mutated to alanine (Fig. 7C; compare the black bar with the dark gray bar). Since ORF59 binds to RRE through RTA, we also quantified the binding of RTA in this overexpression ChIP. Binding of RTA to RRE was comparable (range, 1- to 1.5-fold) in all three samples, suggesting that RTA binding was unaffected by PM3 and the reduction in PM3 binding to oriLyt was solely due its inability to associate with RTA (Fig. 7D). These experiments were performed three independent times, and all of them showed almost similar fold reductions in the binding of PM3 to oriLyt.
Fig 7.
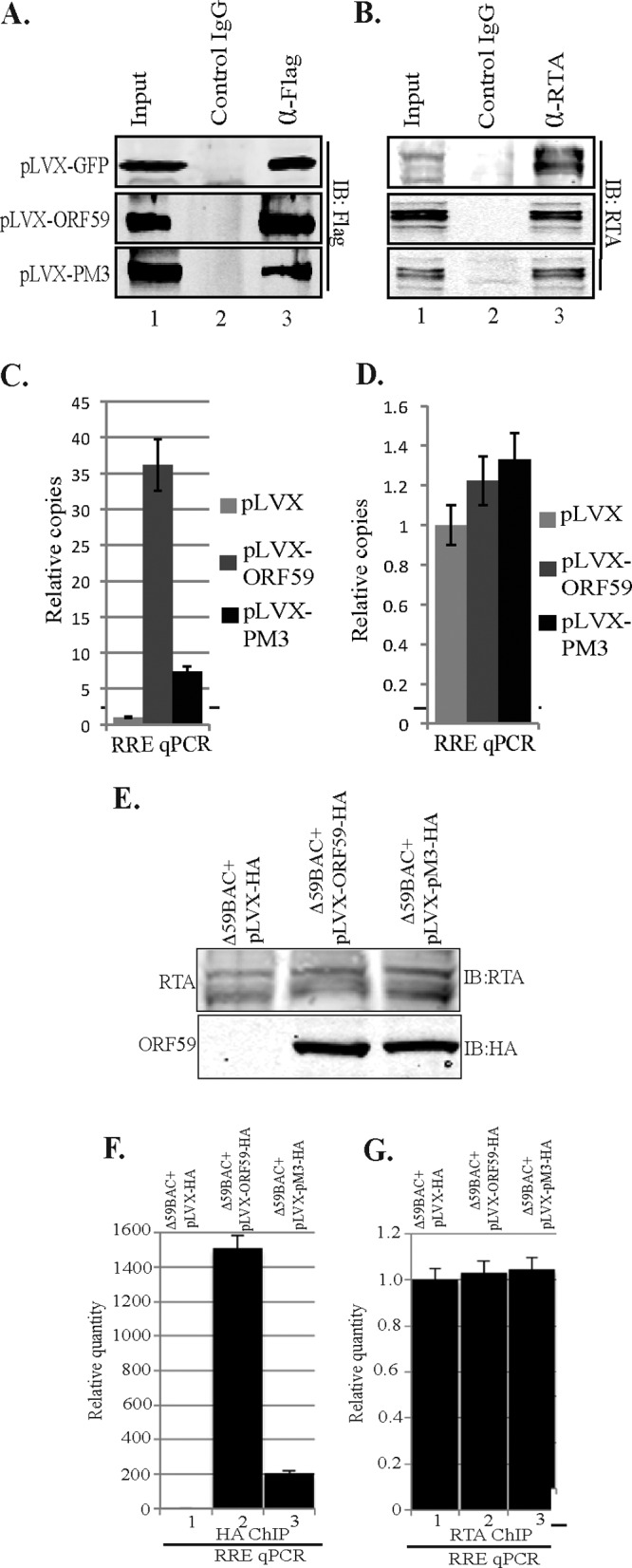
(A) ORF59 PM3 was defective in binding to oriLyt chromatin. 293L cells were transfected with RTA-GFP, the 8088sc-expressing plasmid (which contains oriLyt), and either the GFP-Flag-lentivirus vector, pLVX-ORF59-Flag, or pLVX-PM3-Flag. ChIP was done with anti-Flag and anti-RTA antibodies. Fractions of the precipitated proteins were resolved on an SDS-polyacrylamide gel to check for the immunoprecipitated chromatin with anti-Flag (ORF59) (A) or anti-RTA (B) antibodies. (C) Chromatin-bound oriLyt DNA with ORF59 and PM3 was analyzed by quantitative real-time PCR using RRE primers. The results were analyzed using the ΔΔCT method and are presented as the fold change compared to the result for the vector control (pLVX). A significant decrease of PM3 binding to oriLyt compared to that for wt ORF59 was determined. (D) Relative binding of RTA to oriLyt chromatin was relatively unaffected. (E) Recruitment of ORF59 PM3 to oriLyt chromatin was affected in KSHV BAC36. BAC36 ΔORF59 transfected into 293L cells expressing wt ORF59, PM3, or empty vector (pLVX-HA) was induced for lytic reactivation. (E) Expression of RTA postinduction and detection of ORF59 with anti-HA Western blotting. Δ59BAC36, BAC36 ΔORF59, (F) Detection of chromatin-bound oriLyt with wt ORF59 and the PM3 mutant. (G) Relative number of copies of RTA-bound oriLyt.
We also confirmed the recruitment of ORF59 and PM3 on oriLyt chromatin in BAC36 ΔORF59 stable cells complemented with the respective proteins. Cells were induced to undergo lytic reactivation by a treatment with sodium butyrate and TPA. Induction of lytic reactivation was confirmed by detection of RTA protein in a Western blot (Fig. 7E). Expression of ORF59 and PM3 was also detected using anti-HA Western blotting with cells undergoing lytic reactivation (Fig. 7E). Chromatin was immunoprecipitated with anti-HA and anti-RTA antibodies, and the relative number of copies of oriLyt bound to these two proteins was determined using a vector control as a reference (Fig. 7F and G, lanes 1). The relative number of copies of oriLyt determined by RRE quantitation showed specific binding of ORF59 to oriLyt chromatin, which was significantly reduced in the PM3 mutant (Fig. 7F, lane 1). The levels of RTA binding to oriLyt chromatin were unaffected in the vector control as well as the PM3 mutant, a finding which was similar to the overexpression ChIP data (Fig. 7D and G).
KSHV BAC36 ΔORF59 reconstituted with ORF59 PM3 is defective in virion production.
In order to determine the effect of the ORF59 S376A, S378A, and S379A (PM3) point mutations on the life cycle of KSHV, we used lentivirus vectors, ORF59, or ORF59 PM3 to reconstitute the ORF59 deletion in a BAC36 ΔORF59 BACmid. The BACmid has been proven to be a useful tool for determining the role of individual viral genes or mutations in the gene in the context of the viral genome (42).
BAC36 ΔORF59 BACmid was generated by site-directed homologous recombination using a Kan cassette and FLIP recombination (Fig. 8A), as previously stated (39, 40). The site of Kan cassette insertion was verified by digesting the intermediate BACmid with PstI and analyzing it via Southern blotting using a Kan probe. The resulting fragment containing the Kan cassette yielded a fragment of the expected size (5,883 bp) with the Kan probe (Fig. 8B). FLIP recombinase was then activated to remove the Kan cassette, leaving one FRT site (5′-GAAGTTCCTATTCTCTAGAAAGTATAGGAACTTC-3′) at the mutation site. Although the BACmid contains the entire ORF59 sequence, the insertion of the remaining FRT site interrupted the ORF59 open reading frame (ORF) due to a stop codon and thus blocked ORF59 expression. Furthermore, the absence of the Kan cassette was verified with a Southern blot probing for the Kan cassette. The junction sequence with the FRT insertion was PCR amplified and sequenced to confirm the FRT insertion. The remainder of ORF59 was detected with an ORF59 probe in a Southern blot to further validate the clone (Fig. 8B).
Fig 8.

BAC36 ΔORF59 was generated with Red recombination. (A) General schematic of the Red recombination method used to generate the ΔORF59 BACmid. A PCR-amplified Kan cassette containing homologous ends to ORF59 was transformed into EL350 bacteria containing the BAC36 KSHV genome. FLIP recombinase was then activated in the bacterial culture to remove the Kan cassette using flanking FRT sequences. The final product was sequenced to show the remaining FRT sequence, which contains a stop codon, which thus blocked ORF59 synthesis. nt, nucleotides; AA seq, amino acid sequence. (B) The intermediate clone was screened by Southern blotting, with a band at 5,883 bp expected to be seen when the BACmid was digested with PstI and probed with the Kan probe. As shown, the wild type and the final BACmid did not contain a Kan cassette. Since the ORF59 sequence was not deleted but was interrupted, the presence of the ORF59 sequence in the BACmid was verified with an ORF59 probe.
In order to reconstitute BAC36 ΔORF59, we first transduced 293L cells with the lentivirus pLVX vector pLVX-ORF59-HA or pLVX-ORF59-PM3-HA. Following a puromycin selection for transduced cells, we used the Metafectene Pro reagent to transfect them with the BAC36 ΔORF59 BACmid. A dual selection with puromycin and hygromycin ensured that all surviving cells contained the lentivirus and BAC36 ΔORF59. In addition, by use of a GFP cassette in the BAC backbone, we were able to monitor the presence of BAC by GFP fluorescence (data not shown). We also verified the stable maintenance of the BACmid with anti-LANA and anti-RTA Western blots as well as the expression of ORF59 and PM3 with HA-probed Western blots (Fig. 9A and B).
Fig 9.
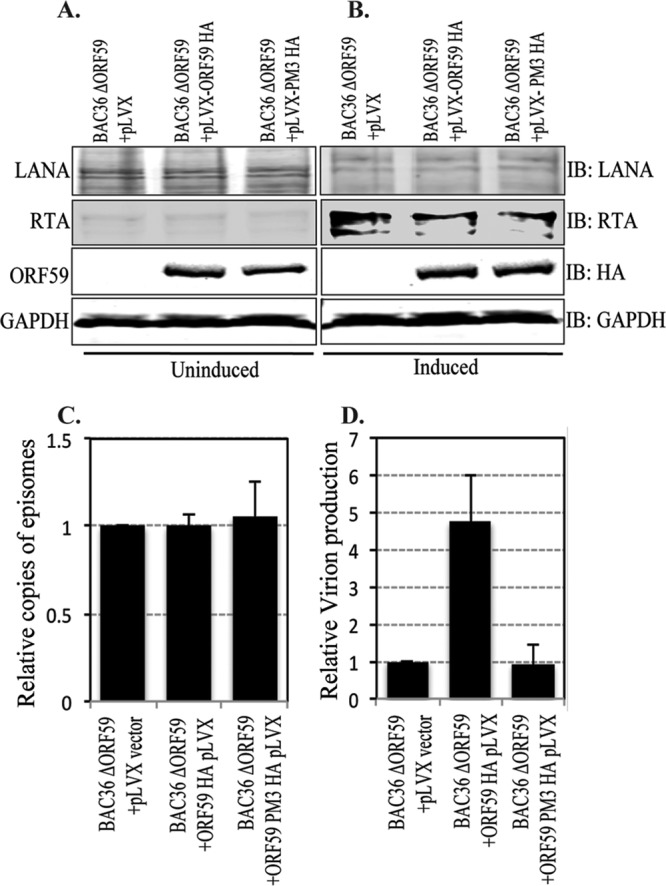
The ORF59 PM3 mutant of KSHV is defective in virion production. (A and B) Cells transfected with BAC36 ΔORF59 into either empty lentivirus vector, wt ORF59-expressing cells, or PM3-expressing cells were tested for LANA-, RTA-, and HA-tagged ORF59 or PM3 expression. Uninduced cells were shown to express LANA (IB: LANA) and ORF59 and PM3 (IB: HA) but to express very little RTA (IB: RTA). The results for GAPDH (IB: GAPDH) are presented to show equal loading. (B) Cells were induced with TPA and sodium butyrate to induce reactivation, which was confirmed by the detection of RTA (IB: RTA) All these cell lines showed similar levels of LANA RTA expressions. (C) DNA was extracted from uninduced cells to examine the latently maintained genome copies in these cell lines. LANA (ORF73) primers were used to calculate the KSHV genome copy number using the ΔΔCT method of quantitation. The relative number of copies of episomally maintained genomes was plotted by using a vector control as a reference. The results showed maintenance of almost identical numbers of viral episomes in these cell lines. (D) Cells were induced with 20 ng TPA and 1.5 mM sodium butyrate for 5 days, after which the supernatant was collected and virion DNA was extracted. The relative numbers of virions produced were determined by real-time qPCR, which showed a significant decrease in virion production in ORF59 with mutated Ser376, Ser378, and Ser379 (PM3) compared to that in cells with wt ORF59.
Approximately 5 × 106 cells with BAC36-ΔORF59-pLVX, BAC36-ΔORF59-pLVX-ORF59-HA, or BAC36-ΔORF59-pLVX PM3-HA were collected for a modified Hirt's extraction to compare the number of copies of the KSHV genome latently maintained in these cells. The purified DNA was used for qPCR with LANA (ORF73) primers, which determined that the genome copy number in the latently persisting genome was almost identical between these three cell lines (Fig. 9C).
In order to determine whether PM3, which lacks the ability to bind to RTA, has any effect on viral DNA replication and, thus, virion production, we compared the numbers of virions produced from BAC36 ΔORF59 reconstituted with either wt or PM3 ORF59. The same numbers of cells were induced with 20 ng of 12-O-tetradecanoylphorbol-13-acetate (TPA) per ml and 1.5 mM sodium butyrate (Sigma-Aldrich, St. Louis, MO) for 5 days. The supernatant was collected to determine the relative number of copies of virions produced from these cells by extracting the DNA from the virions released into the culture supernatants. Purified DNA was used for qPCR with LANA (ORF73) primers, and the relative amounts of virions produced were plotted using a vector control (BAC36-ΔORF59-pLVX) as a reference. The results showed a significant reduction in the amounts of virions produced from the cells complemented with ORF59 mutated at Ser376, Ser378, and Ser379 (PM3) (Fig. 9D). Induction of lytic reactivation by sodium butyrate and TPA was confirmed with BAC36-containing 293L cells, which showed an approximately 9-fold increase in the numbers of virions after induction of lytic DNA replication. Since the virion production levels of PM3-reconstituted BAC36 ΔORF59 was similar to that of empty vector-reconstituted BAC36 ΔORF59, we conclude that the function of ORF59 is dependent on the phosphorylation of the Ser376, Ser378, or Ser379 residue by the viral kinase (ORF36).
DISCUSSION
Kaposi's sarcoma-associated herpesvirus is closely associated with multiple human malignancies, including Kaposi's sarcoma (KS), primary effusion lymphomas (PELs), and multicentric Castleman's disease (MCD). KSHV establishes a lifelong latency following a primary infection with the help of viral and cellular factors. During latency, viral replication is dependent on the cellular replication machinery. Upon reactivation, most of the viral genes are expressed, including those necessary and sufficient for KSHV lytic replication (ORF9, ORF6, ORF40/41, ORF44, ORF56, ORF59, and ORF50) (24). Interestingly, most of the cells in KSHV tumors are latent, and only a subpopulation (approximately 2 to 5% of latent cells) undergoes spontaneous lytic reactivation to produce virions which are implicated in tumorigenesis (10, 11).
Furthermore, similar to other herpesviruses, the replication and transcription activator (RTA) is accepted as one of the most important proteins required for lytic reactivation (43). RTA binds to the C/EBPα and the RTA response element (RRE) binding motifs within the origin of lytic replication (oriLyt) and as a result activates lytic DNA replication through transcriptional activation as well as recruitment of additional factors (6–8). RTA recruits the viral processivity factor ORF59 to oriLyt, where it acts as an accessory factor or sliding clamp, stabilizing the binding between viral polymerase (ORF9) and the DNA. ORF59 forms a homodimer in the cytoplasm, binds to ORF9 directly, and specifically recruits it to the nucleus, which is crucial for long-chain DNA synthesis (32, 36, 44, 45). Rossetto et al. have shown that ORF59's binding to the C/EBPα binding motif within oriLyt is necessary for its function and is dependent on its binding to RTA (31). Furthermore, the processivity function of ORF59 is dependent upon its interaction with RTA; consequently, the function of the viral polymerase is also dependent upon this interaction (31).
Although the structures of processivity factors among different herpesviruses are variable, their functions are fairly conserved. The human cytomegalovirus (hCMV) processivity factor, ppUL44, is phosphorylated by viral Ser/Thr kinase (ppUL97), which modulates its ability to localize to the nucleus (33). BMRF1, the Epstein-Barr virus (EBV) processivity factor, is phosphorylated by the BGLF4 viral kinase within a hinge region-like domain known to be important for inducing conformational changes (34). Consequently, phosphorylation by BGLF4 enhances the transactivation activity of Zta (RTA homolog) and the synergistic activation of lytic replication at oriLyt (35). Similarly, ORF59 is a phosphoprotein which is phosphorylated by the KSHV Ser/Thr kinase, ORF36, but the consequences of phosphorylation on DNA processivity or virion production were not evaluated (36).
Phosphorylation of viral processivity factors is an important modification among human herpesviruses. Our aim was to identify the ORF36 target sites on the KSHV processivity factor (ORF59) and the possible effects of phosphorylation on viral production. In this study, residues Ser376, Ser378, and Ser379 were identified as the major ORF36 kinase targets on ORF59 through use of the KinasePhos program (http://kinasephos.mbc.nctu.edu.tw/) (Fig. 3A). We mapped the binding region of ORF36 to ORF59 between aa 133 and 264 on ORF59 (Fig. 1), which did not correspond with the identified phosphorylation sites, Ser376, Ser378, and Ser379. Nonetheless, as indicated by the ORF59 crystal structure, these regions are in close proximity and may thus allow ORF36 to phosphorylate ORF59 at the C terminus, which harbors the identified phosphorylation sites (46). In fact, detectable binding between kinase and its substrate is not required for phosphorylation to take place. For instance, no binding between the EBV processivity factor (BMRF1) and viral kinase (BGLF4) was detected; however, phosphorylation of BMRF1 by BGLF4 was revealed to be necessary for the initiation of lytic replication at oriLyt (35).
Due to the close proximity of the identified phosphorylation sites with the ORF59 dimerization domain, the ORF9-interacting domain, and nuclear localization signals, we investigated the consequences that these mutations could have on lytic DNA replication by evaluating the effects of S376A, S378A, and S379A (ORF59 PM3) on known functions of ORF59. Binding between ORF9, viral polymerase, and ORF59 is crucial for DNA replication; therefore, we determined that binding between these two proteins and translocation of ORF9 to the nucleus was not affected by the mutation of phosphorylation sites of PM3 (Ser376, Ser378, and Ser379) (Fig. 5C). The binding between ORF59 and ORF9 is dependent upon the ability of ORF59 to dimerize; therefore, we expected that phosphorylation might not have any effect on dimerization of ORF59 (47). Nevertheless, we confirmed that, indeed, the lack of phosphorylation did not affect the dimerization of ORF59 (Fig. 5C).
Notably, upon examination of binding between ORF59 point mutants and RTA, we determined that the S376A, S378A, and S379A mutations of ORF59 PM3 abolished its binding to RTA (Fig. 5E). Fine mapping of the residues required for their interaction suggested that both Ser378 and Ser379 are cooperatively required for binding to RTA and mutants with both of these mutations showed reduced binding. Previous data suggested that dimerization of ORF59 is not necessary for its interaction with RTA; therefore, it is not surprising that despite ORF59 PM3's ability to dimerize, it was unable to bind to RTA. Interestingly, a similar interaction was observed between BMRF1, EBV processivity factor, and BZLF1, which plays an important role in EBV lytic replication (48). These findings are significant because the reduced binding between PM3 and RTA hampered the recruitment of ORF59 to oriLyt, which could subsequently affect the synthesis of viral DNA.
Moreover, our chromatin immunoprecipitation assay and reconstituted BAC36 ΔORF59 demonstrated the downstream effects of the S376A, S378A, and S379A ORF59 mutations on ORF59 recruitment to oriLyt chromatin and synthesis of viral DNA. Although the replacement of ORF59 Ser376, Ser378, and Ser379 with alanines had no effect on its ability to form a homodimer or to bind to viral polymerase (ORF9), the mutations hindered its ability to bind to RTA, which raised the question of whether ORF59 PM3 is able to bind to oriLyt and promote DNA synthesis. We used ChIP to compare the efficiency of ORF59 and its PM3 binding to oriLyt in cells transfected with an oriLyt plasmid in the presence of RTA as well as in cells maintaining the KSHV genome. The results showed a significant decrease in ORF59 PM3 binding at oriLyt compared with that of the wt (Fig. 7C and F). The mutations further proved to have deleterious effects on viral production in BAC ΔORF59 reconstituted with PM3 versus wt ORF59 (Fig. 8). In fact, the virion production of the PM3-reconstituted cell line was very similar to that of the control consisting of a mutant with a full ORF59 deletion, suggesting that the function of ORF59 is largely dependent upon its binding to RTA and phosphorylation of serines at residues 376, 378, and 379.
The precise mechanism by which RTA interacts with ORF59 is not yet clear. The RTA interaction domain of ORF59 has been mapped to aa 266 to 396, and its binding to oriLyt is dependent upon its interaction with RTA (31). These data demonstrate that the binding between RTA and ORF59 is crucial for KSHV DNA synthesis and virion production. We propose that phosphorylation of ORF59 by ORF36 viral kinase at serines 376, 378, and 379 may promote the formation of stabilizing structures which foster the binding of ORF59 and RTA at oriLyt. The phosphorylation of ORF59 at serines 376, 378, and 379 by ORF36 is fundamental to KSHV propagation, as in the absence of this interaction, only limited numbers of virions are produced. Furthermore, this provides a possible model for herpesvirus kinases possibly regulating viral lytic DNA replication at oriLyt.
ACKNOWLEDGMENTS
We thank members of G. S. Pari's laboratory for discussions and Vydehi Kanneganti for microscopy.
This work was supported by CA126182 (K99/R00 to S.C.V.) and the Reno Cancer Foundation.
Footnotes
Published ahead of print 15 May 2013
REFERENCES
- 1. Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332:1186–1191 [DOI] [PubMed] [Google Scholar]
- 2. Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d'Agay MF, Clauvel JP, Raphael M, Degos L, Sigaux F. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86:1276–1280 [PubMed] [Google Scholar]
- 3. Li Q, Zhou F, Ye F, Gao SJ. 2008. Genetic disruption of KSHV major latent nuclear antigen LANA enhances viral lytic transcriptional program. Virology 379:234–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Si H, Verma SC, Lampson MA, Cai Q, Robertson ES. 2008. Kaposi's sarcoma-associated herpesvirus-encoded LANA can interact with the nuclear mitotic apparatus protein to regulate genome maintenance and segregation. J. Virol. 82:6734–6746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boshoff C. 2003. Kaposi virus scores cancer coup. Nat. Med. 9:261–262 [DOI] [PubMed] [Google Scholar]
- 6. Gradoville L, Gerlach J, Grogan E, Shedd D, Nikiforow S, Metroka C, Miller G. 2000. Kaposi's sarcoma-associated herpesvirus open reading frame 50/Rta protein activates the entire viral lytic cycle in the HH-B2 primary effusion lymphoma cell line. J. Virol. 74:6207–6212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lukac DM, Renne R, Kirshner JR, Ganem D. 1998. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 252:304–312 [DOI] [PubMed] [Google Scholar]
- 8. Sun R, Lin SF, Gradoville L, Yuan Y, Zhu F, Miller G. 1998. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. U. S. A. 95:10866–10871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ma W, Galvin TA, Ma H, Ma Y, Muller J, Khan AS. 2011. Optimization of chemical induction conditions for human herpesvirus 8 (HHV-8) reactivation with 12-O-tetradecanoyl-phorbol-13-acetate (TPA) from latently-infected BC-3 cells. Biologicals 39:158–166 [DOI] [PubMed] [Google Scholar]
- 10. Chen L, Lagunoff M. 2005. Establishment and maintenance of Kaposi's sarcoma-associated herpesvirus latency in B cells. J. Virol. 79:14383–14391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Glaunsinger B, Ganem D. 2004. Lytic KSHV infection inhibits host gene expression by accelerating global mRNA turnover. Mol. Cell 13:713–723 [DOI] [PubMed] [Google Scholar]
- 12. Carroll PA, Brazeau E, Lagunoff M. 2004. Kaposi's sarcoma-associated herpesvirus infection of blood endothelial cells induces lymphatic differentiation. Virology 328:7–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ciufo DM, Cannon JS, Poole LJ, Wu FY, Murray P, Ambinder RF, Hayward GS. 2001. Spindle cell conversion by Kaposi's sarcoma-associated herpesvirus: formation of colonies and plaques with mixed lytic and latent gene expression in infected primary dermal microvascular endothelial cell cultures. J. Virol. 75:5614–5626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gao SJ, Deng JH, Zhou FC. 2003. Productive lytic replication of a recombinant Kaposi's sarcoma-associated herpesvirus in efficient primary infection of primary human endothelial cells. J. Virol. 77:9738–9749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hong YK, Foreman K, Shin JW, Hirakawa S, Curry CL, Sage DR, Libermann T, Dezube BJ, Fingeroth JD, Detmar M. 2004. Lymphatic reprogramming of blood vascular endothelium by Kaposi sarcoma-associated herpesvirus. Nat. Genet. 36:683–685 [DOI] [PubMed] [Google Scholar]
- 16. Naranatt PP, Krishnan HH, Svojanovsky SR, Bloomer C, Mathur S, Chandran B. 2004. Host gene induction and transcriptional reprogramming in Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8)-infected endothelial, fibroblast, and B cells: insights into modulation events early during infection. Cancer Res. 64:72–84 [DOI] [PubMed] [Google Scholar]
- 17. Pan H, Zhou F, Gao SJ. 2004. Kaposi's sarcoma-associated herpesvirus induction of chromosome instability in primary human endothelial cells. Cancer Res. 64:4064–4068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qian LW, Greene W, Ye F, Gao SJ. 2008. Kaposi's sarcoma-associated herpesvirus disrupts adherens junctions and increases endothelial permeability by inducing degradation of VE-cadherin. J. Virol. 82:11902–11912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Qian LW, Xie J, Ye F, Gao SJ. 2007. Kaposi's sarcoma-associated herpesvirus infection promotes invasion of primary human umbilical vein endothelial cells by inducing matrix metalloproteinases. J. Virol. 81:7001–7010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sadagopan S, Sharma-Walia N, Veettil MV, Raghu H, Sivakumar R, Bottero V, Chandran B. 2007. Kaposi's sarcoma-associated herpesvirus induces sustained NF-kappaB activation during de novo infection of primary human dermal microvascular endothelial cells that is essential for viral gene expression. J. Virol. 81:3949–3968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sharma-Walia N, Raghu H, Sadagopan S, Sivakumar R, Veettil MV, Naranatt PP, Smith MM, Chandran B. 2006. Cyclooxygenase 2 induced by Kaposi's sarcoma-associated herpesvirus early during in vitro infection of target cells plays a role in the maintenance of latent viral gene expression. J. Virol. 80:6534–6552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang HW, Trotter MW, Lagos D, Bourboulia D, Henderson S, Makinen T, Elliman S, Flanagan AM, Alitalo K, Boshoff C. 2004. Kaposi sarcoma herpesvirus-induced cellular reprogramming contributes to the lymphatic endothelial gene expression in Kaposi sarcoma. Nat. Genet. 36:687–693 [DOI] [PubMed] [Google Scholar]
- 23. Ye FC, Blackbourn DJ, Mengel M, Xie JP, Qian LW, Greene W, Yeh IT, Graham D, Gao SJ. 2007. Kaposi's sarcoma-associated herpesvirus promotes angiogenesis by inducing angiopoietin-2 expression via AP-1 and Ets1. J. Virol. 81:3980–3991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. AuCoin DP, Colletti KS, Cei SA, Papouskova I, Tarrant M, Pari GS. 2004. Amplification of the Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 lytic origin of DNA replication is dependent upon a cis-acting AT-rich region and an ORF50 response element and the trans-acting factors ORF50 (K-Rta) and K8 (K-bZIP). Virology 318:542–555 [DOI] [PubMed] [Google Scholar]
- 25. Carroll KD, Khadim F, Spadavecchia S, Palmeri D, Lukac DM. 2007. Direct interactions of Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 ORF50/Rta protein with the cellular protein octamer-1 and DNA are critical for specifying transactivation of a delayed-early promoter and stimulating viral reactivation. J. Virol. 81:8451–8467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chang PJ, Miller G. 2004. Autoregulation of DNA binding and protein stability of Kaposi's sarcoma-associated herpesvirus ORF50 protein. J. Virol. 78:10657–10673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sakakibara S, Ueda K, Chen J, Okuno T, Yamanishi K. 2001. Octamer-binding sequence is a key element for the autoregulation of Kaposi's sarcoma-associated herpesvirus ORF50/Lyta gene expression. J. Virol. 75:6894–6900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wilson SJ, Tsao EH, Webb BL, Ye H, Dalton-Griffin L, Tsantoulas C, Gale CV, Du MQ, Whitehouse A, Kellam P. 2007. X box binding protein XBP-1s transactivates the Kaposi's sarcoma-associated herpesvirus (KSHV) ORF50 promoter, linking plasma cell differentiation to KSHV reactivation from latency. J. Virol. 81:13578–13586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ye J, Shedd D, Miller G. 2005. An Sp1 response element in the Kaposi's sarcoma-associated herpesvirus open reading frame 50 promoter mediates lytic cycle induction by butyrate. J. Virol. 79:1397–1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang L, Chiu J, Lin JC. 1998. Activation of human herpesvirus 8 (HHV-8) thymidine kinase (TK) TATAA-less promoter by HHV-8 ORF50 gene product is SP1 dependent. DNA Cell Biol. 17:735–742 [DOI] [PubMed] [Google Scholar]
- 31. Rossetto CC, Susilarini NK, Pari GS. 2011. Interaction of Kaposi's sarcoma-associated herpesvirus ORF59 with oriLyt is dependent on binding with K-Rta. J. Virol. 85:3833–3841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen Y, Ciustea M, Ricciardi RP. 2005. Processivity factor of KSHV contains a nuclear localization signal and binding domains for transporting viral DNA polymerase into the nucleus. Virology 340:183–191 [DOI] [PubMed] [Google Scholar]
- 33. Alvisi G, Marin O, Pari G, Mancini M, Avanzi S, Loregian A, Jans DA, Ripalti A. 2011. Multiple phosphorylation sites at the C-terminus regulate nuclear import of HCMV DNA polymerase processivity factor ppUL44. Virology 417:259–267 [DOI] [PubMed] [Google Scholar]
- 34. Tucker PW, Slightom JL, Blattner FR. 1981. Mouse IgA heavy chain gene sequence: implications for evolution of immunoglobulin hinge axons. Proc. Natl. Acad. Sci. U. S. A. 78:7684–7688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang PW, Chang SS, Tsai CH, Chao YH, Chen MR. 2008. Effect of phosphorylation on the transactivation activity of Epstein-Barr virus BMRF1, a major target of the viral BGLF4 kinase. J. Gen. Virol. 89:884–895 [DOI] [PubMed] [Google Scholar]
- 36. Chan SR, Chandran B. 2000. Characterization of human herpesvirus 8 ORF59 protein (PF-8) and mapping of the processivity and viral DNA polymerase-interacting domains. J. Virol. 74:10920–10929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pear WS, Nolan GP, Scott ML, Baltimore D. 1993. Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. U. S. A. 90:8392–8396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Verma SC, Choudhuri T, Kaul R, Robertson ES. 2006. Latency-associated nuclear antigen (LANA) of Kaposi's sarcoma-associated herpesvirus interacts with origin recognition complexes at the LANA binding sequence within the terminal repeats. J. Virol. 80:2243–2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xu Y, Rodriguez-Huete A, Pari GS. 2006. Evaluation of the lytic origins of replication of Kaposi's sarcoma-associated virus/human herpesvirus 8 in the context of the viral genome. J. Virol. 80:9905–9909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, Court DL. 2000. An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 97:5978–5983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Verma SC, Lan K, Choudhuri T, Cotter MA, Robertson ES. 2007. An autonomous replicating element within the KSHV genome. Cell Host Microbe 2:106–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rossetto CC, Pari G. 2012. KSHV PAN RNA associates with demethylases UTX and JMJD3 to activate lytic replication through a physical interaction with the virus genome. PLoS Pathog. 8:e1002680. 10.1371/journal.ppat.1002680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Staudt MR, Dittmer DP. 2007. The Rta/Orf50 transactivator proteins of the gamma-herpesviridae. Curr. Top. Microbiol. Immunol. 312:71–100 [DOI] [PubMed] [Google Scholar]
- 44. Chen X, Lin K, Ricciardi RP. 2004. Human Kaposi's sarcoma herpesvirus processivity factor-8 functions as a dimer in DNA synthesis. J. Biol. Chem. 279:28375–28386 [DOI] [PubMed] [Google Scholar]
- 45. Lin K, Dai CY, Ricciardi RP. 1998. Cloning and functional analysis of Kaposi's sarcoma-associated herpesvirus DNA polymerase and its processivity factor. J. Virol. 72:6228–6232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Baltz JL, Filman DJ, Ciustea M, Silverman JE, Lautenschlager CL, Coen DM, Ricciardi RP, Hogle JM. 2009. The crystal structure of PF-8, the DNA polymerase accessory subunit from Kaposi's sarcoma-associated herpesvirus. J. Virol. 83:12215–12228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bruce AG, Bakke AM, Gravett CA, DeMaster LK, Bielefeldt-Ohmann H, Burnside KL, Rose TM. 2009. The ORF59 DNA polymerase processivity factor homologs of Old World primate RV2 rhadinoviruses are highly conserved nuclear antigens expressed in differentiated epithelium in infected macaques. Virol. J. 6:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang Q, Hong Y, Dorsky D, Holley-Guthrie E, Zalani S, Elshiekh NA, Kiehl A, Le T, Kenney S. 1996. Functional and physical interactions between the Epstein-Barr virus (EBV) proteins BZLF1 and BMRF1: effects on EBV transcription and lytic replication. J. Virol. 70:5131–5142 [DOI] [PMC free article] [PubMed] [Google Scholar]