

Abstract
Mutations in the core protein (HBc) of hepatitis B virus (HBV) are associated with aggressive hepatitis and advanced liver diseases in chronic hepatitis B (CHB). In this study, we identified the L60V variation in HBc that generates a new HLA-A2-restricted CD8+ T cell epitope by screening an overlapping 9-mer peptide pool covering HBc and its variants. The nonameric epitope V60 was determined by structural and immunogenic analysis. The HBc L60V variation is correlated with hepatic necroinflammation and higher viral levels, and it may be associated with a poor prognosis in CHB patients. Immunization with the defined HBV epitope V60 peptide elicited specific cytotoxic T lymphocyte (CTL)-induced liver injury in HLA-A2+ HBV transgenic mice. In addition, in vitro and in vivo experiments both demonstrated that the HBc L60V variation facilitates viral capsid assembly and increases HBV replication. These data suggest that the HBc L60V variation can impact both HBV replication and HBV-specific T cell responses. Therefore, our work provides further dissection of the impact of the HBc L60V variation, which orchestrates HBV replication, viral persistence, and immunopathogenesis during chronic viral infection.
INTRODUCTION
Hepatitis B virus (HBV) is a strictly hepatotropic DNA virus affecting about 350 million people worldwide, especially in Asia. Because HBV is not directly cytopathic, the liver damage induced by chronic hepatitis B (CHB) is thought to be mainly immune mediated. Hepatic infiltration of HBV-specific and nonspecific T cells, NK/NKT cells (1), dendritic cell subsets (2), macrophages (3), polymorphonuclear neutrophils (4), regulatory T cells (5), and intrahepatic Th17 cells (6) contributes to immune-mediated liver pathogenesis and disease progression. Additionally, inflammatory cytokines (7), extracellular matrix proteins (8), chemokines (9), and platelets (10) are important in the complex interplay that orchestrates immune defects and immunopathogenesis during chronic viral infection. Of note, either by direct killing effects or by indirect inflammation-inducible behaviors, virus-specific cytotoxic T lymphocytes (CTLs) play a critical pathogenic role in HBV infection (3, 11, 12).
As a highly prevalent hepadnavirus, HBV can evolve by mutations to increase its adaptation against environmental selection, and these mutations manifest as various effects on viral replication, clinical features, and the pathogenesis of HBV infection. The treatment of CHB with peginterferon and nucleotide analogues (e.g., lamivudine) causes a high risk of selecting for drug-resistant mutants, which primarily occur in association with a high rate of mutation in the basal core promoter and precore (BCP/pre-C) regions, as well as the reverse transcriptase (RT) domain, of the HBV genome (13–15). Mutations in the BCP/pre-C and RT regions are associated with aggressive hepatitis and advanced liver diseases, including severe and fulminant hepatitis B, cirrhosis, and hepatocellular carcinoma (HCC) (16–18). Because T cell responses play a critical role in controlling HBV infection, mutations within T cell-directed epitopes are one of the most common strategies employed by viruses to evade immune detection. In CHB patients who fail to seroconvert to anti-HBV surface protein (anti-HBs), more substitutions within T cell epitopes are observed (19). Due to overlapping HBV genes and restriction of HLA alleles in patients, these viral escape mutations under immune selection are not equally distributed among protein-coding open reading frames (20, 21). Mutations in T cell-immunodominant epitopes are associated with higher rates of liver fibrosis and development of HCC (22), which may be due to inefficient T cell responses and altered protein functions.
In this study, we identified a variation that generates a new CD8+ T cell epitope by screening an overlapping 9-mer peptide pool covering the HBV core protein (HBc) and its variants and showed that the variation in HBc results in significantly facilitated viral capsid assembly and increased HBV replication. Our results indicate the important virological and clinical aspects of this variation, which enhances HBc assembly at the expense of adding an epitope.
MATERIALS AND METHODS
Patients and blood samples.
Analysis of the variation at HBc position 60 was performed with clinical information in the database of records for HBV-infected patients built by the Beijing 302 Hospital (Beijing, China). The associations between the HBc L60V variation and the progression of liver disease or patients' clinical indicators were investigated in this study. The criteria for diagnosis of CHB and acute-on-chronic liver failure (ACLF) complied with those of the Management Scheme of Diagnostic and Therapy of Viral Hepatitis and the Diagnostic and Treatment Guidelines for Liver Failure, respectively, issued by the Chinese Society of Infectious Diseases and Parasitology and the Chinese Society of Hepatology, respectively. CHB patients were defined as those who had chronic HBV infection with serum HBV surface antigen (HBsAg) positivity for >6 months and may have shown symptoms of hepatitis or abnormal hepatic function. CHB is divided into two groups: mild chronic hepatitis B (CHB-M) and severe chronic hepatitis B (CHB-S). CHB-M patients display mild to moderate liver disease activities but do not attain the criteria of CHB-S. CHB-S patients have severe liver disease with evident clinical manifestations and significant alteration of biochemical parameters that includes at least one of the following: serum albumin level of ≤32 g/liter, serum total bilirubin (TBIL) level of >85.5 μmol/liter, plasma prothrombin activity (PTA) of 40 to 60%, and serum cholinesterase level of <4,500 IU/liter (18). The diagnostic standard for ACLF includes a history of CHB with symptoms of extreme fatigue and severe digestive problems with the following criteria: TBIL level >10 times the normal level (171 μmol/liter) and PTA of ≤40% (23). The clinical characteristics of all enrolled patients are described in Table 1.
Table 1.
Clinical and virologic characteristic of CHB-M, CHB-S, and ACLF patientsa
| Characteristic | CHB-M | CHB-S | ACLF | P value |
|---|---|---|---|---|
| No. of patients | 145 | 106 | 122 | |
| Mean ± SD age (yr) | 40.4 ± 13.5 | 38.6 ± 13.8 | 45.2 ± 11.3 | <0.01 |
| Gender (no. of M/no. of F) | 127/18 | 91/15 | 100/21 | >0.05 |
| No. (%) of patients with the following HBV genotype: | ||||
| B | 32 (22.1) | 30 (28.3) | 35 (28.7) | >0.05 |
| C | 110 (75.9) | 75 (70.8) | 87 (71.3) | >0.05 |
| No. (%) of patients with the HBcL60V variation | 16 (11.0) | 24 (22.6) | 31 (25.4) | <0.01 |
| No. (%) of patients HBeAg positive | 98 (67.6) | 45 (42.5) | 31 (25.4) | <0.01 |
| No. of HBV DNA copies/ml | ||||
| Mean | 2.5 × 107 | 1.1 × 107 | 1.2 × 107 | >0.05 |
| Range | 1,282–1.6 × 109 | 500–2.6 × 108 | 500–5.3 × 108 | |
| Mean ± SD concn (IU/liter) | ||||
| ALT | 145.8 ± 218.6 | 457.0 ± 382.3 | 258.5 ± 394.0 | <0.01 |
| AST | 101.8 ± 120.5 | 297.4 ± 280.0 | 307.9 ± 446.3 | <0.01 |
HBV, hepatitis B virus; CHB-M, mild chronic hepatitis B; CHB-S, severe chronic hepatitis B; ACLF, acute-on-chronic liver failure; M, male; F, female; HBeAg, hepatitis B virus e antigen; ALT, alanine aminotransferase; AST, aspartate aminotransferase.
For blood collection, a total of 19 acute hepatitis B (AHB) and 25 CHB patients were enrolled. Diagnostic criteria for AHB were those described previously (9). AHB patients were grouped as follows: HLA-A2-positive patients infected with HBV containing HBc L60 (n = 11) or V60 (n = 8). CHB patients, who were all HLA-A2 positive and in a state of immune activation with high viral loads and with elevated alanine aminotransferase (ALT) levels (>40 IU/liter), were also assigned to two groups: patients infected with HBV containing HBc L60 (n = 15) or V60 (n = 10). All patients were negative for hepatitis C virus, hepatitis D virus, and HIV-1 infection. Blood samples (10 ml) were collected from each of the patients. All patients had been hospitalized in the Beijing 302 Hospital from September 2010 to September 2011. Written informed consent was provided by all study participants. The study was approved by the Ethics Committee of the Beijing 302 Hospital.
Mice.
HLA-A2.1/Kb transgenic mice (24) were kindly provided by W. L. Huang (IMCAS, Beijing, China). HBV transgenic BALB/c mice were purchased from the Transgenic Engineering Lab, Infectious Disease Center (Guangzhou, China). The HBV transgenic mice were generated with a viral DNA construct, pHBV1.3, containing 1.3 copies of the HBV genome. All transgenic mice tested positive for serum HBsAg and viral DNA, as well as HBc expression in hepatocytes in their livers. F1 hybrids of HLA-A2.1/HBV transgenic mice were generated by crossing the HLA-A2.1/Kb mice and HBV transgenic mice. All F1 hybrids were screened for serum HBsAg by enzyme-linked immunosorbent assay (ELISA), viral DNA by real-time PCR, and HLA-A2 by PCR amplification with sequence-specific primers (Protrans, Germany) before experimental manipulations. The study of mice was in strict accordance with the regulations of the Institute of Microbiology, Chinese Academy of Sciences of Research Ethics Committee, and the protocol was approved by the committee.
Plasmid constructs.
The wild-type HBc gene and mutated (L60V) HBc gene were cloned into pcDNA3.1 (Invitrogen), and the recombinant plasmids were designated pcDNA3.1-HBcL60 and pcDNA3.1-HBcV60, respectively. The truncated forms (HBc residues 1 to 149) of the wild-type HBc (HBcL60) and mutated HBc (HBcV60) genes were cloned into pcDNA3.1 and designated pcDNA3.1-HBc149-L60 and pcDNA3.1-HBc149-V60, respectively. The truncated forms of HBc were also cloned into the pET28a plasmid (Invitrogen) and designated pET-HBc149-L60 and pET-HBc149-V60, respectively. pHBV1.3 containing 1.3 copies of the full-length HBV genomic sequence was maintained in the lab. The HBc L60V mutation was introduced into pHBV1.3, and the resulting plasmid was designated pHBV1.3-HBcL60V.
Peptide synthesis.
A careful search of HBc sequences of genotypes B and C that had been detected up to 1 October 2008 was conducted using the protein database from NCBI, including 171 HBc sequences of genotype B and 159 HBc sequences of genotype C. The sequences were compared and used for peptide synthesis. If the rate of variation of a certain amino acid exceeded 10%, a series of peptides associated with this variation was included in the peptide synthesis. We adopted the overlapping method (8-amino-acid [aa] overlap) to synthesize a total of 191 nonapeptides (9-mers) covering HBc aa 1 to 150. An additional 13 peptides (8- to 10-mers) associated with the candidate peptides evaluated in this study are listed in Table 2. All of these peptides were synthesized at Jier Biological (Shanghai, China), and their purity was determined to be >95%. The HBc18-27 (FLPSDFFPSV) and HBc82-90 (RELVVSYVN) peptides were used as positive and negative controls, respectively. The H-2 Kd-restricted epitope HBc87-95 (SYVNTNMGL) was also synthesized as another positive control.
Table 2.
Tests of a series of peptides associated with HBc position 60 by T2 binding assays and in vitro refolding
| Name | Start positiona | Sequenceb | T2 bindingc | Refoldingd |
|---|---|---|---|---|
| IL-10 | 59 | ILCWGELMNL | + | + |
| IN-9 | 59 | ILCWGELMN | − | − |
| IM-8 | 59 | ILCWGELM | − | − |
| L60 | 60 | LCWGELMNL | − | + |
| LN-8 | 60 | LCWGELMN | − | − |
| CL-8 | 61 | CWGELMNL | − | − |
| IL(V)-10 | 59 | IVCWGELMNL | + | + |
| IN(V)-9 | 59 | IVCWGELMN | − | − |
| IM(V)-8 | 59 | IVCWGELM | − | − |
| V60 | 60 | VCWGELMNL | + | + |
| VN-8 | 60 | VCWGELMN | − | − |
The position of the first amino acid located in the HBc protein.
Position 60 in HBc is underlined.
The affinity of peptide binding to HLA-A*0201 was detected according to the method described in the Materials and Methods. If FI is ≥1, it is marked +. Otherwise, it is marked −.
In the refolding assay, if the elution peak of the MHC-peptide complex appears in gel filtration chromatography, the peptide is marked +. Otherwise, it is marked −.
Cell culture and transfection.
The T2 cell line was kindly provided by Bin Gao (IMCAS, Beijing, China) and cultured in RPMI 1640 medium with 10% fetal bovine serum (Gibco BRL, Paisley, United Kingdom). Two human hepatoma cell lines (HepG2 and Huh-7) and the 293T cell line were obtained from ATCC (Manassas, VA). The cell lines were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum. The cells were transfected with the Lipofectamine 2000 (Invitrogen) transfection reagent as instructed by the manufacturer. Supernatants and cells were harvested at different times (24, 48, and 72 h) after transfection.
T2 cell binding assay.
Major histocompatibility complex (MHC) stabilization assays were performed on T2 cells as previously described (25). The binding activity of each peptide was calculated as the fluorescent index (FI), which was determined by the equation (mean fluorescein isothiocyanate [FITC] fluorescence with the given peptide − mean FITC fluorescence without peptide)/(mean FITC fluorescence without peptide). Peptides with FIs of ≥1 were arbitrarily regarded as epitopes with high affinity.
Refolding, protein crystallography, and structure determination.
The HLA-A*0201 heavy chain and β2 microglobulin (β2m) were expressed as recombinant proteins in Escherichia coli (26) and refolded using the gradual dilution method (27). Then, the soluble portion of the refolded complex was concentrated and purified by Superdex 200 10/300 GL gel filtration chromatography, followed by Resource-Q anion-exchange chromatography (GE Healthcare). The purified complexes were crystallized by the hanging-drop vapor diffusion method at 18°C. HLA-A0201/HBc V60 crystals were obtained at a final concentration of 5 mg/ml in 0.1 mol/liter bis-Tris (pH 6.5) and 20% (wt/vol) polyethylene glycol 3350. Crystallographic data were collected at 100 K in-house on a Rigaku MicroMax007 rotating-anode X-ray generator operated at 40 kV and 20 mA (Cu Κα; λ = 1.5418 Å) equipped with an R-AXIS VII++ image-plate detector. The structure of HLA-A*0201/HBc V60 was determined by molecular replacement with the program MolRep using Protein Data Bank (PDB) entry 1JF1 as the search model.
Immunization of mice.
Female HLA-A2.1/Kb transgenic mice (6 to 8 weeks old) were immunized intramuscularly with 50 μg of plasmid pcDNA3.1-HBcL60 or pcDNA3.1-HBcV60 at week 1 and subcutaneously with 50 μg of HBc L60 or HBc V60 peptide bound to 30 μg of heat shock protein gp96 as adjuvant, respectively, at weeks 3 and 4. Mice were sacrificed 1 week after the last immunization. Splenocytes were isolated as previously described (28). The same immunization strategy was performed with HLA-A2.1/HBV transgenic mice and HBV transgenic BALB/c mice.
In a hydrodynamic mouse model (29), 10 μg plasmid pHBV1.3 or pHBV1.3-HBcL60V was injected into the mouse tail vein via hydrodynamic injection. Blood was collected at different time points by retrobulbar puncture. Serum HBV DNA levels were detected by real-time PCR. HBc expression in mouse liver was assayed by immunohistochemical staining (30). Each group contained five to seven HLA-A2.1/Kb transgenic mice.
Gamma interferon (IFN-γ) ELISPOT assay.
Enzyme-linked immunosorbent spot (ELISPOT) assay was performed to detect epitope-specific T cells according to the manufacturer's instruction. Briefly, 96-well polyvinylidene difluoride plates (BD-Pharmingen, San Diego, CA) were precoated with the coating antibody overnight at 4°C and blocked for 1 h at 37°C. Murine splenocytes (106) or human peripheral blood mononuclear cells (PBMCs; 2 × 105) together with 50 μg/ml of peptide were added to each well and incubated at 37°C for 24 to 48 h. Phytohemagglutinin (PHA)-stimulated T cells were employed as positive controls. Each test was performed in triplicate. The spots were counted and analyzed with an ELISPOT assay reader (Cellular Technology Ltd.). Results were reported as the mean number of spot-forming cells (SFCs)/106 murine splenocytes or 2 × 105 PBMCs.
Cytotoxicity assay.
293T cells or HepG2 cells transfected with pHBV1.3 or pHBV1.3-HBcL60V were labeled with 2 μM carboxyfluorescein succinimidyl ester (CFSE) as target cells and seeded into a 96-well plate. CTLs were then added at different ratios: 1:1, 10:1, and 20:1. Plates were incubated at 37°C for 4 to 6 h, and target cells were stained with propidium iodide (PI) using a Vybrant apoptosis assay kit (Invitrogen).
HBc expression, purification, and in vitro assembly.
The pET-HBc149-L60 or pET-HBc149-V60 vector was transformed into E. coli BL21 cells, and the recombinant HBc proteins were expressed and purified as described previously (31). HiPrep 16/60 Sephacryl S-400 HR and Superdex 75 10/300 GL columns were used to purify HBc capsids and dimers, respectively. Capsids were assembled at 4°C by adding NaCl to a final concentration of 0.5 M in 50 mM HEPES (pH 7.5)–2 mM dithiothreitol. The assembled HBc capsids were detected by native agarose gel electrophoresis as previously described (32).
Detection of HBsAg and HBeAg by ELISA.
The amounts of HBsAg and HBV e antigen (HBeAg) were measured according to the manufacturer's instructions (Kehua Biotech, Shanghai, China).
Southern blotting and real-time PCR.
Intracellular HBV DNA was extracted and analyzed by Southern blotting as described previously (23). Alkaline phosphatase-labeled HBV DNA (the probe) was generated using an Amersham AlkPhos direct labeling and detection system (GE Healthcare) for Southern blot analysis. Core particles associated with HBV DNA from cell supernatants were quantified by real-time PCR as previously described (33). Real-time PCR was performed using the SYBR green premix reagent (TaKaRa Bio Inc., Shiga, Japan).
Statistical analysis.
Student's t test was performed to compare differences between independent groups. The correlation between the rate of variation and disease progression and the correlation between HBV DNA loads and serum ALT levels in CHB patients were detected using Pearson's χ2 test. Clinical statistical analyses were performed with SPSS (version 16.0) software (SPSS Inc., Chicago, IL). A P value of <0.05 was considered significant.
Protein structure accession number.
The accession number of the structure of V60-pMHC in the Protein Data Bank is 4K7F.
RESULTS
Identification of a potential HLA-A*0201-restricted epitope associated with the L60V variation.
As the most conservative and immunogenic component of HBV (34), HBc was chosen as the focus of this study. We first used an overlapping (by 8 aa) 9-mer peptide pool covering HBc (aa 1 to 150) and its variants for a T2 cell binding assay to screen genotype B- and C-derived HBcAg-specific T cell epitopes. A variant peptide (V60, VCWGELMNL) spanning from HBc aa 60 to 68 was found to have a high affinity for binding to HLA-A*0201 molecules, whereas its corresponding wild-type peptide (L60, LCWGELMNL) displayed little binding to HLA-A*0201, as evidenced by the FI (3.38 for the mutant V60 peptide but only 0.05 for the wild-type L60 peptide) (Fig. 1A). The positive-control HBV peptide HBc18-27 displayed a high affinity (FI = 3.9), whereas the negative-control HBc82-90 peptide had an FI for affinity of 0.04. Panels of N- or C-terminally truncated or extended peptides of L60 or V60 were synthesized to determine the minimal sequence length. Of these, only the decapeptides ILCWGELMNL and its variant, IVCWGELMNL, could moderately increase the surface level of HLA-A*0201 molecules (Table 2). The FIs of these two decapeptides were 1.33 and 1.72, respectively, indicating that the V60 peptide is the optimized epitope. A refolding assay was performed to further investigate the binding affinity of the V60 and L60 peptides (Fig. 1B). The V60 peptide possessed a high affinity for binding to HLA-A*0201, whereas L60 did not.
Fig 1.

Affinity of binding of the HBc L60 and V60 peptides to HLA-A*0201 and the structure of the HLA-A*0201/V60 complex. (A) The affinity of peptide binding to HLA-A*0201 was quantified using MHC stabilization assays with T2 cells. The HBc18-27 and HBc82-90 peptides served as positive and negative controls, respectively. The results shown are representative of three independent experiments. (B) In vitro refolding of L60 or V60 peptide with HLA-A*0201 heavy chain and β2m. The refolded complexes were analyzed by gel filtration chromatography on a Superdex 200 16/60 column. The peaks of HLA complexes with the expected molecular mass of 45 kDa eluted at the estimated volume of 15.9 ml. mAU, milli-absorbance units (C) Overview of the structure of peptide V60 presented by HLA-A*0201; V60 is presented in the peptide-binding cleft. (D) V60 is presented in the peptide-binding clefts with electron density at the 1σ contour level. (E) Residues Val1 and Cys2 (green sticks) are located in pocket A and pocket B of HLA-A*0201, respectively. The heavy chain of HLA-A*0201 is show in cyan surfaces. The large S atom (yellow sphere) of V60 is shown in the B pocket, as confirmed by its electron density. (F) Alignment of pockets A and B of HLA-A*0201/V60 with the previously determined structure with PDB accession number 1QRN. Leu1 and Leu2 (purple sticks) of the peptide presented by the 1QRN structure are located in the A and B pockets of the 1QRN structure heavy chain (purple surfaces), respectively.
To further confirm that V60 is a typical HLA-A2-restricted epitope, a complex of HLA-A2 and V60 was prepared for crystallization. The crystal structure of the complex was determined to 2.0-Å resolution, and the statistics of the complex data are shown in Table 3. The structure of HLA-A*0201/V60 shows that V60 is a typical HLA-A2-binding peptide which possesses a classical conformation of an HLA-A2-restricted 9-mer epitope. The unambiguous electron densities of V60 clearly demonstrate that position 2 (C2) and position 9 (L9) are buried in pockets B and F, respectively, which are identified as anchor residues (Fig. 1C and D). All of these anchor residues play an important role in the stability of the entire complex. The side chains of amino acids E5 and M7 protrude out from the HLA-A2 surface and may be involved in TCR attachment.
Table 3.
X-ray diffraction data processing and refinement statistics
| Parametera | Value(s) for HLA-A*0201/V60b |
|---|---|
| Data processing statistics | |
| Space group | P21 |
| Cell parameters | |
| a (Å) | 83.86 |
| b (Å) | 58.55 |
| c (Å) | 89.58 |
| α (°) | 90.00 |
| β (°) | 109.86 |
| γ (°) | 90.00 |
| Resolution range (Å) | 50.00–2.00 (2.07–2.00) |
| No. of total reflections | 54,699 (5,011) |
| No. of unique reflections | 14,025 (1,474) |
| Completeness (%) | 97.9 (90.8) |
| Rmerge (%) | 12.5 (34.0) |
| I/σ | 11.0 (3.2) |
| Refinement statistics | |
| Rwork (%) | 23.8 |
| Rfree (%) | 28.5 |
| RMSD | |
| Bond lengths (Å) | 0.006 |
| Bond angles (°) | 0.912 |
Rmerge = Σ|I − 〈I〉|/ΣI × 100, where I is the intensity of a reflection and 〈I〉 is the average intensity; Rfree is calculated for a randomly chosen 5% of reflections and Rwork is calculated for the remaining 95% of reflections used for structure refinement. RMSD, root mean square deviation.
Values in parentheses are for the highest-resolution shell.
Next, we comparatively analyzed the structure of the HLA-A*0201-V60 complex and the previously determined structure with PDB accession number 1QRN, which has L1 and L2 in pockets A and B of the heavy chain, respectively. First, our analysis indicated that for peptide L60, the longer side chain of L1 pointing away from the HLA-A2 surface prevents C2 from properly inserting into pocket B (Fig. 1E), which disrupts L60 binding to the HLA-A2 heavy chain and reduces the stability of the entire complex. Second, we observed that pocket B in the structure with PDB accession number 1QRN is smaller than that in HLA-A*0201-V60, although the structure with PDB accession number 1QRN has a Leu at position 2 in pocket B (Fig. 1F). This may indicate that (i) the large S atom of C2 in the V60 peptide requires more space or (ii) L1 may be correlated with a smaller B pocket. Thus, the conformational differences between L60 and V60 by replacement of Leu by Val at position 1 may contribute to the variance in binding affinity of the two peptides to HLA-A2 molecules.
In vivo generation of a V60 peptide-specific CTL response.
Female HLA-A2.1/Kb transgenic mice were immunized with an HBc DNA prime/peptide boost regimen three times with heat shock protein gp96 as the adjuvant. A peptide-specific CTL response to L60 or V60 was detected by ELISPOT assay 1 week after the last immunization. As can be seen in Fig. 2A, a dramatically higher CTL response was observed in splenocytes from V60-immunized than L60-immunized mice (18.00 ± 4.04 versus 147.5 ± 10.37; P < 0.01). No peptide-specific CD8+ T cell responses were detected from HBc82-90 peptide-immunized mice (negative control), and the positive-control peptide-immunized mice exhibited a high CD8+ T cell response. Similarly, a significant difference in the antiviral cytotoxicity of CTLs was observed between mice immunized with L60 peptide and those immunized with V60 peptide by the killing assay using HBV plasmid-transfected 293T cells as the target (P < 0.05) (Fig. 2B). In addition, peptide-specific CD8+ T cells were detected by IFN-γ ELISPOT assays ex vivo using fresh HLA-A2-positive (HLA-A2+) AHB patient PBMCs stimulated with the respective peptides. As shown in Fig. 2C, a much higher specific CTL response was observed in patients infected with the HBc L60V variants than patients infected with wild-type HBV (140.88 ± 85.5 versus 22.55 ± 17.51; P < 0.01). Similar numbers of V60 peptide-specific and immunodominant epitope HBc18-27 peptide-specific CTLs were observed in AHB patients. Taken together, the data indicate that 60V is an HLA-A2-restricted CTL epitope that naturally exists in HBV-infected patients.
Fig 2.
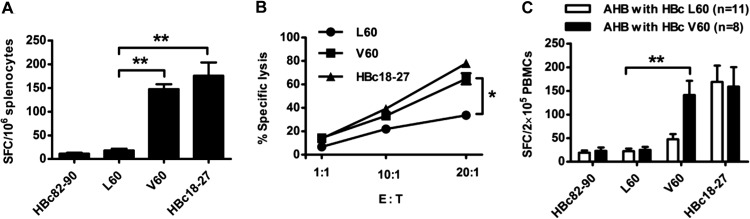
Detection of the HBc L60 peptide- and V60 peptide-specific CD8+ T cell response in HLA-A2.1/Kb mice and AHB patients. Female HLA-A2.1/Kb transgenic mice were immunized with a DNA prime/peptide boost regimen at weeks 1, 3, and 4. The HBc82-90 and HBc18-27 peptides served as negative and positive controls, respectively. Mice were sacrificed 1 week after the last immunization. (A) Fresh splenocytes (1 × 106) were stimulated with L60 peptide, V60 peptide, or HBc82-90 or HBc18-27 as negative and positive controls, respectively, and peptide-specific CTLs were detected by IFN-γ ELISPOT assay. (B) 293T cells labeled with CFSE were transfected with pHBV1.3 or pHBV1.3-HBcL60V as target cells and mixed with L60-, V60-, or HBc18-27-stimulated splenocytes at different ratios: 1:1, 1:10, and 1:20. After 4 h, the mixed samples were stained with PI and analyzed by fluorescence-activated cell sorting. The data shown are the means ± SDs of five mice. E:T, effector-to-target cell ratio. (C) Detection of peptide-specific CTLs in AHB patients by ELISPOT assay. PBMCs (2 × 105/well) from patients infected with wild-type (HBcL60) or variant (HBcV60) virus were stimulated with L60 peptide, V60 peptide, or HBc82-90 or HBc18-27 as negative and positive controls, respectively, and analyzed by ELISPOT assay. *, P < 0.05; **, P < 0.01. Data are representative of two independent experiments.
The HBc L60V variation is associated with severe hepatitis and disease progression in HBV-infected patients.
To address the clinical relevance of the HBc L60V variation in CHB, clinical and virological assays were performed for 251 CHB and 122 ACLF patients (Table 1). Among the patients with CHB, in comparison to patients infected with wild-type isolates (L60), patients infected with HBc L60V variants had much higher ALT levels (253.9 ± 22.67 versus 402.8 ± 55.47 IU/liter; P < 0.01) and aspartate aminotransferase (AST) levels (176.2 ± 16.68 versus 272.4 ± 37.11 IU/liter; P < 0.05) (Fig. 3A). Moreover, CHB patients infected with HBc L60V variants had much higher HBV DNA loads (approximately 10-fold higher) than those infected with wild-type isolates (P < 0.01) (Fig. 3B). As expected, patients infected with HBc L60V variants had a significantly higher V60-specific CTL response by ELISPOT assay than those infected with wild-type isolates (57.00 ± 35.53 versus 23.50 ± 13.61; P < 0.05). Moreover, HBc L60V variant-infected patients with higher ALT levels (>80 IU/liter) demonstrated much higher V60-specific CTL activity than patients with lower ALT levels (<80 IU/liter) (57.00 ± 35.53 versus 14.60 ± 11.15; P < 0.05) (Fig. 3C). There were no statistically significant differences in sex and age between patients with the wild-type and variant isolates. The prevalence of the L60V variation increased with the disease progression in HBV-infected patients (Fig. 3D). We further analyzed the association between ALT levels and HBV DNA loads in CHB patients. Pearson analysis showed that there is a positive correlation between HBV DNA loads and ALT levels (Fig. 3E). Taken together, these results suggest that the HBc L60V variation is associated with necroinflammation and higher viral levels, and it may also be associated with a poor prognosis and a risk of ACLF.
Fig 3.

The HBc L60V variation is correlated with ALT levels, viral DNA loads, and disease progression in HBV-infected patients. (A and B) Distribution of serum ALT and AST levels (A) and HBV DNA loads (B) in CHB patients infected with HBV containing HBcL60 or HBcV60. (C) PBMCs were collected from HLA-A2-positive CHB patients with serum ALT levels of >40 IU/liter and were stimulated with the indicated peptides for detection of peptide-specific CTLs by ELISPOT assay. These patients were further divided into two groups by ALT levels: low (Lo) ALT levels (<80 IU/liter) and high (Hi) ALT levels (>80 IU/liter). The HBc82-90 peptide served as a negative control for background evaluation. Student's t test was used to determine P values. *, P < 0.05; **, P < 0.01. (D) Distribution of the rate of the HBcL60V variation in CHB and ACLF patients. Pearson's χ2 test was used to determine P values. (E) Correlation between HBV DNA loads and serum ALT levels in CHB patients. Pearson's χ2 test was used to determine P values.
The HBc L60V variation results in liver injury in HLA-A2.1/HBV transgenic mice.
Next, we examined whether the HBc L60V variation in HBV was able to induce immune-mediated liver injury using F1 hybrids of HBV transgenic BALB/c mice and HLA-A2.1/Kb transgenic mice as the experimental model. HLA-A2.1/HBV transgenic mice were immunized with an HBc DNA prime/peptide boost formulation. Serum ALT levels were measured at 1-week intervals after the first immunization. Mice were sacrificed at week 8, and splenocytes were isolated for ELISPOT and cytotoxicity assays. As shown in Fig. 4A, in comparison to mice immunized with HBc L60 DNA and L60 peptide, HBc V60 DNA- and V60 peptide-treated mice displayed approximately 50% greater serum ALT activity, which peaked at weeks 4 to 6. As expected, compared to L60 peptide-immunized mice, the number of SFCs with reactivity to V60 increased by 5-fold in V60-immunized mice (Fig. 4B). Interestingly, moderate cross-reactivity was also observed between the L60- and V60-specific T cell responses, as the number of SFCs with reactivity to V60 was moderately higher than that with reactivity to L60 in L60-immunized mice (P < 0.05). Similar results were observed by the cytotoxicity assay of splenocytes (Fig. 4C). In addition, HBV transgenic BALB/c mice (as a negative control) were immunized with the HBc DNA prime/peptide boost formulation, and no elevated ALT levels (Fig. 4D) or peptide-specific T cell responses (Fig. 4E) were observed in V60-treated mice. These results indicate that the epitope-specific CTL responses induced by the HBc L60V variation may contribute to liver injury.
Fig 4.

The HBc L60V variation causes CTL-mediated liver injury in HBV transgenic mice. F1 hybrids of HLA-A2.1/Kb transgenic mice and HBV transgenic BALB/c mice were immunized three times, as described in the legend to Fig. 2. (A) Serum ALT activity was measured at 2-week intervals after the first immunization. (B) ELISPOT assay. Splenocytes from immunized mice were stimulated with L60 or V60 peptide or with HBc82-90 or HBc18-27 as negative and positive controls, respectively. (C) Cytotoxicity assay. HepG2 cells labeled with CFSE were transfected with pHBV1.3 or pHBV1.3-HBcL60V as target cells and mixed with L60, V60, HBc18-27, or HBc82-90 peptide-stimulated splenocytes at the indicated ratios. HBV transgenic BALB/c mice were immunized with the DNA prime/peptide boost formula as described in the text. (D) Serum ALT activity was measured every week. (E) Splenocytes from immunized mice were stimulated with the L60 or V60 peptide or with HBc82-90 or HBc87-95 as the negative and positive controls, respectively. The data show the means ± SDs of five mice. *, P < 0.05; **, P < 0.01.
The HBc L60V variation facilitates viral capsid assembly and enhances HBV replication.
Because much higher HBV DNA loads were observed in CHB patients infected with HBc L60V variants than in those infected with wild-type virus (Fig. 3B), we examined if the HBc L60V variation affects viral replication. A leucine-to-valine mutation at HBc position 60 was introduced in the HBV replication vector pHBV1.3. Transfection of mutant HBV plasmid pHBV1.3-L60V in Huh7 cells resulted in moderately higher levels of HBsAg and HBeAg expression (Fig. 5A) (P < 0.01 for both at all time points) and approximately 10-fold higher HBV DNA levels in the supernatant compared to transfection of the wild-type HBV plasmid pHBV1.3 (Fig. 5B). Similar results were observed by Southern blotting of HBV replicative intermediates (Fig. 5C).
Fig 5.

The HBc L60V variation results in increased core capsid assembly and HBV replication. Huh7 cells were transfected with pHBV1.3 or pHBV1.3-HBcL60V. (A) Secretion of HBsAg and HBeAg was detected by ELISA at 24, 48, and 72 h after transfection. (B) HBV DNA levels in supernatants were quantified by real-time PCR at 48 h after transfection. (C) Southern blot analysis was performed to detect intracellular HBV levels at 48 h posttransfection. rcDNA, relaxed circulated HBV DNA; dsDNA, double-stranded HBV DNA; and ssDNA, single-stranded HBV DNA. HLA-A2.1/Kb transgenic mice were injected with 10 μg plasmid pHBV1.3 or pHBV1.3-HBcV60 via the tail vein using the hydrodynamic method. (D) Assays for the number of serum HBV DNA copies at day 4 were performed by real-time PCR. (E) HBcAg expression in the liver of transgenic mice was analyzed at day 8 by immunohistochemical staining. Data show the means ± SDs of five mice. (F) Huh7 cells were transfected with the pcDNA3.1-HBc149-L60 or pcDNA3.1-HBc149-V60 plasmid, and the cell lysates were harvested at 48 h after transfection. The assembly of HBc particles was detected by native agarose gel electrophoresis. The levels of total core protein and actin were determined by 15% SDS-PAGE and used as expression efficiency and loading controls, respectively. (G) The truncated forms (aa 1 to 149) of HBcL60 and HBcV60 were produced in E. coli; purified core particles were assembled in vitro. The levels of total core protein were used as a loading control. Data are representative of two independent experiments. *, P < 0.05; **, P < 0.01; OD, optical density.
We further examined the effect of the HBc L60V variation on HBV replication in vivo in HLA-A2.1/Kb transgenic mice that were hydrodynamically injected with the wild-type or the mutant HBV plasmid. In concert with the in vitro results, injection of the mutant HBV plasmid resulted in an approximately 3-fold increase in HBV DNA load (Fig. 5D) in mouse serum compared to that after injection of the wild-type HBV plasmid. Similarly, immunohistochemical staining results revealed that the number of HBcAg-positive cells in the mutant HBV plasmid-treated mice was greater (about 2-fold) than that in the wild-type plasmid-treated mice (Fig. 5E).
Because the HBc L60V variation site is not located in overlapping regions of the HBV genome or in the enhancer/promoter regions, we next investigated the effect of the HBc L60V variation on the expression of HBc and the formation of the core capsid. Huh7 cells were transfected with HBc expression vectors encoding the HBcL60 or HBcV60 protein. As shown in Fig. 5F, although comparable HBc expression levels were observed in pcDNA3.1-HBc149-L60- and pcDNA3.1-HBc149-V60-transfected cells, an elevated level of capsid assembly was detected in pcDNA3.1-HBc149-V60-transfected cells. Moreover, truncated HBcL60 and HBcV60 proteins were expressed in E. coli, and the purified proteins were assembled into capsids in vitro (31). As shown in Fig. 5G, a dramatic increase in capsid assembly was observed for HBcV60 compared to HBcL60. These results indicate that the leucine-to-valine mutation at position 60 of HBc has a major effect on capsid assembly, which may cause increased HBV replication.
DISCUSSION
Mutations in the pre-C/C region of HBV occur more frequently in HBV-infected patients with aggressive hepatitis and advanced liver diseases than inactive carriers or patients with acute self-limited hepatitis (35). In this study, we investigated the clinical and functional relevance of the hot variation site HBc60 for HBV infection. By screening the potential HLA-A2-restricted epitopes using an overlapping 9-mer peptide pool covering 1 to 150 aa of the HBc protein, we identified the epitope-generating variation HBc L60V. Further, we demonstrated that the HBc L60V variation correlates with clinically relevant parameters in patients with HBV infection, such as higher ALT/AST levels, higher viral DNA loads, a poor prognosis, and a greater likelihood of ACLF. Moreover, we demonstrated that the HBc L60V variation led to specific CTL responses and liver damage in vivo. Meanwhile, the HBc L60V variation caused a dramatic increase in viral capsid assembly and enhanced HBV replication. Our work may therefore provide further dissection of the impact of the HBc L60V variation on both HBV replicative competence and HBV-specific CD8+ T cell responses, suggesting a possible adaptation for the maintenance of viral persistence and the pathogenesis of liver disease.
Currently, the most popular strategy for T cell epitope screening is to begin with in silico prediction of the potential epitopic peptides using computer algorithms according to the mode of binding of the peptides to MHC molecules or assessing T cell responses with panels of overlapping peptides. However, a considerable number of epitopes are ignored by these methods (36). Using synthesized 9-mer peptides that overlap each other, we identified the HBc L60V variation-generated epitope V60 (VCWGELMNL), which is not a typical HLA-A2-restricted epitope, with Cys in position 2 at the N terminus, rather than Leu, Val, or Met (36). The molecular basis of the V60 epitope presented by HLA-A*0201 was also investigated through structural determination of the HLA-A*0201/V60 complex. We also verified that the epitope is generated through natural processing by detection of epitope-specific T cells in HBV-infected patients (Fig. 2C and 3C).
HBV evolves to accumulate epitope-reducing mutations to evade immune detection and destruction, including escape from T cell recognition (21, 37) and neutralization by anti-HBs antibodies (38), which may cause viral persistence. In addition, nonsynonymous mutations in HBV epitopes are associated with hepatitis activity and disease progression in HBV-infected patients (22, 39). In contrast to these results, in this study, we found that the HBc L60V variation actually added a new HLA-A2-restricted CD8+ T cell epitope that was associated with severe hepatitis and disease progression in CHB patients. This intrigued us and encouraged to explore the effect of the HBc L60V variation on viral replication and immune-mediated hepatic injury. Immunization with the V60 peptide elicited T cell-induced liver injury, as evidenced by increased ALT levels (Fig. 4A). HBV-specific T cells and hepatic infiltration of nonspecific inflammatory T cells play a key role in liver pathogenesis and disease progression (12). Furthermore, the HBc L60V variation increased viral capsid assembly and, thus, possibly enhanced HBV replication. CHB patients infected with HBc L60V variants exhibited much higher HBV DNA loads (approximately 10-fold) than those infected with wild-type virus (Fig. 3B), which is likely due to enhanced viral replicative competence by the HBc L60V variant protein. HBc capsid assembly is a critical step for the formation of HBV particles. Hydrophobic interactions and extensive interactions between α3 helices from the two HBc monomers where the L60V mutation is located have been reported to play an important role in capsid formation (40). Based on these results, we hypothesize that replacement of leucine by valine, which is more hydrophobic and has a shorter side chain, makes the entire complex more compact, enhances the stability of the HBc capsid, and facilitates the formation of virus particles (Fig. 6). Currently, it is difficult to mechanistically distinguish the exact roles of the new epitope-specific T cell response and the increased HBV replication in the necroinflammation and pathogenesis of liver disease in patients infected with HBc L60V variants. As HBV DNA loads are positively correlated with serum ALT levels in CHB (Fig. 3E) and the same L60V substitution has been reported by Hosono et al. (41) to affect an HLA class II-restricted T cell epitope and possibly result in immune escape, we speculate that the increased HBV replication induced by the HBc L60V variation may play a major role in the necroinflammation and pathogenesis in CHB. Taken together, these results reveal the mechanism of the impact of a single nonsynonymous variation in the HBV core region on HBV replication and T cell-mediated hepatic injury, which may help to predict HBV disease progression.
Fig 6.
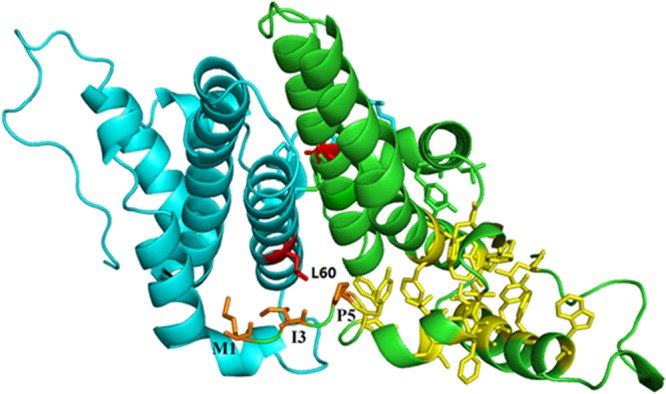
Structure of the HBV capsid dimer. Hydrophobic residues Met1, Ile3, and Pro5 (orange sticks) and the hydrophobic core (yellow sticks) of one monomer are shown as previously described (40). The two Leu60 residues are shown as red sticks. The structure with PDB accession number 1QGT was used to generate this stereo model using the PyMOL program.
Interestingly, Le Pogam et al. (42) report that the HBc L60V variation results in decreased HBsAg-coated virion secretion and increased naked core particles, indicating that such a variant may be less prone to infectiveness than its wild-type counterpart. HBV nucleoside analog-resistant variants also exhibit lower infectivity (13). In addition, the HBc L60A mutation blocks virion formation and secretion to undetectable levels but allows nucleocapsid formation (43). Based on these results and together with our data showing that the L60V variation both generated a new CTL epitope and increased viral replication, we speculate that the L60V variant may mainly produce subviral naked capsid particles with less infectious virions. Increased HBc capsid formation and direct exposure of the HBcAg to lymphocytes may induce a potent V60-specific CTL response, as evidenced by the similar number of V60 peptide-specific and the immunodominant epitope HBc18-27-specific CTLs detected in HBV-infected patients (Fig. 2C). Conceivably, the HBc L60V variant may be less infective than its wild-type counterpart, and the potential impact of the naked capsid on the infected host and the virus in the long run remains to be addressed.
The HBc L60V variation leads to relatively moderate but apparent increases of HBsAg and HBeAg secretion by about 20 to 30% (Fig. 5A). We speculate that the dramatically increased capsid package and viral replication may indirectly affect HBsAg folding, translocation, and/or secretion and HBeAg maturation and/or secretion. As the HBV capsid assembly is an immensely complicated molecular machine which many host factors are involved in (44), it is possible that the altered capsid assembly may affect the host interaction partners of HBV envelop proteins, and the effect may lead to altered HBsAg translocation and integration to the endoplasmic reticulum, as well as altered assembly and secretion of the envelope particles. Similarly, as a sign of viral activity, HBeAg maturation and secretion are complicated procedures and are regulated by many factors which could be influenced by HBV capsid assembly. This needs further investigation.
In summary, we investigated the clinical significance, variable T cell immune response, and virologic characteristics associated with a hot spot variation (L60V) located in the HBc protein, and we demonstrated a unique pattern of virus mutation in favor of viral persistence and necroinflammation. Our data suggest a reciprocal effect of the HBc L60V variation on new epitope-induced CTL responses and increased viral replication. Understanding the impact of the HBc L60V variation in the complex regulatory networks that orchestrate HBV replication, T-cell immune responses, and immunopathogenesis in chronic viral infections will allow us to predict the progression of the liver disease.
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (31230026, 30970146, 91029724, 81021003, and 81102018) and a grant from Key Projects in the National Science & Technology Program (2013ZX10002001-003-003).
Footnotes
Published ahead of print 15 May 2013
REFERENCES
- 1. Dunn C, Brunetto M, Reynolds G, Christophides T, Kennedy PT, Lampertico P, Das A, Lopes AR, Borrow P, Williams K, Humphreys E, Afford S, Adams DH, Bertoletti A, Maini MK. 2007. Cytokines induced during chronic hepatitis B virus infection promote a pathway for NK cell-mediated liver damage. J. Exp. Med. 204:667–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhang Z, Chen D, Yao J, Zhang H, Jin L, Shi M, Zhang H, Wang FS. 2007. Increased infiltration of intrahepatic DC subsets closely correlate with viral control and liver injury in immune active pediatric patients with chronic hepatitis B. Clin. Immunol. 122:173–180 [DOI] [PubMed] [Google Scholar]
- 3. Kakimi K, Lane TE, Wieland S, Asensio VC, Campbell IL, Chisari FV, Guidotti LG. 2001. Blocking chemokine responsive to gamma-2/interferon (IFN)-gamma inducible protein and monokine induced by IFN-gamma activity in vivo reduces the pathogenetic but not the antiviral potential of hepatitis B virus-specific cytotoxic T lymphocytes. J. Exp. Med. 194:1755–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sitia G, Isogawa M, Kakimi K, Wieland SF, Chisari FV, Guidotti LG. 2002. Depletion of neutrophils blocks the recruitment of antigen-nonspecific cells into the liver without affecting the antiviral activity of hepatitis B virus-specific cytotoxic T lymphocytes. Proc. Natl. Acad. Sci. U. S. A. 99:13717–13722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stross L, Gunther J, Gasteiger G, Asen T, Graf S, Aichler M, Esposito I, Busch DH, Knolle P, Sparwasser T, Protzer U. 2012. Foxp3+ regulatory T cells protect the liver from immune damage and compromise virus control during acute experimental hepatitis B virus infection in mice. Hepatology 56:873–883 [DOI] [PubMed] [Google Scholar]
- 6. Zhang JY, Zhang Z, Lin F, Zou ZS, Xu RN, Jin L, Fu JL, Shi F, Shi M, Wang HF, Wang FS. 2010. Interleukin-17-producing CD4(+) T cells increase with severity of liver damage in patients with chronic hepatitis B. Hepatology 51:81–91 [DOI] [PubMed] [Google Scholar]
- 7. Leifeld L, Cheng S, Ramakers J, Dumoulin FL, Trautwein C, Sauerbruch T, Spengler U. 2002. Imbalanced intrahepatic expression of interleukin 12, interferon gamma, and interleukin 10 in fulminant hepatitis B. Hepatology 36:1001–1008 [DOI] [PubMed] [Google Scholar]
- 8. Sitia G, Isogawa M, Iannacone M, Campbell IL, Chisari FV, Guidotti LG. 2004. MMPs are required for recruitment of antigen-nonspecific mononuclear cells into the liver by CTLs. J. Clin. Invest. 113:1158–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tan AT, Koh S, Goh W, Zhe HY, Gehring AJ, Lim SG, Bertoletti A. 2010. A longitudinal analysis of innate and adaptive immune profile during hepatic flares in chronic hepatitis B. J. Hepatol. 52:330–339 [DOI] [PubMed] [Google Scholar]
- 10. Iannacone M, Sitia G, Ruggeri ZM, Guidotti LG. 2007. HBV pathogenesis in animal models: recent advances on the role of platelets. J. Hepatol. 46:719–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thimme R, Wieland S, Steiger C, Ghrayeb J, Reimann KA, Purcell RH, Chisari FV. 2003. CD8(+) T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J. Virol. 77:68–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Maini MK, Boni C, Lee CK, Larrubia JR, Reignat S, Ogg GS, King AS, Herberg J, Gilson R, Alisa A, Williams R, Vergani D, Naoumov NV, Ferrari C, Bertoletti A. 2000. The role of virus-specific CD8(+) cells in liver damage and viral control during persistent hepatitis B virus infection. J. Exp. Med. 191:1269–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Billioud G, Pichoud C, Parent R, Zoulim F. 2012. Decreased infectivity of nucleoside analogs-resistant hepatitis B virus mutants. J. Hepatol. 56:1269–1275 [DOI] [PubMed] [Google Scholar]
- 14. Amini-Bavil-Olyaee S, Herbers U, Sheldon J, Luedde T, Trautwein C, Tacke F. 2009. The rtA194T polymerase mutation impacts viral replication and susceptibility to tenofovir in hepatitis B e antigen-positive and hepatitis B e antigen-negative hepatitis B virus strains. Hepatology 49:1158–1165 [DOI] [PubMed] [Google Scholar]
- 15. Sonneveld MJ, Rijckborst V, Zeuzem S, Heathcote EJ, Simon K, Senturk H, Pas SD, Hansen BE, Janssen HL. 2012. Presence of precore and core promoter mutants limits the probability of response to peginterferon in hepatitis B e antigen-positive chronic hepatitis B. Hepatology 56:67–75 [DOI] [PubMed] [Google Scholar]
- 16. Yuen MF, Tanaka Y, Fong DY, Fung J, Wong DK, Yuen JC, But DY, Chan AO, Wong BC, Mizokami M, Lai CL. 2009. Independent risk factors and predictive score for the development of hepatocellular carcinoma in chronic hepatitis B. J. Hepatol. 50:80–88 [DOI] [PubMed] [Google Scholar]
- 17. Guo X, Jin Y, Qian G, Tu H. 2008. Sequential accumulation of the mutations in core promoter of hepatitis B virus is associated with the development of hepatocellular carcinoma in Qidong, China. J. Hepatol. 49:718–725 [DOI] [PubMed] [Google Scholar]
- 18. Xu Z, Ren X, Liu Y, Li X, Bai S, Zhong Y, Wang L, Mao P, Wang H, Xin S, Wong VW, Chan HL, Zoulim F, Xu D. 2011. Association of hepatitis B virus mutations in basal core promoter and precore regions with severity of liver disease: an investigation of 793 Chinese patients with mild and severe chronic hepatitis B and acute-on-chronic liver failure. J. Gastroenterol. 46:391–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Carey I, D'Antiga L, Bansal S, Longhi MS, Ma Y, Mesa IR, Mieli-Vergani G, Vergani D. 2011. Immune and viral profile from tolerance to hepatitis B surface antigen clearance: a longitudinal study of vertically hepatitis B virus-infected children on combined therapy. J. Virol. 85:2416–2428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Abbott WGH, Tsai P, Leung E, Trevarton A, Ofanoa M, Hornell J, Gane EJ, Munn SR, Rodrigo AG. 2010. Associations between HLA class I alleles and escape mutations in the hepatitis B virus core gene in New Zealand-resident Tongans. J. Virol. 84:621–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maman Y, Blancher A, Benichou J, Yablonka A, Efroni S, Louzoun Y. 2011. Immune-induced evolutionary selection focused on a single reading frame in overlapping hepatitis B virus proteins. J. Virol. 85:4558–4566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim D, Lyoo KS, Smith D, Hur W, Hong SW, Sung PS, Yoon SK, Mehta S. 2011. Number of mutations within CTL-defined epitopes of the hepatitis B virus (HBV) core region is associated with HBV disease progression. J. Med. Virol. 83:2082–2087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang S, Qiu L, Yan X, Jin W, Wang Y, Chen L, Wu E, Ye X, Gao GF, Wang F, Chen Y, Duan Z, Meng S. 2012. Loss of microRNA 122 expression in patients with hepatitis B enhances hepatitis B virus replication through cyclin G(1)-modulated P53 activity. Hepatology 55:730–741 [DOI] [PubMed] [Google Scholar]
- 24. Vitiello A, Marchesini D, Furze J, Sherman LA, Chesnut RW. 1991. Analysis of the HLA-restricted influenza-specific cytotoxic T lymphocyte response in transgenic mice carrying a chimeric human-mouse class I major histocompatibility complex. J. Exp. Med. 173:1007–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhou M, Xu D, Li X, Li H, Shan M, Tang J, Wang M, Wang FS, Zhu X, Tao H, He W, Tien P, Gao GF. 2006. Screening and identification of severe acute respiratory syndrome-associated coronavirus-specific CTL epitopes. J. Immunol. 177:2138–2145 [DOI] [PubMed] [Google Scholar]
- 26. Garboczi DN, Hung DT, Wiley DC. 1992. HLA-A2-peptide complexes: refolding and crystallization of molecules expressed in Escherichia coli and complexed with single antigenic peptides. Proc. Natl. Acad. Sci. U. S. A. 89:3429–3433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang N, Qi J, Feng S, Gao F, Liu J, Pan X, Chen R, Li Q, Chen Z, Li X, Xia C, Gao GF. 2011. Crystal structure of swine major histocompatibility complex class I SLA-1 0401 and identification of 2009 pandemic swine-origin influenza A H1N1 virus cytotoxic T lymphocyte epitope peptides. J. Virol. 85:11709–11724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu Z, Li XH, Qiu LP, Zhang XJ, Chen LZ, Cao S, Wang FS, Meng SD. 2009. Treg suppress CTL responses upon immunization with HSP gp96. Eur. J. Immunol. 39:3110–3120 [DOI] [PubMed] [Google Scholar]
- 29. Yang PL, Althage A, Chung J, Chisari FV. 2002. Hydrodynamic injection of viral DNA: a mouse model of acute hepatitis B virus infection. Proc. Natl. Acad. Sci. U. S. A. 99:13825–13830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang SF, Qiu LP, Liu GZ, Li Y, Zhang XJ, Jin WS, Gao GF, Kong XP, Meng SD. 2011. Heat shock protein gp96 enhances humoral and T cell responses, decreases Treg frequency and potentiates the anti-HBV activity in BALB/c and transgenic mice. Vaccine 29:6342–6351 [DOI] [PubMed] [Google Scholar]
- 31. Zlotnick A, Lee A, Bourne CR, Johnson JM, Domanico PL, Stray SJ. 2007. In vitro screening for molecules that affect virus capsid assembly (and other protein association reactions). Nat. Protoc. 2:490–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Choi YW, Park SG, Yoo J, Jung GH. 2005. Calcium ions affect the hepatitis B virus core assembly. Virology 332:454–463 [DOI] [PubMed] [Google Scholar]
- 33. Turelli P, Mangeat B, Jost S, Vianin S, Trono D. 2004. Inhibition of hepatitis B virus replication by APOBEC3G. Science 303:1829. [DOI] [PubMed] [Google Scholar]
- 34. Desmond CP, Bartholomeusz A, Gaudieri S, Revill PA, Lewin SR. 2008. A systematic review of T-cell epitopes in hepatitis B virus: identification, genotypic variation and relevance to antiviral therapeutics. Antivir. Ther. 13:161–175 [PubMed] [Google Scholar]
- 35. Malik A, Singhal DK, Albanyan A, Husain SA, Kar P. 2012. Hepatitis B virus gene mutations in liver diseases: a report from New Delhi. PLoS One 7:e39028. 10.1371/journal.pone.0039028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu J, Zhang SH, Tan SG, Zheng BW, Gao GF. 2011. Revival of the identification of cytotoxic T-lymphocyte epitopes for immunological diagnosis, therapy and vaccine development. Exp. Biol. Med. 236:253–267 [DOI] [PubMed] [Google Scholar]
- 37. Westover KM, Hughes AL. 2007. Evolution of cytotoxic T-lymphocyte epitopes in hepatitis B virus. Infect. Genet. Evol. 7:254–262 [DOI] [PubMed] [Google Scholar]
- 38. Lai MW, Lin TY, Tsao KC, Huang CG, Hsiao MJ, Liang KH, Yeh CT. 2012. Increased seroprevalence of HBV DNA with mutations in the S gene among individuals greater than 18 years old after complete vaccination. Gastroenterology 143:400–407 [DOI] [PubMed] [Google Scholar]
- 39. Frelin L, Wahlstrom T, Tucker AE, Jones J, Hughes J, Lee BO, Billaud JN, Peters C, Whitacre D, Peterson D, Milich DR. 2009. A mechanism to explain the selection of the hepatitis e antigen-negative mutant during chronic hepatitis B virus infection. J. Virol. 83:1379–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wynne SA, Crowther RA, Leslie AG. 1999. The crystal structure of the human hepatitis B virus capsid. Mol. Cell 3:771–780 [DOI] [PubMed] [Google Scholar]
- 41. Hosono S, Tai PC, Wang W, Ambrose M, Hwang DG, Yuan TT, Peng BH, Yang CS, Lee CS, Shih C. 1995. Core antigen mutations of human hepatitis B virus in hepatomas accumulate in MHC class II-restricted T cell epitopes. Virology 212:151–162 [DOI] [PubMed] [Google Scholar]
- 42. Le Pogam S, Yuan TT, Sahu GK, Chatterjee S, Shih C. 2000. Low-level secretion of human hepatitis B virus virions caused by two independent, naturally occurring mutations (P5T and L60V) in the capsid protein. J. Virol. 74:9099–9105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ponsel D, Bruss V. 2003. Mapping of amino acid side chains on the surface of hepatitis B virus capsids required for envelopment and virion formation. J. Virol. 77:416–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Prange R. 2012. Host factors involved in hepatitis B virus maturation, assembly, and egress. Med. Microbiol. Immunol. 201:449–461 [DOI] [PubMed] [Google Scholar]