

Abstract
HIV-1 infections cannot be completely eradicated by drug therapy, as the virus persists in reservoirs. Low-level plasma viremia has been detected in patients treated for over 7 years, but the cellular compartments that support this low-level viremia have not been identified. The decay of HIV-1 during treatment appears to occur in four phases, with the 3rd and 4th phases occurring when the virus is below the limit of detection of conventional assays. Here, we focus on the 3rd phase of decay, which has been estimated to have a half-life of 39 months. We show that follicular dendritic cells (FDC), which have been identified as an HIV reservoir, can be the main source of the low-level viremia detected during the 3rd phase of decay and contribute to viremia at even longer times. Our calculations show that the kinetics of leakage of virus from FDC is consistent with three types of available clinical data.
INTRODUCTION
Highly active antiretroviral therapy (HAART) is effective in suppressing progression of HIV infection. In the first few months of suppressive therapy, the plasma HIV-1 RNA load experiences a biphasic decay (1–5). The first phase has a short half-life (1 to 2 days) and is attributed to the loss of infected activated CD4+ T cells. The second phase has a half-life of 1 to 4 weeks and has been attributed to the loss of long-lived infected cells such as resting CD4+ T cells or macrophages (4, 6). The presence of other decay phases has been suggested (5, 7), and a recent study by Palmer et al. (8), using a very sensitive assay with a lower limit of quantification of 0.63 HIV RNA copies/ml (9), identified a third and fourth decay phase in patients on suppressive therapy for 7 years (Fig. 1). Phase 3 had a half-life, τ3, of 39 weeks (supported by a recent study [10]), and phase 4 showed a nearly constant plasma viral load for the period studied out to 7 years. Overall, the median plasma viral load, V(t), for patients on suppressive therapy can be represented by the following equation:
| (1) |
where t is the time on therapy, V1(t) through V4(t) designate the contributions to the plasma viral load of the virus generated by the compartments 1 through 4, V1(0) through V4(0) designate the contributions of the virus generated by compartments 1 through 4 to the baseline viral load, λi = ln2/τi is the decay rate constant of phase i, and τi is the half-life of decay phase i, i = 1, …, 3.
Fig 1.
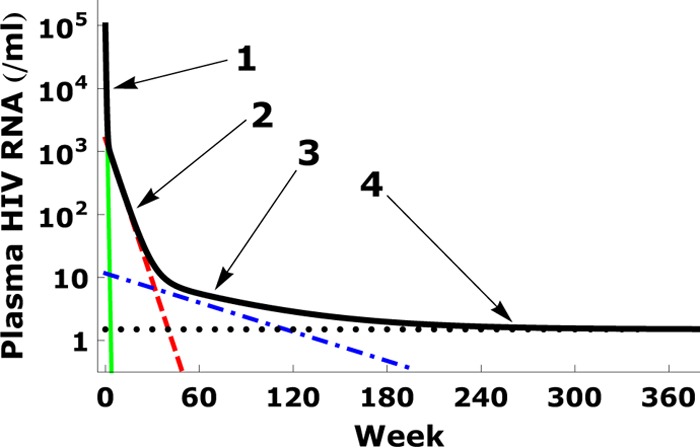
Four-phase decay of plasma viral load. The median viral load decay observed by Palmer et al. (8) is shown by the thick black line. The four phases are indicated by the arrows, and the four corresponding terms in equation 1 are shown by solid (green), dashed (red), dash-dotted (blue), and dotted (black) lines, respectively, where we use V1(0) = 105 copies/ml, V2(0) = 103.2 copies/ml, V3(0) = 11.6 copies/ml, and V4(0) = 1.5 copies/ml, as well as half-lives τ1 = 1.5 days, τ2 = 4 weeks, and τ3 = 39 weeks, as reported in the work of Palmer et al. (8).
The cellular compartments responsible for decay phases 3 and 4 have not been identified. Palmer et al. (8) suggest that candidates for “compartment 3” and “compartment 4” are sanctuary sites with suboptimal drug penetration, long-lived productively infected cells, and latently infected CD4+ cells. Another candidate is follicular dendritic cells (FDC), which can bind virus and later can release that virus into the circulation (11, 12). Among these candidates, latently infected CD4+ cells have attracted the most attention (13). However, the residual viremia in plasma samples from patients on long-term suppressive therapy can be genetically distinct from viral sequences identified in latently infected CD4+ cells (14), suggesting that the source of the third or fourth decay phases may be not only latently infected CD4+ cells.
Here, we probe the possibility that FDC-associated virus is the main biological source of decay phase 3 virus. FDC reversibly bind virions on their surface and can slowly release them back into the circulation (12). In the asymptotic phase of HIV infection, the pool of virus stored on FDC is estimated to be ∼1011 HIV RNA copies (i.e., 5 × 1010 virions), greatly exceeding the viral load in infected cells or in plasma (15). During HAART, the viral load is reduced significantly, and most trapped virions dissociate from FDC. Nevertheless, due to the multivalent nature of FDC-HIV binding, i.e., the ability of each virion to bind multiple surface receptors on FDC, abundant virions remain trapped for a prolonged time, serving as an HIV-1 reservoir that hinders the eradication of HIV-1 (16–22).
Using a modeling approach, we study the dynamics of HIV-1 virions on FDC and quantify the contribution of FDC to the plasma viral load. Taking into account variations in the FDC surface receptor density and in the number of FDC binding sites per virion, we obtain a model for the time-dependent decay of the FDC viral pool and show that FDC may serve as compartment 3. We also show that our model is consistent with data from an older study in which the amount of FDC-associated virus was quantified in patients on HAART through longitudinal tonsillar biopsies (12). Knowing the sources of virus contributing to persistent viremia is an important step toward the goal of viral eradication.
MATERIALS AND METHODS
Clinical specimens.
The data from the work of Palmer et al. (8) that we analyze were previously published and kindly provided by F. Maldarelli. The data came from the Abbott M97-720 trial (8), where 100 antiretroviral-naive patients received lopinavir-ritonavir (400/100 mg twice daily) with stavudine and lamivudine twice daily for up to 7 years (8, 23). Among the 62 patients who completed 7 years of treatment, 47 patients controlled viremia without “blips.” To exclude blips, plasma HIV-1 RNA had to remain at <50 copies/ml by standard Amplicor assay at all visits after week 96. When the 47 patients were further tested by single-copy assay (SCA) (9), 6 participants with inefficient amplification of pretherapy HIV-1 RNA by SCA were excluded for further analysis, and one additional patient had invalid SCA results with internal standard values unacceptably low for all samples. As a result, 40 patients with 293 samples were used for longitudinal analyses (8).
The data from the work of Cavert et al. (12) that we fitted were previously published and kindly provided by A. Haase. They were collected using in situ hybridization and computer-assisted quantitative image analysis to measure HIV-1 RNA in virion-antibody immune complexes deposited on FDC (15). Tonsil biopsy specimens were obtained from 34 patients in a treatment combining ritonavir, zidovudine, and lamivudine. Participants underwent tonsil biopsy 2 weeks before and 2 days, 22 days, and 24 weeks after treatment commenced. Each tonsil biopsy specimen was cut in half. One portion was fixed and embedded in paraffin; the other portion was flash-frozen for later extraction and assay of viral nucleic acids. Paraffin blocks were then sectioned and hybridized in situ to a 35S-labeled RNA probe complementary to >90% of the sequences in HIV-1 RNA. After autoradiography, the hybridization signal overlying FDC or monocytes (MNCs) was quantitated by computer-assisted image analysis (15). Sequential tissue samples suitable for evaluation from 10 individuals were obtained.
While the HIV RNA densities on FDC were measured (12), the total amount of HIV on FDC in a patient can be only estimated. To assess the general decay behavior of HIV on FDC, we assume that the measured baseline HIV RNA density on FDC of a patient when multiplied by the surface area of the FDC in the body corresponds to a total of 1011 HIV RNA copies in the patient, as previously estimated (15).
Model of virion-FDC binding.
HIV-1 virions are held on the FDC surface through interactions between the proteolytic fragments of complement C3 (C3dg and C3d) and complement receptors, especially CR2, on FDC, as well as antibodies on HIV-1 and Fc receptors on FDC (11, 24–26). Although there may be multiple types of bonds holding a virion to the FDC surface, for simplicity we shall refer only to the C3-CR2 interaction. The binding between virions and FDC surfaces is multivalent. A virion can have many bound C3 fragments and therefore can bind to i = 1, 2, …, n CR2 receptors at the same time. The total number of possible bonds between a virion and FDC is called the valence, and hence, in our model, HIV is treated as an n-valent particle.
To account for multivalent binding and dissociation of HIV-1 virions, Hlavacek et al. (11, 24–26) developed the following mass-action model:
| (2) |
and
| (3) |
where Bi is the surface density of virions bound to i receptors, RT is the total surface density of receptors that HIV can bind (e.g., the average number of CR2 per unit area on FDC), R is the surface density of unbound receptors, V is the concentration of free virions in plasma and other extracellular body fluids, kx is the single site forward rate constant for formation of a new bond between a virion and the FDC surface, k−x is the single site reverse rate constant for a bond to break, kr is the effective rate constant for the detachment of a virion from FDC, and α is the forward rate constant describing the binding of virion to the FDC surface (Fig. 2). The association and dissociation terms in equation 2 include factors that account for the number of free and bound sites on a virion. Free virions can get trapped on FDC even during therapy, as reflected by the αVR term in the first line of equation 2.
Fig 2.
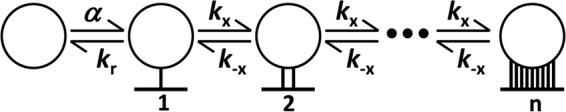
Multivalent binding and unbinding steps between an HIV-1 virion (circle) and surface receptors on FDC. Free virions attach to the FDC surface and first form a single bond with rate constant α. This bond can break with rate constant kr, releasing the virion back into solution. Once the virion is bound to the surface, the rate constant to form an additional bond is kx, and the reverse rate constant is k−x. Equation 2 in Materials and Methods describes the kinetics of these reactions and includes statistical factors that incorporate the number of ways that bonds can form and break.
The bound states of virions {Bi} on FDC together could be viewed as an energy well trapping the virions. Hlavacek et al. (26) found that the rate that virions leave FDC depends sensitively on the density of unbound receptors, R, to which they can reattach. Indeed, a higher R leads to a stronger attraction and/or a deeper energy well, making it more difficult for trapped virions to leave.
Model of virion leakage to plasma.
Let VFDC stand for the concentration of virions in plasma that have dissociated from FDC. When virions dissociate from FDC, we assume that they become diluted in the extracellular body fluid and become subject to clearance at rate c per virion. Then according to our model
| (4) |
where the volume of extracellular fluid in a person is taken as 15 liters, and the total FDC surface area is estimated such that the total FDC viral load before therapy is virions (12, 15). In our calculations, the representative surface area of FDC in the whole human body is a few thousand cm2, corresponding to ∼109 FDC with a surface area of a few hundred μm2 per cell (24, 27).
Parameter values.
The parameter values used in our calculations are summarized in Table 1. Following the work of Hlavacek et al. (26), the average CR2 density is <RT> = 9.6 × 109 cm−2 (28–30); the number of sites available on a virion to bind CR2 is estimated to be between 10 and 100 (31–33), and we explore using an average value <n> = 20. The estimates for the dissociation rate constants, kr = k−x = 0.1 s−1 (34), and the attachment rate constant, α = 3.6 × 10−11 ml s−1, are also taken from the work of Hlavacek et al. (26). We choose the surface association constant, i.e., the cross-linking rate constant kx, to be 10−12 − 10−11 cm2 s−1, which is in the range of values estimated by Hlavacek et al. (26, 34–37). By trying different values of kx, we find that the viremia data in the work of Palmer et al. (8) and Maldarelli et al. (38) can be simultaneously fitted using kx = 0.38 k−x/<RT>, i.e., kx = 3.96 × 10−12 cm2/s. The clearance rate of free virions is chosen to be c = 23 day−1, as previously estimated (39).
Table 1.
Parameter values used in the calculations
| Parameter | Definition | Value |
|---|---|---|
| <n> | Mean no. of C3 fragments per virion | 20 |
| σn | SD of n | 3.0–7.0 |
| <RT> | Mean surface density on CR2 receptors on FDC | 9.6 × 109 cm−2 |
| σ | SD of RT | 0.1 |
| k−x | Dissociation rate constant | 0.1/s |
| kx | Association rate constant | 10−12-10−11 cm2/s |
| kr | Detachment rate constant | 0.1/s |
| α | Attachment rate constant | 3.6 × 10−11 ml/s |
| V0 | Baseline virion RNA concn (median) | 105 |
| c | Clearance rate constant of free virions | 23/day |
In the patients studied by Palmer et al., the median baseline viral load is V = 105 copies/ml (8). When exploring the effect of other baseline viral loads with our model, we need to rescale all four terms in equation 1. Since V1 dominates the viral load at the beginning of therapy, we set V1 = V(0). To determine how V2, V3, and V4 are rescaled, we examine the viral load at week 4 (dominated by V2), week 60 (dominated by V3 + V4), and week 350 (dominated by V4). From the linear regression lines fitted to the data in the work of Palmer et al. (8), shown in Fig. A1 in the Appendix, we find that a 10-fold change in the baseline viral load translates into a 10-fold change of V1, a 3.2-fold change in V2, a 2.2-fold change in V3, and a 1.6-fold change in V4.
Variation of parameters.
The number of binding sites per virion, i.e., its valence n, most likely varies from virion to virion, as it depends on the number of C3 fragments deposited on a virion. Similarly, the CR2 surface density on FDC, RT, is expected to vary from cell to cell. We assume that RT is lognormally distributed, which is the characteristic distribution of many types of cell surface receptors, such as receptors on T lymphocytes (40–42). Thus, the variation of CR2 surface density RT on FDC is given by
| (5) |
where ln RT has a normal distribution with mean <RT> and standard deviation σ. The number of binding sites on a virion is estimated to be between 10 and 100 (26), and we assume that the number follows a normal distribution,
| (6) |
with mean <n> and standard deviation σn. The parameter values σ = 0.1 and σn = 7.0 (see Fig. 8) are chosen as explained below.
Fig 8.
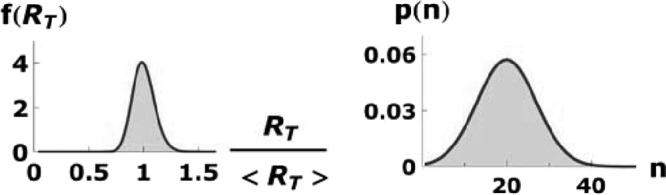
Lognormal distribution of RT and normal distribution of n. The standard deviations are σ = 0.1 and σn = 7.0, respectively.
Numerical calculations and data fitting.
Equations 2 to 4 assume that there is a single value of RT for all FDC and a single value of n for all virions. To include a distribution of values for these parameters in model calculations (see Fig. 8), we include 80 discrete values of n (1 to 80), and 80 discrete bins of RT (from 0.02 <RT> to 1.6 <RT>). Thus, we generalize equation 2 into 80 × 80 = 6,400 similar equations as described by equation A2 in the Appendix and revise equations 3 and 4 to equations A3 and A4 accordingly. The generalized forms of equations 2, 3, and 4, i.e., equations A2, A3, and A4, respectively, are used in all our numerical calculations.
Using these 6,400 equations to perform calculations, we tune parameters σn, σ, and kx to fit three kinds of clinical data, the viral decay on therapy from the work of Palmer et al. (8) (see Fig. 4), the decay of HIV on FDC from the work of Cavert et al. (12) (see Fig. 6), and the relationship between baseline viremia and viremia at week 60 from the work of Maldarelli et al. (38) (see Fig. 7). Since Fig. 4 and 7 show measurements on the same cohort of patients, we fit them with identical parameter values and use the values as the default parameter values in other calculations such as those for the amount of HIV bound to FDC at baseline (see Fig. 3). As the data from the work of Cavert et al. (see Fig. 6) come from a different cohort of patients treated with a different drug regimen, the parameter values could differ from the default values; however, we try to make the parameter differences as small as possible.
Fig 4.
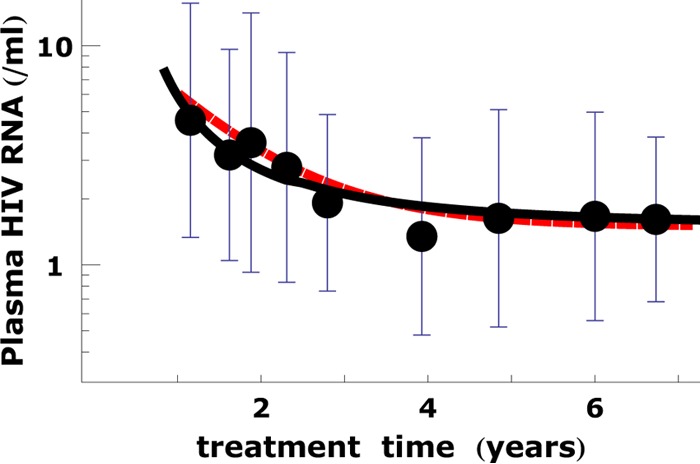
Calculated VFDC + V4 can fit the long-term plasma viral load data. The calculated values of VFDC + V4 are shown by the black solid line. The plasma viral load data for the patients were reported by Palmer et al. (8). Each dot and bar represent the average and standard deviation, respectively, of the logarithmic HIV RNA loads of all patient measured at a given treatment time. The ability of VFDC + V4 to fit the data from the work of Palmer et al. (8) is comparable to that of V3 + V4 (red dashed line). This indicates that the FDC reservoir can be the dominating source of viremia during the 3rd phase of decay. The parameters in the calculation are the same as in Fig. 3.
Fig 6.
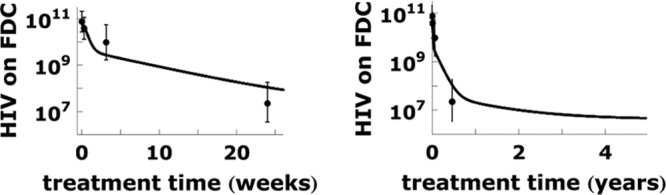
The decay of the FDC viral pool in the first months and years of treatment. The solid lines show the model calculation. The averages and standard deviations of the logarithm of the measured HIV RNA on FDC (12) are shown by dots and bars, respectively. We use parameter values σn = 3.0 and association rate constant kx = 0.55 k−x/<RT> in the calculation. The other parameters used in calculations are the same as those in Fig. 4. The decay of the FDC viral pool slows down steadily over 5 years (right).
Fig 7.
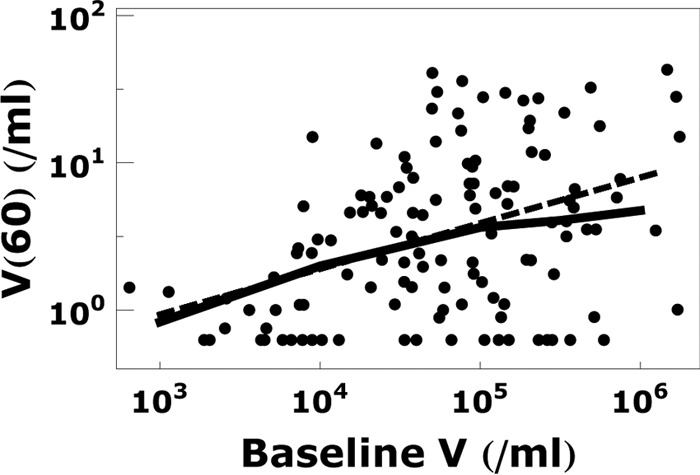
Relationship between the plasma viral load at baseline and week 60 of treatment. The calculated plasma viral load at week 60 (solid line) assuming that V3 is contributed by FDC is consistent with the data (dots) and linear regression (dashed line) in the work of Maldarelli et al. (38). The parameters in the calculation are the same as those in Fig. 4.
Fig 3.
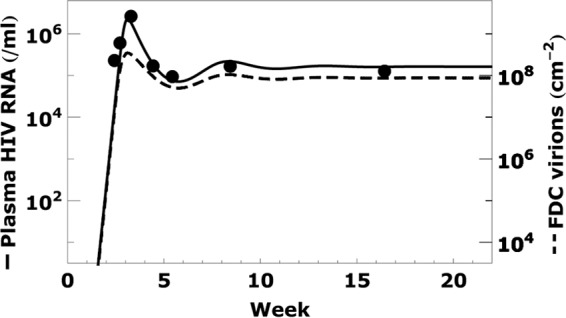
Acute infection dynamics. The plasma viral RNA load in an acutely infected patient (dots) and best fit curve (solid line) are taken from the work of Stafford et al. (43). The density of virions bound to FDC (dashed line), calculated using the generalized form of equations 2 and 3, changes with the plasma viral load. The parameter values used in the calculation, chosen to fit the clinical data (8, 38), are σn = 7, association rate constant kx = 0.38 k−x/<RT>, and those listed in Table 1.
We use a grid search method with parameters {σn, σ}. The observed viremia data (see Fig. 4) can be fitted by different {σn, σ} combinations (see Table A1 in the Appendix), as long as kx is tuned to minimize
| (7) |
where V(ti) is the calculated viral load at time ti, log10 V(ti) and εi are the mean and standard deviation of the observed log10 V values at time ti, and N is the total number of measurement time points.
After finding kx for a {σn, σ} combination, we examine whether these parameters give rise to a significant correlation between baseline and week 60 viremia (see Fig. 7). We find that 7 ≤ σn ≤ 11 is necessary for good agreement with the data in Fig. 7. On the other hand, if our only criterion were fitting the data in Fig. 6 well, we would choose σn ≤ 3 and σ ≤ 0.1. In order to get reasonable agreement with both the viral decline kinetics and the correlation between baseline and week 60 viremia, we chose as default parameter values {σn = 7, σ = 0.1}. Meanwhile kx = 0.38 k−x/<RT> minimizes the error between model predictions and the data from the work of Palmer et al. (8). We also choose {σn = 3, σ = 0.1} to fit to data from the work of Cavert et al. (12) in Fig. 6 in order to make the difference from the default values minimal.
When fitting to the viremia data of individual patients in Fig. 5, we use the default values {σn = 7, σ = 0.1} but allow small deviations from the default kx value. We also find it necessary to tune V4 to capture the viral load at >5 years of treatment for each patient.
Fig 5.
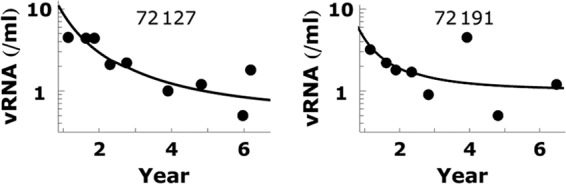
Plasma viral load data for two individual patients are fitted by VFDC + V4. The calculated values of VFDC + V4 are shown by the solid line. Fitting parameters are kx = 0.38 k−x/<RT> and V4 = 0.5 for patient 72127 and kx = 0.35 k−x/<RT> and V4 = 1.0 for patient 72191. The other parameters used in calculations are the same as in Fig. 4. Note that the parameter values in the fits are close to those used in Fig. 4.
RESULTS
Model.
In order to analyze the contribution of virus dissociating from FDC to long-term plasma viremia, we used the model shown in Fig. 2 for the reversible binding of HIV to FDC. The mathematical details of the model and its underlying equations are given in Materials and Methods. The model is a generalization of one developed by Hlavacek et al. (11, 24–26) that incorporates the fact that different HIV virions may contain different numbers of sites that can attach to FDC and that different FDC may have on their surfaces different numbers of receptors capable of binding HIV-1 virions.
HIV-1 bound on FDC before therapy.
During primary HIV-1 infection, the plasma viral load reaches a peak and then falls and establishes a constant set point (baseline) level. Because no viral load data were available for this cohort of patients during acute infection, we used the data [and best fit curve of V(t)] for an acutely infected patient studied in the work of Stafford et al. (43), i.e., patient 5, who had a set point viral load of ∼105/ml (the median set point viral load in the data from the work of Palmer et al. [8] data), and use the generalized form of equations 2 and 3, i.e., equations A2 and A3, to calculate the corresponding number of virions bound to FDC (dashed line in Fig. 3). We find that the FDC viral load grows proportionally to the plasma viral load until V(t) exceeds 105 copies/ml. After that, the FDC viral load is limited by the available surface receptors on FDC, and the change of FDC viral load is less significant than that of V(t). When V(t) decreases after the viral load peak, the FDC viral load is predicted to also decrease. Overall, the FDC viral load remains in equilibrium with the plasma viral load before therapy.
During therapy, FDC may serve as compartment 3.
After therapy is initiated, virions gradually dissociate from FDC and enter the extracellular body fluid. We reproduce two clinical observations with our calculations. First, we show that our model can reproduce the plasma viral loads measured by Palmer et al. (8) during the 3rd phase of viral decay, which has a half-life of 39 weeks. Second, we show that our model is consistent with the decay of the viral pool on FDC in tonsillar lymphoid tissue, which was shown to decrease by 2 to 4 orders of magnitude over the first 6 months of treatment (12).
To fit the plasma viral load data from week 60 to week 394 in the work of Palmer et al., we note that for t > 60 weeks, V1(t) and V2(t) are negligible so that V(t) in equation 1 becomes V(t) = V3(t) + V4(t). As described in Materials and Methods, we use equations A2 to A4, i.e., the generalized forms of equations 2 to 4, respectively, to calculate VFDC(t), the contribution of virus that dissociates from FDC to the plasma virion load, and find that VFDC(t) can replace V3(t) in equation 1 in describing the long-term plasma viral load data (Fig. 4). Therefore, the FDC reservoir could be compartment 3 in the work of Palmer et al. (8) and hence the only source or the major source of the low-level viremia during the 3rd phase.
The V3 term is the largest among the four terms in Fig. 1 from 8 months to 2 years of therapy, suggesting that compartment 3 is the major source of virus during this period. Due to the significant variance of the plasma HIV RNA loads between individual patients, the time range of the 3rd phase is expected to vary strongly between individual patients.
We can also fit the viral load data for individual patients (Fig. 5). However, the statistical noise near the detection limit of the single-copy assay is significant. Since the data for individual patients are noisy, it is difficult to see the viral load trend for most patients. Therefore, the collected data for all patients in Fig. 4 are more informative. In Fig. 5, we show the fitting of two patients whose viral load data are less noisy than others. Here, we adopt the parameters used in Fig. 4, tuned kx slightly, and adjusted V4 to obtain Fig. 5.
Next, we calculated the total viral load on FDC and compared that with the data from the work of Cavert et al. (12), in which viral loads on FDC were measured longitudinally by tonsillar biopsy. We used the model parameters from Fig. 4, only tuning kx and σn to obtain the fit in Fig. 6. Noticing that Fig. 4 and 6 describe different quantities measured in different patient groups under different drug therapies (lopinavir-ritonavir, stavudine, and lamivudine for Fig. 4 [8] and ritonavir, zidovudine, and lamivudine for Fig. 6 [12]), the consistency between parameters in Fig. 4 and 6 is quite significant. The fitting quality of Fig. 6 could be improved if we do not require the parameters to be comparable to those used in Fig. 4 and 5.
As illustrated in Fig. 6, the decay of HIV from FDC is rapid during the first 2 weeks and then slows down steadily over the subsequent several years of treatment. The decrease of the decay rate is due to the variations of the FDC receptor densities and the number of binding sites on virions. According to our model, a virion with fewer binding sites can make fewer bonds with FDC than can virions with more binding sites and hence is trapped on the FDC network for a shorter time than are virions with more binding sites. Thus, as the virions with fewer binding sites dissociate, the virion population becomes dominated by virions with more binding sites and virion decay slows. Similarly, FDC with lower CR2 surface densities trap virions for a shorter time than do FDC with higher CR2 densities. Thus, as virions dissociate from FDC with low CR2 densities, the decay slows as the dissociation becomes more dependent on dissociation from FDC with high receptor densities. The time-dependent virion decay allows us to fit both the FDC viral pool data from the work of Cavert et al. (12), in which virus has an average half-life of 2 to 3 weeks during the first 6 months of treatment, and the plasma viral load data from the work of Palmer et al. (8), in which virus decays with a half-life of 39 weeks at treatment times greater than 60 weeks.
Dependence of viral load at week 60 on baseline viral load: agreement with data.
Maldarelli et al. (38) reported that the plasma viremia at week 60 of treatment is correlated with the baseline viremia. Our model explains this correlation. Patients with higher baseline viral loads deposit more virus on FDC, and hence, as virus dissociates during treatment, more virus remains at week 60 in patients with high baseline viral loads. A comparison of our model predictions with the data from the work of Maldarelli et al. is shown in Fig. 7. Since according to our model FDC are the dominant contributor to the plasma viral load at week 60, we use the generalized form of equations 2 to 4 to find VFDC and replace V3(60) with VFDC(60) in the predicted viral load V(t) (see equation 1 in Materials and Methods and also see Fig. A1 in the Appendix). The predicted relationship between the viral load at 60 weeks and the baseline viral load shown in Fig. 7 further supports the hypothesis that FDC are a long-term reservoir for HIV-1.
The positive correlation is not surprising since higher baseline viremia results in a larger FDC viral pool throughout treatment. Notice, however, that a difference of 2 log units in baseline viremia translates to a difference of only <1 log unit in week 60 viremia. This is due to the fact that a higher baseline viral load leads to a higher density of occupied CR2 receptors and this in turn leads to a higher leakage rate of FDC virions from patients with higher baseline viral loads. By week 60, this partly reduces the difference in FDC viral loads between patients and hence leads to relatively small differences in plasma viral loads among patients.
Sensitivity analysis.
Parameters σn and σ characterize the widths of the distributions of the number of sites on HIV that can bind FDC and the receptor density on FDC, respectively. If their values are changed, we can tune the association rate constant kx and still fit the viremia data in Fig. 4 with similar fitting quality. We use various values of σn and σ and find the best fit value of kx for each case (see Table A1 in the Appendix). Note that the kx values in Table A1 in the Appendix all agree with the estimated lower limit kx > 0.05 k−x/<RT> determined by Hlavacek et al. (26).
DISCUSSION
The low-level plasma viremia observed to persist during more than 7 years of therapy was suggested to come from two unidentified sources: “compartment 3” and “compartment 4” (8). Here, we quantitatively confirmed the possibility that FDC serve as compartment 3. In agreement with clinical data, our calculations predict an early fast decay of the FDC viral pool during the first 6 months of treatment, followed by a slower leakage of virions from FDC to produce an extended low-level plasma viremia. Our calculations also agree with the observed correlation between plasma viral loads at baseline and at week 60 of therapy.
There are several tunable parameters in our model. The distributions of binding sites on HIV and of receptors on FDC shown in Fig. 8 contain two parameters, and the association rate constant is another tunable parameter. In addition, several other parameters are based on rough estimates. Although we found a set of parameters compatible with currently available clinical data, we have not performed an exhaustive study to determine the range of parameters that agree best with the data. In this paper, we focused on the average trend of plasma viral loads for 40 patients. The individual patient data at these low viremia levels are too noisy to be informative. Moreover, our model has certain simplifications. For instance, we ignored B cells, which may also reversibly bind HIV virions (44). We also used a virion clearance rate of 23 day−1, which was estimated in plasma (39). It is possible that the viral clearance rates are different in lymph nodes and blood (45), so that the overall average clearance rate may differ from the value that we used. To accommodate this, other parameters can be tuned to fit the data. For example, if the virion clearance rate is changed to 100 day−1 (45), by changing the standard deviation of the receptor density distribution from 0.1 to 0.25 we obtain a fit to the long-term viremia data with this higher virion clearance rate comparable to that shown in Fig. 4.
HIV virions trapped on FDC have been found to remain infectious for at least 9 months in vivo (19). However, the fraction of virions that remain infectious has not been quantified. As multidrug therapy may effectively suppress viral replication, our model calculates the plasma viral load that comes from FDC-trapped virions, assuming that virions do not cause infection or replication. Thus, our calculation results on levels of persistent viremia do not depend on the fraction of infectious virions.
An open question is which compartment is the main contributor to the fourth stage of viral decay. Even if we change parameters in various ways, our calculations using the FDC reservoir as the only viral source could not reproduce the fourth decay stage. It is possible that a reservoir, such as latently infected CD4+ cells, is the source of fourth-phase virus. One way to evaluate this possibility is to determine whether the half-life of latently infected CD4+ cells matches that of the viremia decay stage 3 or 4. The half-life of latently infected cells has been estimated to be 44 months (46). On the other hand, there are also reports of shorter half-lives of latently infected cells, ranging from 4.6 months (47), to ≥11 months (48), to 6 months (49). Strain et al. (50) and Fischer et al. (51) suggest that the decay of latently infected cells may progressively slow, and Archin et al. (52) suggested that there may be a pool of latently infected cells that are extremely stable and hardly decay at all. If the half-life of latently infected CD4+ cells reaches a few years, they may serve as the source of persistent viremia. Such a long half-life might be realized if the pool of latently infected CD4+ cells is replenished or maintained by division of latently infected CD4+ cells without activation (53–56), as has been shown to occur by Chomont et al. (57). Thus, latently infected cells may well be compartment 4. To clarify this question, it will be important to directly measure the decay of viral RNA on FDC in lymphoid tissue biopsy specimens and the decay of latently infected cells in the same patients over a period of years on potent therapy. Also, sequencing the virus on FDC and the virus released after activation of latently infected cells from these patients may shed light on the source of persistent viremia.
We cannot rule out other possible explanations for the source of third-phase viremia. For example, Althaus and De Boer (58) proposed that there exist subpopulations of latently infected cells with different activation rates. Latently infected cells that have very low activation rates could then contribute to long-term viremia. Via simulations, Althaus and De Boer show that their model can give rise to viral decays on treatment that slow and hence resemble both second- and third-phase decays. To maintain a large enough latent reservoir in the face of loss by activation, Althaus and De Boer need to hypothesize that activated CD4 T cells that are producing virus revert to a latent state (58) and repopulate all of the different latently infected subpopulations. Whether some productively infected cells live long enough for this to be a sufficient source of latently infected cells in patients on ART for over 7 years is unclear, but nonetheless, this work highlights that other hypothesized mechanisms can predict third-phase decay. Experiments, such as those proposed in the paragraph above, will be needed to distinguish among hypotheses. Another way to test our model is to measure the FDC viral pool at longer treatment times than those in the work of Cavert et al. (12), e.g., after 1 or 2 years. According to our prediction (Fig. 6), the measured value should be much higher than that obtained by naive extrapolation from the earlier data points. If the measured data disagree with this prediction, our model can be rejected.
Long-term viral decay data were collected during therapy weeks 60 to 110 in the work of Maldarelli et al. (38), where the decay third-phase half-life was estimated as 69 weeks (95% confidence interval, 38 to 408 weeks) and hence was slower than that in the work of Palmer et al. (8) (half-life, 39 weeks; 95% confidence interval, 25 to 90 weeks), although the confidence intervals overlapped. Given the noisy nature of the single-copy assay data, we focused on the decay rate obtained by Palmer et al. (8) because they collected more data that covered a significantly longer time period (60 to 380 weeks). To reproduce a lower decay rate, as in the work of Maldarelli et al. (38), our parameter values would need to be revised. Also, because of the large 95% confidence intervals for the experimentally determined third-phase decay rates, one should recognize that our best-fit parameter estimates should not be taken too literally but rather are used to exemplify the ability of this theory of HIV interaction with FDC to account for these slow third-phase viral decays.
Since the decay of the FDC viral pool slows down as virus is eliminated from it, it could remain as a source for many years (26) and rekindle the infection when treatment is interrupted if virions remain infectious for these long periods. Thus, it may be important to develop new drugs that drive HIV-1 virions out of the FDC reservoir. Developing such drugs could be an important step toward eliminating HIV infection. A potential drug for depleting FDC virus may be one that blocks formation of the bond between terminal C3 fragments and CR2 by binding to one or the other protein. Possible candidates for this type of drug include proteins that bind to C3, such as Sbi (59), Efb (60), Ehp (61), prion protein (PrPSc) (62), and their engineered modifications, as well as proteins that bind to CR2. These proteins provide another way to verify our model. For example, in a nonhuman primate model, if the 3rd decay phase can be significantly perturbed by the use of any of these proteins as drugs, the role of FDC in long-term viremia will be confirmed.
ACKNOWLEDGMENT
We are grateful to William S. Hlavacek for stimulating discussions and helpful suggestions.
Appendix
Complete model equations, including a distribution of values for the parameters RT and n.
Equations 2 to 4 assume that there is a single value of RT and n. To include a distribution of their values, we need to rewrite equations 2 to 4. Whereas the distribution of RT in Fig. 3 is continuous, for numerical calculations we choose to use small discrete bins of surface receptor densities. Thus, we write separate equations for HIV binding to cells with receptor density
| (A1) |
where R0 = 0.02 <RT>. Then, equations 2 and 3 are changed to
| (A2) |
and
| (A3) |
where Bijn is the surface density of virions with n binding sites bound to i receptors on FDC that have a receptor surface density RT(j). R(j) is the surface density of unbound CR2 on FDC with total surface receptor density RT(j). V(n) is the concentration of virions with n binding sites in body fluid and is determined using the discrete form of equation 6:
Similarly, using the discrete form of equation 5, equation 4 becomes
| (A4) |
where area is the surface area of all FDC in the body and volume is the total extracellular fluid volume in which HIV may disperse. In our numerical calculations, both n and j run from 1 to 80. As a generalized form of equation 2, equation A2 contains 80 × 80 = 6,400 equations, each equation being similar to equation 2.
Correlations.
To explore the correlation between the baseline viral load and VFDC (Fig. 8), we need to rescale Vi(0), i = 1, …, 4 in equation 1 when baseline viral concentration differs from V(0) = 105 ml−1. Since V1 dominates the viral load at the beginning of therapy, we make V1(0) the same as V(0). To find V2(0), V3(0), and V4(0), we look at several time points where the contribution by another compartment dominates the total viral load in equation 1, including week 4 (dominated by V2), week 60 (dominated by V3 + V4), and week 350 (dominated by V4). Figure A1 shows the correlations between the viral loads at these time points and the baseline viral load using the data in the work of Palmer et al. (8) and the linear regression lines. Note that V3(60) = V(60) − V4(60), where V4(60) = V4(350). Assuming an exponential decay for each Vi(t), an f-fold change in Vi(t) implies an f-fold change in Vi(0). From the regression lines, we find that a 10-fold change in the baseline viral concentration translates into a 10-fold change of V1(0), a 3.2-fold change in V2(0), a 2.2-fold change in V3(0), and a 1.6-fold change in V4(0).
Fig A1.
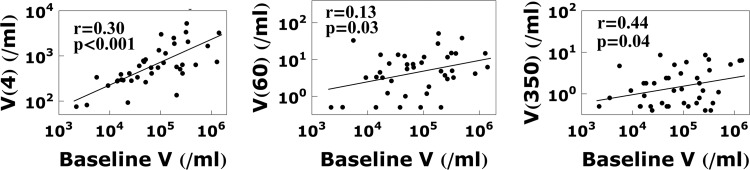
Correlation between baseline viral load and the viral load at weeks 4, 60, and 350.
Sensitivity analysis.
If the values of the parameters σn and σ are changed, we can tune the association rate constant kx and still fit the viremia data in Fig. 4 with similar fitting quality. We use various values of σn and σ and find the best fit value of kx for each case (Table A1). Note that the kx values in Table A1 are all above the estimated lower limit kx > 0.05 k−x/<RT> determined by Hlavacek et al. (26). J, given by equation 7, is the fitting quality, and our fitting procedure aims to minimize J.
Table A1.
Estimated value of kx for different values of σn and σ
| σn | σ | kx <RT>/k−x | J |
|---|---|---|---|
| 2 | 0.05 | 0.91 | 0.017 |
| 0.1 | 0.86 | 0.016 | |
| 0.2 | 0.82 | 0.016 | |
| 0.3 | 0.57 | 0.018 | |
| 0.4 | 0.42 | 0.018 | |
| 4 | 0.05 | 0.63 | 0.016 |
| 0.1 | 0.60 | 0.017 | |
| 0.2 | 0.50 | 0.020 | |
| 0.3 | 0.38 | 0.022 | |
| 0.4 | 0.27 | 0.023 | |
| 7 | 0.05 | 0.40 | 0.017 |
| 0.1 | 0.38 | 0.018 | |
| 0.2 | 0.30 | 0.021 | |
| 0.3 | 0.22 | 0.025 | |
| 0.4 | 0.16 | 0.024 | |
| 10 | 0.05 | 0.29 | 0.017 |
| 0.1 | 0.27 | 0.018 | |
| 0.2 | 0.21 | 0.022 | |
| 0.3 | 0.15 | 0.027 | |
| 0.4 | 0.11 | 0.025 | |
| 15 | 0.05 | 0.23 | 0.024 |
| 0.1 | 0.21 | 0.016 | |
| 0.2 | 0.16 | 0.018 | |
| 0.3 | 0.11 | 0.022 | |
| 0.4 | 0.07 | 0.018 |
Footnotes
Published ahead of print 8 May 2013
REFERENCES
- 1. Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. 1995. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 373:123–126 [DOI] [PubMed] [Google Scholar]
- 2. Perelson AS, Neumann AU, Markowitz M, Leonard JM, Ho DD. 1996. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science 271:1582–1586 [DOI] [PubMed] [Google Scholar]
- 3. Bonhoeffer S, Coffin JM, Nowak MA. 1997. Human immunodeficiency virus drug therapy and virus load. J. Virol. 71:3275–3278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Perelson AS, Essunger P, Cao Y, Vesanen M, Hurley A, Saksela K, Markowitz M, Ho DD. 1997. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature 387:188–191 [DOI] [PubMed] [Google Scholar]
- 5. Finzi D, Siliciano RF. 1998. Viral dynamics in HIV-1 infection. Cell 93:665–671 [DOI] [PubMed] [Google Scholar]
- 6. Zhang ZQ, Wietgrefe SW, Li Q, Shore MD, Duan L, Reilly C, Lifson JD, Haase AT. 2004. Roles of substrate availability and infection of resting and activated CD4+ T cells in transmission and acute simian immunodeficiency virus infection. Proc. Natl. Acad. Sci. U. S. A. 101:5640–5645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Di Mascio M, Dornadula G, Zhang H, Sullivan J, Xu Y, Kulkosky J, Pomerantz RJ, Perelson AS. 2003. In a subset of subjects on highly active antiretroviral therapy, human immunodeficiency virus type 1 RNA in plasma decays from 50 to <5 copies per milliliter, with a half-life of 6 months. J. Virol. 77:2271–2275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Palmer S, Maldarelli F, Wiegand A, Bernstein B, Hanna GJ, Brun SC, Kempf DJ, Mellors JW, Coffin JM, King MS. 2008. Low-level viremia persists for at least 7 years in patients on suppressive antiretroviral therapy. Proc. Natl. Acad. Sci. U. S. A. 105:3879–3884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Palmer S, Wiegand AP, Maldarelli F, Bazmi H, Mican JM, Polis M, Dewar RL, Planta A, Liu S, Metcalf JA, Mellors JW, Coffin JM. 2003. New real-time reverse transcriptase-initiated PCR assay with single-copy sensitivity for human immunodeficiency virus type 1 RNA in plasma. J. Clin. Microbiol. 41:4531–4536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Andrade A, Rosenkranz SL, Cillo AR, Lu D, Daar ES, Jacobson JM, Lederman M, Acosta EP, Campbell T, Feinberg J, Flexner C, Mellors JW, Kuritzkes DR, AIDS Clinical Trials Group A5248 Team Three distinct phases of HIV-1 RNA decay in treatment-naïve patients receiving r-based antiretroviral therapy: ACTG A5248 study. J. Infect. Dis., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hlavacek WS, Stilianakis NI, Perelson AS. 2000. Influence of follicular dendritic cells on HIV dynamics. Philos. Trans. R. Soc. Lond. B Biol. Sci. 355:1051–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cavert W, Notermans DW, Staskus K, Wietgrefe SW, Zupancic M, Gebhard K, Henry K, Zhang ZQ, Mills R, McDade H, Schuwirth CM, Goudsmit J, Danner SA, Haase AT. 1997. Kinetics of response in lymphoid tissues to antiretroviral therapy of HIV-1 infection. Science 276:960–964 [DOI] [PubMed] [Google Scholar]
- 13. Eisele E, Siliciano RF. 2012. Redefining the viral reservoirs that prevent HIV-1 eradication. Immunity 37:377–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bailey JR, Sedaghat AR, Kieffer T, Brennan T, Lee PK, Wind-Rotolo M, Haggerty CM, Kamireddi AR, Liu Y, Lee J, Persaud D, Gallant JE, Cofrancesco J, Jr, Quinn TC, Wilke CO, Ray SC, Siliciano JD, Nettles RE, Siliciano RF. 2006. Residual human immunodeficiency virus type 1 viremia in some patients on antiretroviral therapy is dominated by a small number of invariant clones rarely found in circulating CD4+ T cells. J. Virol. 80:6441–6457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Haase AT, Henry K, Zupancic M, Sedgewick G, Faust RA, Melroe H, Cavert W, Gebhard K, Staskus K, Zhang ZQ, Dailey PJ, Balfour HH, Jr, Erice A, Perelson AS. 1996. Quantitative image analysis of HIV-1 infection in lymphoid tissue. Science 274:985–989 [DOI] [PubMed] [Google Scholar]
- 16. Cohen J. 2011. HIV/AIDS research. Tissue says blood is misleading, confusing HIV cure efforts. Science 334:1614. [DOI] [PubMed] [Google Scholar]
- 17. Fox CH, Tenner-Racz K, Racz P, Firpo A, Pizzo PA, Fauci AS. 1991. Lymphoid germinal centers are reservoirs of human immunodeficiency virus type 1 RNA. J. Infect. Dis. 164:1051–1057 [DOI] [PubMed] [Google Scholar]
- 18. Laurence J. 1993. Reservoirs of HIV infection or carriage: monocytic, dendritic, follicular dendritic, and B cells. Ann. N. Y. Acad. Sci. 693:52–64 [DOI] [PubMed] [Google Scholar]
- 19. Smith BA, Gartner S, Liu Y, Perelson AS, Stilianakis NI, Keele BF, Kerkering TM, Ferreira-Gonzalez A, Szakal AK, Tew JG, Burton GF. 2001. Persistence of infectious HIV on follicular dendritic cells. J. Immunol. 166:690–696 [DOI] [PubMed] [Google Scholar]
- 20. Keele BF, Tazi L, Gartner S, Liu Y, Burgon TB, Estes JD, Thacker TC, Crandall KA, McArthur JC, Burton GF. 2008. Characterization of the follicular dendritic cell reservoir of human immunodeficiency virus type 1. J. Virol. 82:5548–5561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pantaleo G, Graziosi C, Demarest JF, Butini L, Montroni M, Fox CH, Orenstein JM, Kotler DP, Fauci AS. 1993. HIV infection is active and progressive in lymphoid tissue during the clinically latent stage of disease. Nature 362:355–358 [DOI] [PubMed] [Google Scholar]
- 22. Spiegel H, Herbst H, Niedobitek G, Foss HD, Stein H. 1992. Follicular dendritic cells are a major reservoir for human immunodeficiency virus type 1 in lymphoid tissues facilitating infection of CD4+ T-helper cells. Am. J. Pathol. 140:15–22 [PMC free article] [PubMed] [Google Scholar]
- 23. Hicks C, King MS, Gulick RM, White AC, Jr, Eron JJ, Jr, Kessler HA, Benson C, King KR, Murphy RL, Brun SC. 2004. Long-term safety and durable antiretroviral activity of lopinavir/ritonavir in treatment-naive patients: 4 year follow-up study. AIDS 18:775–779 [DOI] [PubMed] [Google Scholar]
- 24. Hlavacek WS, Percus JK, Percus OE, Perelson AS, Wofsy C. 2002. Retention of antigen on follicular dendritic cells and B lymphocytes through complement-mediated multivalent ligand-receptor interactions: theory and application to HIV treatment. Math. Biosci. 176:185–202 [DOI] [PubMed] [Google Scholar]
- 25. Hlavacek WS, Stilianakis NI, Notermans DW, Danner SA, Perelson AS. 2000. Influence of follicular dendritic cells on decay of HIV during antiretroviral therapy. Proc. Natl. Acad. Sci. U. S. A. 97:10966–10971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hlavacek WS, Wofsy C, Perelson AS. 1999. Dissociation of HIV-1 from follicular dendritic cells during HAART: mathematical analysis. Proc. Natl. Acad. Sci. U. S. A. 96:14681–14686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mehrishi JN, Zeiller K. 1974. T and B lymphocytes: striking differences in surface membranes. Br. Med. J. 1:360–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rasmussen JM, Marquart HV, Rask R, Jepsen HH, Svehag SE. 1988. Quantification of C3dg/Epstein-Barr virus receptors on human B cells and B cell lines. Complement 5:98–107 [DOI] [PubMed] [Google Scholar]
- 29. Tooze JA, Bevan DH. 1991. Decreased expression of complement receptor type 2 (CR2) on neoplastic B cells of chronic lymphocytic leukaemia. Clin. Exp. Immunol. 83:423–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Reynes M, Aubert JP, Cohen JH, Audouin J, Tricottet V, Diebold J, Kazatchkine MD. 1985. Human follicular dendritic cells express CR1, CR2, and CR3 complement receptor antigens. J. Immunol. 135:2687–2694 [PubMed] [Google Scholar]
- 31. Thieblemont N, Haeffner-Cavaillon N, Weiss L, Maillet F, Kazatchkine MD. 1993. Complement activation by gp160 glycoprotein of HIV-1. AIDS Res. Hum. Retroviruses 9:229–233 [DOI] [PubMed] [Google Scholar]
- 32. Gelderblom HR, Hausmann EH, Ozel M, Pauli G, Koch MA. 1987. Fine structure of human immunodeficiency virus (HIV) and immunolocalization of structural proteins. Virology 156:171–176 [DOI] [PubMed] [Google Scholar]
- 33. Wyatt R, Sodroski J. 1998. The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science 280:1884–1888 [DOI] [PubMed] [Google Scholar]
- 34. Fremeaux-Bacchi V, Bernard I, Maillet F, Mani JC, Fontaine M, Bonnefoy JY, Kazatchkine MD, Fischer E. 1996. Human lymphocytes shed a soluble form of CD21 (the C3dg/Epstein-Barr virus receptor, CR2) that binds iC3b and CD23. Eur. J. Immunol. 26:1497–1503 [DOI] [PubMed] [Google Scholar]
- 35. Kalli KR, Ahearn JM, Fearon DT. 1991. Interaction of iC3b with recombinant isotypic and chimeric forms of CR2. J. Immunol. 147:590–594 [PubMed] [Google Scholar]
- 36. Diefenbach RJ, Isenman DE. 1995. Mutation of residues in the C3dg region of human complement component C3 corresponding to a proposed binding site for complement receptor type 2 (CR2, CD21) does not abolish binding of iC3b or C3dg to CR2. J. Immunol. 154:2303–2320 [PubMed] [Google Scholar]
- 37. Gentile M, Adrian T, Scheidler A, Ewald M, Dianzani F, Pauli G, Gelderblom HR. 1994. Determination of the size of HIV using adenovirus type 2 as an internal length marker. J. Virol. Methods 48:43–52 [DOI] [PubMed] [Google Scholar]
- 38. Maldarelli F, Palmer S, King MS, Wiegand A, Polis MA, Mican J, Kovacs JA, Davey RT, Rock-Kress D, Dewar R, Liu S, Metcalf JA, Rehm C, Brun SC, Hanna GJ, Kempf DJ, Coffin JM, Mellors JW. 2007. ART suppresses plasma HIV-1 RNA to a stable set point predicted by pretherapy viremia. PLoS Pathog. 3:e46. 10.1371/journal.ppat.0030046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ramratnam B, Bonhoeffer S, Binley J, Hurley A, Zhang L, Mittler JE, Markowitz M, Moore JP, Perelson AS, Ho DD. 1999. Rapid production and clearance of HIV-1 and hepatitis C virus assessed by large volume plasma apheresis. Lancet 354:1782–1785 [DOI] [PubMed] [Google Scholar]
- 40. Gett AV, Hodgkin PD. 2000. A cellular calculus for signal integration by T cells. Nat. Immunol. 1:239–244 [DOI] [PubMed] [Google Scholar]
- 41. Devereux G, Hall AM, Barker RN. 2000. Measurement of T-helper cytokines secreted by cord blood mononuclear cells in response to allergens. J. Immunol. Methods 234:13–22 [DOI] [PubMed] [Google Scholar]
- 42. Bains I, Antia R, Callard R, Yates AJ. 2009. Quantifying the development of the peripheral naive CD4+ T-cell pool in humans. Blood 113:5480–5487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stafford MA, Corey L, Cao Y, Daar ES, Ho DD, Perelson AS. 2000. Modeling plasma virus concentration during primary HIV infection. J. Theor. Biol. 203:285–301 [DOI] [PubMed] [Google Scholar]
- 44. Moir S, Malaspina A, Li Y, Chun TW, Lowe T, Adelsberger J, Baseler M, Ehler LA, Liu S, Davey RT, Jr, Mican JA, Fauci AS. 2000. B cells of HIV-1-infected patients bind virions through CD21-complement interactions and transmit infectious virus to activated T cells. J. Exp. Med. 192:637–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. De Boer RJ, Ribeiro RM, Perelson AS. 2010. Current estimates for HIV-1 production imply rapid viral clearance in lymphoid tissues. PLoS Comput. Biol. 6:e1000906. 10.1371/journal.pcbi.1000906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Siliciano JD, Kajdas J, Finzi D, Quinn TC, Chadwick K, Margolick JB, Kovacs C, Gange SJ, Siliciano RF. 2003. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 9:727–728 [DOI] [PubMed] [Google Scholar]
- 47. Chun TW, Justement JS, Moir S, Hallahan CW, Maenza J, Mullins JI, Collier AC, Corey L, Fauci AS. 2007. Decay of the HIV reservoir in patients receiving antiretroviral therapy for extended periods: implications for eradication of virus. J. Infect. Dis. 195:1762–1764 [DOI] [PubMed] [Google Scholar]
- 48. Gandhi RT, Bosch RJ, Aga E, Albrecht M, Demeter LM, Dykes C, Bastow B, Para M, Lai J, Siliciano RF, Siliciano JD, Eron JJ. 2010. No evidence for decay of the latent reservoir in HIV-1-infected patients receiving intensive enfuvirtide-containing antiretroviral therapy. J. Infect. Dis. 201:293–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang L, Ramratnam B, Tenner-Racz K, He Y, Vesanen M, Lewin S, Talal A, Racz P, Perelson AS, Korber BT, Markowitz M, Ho DD. 1999. Quantifying residual HIV-1 replication in patients receiving combination antiretroviral therapy. N. Engl. J. Med. 340:1605–1613 [DOI] [PubMed] [Google Scholar]
- 50. Strain MC, Gunthard HF, Havlir DV, Ignacio CC, Smith DM, Leigh-Brown AJ, Macaranas TR, Lam RY, Daly OA, Fischer M, Opravil M, Levine H, Bacheler L, Spina CA, Richman DD, Wong JK. 2003. Heterogeneous clearance rates of long-lived lymphocytes infected with HIV: intrinsic stability predicts lifelong persistence. Proc. Natl. Acad. Sci. U. S. A. 100:4819–4824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fischer M, Joos B, Niederost B, Kaiser P, Hafner R, von Wyl V, Ackermann M, Weber R, Gunthard HF. 2008. Biphasic decay kinetics suggest progressive slowing in turnover of latently HIV-1 infected cells during antiretroviral therapy. Retrovirology 5:107. 10.1186/1742-4690-5-107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Archin NM, Vaidya NK, Kuruc JD, Liberty AL, Wiegand A, Kearney MF, Cohen MS, Coffin JM, Bosch RJ, Gay CL, Eron JJ, Margolis DM, Perelson AS. 2012. Immediate antiviral therapy appears to restrict resting CD4+ cell HIV-1 infection without accelerating the decay of latent infection. Proc. Natl. Acad. Sci. U. S. A. 109:9523–9528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim H, Perelson AS. 2006. Viral and latent reservoir persistence in HIV-1-infected patients on therapy. PLoS Comput. Biol. 2:e135. 10.1371/journal.pcbi.0020135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rong LB, Perelson AS. 2009. Asymmetric division of activated latently infected cells may explain the decay kinetics of the HIV-1 latent reservoir and intermittent viral blips. Math. Biosci. 217:77–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rong LB, Perelson AS. 2009. Modeling latently infected cell activation: viral and latent reservoir persistence, and viral blips in HIV-infected patients on potent therapy. PLoS Comput. Biol. 5:e1000533. 10.1371/journal.pcbi.1000533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jones LE, Perelson AS. 2007. Transient viremia, plasma viral load, and reservoir replenishment in HIV-infected patients on antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 45:483–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chomont N, El-Far M, Ancuta P, Trautmann L, Procopio FA, Yassine-Diab B, Boucher G, Boulassel MR, Ghattas G, Brenchley JM, Schacker TW, Hill BJ, Douek DC, Routy JP, Haddad EK, Sekaly RP. 2009. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 15:893–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Althaus CL, De Boer RJ. 2010. Intracellular transactivation of HIV can account for the decelerating decay of virus load during drug therapy. Mol. Syst. Biol. 6:348. 10.1038/msb.2010.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Isenman DE, Leung E, Mackay JD, Bagby S, van den Elsen JM. 2010. Mutational analyses reveal that the staphylococcal immune evasion molecule Sbi and complement receptor 2 (CR2) share overlapping contact residues on C3d: implications for the controversy regarding the CR2/C3d cocrystal structure. J. Immunol. 184:1946–1955 [DOI] [PubMed] [Google Scholar]
- 60. Hammel M, Sfyroera G, Ricklin D, Magotti P, Lambris JD, Geisbrecht BV. 2007. A structural basis for complement inhibition by Staphylococcus aureus. Nat. Immunol. 8:430–437 [DOI] [PubMed] [Google Scholar]
- 61. Hammel M, Sfyroera G, Pyrpassopoulos S, Ricklin D, Ramyar KX, Pop M, Jin Z, Lambris JD, Geisbrecht BV. 2007. Characterization of Ehp, a secreted complement inhibitory protein from Staphylococcus aureus. J. Biol. Chem. 282:30051–30061 [DOI] [PubMed] [Google Scholar]
- 62. Mitchell DA, Kirby L, Paulin SM, Villiers CL, Sim RB. 2007. Prion protein activates and fixes complement directly via the classical pathway: implications for the mechanism of scrapie agent propagation in lymphoid tissue. Mol. Immunol. 44:2997–3004 [DOI] [PubMed] [Google Scholar]