

Abstract
Serine protease inhibitor elafin (E) and its precursor, trappin-2 (Tr), have been associated with mucosal resistance to HIV-1 infection. We recently showed that Tr/E are among principal anti-HIV-1 molecules in cervicovaginal lavage (CVL) fluid, that E is ∼130 times more potent than Tr against HIV-1, and that Tr/E inhibited HIV-1 attachment and transcytosis across human genital epithelial cells (ECs). Since herpes simplex virus 2 (HSV-2) is a major sexually transmitted infection and risk factor for HIV-1 infection and transmission, we assessed Tr/E contribution to defense against HSV-2. Our in vitro studies demonstrated that pretreatment of endometrial (HEC-1A) and endocervical (End1/E6E7) ECs with human Tr-expressing adenovirus (Ad/Tr) or recombinant Tr/E proteins before or after HSV-2 infection resulted in significantly reduced virus titers compared to those of controls. Interestingly, E was ∼7 times more potent against HSV-2 infection than Tr. Conversely, knockdown of endogenous Tr/E by small interfering RNA (siRNA) significantly increased HSV-2 replication in genital ECs. Recombinant Tr and E reduced viral attachment to genital ECs by acting indirectly on cells. Further, lower viral replication was associated with reduced secretion of proinflammatory interleukin 8 (IL-8) and tumor necrosis factor alpha (TNF-α) and decreased NF-κB nuclear translocation. Additionally, protected Ad/Tr-treated ECs demonstrated enhanced interferon regulatory factor 3 (IRF3) nuclear translocation and increased antiviral IFN-β in response to HSV-2. Lastly, in vivo studies of intravaginal HSV-2 infection in Tr-transgenic mice (Etg) showed that despite similar virus replication in the genital tract, Etg mice had reduced viral load and TNF-α in the central nervous system compared to controls. Collectively, this is the first experimental evidence highlighting anti-HSV-2 activity of Tr/E in female genital mucosa.
INTRODUCTION
The estimated seroprevalence of herpes simplex virus 2 (HSV-2) in North America is nearly 20% and is even higher, around 30 to 80%, in some developing countries and sub-Saharan Africa (1, 2). These numbers make genital herpes one of the leading and most prevalent sexually transmitted infections (STIs) worldwide. Most sexual and perinatal transmissions of HSV-2 occur during asymptomatic, or “mute,” mucocutaneous viral shedding (3), when a person is unaware of transmitting the pathogen to others. Even more alarming is the fact that HSV-2 is closely linked to HIV-1 infections, by being a risk factor for HIV-1 acquisition (4) and transmission (5, 6).
As a natural consequence of attachment, entry, and infection, viruses, including HSV-2, become exposed to a variety of innate sensors, or pathogen recognition receptors (PRRs), including Toll-like receptors (TLRs), RNA helicases, and inflammasomes (7, 8). Subsequently, viral recognition triggers a series of intracellular signal transduction events that activate key transcription factors involved in antiviral and immune-inflammatory responses. Specifically, upon activation, mitogen-activated protein kinase (MAPK), NF-κB (9), and the interferon (IFN) regulatory factors (IRF) (10) coordinate the expression of genes with antiviral and inflammatory activity. Type I IFNs (11), with IFN-β leading the way in defense against HSV-2 (12, 13), and interferon-stimulated genes (ISGs) (14, 15) are only a few examples of factors contributing to antiviral defense.
Exposure to HSV-2 also triggers the release of proinflammatory mediators, including tumor necrosis factor alpha (TNF-α) (16), interleukin 1β (IL-1β), and IL-6 (9, 12). Such factors contribute not only to the induction of protective innate and adaptive immune responses (12, 17) but also, if poorly controlled, to the development of systemic inflammatory reactions, as seen in neonatal sepsis (18), or in breeching the blood-brain barrier and HSV translocating into the central nervous system (CNS) (16, 19). HSV-2 enters the nervous system through the sensory nerve fibers within the stratified squamous epithelium into the dorsal root ganglion. Following the episode of acute infection, HSV-2 establishes a lifelong and latent infection, arguably in sensory ganglia, with recurrent episodes of reactivation and symptomatic or asymptomatic viral shedding at the original sites of viral entrance at the dermal and mucosal surfaces (20). These processes, however, remain poorly understood in humans. While HSV-2 infection in humans is not usually life-threatening, unless generalized, murine models demonstrate high morbidity and mortality associated with viral CNS dissemination, limb paralysis, and extensive mucosal and skin lesions, often requiring animal euthanasia (16). Hence, a murine model of HSV-2 infection may not be the most appropriate to mimic HSV-2 in humans. Regardless of the severity and presentation of herpetic lesions, the “mute” transmission of HSV-2, its lifelong latency, and the interplay between genital herpes and HIV-1 infections (21) place HSV-2 among high-priority infections, requiring the development of novel and efficient therapeutic and preventative measures, especially those protecting the genital mucosal.
Human genital epithelial cells (ECs), being the principle target in HSV-2 infection, respond to viral encounter by releasing innate antimicrobial factors in efforts to eradicate or contain viral replication (22, 23). Serine protease inhibitor elafin (E) and its precursor, trappin-2 (Tr), along with secretory leukocyte protease inhibitor (SLPI) and defensins, belong to a large family of cationic antimicrobials that have been linked to endogenous mucosal protection against sexually transmitted pathogens, including HSV-2 and HIV-1 (23–26). Trappin-2 and elafin (Tr/E), as well as SLPI, share the cysteine-rich fold with four disulfide bonds called the whey acidic protein (WAP) domain, involved in protease inhibition (27). Secreted as unglycosylated protein, Tr (9.9 kDa) (28) (or pre- or full-length elafin) contains an N-terminal cementoin domain (38 amino acids [aa]) (29), and elafin (5.9 kDa) contains a C-terminal 57-residue domain with a WAP structure (30, 31). Inhibition of human neutrophil elastase and proteinase 3 through the WAP domain allows Tr/E to control excessive inflammation and tissue damage. Additionally, cross-linking between the transglutaminase-binding motifs located on the N terminus of each Tr/E (29, 32) and extracellular matrix proteins like heparin and fibronectin makes Tr/E indispensable for repairing compromised tissue integrity (33).
Both Tr and E have also been shown to possess broad-spectrum antibacterial (34) and antifungal properties (32). Furthermore, we and others have shown that Tr/E exert immunomodulatory activity, where depending on the environment, they can either dampen inflammation or promote immunostimulatory events and prime the immune system (35–37). Tr/E are found at mucosal surfaces and in secretions, including cervicovaginal lavage fluid (CVL) (22, 23, 38); in tissues, like skin, placenta, and genital and gastrointestinal tracts (38–44); and in multiple cell types, such as neutrophils, macrophages, keratinocytes, and epithelial cells (12, 22, 45), including genital ECs (22, 38). Tr/E are regarded as alarm antiproteases, since they are produced mainly in response to proinflammatory stimuli, such as lipopolysaccharide (LPS) (46), TNF-α (47), and IL-1β (38, 48). Additionally, Tr/E were recently shown to be induced and secreted by genital ECs in response to poly(I·C), a surrogate of a viral double-stranded RNA (dsRNA) (22, 35, 49), thus further supporting their importance in the inflammatory environment.
We and others recently demonstrated the importance of Tr/E in increased antiviral protection against HIV-1 as well as enhanced poly(I·C)-induced antiviral immune responses (22, 23, 30, 35, 49). Specifically, elevated Tr/E levels in CVLs of HIV-exposed seronegative commercial sex workers (CSWs) were found to be associated with mucosal HIV-1 resistance (23) and to significantly contribute to CVL's natural anti-HIV-1 activity in vitro (49). However, the contribution of Tr/E to defense against HSV-2 genital infections remains undefined. The objective of this study was to elucidate whether and how each Tr/E individually contributes to anti-HSV-2 defense mechanisms in female genital mucosa.
MATERIALS AND METHODS
Reagents.
Two commercial Tr and E proteins were tested following protein reconstitution as per the manufacturer's instruction: (i) human recombinant 6×His-Tr (with a C terminus His tag) (R&D Systems, Burlington, ON, Canada) (35, 49); (ii) commercial human recombinant E (without a tag) HC4011 (Hycult Biotech, Uden, Netherlands) (49). Poly(I·C) (P1530; Sigma-Aldrich, Oakville, ON, Canada) was reconstituted in the phosphate-buffered saline (PBS).
Cell lines and viruses.
Human endometrial carcinoma 1A (HEC-1A; ATCC HTB-112; Rockville, MD) and Vero African green monkey kidney cells (ATCC CCL81) were maintained in McCoy's 5A modified medium (Invitrogen Life Technologies, Burlington, ON, Canada) and in alpha minimum essential medium (α-MEM) (Invitrogen Life Technologies), correspondingly, supplemented with 10% fetal bovine serum, 1% HEPES, 1% l-glutamine (Invitrogen Life Technologies), and 1% penicillin-streptomycin (Sigma-Aldrich, Oakville, ON, Canada). The endocervical End1/E6E7 cell line (kind gift from R. Fichorova, Brigham & Women's Hospital, Boston, MA) was generated as described before (50) and maintained in keratinocyte serum-free medium (GIBCO/BRL; Life Technologies) supplemented with 50 μg/ml bovine pituitary extract, 0.1 ng/ml epidermal growth factor, 100 units/ml penicillin, 100 μg/ml streptomycin, and CaCl2 to a final calcium concentration of 0.4 mM. All cells were cultured at 37°C in 5% CO2. Stocks of HSV-2 (strain 333) were generated using Vero cells and stored at −70°C until used.
HSV-2 in vitro model.
For viral infections, ECs were first pretreated with Tr/E for 1 h in serum-free medium, followed by HSV-2 inoculum in the same serum-free medium for an additional 2 h, after which virus was removed, cells were repeatedly washed with PBS, and complete growth medium was added for 24 or 48 h. In some experiments, Tr and E proteins were added not before but rather 2 h after HSV-2 infection and cell washings. To this end, HEC-1A cells were incubated with Tr/E for 1 h at 37°C in a serum-free medium, to which a complete growth medium was subsequently added for 24 h. After the infection, supernatants were collected and stored cell free at −70°C until further use. Viral titers were determined by plaque assay using Vero cells grown on 12-well plates to 70 to 80% confluence. Virus-containing samples were serially diluted in serum-free medium and added to Vero cell monolayers. Infected monolayers were incubated at 37°C for 2 h, being rocked every 15 min for viral absorption, and subsequently overlaid with complete α-MEM culture medium further supplemented with 0.05% human immune serum globulin (Canadian Blood Services). Infection was allowed to occur for 48 h at 37°C. Monolayers were then fixed and stained with crystal violet, and viral plaques were counted under a light microscope and expressed as PFU per milliliter of HSV-2.
Adenoviral constructs and their delivery into cell culture.
The replication-deficient adenoviral constructs (Ad) used in this study have been described in detail elsewhere (35, 51–53). To express human Tr, the Ad/Tr vector, encoding a gene for 95-aa human Tr, was used (52, 53). This adenoviral construct was previously called Ad/E. E1,E3-deleted empty adenovirus Ad-dl703 (Ad/dl), coding for no transgene, was used as a control for Ad/Tr (51). Both Ad vectors were prepared at the Centre for Gene Therapeutics at McMaster University (Hamilton, ON, Canada). ECs were either treated with Opti-MEM I reduced serum medium (Invitrogen Life Technologies) alone (untreated [UT]) or with a multiplicity of infection (MOI) of 50 PFU of Ad/dl or Ad/Tr at 37°C overnight. After repeated PBS washes and rest for 4 h, cells were exposed to various MOIs of HSV-2 in a serum-free medium for 2 h and then repeatedly washed with PBS, and complete growth medium was added for an additional 24 to 48 h. Viral titers were determined in cell-free supernatants as described above, pertaining to cell lines and viruses.
Viral attachment.
Viral attachment was determined as described previously (24), but with slight modifications. Briefly, cells and virus were first pretreated with either medium or Tr/E for 1 h at 37°C. Cells pretreated with medium received either virus alone or virus pretreated with proteins for 1 h at 37°C. Cells that were pretreated with proteins first were either repeatedly washed or not and exposed to HSV-2 virus with an MOI of 1 (1 PFU per 1 cell) for 5 h at 4°C to allow for viral attachment. After repeated washes with PBS to remove unbound virus, cell-associated virus was detected by probing Western blots (WB) of whole-cell lysates with anti-gD primary antibody (P1103; Virusys, Atlanta, GA) and anti-GAPDH antibody (ab9485; Abcam) to control for protein loading.
MTT viability assay.
MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] assay (Biotium Inc., Hayward, CA) was used as per the manufacturer's instruction to determine viability of HEC-1A cells after Ad and HSV-2 exposure and was described elsewhere (13, 35).
Tr/E knockdown by RNA interference.
A small interfering RNA (siRNA) molecule (Invitrogen Life Technologies) was used to target human Tr/E (GenBank accession number NM_002638) within positions 67 to 420 with a single open reading frame (ORF) through the following nucleotide sequence, starting from Tr/E gene position 202: 5′-CCCGUUAAAGGACAAGUUU-3′. RNA interference (RNAi) negative control (medium GC content), catalog no. 12935-300 (Invitrogen Life Technologies), was used as a nontargeting siRNA control. Reverse and scaled-down for a 96-well plate, transfections of siRNA (8 pmol) were done using Lipofectamine RNAiMAX and Opti-MEM medium (Invitrogen Life Technologies) as per the supplier's instructions. HEC-1A cells, 3 × 104 in a 100-μl total volume of complete growth medium, were transfected in a 96-well BD Falcon culture plate (BD Biosciences) for 48 to 72 h before challenging with HSV-2 at an MOI of 0.1 and 1. Knockdown efficiency was monitored by assessing Tr/E content in supernatants of HEC-1A cells using Tr/E enzyme-linked immunosorbent assay (ELISA) 24 h after viral challenge.
ELISAs.
Cell-free samples were stored at −70°C until assayed for human Tr/E, IL-8, IL-6, and TNF-α with an ELISA Duoset kit (R&D Systems) and for human IFN-β by an ELISA kit from Antigenic America Inc. (Huntington Station, NY), according to the supplier's protocol. For animal experiments, murine TNF-α, IL-6, MIP-2, and IFN-γ levels were measured using ELISA Duoset kits (R&D Systems), and for IFN-β detection, ELISA was conducted using the PBL biomedical kit from PBL (Piscataway, NJ). Analytes were quantified based on standard curves obtained using a Tecan Safire ELISA reader (MTX Labs Systems Inc.) as per the supplier's protocol.
Preparation of cell extracts and WB analysis.
Whole-cell extracts were prepared by using whole-cell extract buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1% NP-40, 1% SDS, and 1× protease inhibitor [Roche, Mississauga, ON, Canada]) as per standard protocol. Protein amount was quantified using a Bradford assay with bovine serum albumin (Sigma-Aldrich) as a standard and Bio-Rad dye reagent concentrate as a protein stain (Bio-Rad Laboratories, Mississauga, ON, Canada). WB was performed on a 10% polyacrylamide denaturing SDS-PAGE gel and polyvinylidene difluoride (PVDF) membranes (Amersham, Arlington Heights, IL) as per standard protocol, using anti-gD primary antibody (P1103; Virusys, Atlanta, GA) and anti-GAPDH primary antibody (ab9485; Abcam) to control for protein loading. After incubation with the corresponding horseradish peroxidase (HRP)-conjugated secondary antibody (Bio-Rad Laboratories), blots were visualized using a SuperSignal West Femto or Pico chemiluminescent substrate kit (Thermo Scientific, Rockford, IL). Quantification of band intensities was done using MBF ImageJ for Microscopy Software.
Immunofluorescence staining.
Immunofluorescence staining was performed as described before, but with minor modifications. HEC-1A cells grown on an 8-well BD Falcon culture slide (BD Biosciences) were pretreated with Tr and E proteins for 1 h before receiving either medium or HSV-2 at an MOI of 1 per well for 4 h. Following treatment, cells were fixed and blocked as described elsewhere (13, 14). IRF3 was detected using a 1:100 dilution (in blocking solution) of primary antibody IBL18781 (IBL, Gunma, Japan) for 1 h; NF-κB p65 sc-372 (Santa Cruz Biotechnologies, Santa Cruz, CA) (1:500) was used to detect nuclear translocation of NF-κB p65; gD was detected using anti-gD primary antibody (1:100) (P1103; Virusys, Atlanta, GA). Negative-control rabbit immunoglobulin fraction (DakoCytomation, Glostrup, Denmark) served as an isotype control and was diluted to match the protein content of the primary antibodies. Secondary antibodies, Alexa Fluor 488-conjugated goat anti-rabbit IgG, Alexa Fluor 488-conjugated goat anti-mouse, and Alexa Fluor 647-conjugated goat anti-rabbit antibodies (Molecular Probes, Eugene, OR), were added to cells in a blocking solution for 1 h. Nuclei were visualized by staining with propidium iodide. Images were acquired using an inverted laser-scanning confocal microscope (LSM 510; Zeiss, Oberkochen, Germany).
RNA extraction and real-time quantitative PCR analysis.
The protocol for RNA isolation was described elsewhere (35, 54). Briefly, total RNA was isolated from HEC-1A-Ad-infected cells cultured with medium alone or HSV-2 for 6 h, using TRIzol reagent (Invitrogen Life Technologies), and DNase treated (Ambion, Austin, TX), as per the supplier's recommendations. RNA was quantified using the Agilent 2100 Bio-Analyzer (Agilent, Santa Clara, CA). Total RNA was reverse transcribed into cDNA with SuperScript reverse transcriptase III (Invitrogen Life Technologies). Real-time quantitative PCR was performed in a total volume of 25 μl using 1× universal PCR master mix (Applied Biosystems, Foster City, CA), 5 μl of diluted cDNA, 500 nmol forward primer, 500 nmol reverse primer (Mobix; McMaster University, ON, Canada), and 200 nmol probe in a 96-well plate. TaqMan oligonucleotide primers and probes, listed below and labeled with 6-carboxyfluorescein (FAM) at the 5′ end and a nonfluorescent quencher at the 3′ end, were designed using Primer Express 1.5 and purchased from Applied Biosystems: IFN-β, 5′-CGCCGCAGTGACCATCTA-3′ (forward) and 5′-CCAGGAGGTTCTCAACAATAGTC-3′ (reverse); 18S rRNA, 5′-CGGAATTAACCAGACAAATCGCTCCA-3′ (probe), 5′-GTGCATGGCCGTTCTTAGTT-3′ (forward), and 5′-TGCCAGAGTCTCGTTCGTTAT-3′ (reverse). The expression of 18S ribosomal RNA was used as an internal control. PCR was run with the standard program: 95°C for 10 min and 40 times of cycling at 95°C for 15 s and 60°C for 1 min in a 96-well plate with an ABI PRIZM 7900HT sequence detection system using Sequence Detector Software 2.2 (Applied Biosystems).
Animals.
Female mice used in this study included wild-type (WT) C57BL/6 mice (Charles River Canada, St. Constant, Quebec, Canada) and EcDNA mice, also on a C57BL/6 background, that were generated to express the gene for human full-length elafin (pre-elafin), or trappin-2, under the MCMV promoter as described elsewhere (55). Presence of the Tr gene was routinely confirmed with genotypic analysis as per standard protocol. All mice were 8 to 16 weeks old and maintained in level B housing conditions in a 12-hour light-dark cycle. All experimental protocols involving mice were approved by the Animal Research Ethics Board of McMaster University.
HSV-2 in vivo model.
Five days before viral challenge, mice were injected subcutaneously with 2 mg of Depo-Provera (Depo) (Upjohn, Don Mills, ON, Canada) to facilitate vaginal infection, since Depo is a long-acting progesterone formulation that induces a diestrus-like state in the genital tract. Mice were anesthetized with ketamine (150 mg/kg) and xylazine (10 mg/kg) and injected intraperitoneally (i.p.), their tails were lifted, and 1 × 104 PFU/mouse was administered intravaginally (IVAG) in a total volume of 10 μl in PBS. Mice were kept on their backs under the influence of anesthesia between 45 min and 1 h to allow the inoculum to be retained. Mice were monitored daily for survival and disease progression using the following five-point scale: 0, no apparent inflammation and infection; 1, slight redness of external vaginal os; 2, swelling and redness of external vaginal os; 3, severe swelling and redness of both vaginal os and surrounding tissue and hair loss in genital area; 4, genital ulceration with severe redness, swelling, and hair loss of genital and surrounding tissue; 5, severe genital ulceration extending to surrounding tissue or signs of paralysis. Animals were euthanized at stage 5. Vaginal washes were collected daily over 8 days by pipetting twice consecutively with 30 μl of PBS in and out of the vagina of unanesthetized mice several times. On days 2, 6, and 8 postinoculation, whole genital tracts as well as spinal cords and brainstems were removed, placed in 1 ml of PBS, and processed individually via homogenization as described earlier (56). Viral titers were determined in supernatants using plaque assay as described earlier in this section.
Statistical analysis.
Statistical analysis was performed with either nonpaired Student's t test for differences between two groups or a one-way analysis of variance (ANOVA) for more than two groups, using Sigma Stat 2.0. Survival curves were compared with a log rank test using GraphPad Prism version 4.0. In all cases, data were expressed as means ± standard deviations (SD), and P values of <0.05 were considered significant.
RESULTS
Tr/E significantly reduce HSV-2 replication in genital ECs.
To elucidate if and how Tr/E contribute to defense against HSV-2 challenge in genital ECs, the role of exogenous Tr/E proteins was assessed. A replication-deficient adenovirus construct expressing the human Tr gene (Ad/Tr) (35, 52, 53) was used to express human Tr in endometrial HEC-1A and endocervical End1/E6E7 EC lines. To elucidate whether each Tr/E individually exhibits anti-HSV-2 activity, as well as to counteract side effects of adenovirus-expressed Tr, we also utilized recombinant Tr and E with HEC-1A cells. Figure 1A shows that supernatants from HEC-1A cells infected with Ad/Tr (Ad/Tr cells), prior to HSV-2 infection, had significantly (P < 0.05) lower HSV-2 titers than HEC-1A cells infected with a control Ad/dl vector (51) (Ad/dl cells). Although viral load in Ad/dl cells alone appeared to be reduced compared to untreated (UT) cells, the reduction in HSV-2 shedding in Ad/Tr cells was significantly greater, indicating that exogenous Tr further reduced viral replication and augmented anti-HSV-2 protection in ECs. A similar inhibitory pattern of Tr on HSV-2 replication was also demonstrated in endocervical End/E6E7 ECs (Fig. 1B), albeit it was not as marked as in HEC-1A cells. To determine if each Tr/E exhibits anti-HSV-2 activity, recombinant Tr/E (rTr/E) proteins were used to pretreat HEC-1A cells. Data in Fig. 1C and D demonstrate that each rTr/E independently significantly (P < 0.05) reduced viral titers in HEC-1A cells, compared to medium alone. The inhibition of HSV-2 replication was dose dependent and with physiologically relevant (0.04 to 1 μg/ml) protein concentrations for the female genital tract (22). Additionally, we ruled out impaired cell viability as a potential cause of reduced susceptibility to viral infection by performing an MTT viability assay (data not shown). Taken together, our results demonstrate that Tr and E, each individually, in physiologic concentrations not toxic to cells, have the capacity to inhibit HSV-2 replication and enhance antiviral protection.
Fig 1.
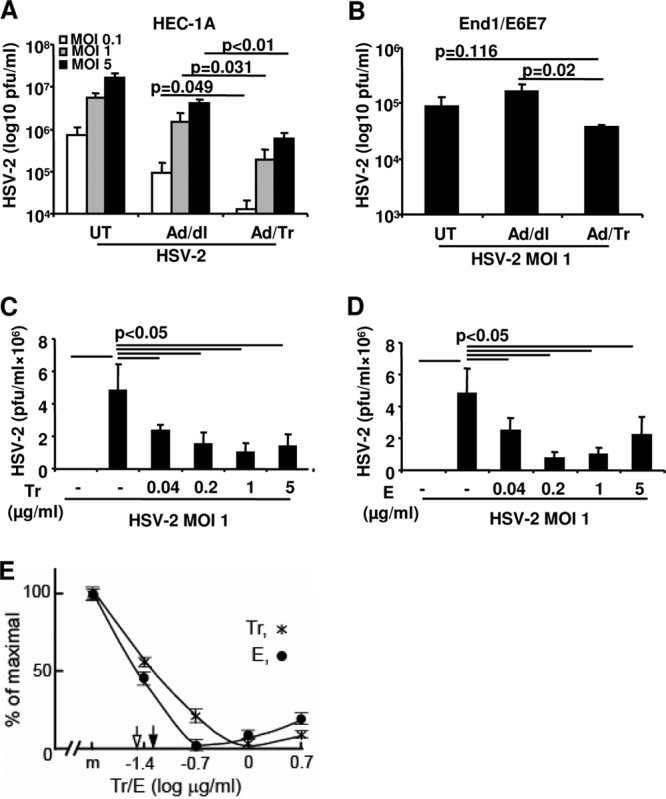
Tr and E reduce HSV-2 replication in genital ECs, and E has greater anti-HSV-2 activity than Tr. Genital ECs were left untreated (UT) or treated with an MOI of 0 to 50 of Ad/dl or Ad/Tr and inoculated with HSV-2 at an MOI of 0.1 to 5. Viral titers in supernatants were determined 24 h postinfection by standard plaque assay and presented as log-transformed PFU/ml for HEC-1A cells (A) and for End1/E6E7 (B). In panels C and D, HEC-1A cells were pretreated with Tr (C) and E (D) for 1 h and subsequently exposed to HSV-2 at an MOI of 1 for 2 h and, after repeated washes, cultured for an additional 24 h. Viral titers were determined in supernatants by plaque assay. For all, the data are shown as the means ± SD and are representative of three independent experiments performed in triplicate. Statistical analysis was performed using Student's t test (A, B) or ANOVA with Tukey's post hoc test (C, D), with significance indicated in graphs. For determining IC50s of Tr and E anti-HSV-2 activity, readouts from dose-dependent functional studies were used for determining efficiency parameters of relevant experimental points. The maximum inhibitory effect was considered 100% effect, and the other values were computed accordingly and plotted against relevant doses of Tr (*) and E (●) as percentages of maximal inhibitory effect. (E) The relevant IC50s for Tr and E are indicated with a filled arrowhead and an open arrowhead, respectively, pertaining to inhibitory effects of Tr and E on HSV-2 infectivity of HEC-1A cells. Error bars, SD.
E has greater anti-HSV-2 activity than Tr.
We next determined the comparative potency of each Tr/E against HSV-2 by identifying the 50% inhibitory concentration (IC50) values of Tr/E, given that tested recombinant Tr/E proteins possessed similar antiprotease activity (49). The IC50s for Tr/E with respect to infectivity of HEC-1A cells were estimated to be 0.07 ± 0.01 μg/ml and 0.01 ± 0.008 μg/ml (Fig. 1E). Comparison of IC50s for each Tr/E revealed that anti-HSV-2 efficacy of E was about 7 times greater than for Tr. Collectively, these results revealed that E is superior to its precursor, Tr, with respect to anti-HSV-2 activity.
Knockdown of endogenous Tr/E by siRNA significantly increased HSV-2 replication in HEC-1A cells.
To corroborate results shown in Fig. 1, endogenous Tr/E were knocked down in HEC-1A cells by Tr/E-specific small interfering RNA (siRNA), and cells were subsequently challenged with HSV-2. Figure 2A shows that levels of secreted Tr/E detected by ELISA in HEC-1A cell supernatants were significantly (P < 0.05) reduced after Tr/E siRNA knockdown and HSV-2 treatment, compared to those of control siRNA- and HSV-2-treated cells. Interestingly, the levels of Tr/E in the siRNA control group appeared to be moderately induced by HSV-2 challenge compared to medium-treated cells. The stimulatory effect, however, was more pronounced with the lower viral MOI challenge, and Tr/E expression was even completely abrogated following the high viral challenge (MOI of 5) (data not shown), suggesting a regulatory mechanism exploited by HSV-2 against antimicrobial innate factors. Further, cells that received Tr/E-specific siRNA and were challenged with HSV-2 had significantly increased amounts of infectious virus detected in supernatants (P = 0.02) compared to control siRNA-treated cells that maintained their endogenous Tr/E production and were better protected against HSV-2 challenge at an MOI of 0.1 (Fig. 2B). However, this protection in control siRNA-treated cells was overcome when a higher dose of HSV-2, an MOI of 1, was used (P < 0.01), which could be a consequence of viral downregulatory mechanisms against Tr/E. All siRNA-untreated cells had Tr/E levels within 50 pg/ml and viral loads similar to control siRNA-treated cells (data not shown). Overall, these results indicate that endogenous Tr/E play an important role in anti-HSV-2 defense in genital ECs, but their protective effect might be limited to lower viral doses.
Fig 2.
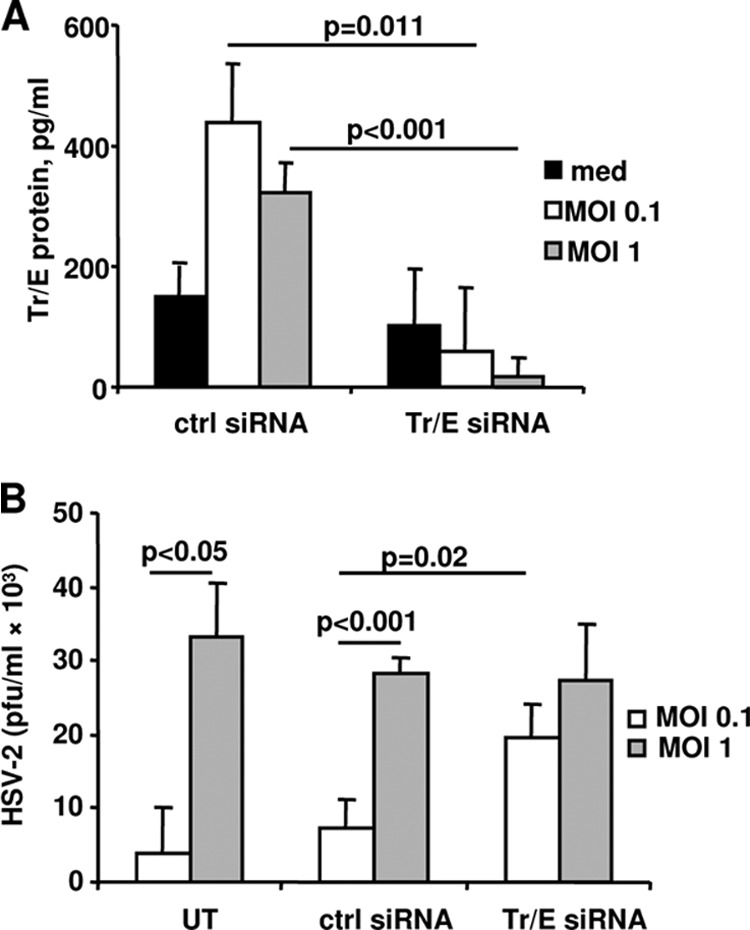
Knockdown of endogenous Tr/E by siRNA significantly increased HSV-2 replication in HEC-1A cells. HEC-1A cells were either left untreated (UT) or transfected with a nontargeting control siRNA (ctrl siRNA) or Tr/E siRNA for 48 to 72 h with subsequent challenge with HSV-2 at an MOI of 0.1 and 1 for 2 h. After repeated washes to remove viral inoculum, cells were cultured for an additional 24 h. Levels of Tr/E in supernatants were measured by ELISA (A), and viral titers were determined by plaque assay (B). The data are representative of at least two independent experiments performed in triplicate and are shown as the means ± SD. Statistical analysis was performed using Student's t test, with significance indicated in the graphs.
Tr and E reduce binding/attachment of HSV-2 to HEC-1A cells, preferentially acting through cells rather than the virus, and lower viral replication, even when added after HSV-2 infection.
Binding between HSV-2 glycoprotein D (gD) and its cognate cell surface receptor(s) is important for viral entry into cells (57). We hypothesized that reduced HSV-2 replication HEC-1A cells could be attributed to Tr/E-mediated alteration in viral binding/attachment. Thus, we determined if each Tr/E interfered with viral attachment to the cell surface at the entry level. Western blotting (WB) was performed to assess the amount of HSV-2 gD present in cell lysates as a representative of HSV-2 attached to HEC-1A cells. Figure 3A and B demonstrate that no gD was detected in cells pretreated with medium alone (Fig. 3A, lanes 1 and 2). Compared to cells exposed to HSV-2 alone (Fig. 3A, lanes 3 and 4), cells pretreated with Tr/E, and especially with E, before virus addition showed significantly reduced levels of gD detected in cell lysates (Fig. 3A, lanes 5, 6, 7, 8), indicating that Tr/E interfered with viral binding/attachment to genital ECs. We next elucidated whether Tr/E act on cells or the virus. Figure 3C and D show that no gD was detected in medium-treated cells, in contrast to cells treated with virus alone (Fig. 3C, compare lanes 1 and 2). Further, when cells were first pretreated with Tr or E (Fig. 3C, lane 3 or 5, respectively), repeatedly washed, and exposed to HSV-2, reduced amounts of gD were detected, compared to the virus-only group (Fig. 3C, lane 2). Interestingly, when the virus, and not the cells, was first pretreated with Tr or E (Fig. 3C, lane 4 or 6, respectively) and then added onto cells, similarly reduced amounts of gD were detected. Figure 3B and D show quantifying histograms of corresponding bands depicted in Fig. 3A and C. Notably, the inhibitory effect of E (Fig. 3A, lanes 7 and 8, and C, lanes 5 and 6) was more pronounced compared to the effect of Tr (Fig. 3A, lanes 5 and 6, and C, lanes 3 and 4). Interestingly, though, in separate repeated experiments using African green monkey kidney epithelial (Vero) cells, a preincubation of HSV-2 or cells with recombinant Tr and E did not have any inhibitory effect on viral infection of Vero cells (Fig. 3E and F), suggesting that Tr and E did not act directly on HSV-2 but rather on cells. Further, these results also suggested a selective antiviral mechanism of Tr and E in the female human genital ECs, compared to in African green monkey kidney ECs. Finally, since we earlier demonstrated that anti-HIV-1 activity of Tr/E was associated with nuclear localization (31), we next tested if Tr/E could act against HSV-2 beyond binding/attachment, by adding the proteins to HEC-1A cells 2 h after viral infection, as described in Materials and Methods. Figure 3G shows that both Tr and E were able to significantly reduce viral replication in HEC-1A cells even when added after viral infection, indicating that they can also act at the postentry level. Collectively, these results clearly indicate that Tr and E inhibited HSV-2 at the entry and postentry levels, and this was associated with reduced viral attachment to genital ECs, which was mediated through indirect interaction of Tr and E with the cells.
Fig 3.

Tr and E reduce HSV-2 binding/attachment, preferentially acting through cells rather than the virus, and lower viral replication, even when added after HSV-2 infection. In panel A, HEC-1A cells and HSV-2 were either treated with medium alone (−) or with 1 μg/ml of Tr or E protein for 1 h at 37°C before HSV-2 at an MOI of 1 was added to cells and allowed to attach at 4°C for 5 h. After viral challenge, cells were repeatedly washed, and whole-cell lysates were prepared and assessed for attached virus by immunoblotting using anti-gD antibodies and GAPDH as a loading control. (B) Relative intensities of WB bands were quantified using MBF_ImageJ for Microscopy Software. In panel C, HEC-1A cells (C lanes) were either pretreated with medium (med) or Tr and E as described above, repeatedly washed, and received HSV-2 at an MOI of 1 (V) for 5 h at 4°C as described above. Alternatively, virus was either left untreated or pretreated for 1 h with Tr or E before being added to medium-treated cells for 5 h at 4°C. WB in panel C and its quantification in panel D were performed as described above. In panels E and F, virus was either left untreated or preincubated with Tr or E for 1 h either at 4°C (E) or 37°C (F) and then added onto Vero cells for plaque assay as described in Materials and Methods. Data show viral titers as PFU/ml and are representative of two experiments, means ± SD. In panel G, Tr and E (1 μg/ml) were added in serum-free medium for 1 h at 37°C to HEC-1A cells 2 h after infection with HSV-2 at an MOI of 1 and cell washing. After 1 h, serum-containing medium was added, and cells were incubated for 24 h, after which viral titers were determined in cell supernatants by plaque assay as described in Materials and Methods. Data represent an average of three experiments with similar results, done in triplicate; means ± SD of PFU/ml, with significance shown in the graph.
Ad/Tr cells respond to HSV-2 challenge with increased IFN-β secretion.
The protective role of IFN-β against HSV-2 infection is well established (12–14, 58). We determined whether Tr/E pretreatment of genital ECs could increase IFN-β expression in response to HSV-2 challenge. Figure 4A shows that mRNA levels of IFN-β in Ad/Tr cells were significantly (P = 0.007) increased at 6 h after low-dose HSV-2 challenge of HEC-1A cells, compared to those in Ad/dl cells. Further, the stimulatory effect was also evident from ELISA data, showing significantly (P = 0.039) increased levels of IFN-β protein in supernatants of Ad/Tr cells compared to those in Ad/dl controls at 24 h postinfection (Fig. 4B). Overall, these data indicate that genital ECs exposed to Ad-expressed Tr before HSV-2 challenge secrete higher levels of IFN-β upon HSV-2 exposure.
Fig 4.

Increased IFN-β secretion and enhanced IRF3 nuclear translocation in Tr/E-pretreated HEC-1A cells after HSV-2 challenge. IFN-β release from HEC-Ad/Tr cells following exposure to HSV-2 at an MOI of 1. (A) HEC-1A cells were left untreated or treated with an MOI of 0 to 50 of Ad/dl or Ad/Tr and incubated for 6 to 24 h in the presence of media or HSV-2 at an MOI of 1. At 6 h posttreatment, total RNA was harvested, and relative expression of IFN-β mRNA by real-time quantitative reverse transcription (RT)-PCR was assessed. Values were normalized to internal control 18S. (B) At 6 and 24 h posttreatment with HSV-2 at an MOI of 1, secreted IFN-β was measured by ELISA in cell-free supernatants and presented as pg/ml. The data are representative of three independent experiments performed in triplicate and are shown as the means ± SD. Statistical analysis was performed using Student's t test, with significance indicated in the graphs. In panels C and D, immunofluorescence analysis of IRF3 nuclear translocation is demonstrated for HEC-1A cells that were left untreated (UT) or treated with an MOI of 0 to 50 of Ad/dl or Ad/Tr (C) or pretreated with Tr or E for 1 h (D) and subsequently exposed to either medium alone or HSV-1 at an MOI of 1 or 5 for 4 h. Representative staining is shown for IRF3 (green), nuclear stain propidium iodide (red), HSV-2 gD (blue), and composite (yellow) at magnifications of ×2520 or ×1260 (D, left column). The data are representative of two independent experiments with similar results.
Tr/E-treated ECs exhibit increased HSV-2-induced nuclear translocation of IRF3.
Interferon regulatory factor 3 (IRF3) is one of the key factors contributing to poly(I·C)-induced antiviral protection via activation of ISGs either with or without the induction of IFN-β (14, 35, 59, 60). Here, we determined whether Ad/Tr cells, with reduced viral load and induced IFN-β secretion, would exhibit increased nuclear translocation of IRF3 following HSV-2 challenge. Immunofluorescence staining for IRF3 nuclear translocation demonstrates that in the presence of medium alone, adenovirus-untreated HEC-1A cells (UT), or Ad/dl and Ad/Tr cells did not have significant translocation of IRF3 (green) into the nucleus (red), as no merged (yellow) color was present within the nucleus (Fig. 4C and D). In contrast, in HSV-2-treated rows, for both an MOI of 1 and 5, Ad/Tr cells showed a markedly increased number of cells with IRF3 nuclear translocation compared to control cells (Fig. 4C). Additionally, HEC-1A cells pretreated with recombinant Tr/E proteins also showed enhanced IRF3 nuclear translocation (Fig. 4D, white arrows), compared to virus-only-treated cells (Fig. 4D, HSV-2 MOI 1). Interestingly, Tr/E-treated cells with nuclear IRF3 also demonstrated the presence of HSV-2 gD in the cytoplasm, indicating that increased IRF3 nuclear translocation in these cells was not a result of reduced viral exposure, given that viruses are known to downregulate IRF3 activation. Taken together, these results indicate that preexposure of HEC-1A cells to either Ad-expressed Tr or recombinant Tr/E proteins enhanced nuclear translocation of IRF3 that was also associated with increased anti-HSV-2 protection of cells.
Tr/E-treated ECs secrete significantly reduced HSV-2-induced proinflammatory cytokines.
A physiologically relevant outcome of the attachment, entry, and infection of cells with HSV-2 is the release of immune-inflammatory factors (9, 12, 16), which may contribute to the pathogenesis and inflammation associated with genital herpes (61, 62). Given that Tr/E were previously shown to exert anti-inflammatory effects against bacterial (63, 64) and viral (30, 35, 49) ligands, we next determined whether pretreatment of genital ECs with Tr/E affected HSV-2-induced levels of proinflammatory IL-8 and TNF-α. Figure 5 shows that Ad/Tr cells (Fig. 5A) or HEC-1A cells pretreated with 1 μg/ml of each recombinant Tr/E before viral challenge (Fig. 5B and C) responded to HSV-2 infection with significantly (P < 0.05) reduced levels of IL-8 and TNF-α at 24 h, compared to the Ad/dl or medium-only groups, respectively. The secreted TNF-α in Ad-treated cell supernatants, however, was below the ELISA detection level (data not shown). All together, these data demonstrate that exogenous Tr/E modulated the secretion of inflammatory factors by ECs in response to HSV-2 challenge, which was also associated with reduced viral replication and attachment to cells.
Fig 5.

Tr/E-treated HEC-1A cells secrete significantly reduced proinflammatory cytokines and have reduced nuclear translocation of NF-κB in response to HSV-2 challenge. HEC-1A cells were left untreated (UT) or treated with an MOI of 0 to 50 of Ad/dl or Ad/Tr (A, D) or 1 μg/ml of Tr or E (B, C, E, F) and incubated for 4 to 24 h in the presence of media or HSV-2 at an MOI of 1. At 24 h post-viral challenge, secreted IL-8 and TNF-α were measured by ELISA in cell-free supernatants and presented as pg/ml (A, B, C). The data are representative of at three independent experiments performed in triplicate and are shown as the means ± SD. Statistical analysis was performed using Student's t test, with significance indicated in graphs. In panels D, E, and F, immunofluorescence analysis of NF-κB p65 subunit nuclear translocation is demonstrated for HEC-1A cells that were left untreated (UT) or treated with an MOI of 0 to 50 of Ad/dl or Ad/Tr (D) or pretreated with Tr or E for 1 h (E, F) and subsequently exposed to either medium alone or HSV-1 at an MOI of 1 for 4 h. Representative staining is shown for NF-κB p65 (green), nuclear stain propidium iodide (red), HSV-2 gD (blue), and composite (yellow) at magnifications of ×2520 or ×1260 (F, top). The data are representative of two independent experiments with similar results.
Tr/E-treated ECs exhibit reduced HSV-2-triggered NF-κB nuclear translocation.
NF-κB is one of the key transcription factors that regulate production of antiviral and proinflammatory factors, like IL-8 and TNF-α, upon encounter with viral ligands (9, 65), and Tr/E were previously shown to reduce NF-κB activation in response to bacterial (66) and viral ligand stimulation (35). Thus, given the reduced secretion of IL-8 and TNF-α observed in this study, we next determined if Tr/E pretreatment of HEC-1A cells results in attenuated NF-κB activation after HSV-2 exposure. Immunofluorescence staining was performed, assessing the localization of the p65 subunit of NF-κB (green) in HEC-1A cells, with nuclei visualized with propidium iodide (PI) (red), HSV-2 detected with anti-gD antibodies (blue), and nuclear translocation of p65 visualized as yellow. Qualitative analysis of representative confocal images (Fig. 5D to F) shows that treatment with medium alone did not trigger significant activation and nuclear translocation of p65 in either HEC-1A cells untreated or treated with Ad vector (Fig. 5D) or Tr/E proteins alone (Fig. 5F; data not shown for cells treated with Tr/E alone), as there was no yellow visualized in the nuclear area of the cells across the groups (Fig. 5D and F, med). Conversely, HSV-2 treatment alone triggered significant p65 nuclear translocation, which was reduced in Ad/Tr cells and in Tr/E-treated cells but not in Ad/dl cells (Fig. 5D to F, HSV-2). Although some nuclear localization of p65 was noted in Ad/Tr- and Tr/E-treated and virus-exposed groups, the intensity of the observed signal was substantially reduced compared to that of virus-only- or Ad/dl-HSV-2-treated groups (Fig. 5E and F). This reduction in nuclear NF-κB staining in Tr/E-treated virus-exposed groups was not due to a lack of virus exposure, since gD staining was observed (Fig. 5F). Collectively, these data indicate that pretreatment with both Tr and E before viral challenge reduces HSV-2-triggered nuclear translocation of the p65 subunit of NF-κB, which was also associated with reduced secretion of proinflammatory IL-8 and TNF-α. Next, we elucidated whether attenuated NF-κB activity in Tr/E-pretreated cells was associated with altered levels of innate sensors TLR3, RIG-I, and MDA-5, since we recently showed them to be modulated in the presence of Tr/E in response to poly(I·C) (35) and HIV-1 stimulation (31). However, no changes between the groups were detected (data not shown), suggesting a differential mechanism of Tr/E antiviral activity with different viral pathogens.
Tr-transgenic (Etg) mice have significantly lower viral load and reduced TNF-α protein levels in the CNS following intravaginal HSV-2 challenge compared to control mice.
Considering our in vitro findings in human genital ECs, next we tested antiviral properties of Tr in a murine model of genital HSV-2 infection. Since mice do not produce Tr/E, Tr-transgenic mice (earlier referred to as the full-length-elafin-transgenic mice, thus the name Etg) (55) were used, along with their corresponding wild-type (WT) C57BL/6 counterparts. Mice were intravaginally inoculated with a lethal 1 × 104 PFU/mouse dose of HSV-2. Our data show that Etg and WT control mice had no significant differences in viral replication in the genital area, including in vaginal washes (Fig. 6A) and the genital tract (GT) (Fig. 6B), throughout the time course of the genital HSV-2 infection. Interestingly, despite no differences in viral load in the GT between the groups, Etg mice appeared to have reduced viral loads in the CNS at day 2 (Fig. 6C), possibly due to delayed or less efficient viral translocation from the GT of Etg mice into the CNS. Indeed, despite earlier viral clearance from the GT, virus appeared in WT CNS 4 days earlier, on day 2, than in Etg mice, on day 6, and remained at higher levels on days 6 and 8, albeit reaching statistical significance only on day 8 (Fig. 6C). Moreover, a pattern of reduced viral load in the CNS of Etg mice was continuously observed throughout the infection, reaching statistical significance by day 8 (Fig. 6C). Furthermore, reduced viral replication in the CNS in Etg mice compared to that in control mice was also associated with significantly decreased levels of TNF-α in the GT and CNS at day 8 (Fig. 6D), as well as significantly attenuated pathology scores at day 5 (Fig. 6E) and a trend toward a better survival starting at day 8 (Fig. 6F). Taken together, these findings indicate that even though viral load in the GT of Etg animals was not more efficiently cleared compared to that in controls, viral spread and replication in the CNS was less efficient and better controlled in Etg mice, likely related to decreased levels of TNF-α detected in the GT and CNS. The latter would suggest a link between reduced inflammation and less efficient spread of HSV-2. Levels of other proinflammatory factors, namely, MIP-2, IL-6, IFN-β, and IFN-γ, were also measured, but only MIP-2 was consistently elevated in Etg GT at day 0 before viral inoculation and later reduced at day 8 in the CNS similar to TNF-α, albeit not reaching statistical significance (data not shown). Overall, these data indicate that in the presence of human Tr, mice had better virus containment and control of its spread to the CNS, associated with lower levels of proinflammatory factors locally and systemically.
Fig 6.
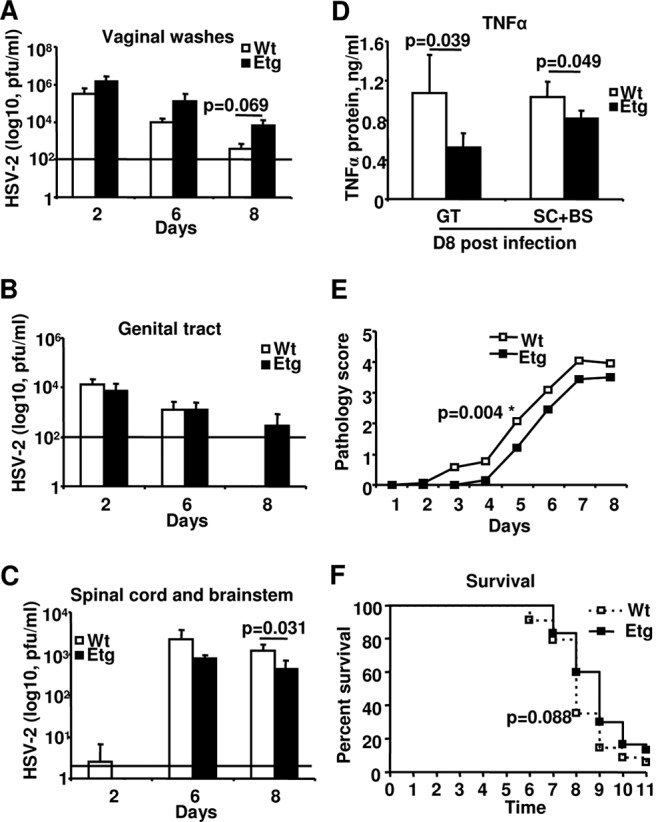
Tr-transgenic (Etg) mice have significantly lower viral load and reduced TNF-α protein levels in the CNS compared to control mice. Wild-type (Wt) control mice or transgenic mice expressing the gene for human Tr (Etg) were Depo treated and 5 days later intravaginally inoculated with 1 × 104 PFU per mouse of wild-type HSV-2 strain 333. Viral titers determined by plaque assay in vaginal washes (A) and in supernatants of homogenized genital tracts (GT) (B) and spinal cord with brainstem (SC+BS) (C), as well as protein levels of TNF-α determined by ELISA in supernatants of GT and SC+BS (D), pathology score (E), and survival (F) are shown. For survival and disease score, cumulative data (n = 30 to 34 mice) are presented. For viral titers and TNF-α levels, the representative data are from three independent experiments (n = 3 or 4 mice per group, per time point) and are shown as the means ± SD. Statistical analyses of survival curves were performed with a log rank test using GraphPad Prism. Statistical analyses of disease scores, viral titers, and TNF-α levels were performed using Student's t test, with significance indicated in graphs.
DISCUSSION
High prevalence of HSV-2 infection (45, 67), its serious complications in neonates and immunocompromised individuals (68), and the close interplay with HIV-1 (5) are worrisome and continue to remind us of the lack of efficient control of this STI. Identification of novel molecules capable of controlling primary HSV-2 infection and inflammatory responses at mucosal sites may advance our efforts in designing effective measures to curb both HSV-2 and HIV-1 infections.
We evaluated the contribution of each Tr/E to host defense against HSV-2 by utilizing human genital ECs in vitro and a model of genital HSV-2 infection in Tr-transgenic mice in vivo. Our results demonstrated that Tr/E likely influenced HSV-2 infection by acting indirectly on cells, rather than through virus, and targeting viral attachment and potentially postentry cellular antiviral and inflammatory responses, although the precise mechanism remains unknown. Cells pretreated with Tr/E secreted significantly lower levels of proinflammatory factors IL-8 and TNF-α and had attenuated nuclear translocation of NF-κB following HSV-2 challenge, which was not due to the lack of viral exposure or reduced viral attachment. Moreover, reduced viral replication in Tr/E-treated cells was also associated with increased IFN-β secretions and nuclear translocation of IRF3; the latter, once again, was not associated with reduced viral exposure of HEC-1A cells. Interestingly, we found that recombinant E was more potent against HSV-2 infection than its precursor, Tr. Finally, our results from in vivo experiments showed that Tr-transgenic mice, compared to WT controls, showed a consistent trend of increased survival; and a better disease outcome was also associated with lower viral translocation from the GT into the CNS, as well as reduced levels of TNF-α at the target organs. Collectively, these observations likely describe multiple antiviral activities of Tr/E in defense against HSV-2 infection in the female genital mucosa.
Although it is unclear at the moment how specifically Tr/E inhibited viral attachment to HEC-1A cells, our results propose that Tr/E may act through the target cells. Despite that we and others have recently demonstrated direct antiviral activities of Tr/E against vesicular stomatitis virus (VSV) (35) as well as HIV-1 (22, 31, 49), our findings could indicate that in defense against HSV-2, Tr/E acts indirectly through cells but not through the virus. Considering that Tr/E can bind to heparin and fibronectin (33), the interaction between Tr/E and cell surface HSV-2 binding/entry receptors, including heparan sulfate (HS) chains and nectin-1 and nectin-2 members of the immunoglobulin superfamily (57), can be one of the proposed modes of Tr/E antiviral activity. This suggested mechanism could explain why we observed decreased viral titers and attachment of HSV-2 to HEC-1A cells that resemble primary genital ECs and highly express HS moieties (69). An indirect inhibitory anti-HSV-2 effect of SLPI would support our data, since SLPI was found to exert its protective antiviral effect by interacting through ECs and not virus (70). However, the involvement of annexin II in anti-HSV-2 defense, which was important for anti-HIV-1 activity of SLPI, was ruled out (25). The observed inhibitory effect of Tr/E on HSV-2 attachment may have significant implications, since viral attachment to ECs is one of the critical steps required for subsequent fusion and entry of HSV-2 into ECs. Thus, our results indicate that Tr/E may affect the pathogenesis of HSV-2 by interfering with viral entry and consequently the establishment of primary HSV-2 infection mucosally, as well as by hindering entry/access and spread of virus in the nervous system.
Silencing of endogenous Tr/E with siRNA further corroborated the importance of Tr/E in anti-HSV-2 defense in the genital ECs. Our results also showed that the inhibitory effect of endogenous Tr/E was overcome with higher viral inoculum, suggesting a context-dependent nature of anti-HSV-2 activity of endogenous Tr/E that can be limited by HSV-2 evasion mechanisms. One such evasion mechanism may involve the regulation of Tr/E production, since we found significantly reduced mRNA and protein levels of Tr/E in cells challenged with a high dose (MOI of 5) of HSV-2 (data not shown), in contrast to a somewhat stimulatory effect from the low-dose viral challenge (Fig. 2A), as well as from the stimulation of genital ECs with a mimic of viral dsRNA, poly(I·C) (22, 35). The specific mechanisms of either stimulatory or inhibitory effects remain undefined at the moment but clearly reflect the complex dialogue between the innate immune system of the host and the pathogen.
Interestingly, a similar downregulatory evasion strategy was described for the papillomavirus protein E6 and Tr expression (71), as well as for HSV-2 and SLPI expression due to viral early-gene expression (47) and for HSV-2 and type I IFNs due to the expression of the HSV-2 virion host shutoff (Vhs) protein (58, 72). The latter two studies support our earlier findings showing that Vhs, in a dose-dependent way, inhibited innate immune sensing, IRF3 activation, and IFN-β expression in human vaginal ECs (73). All together, these data indicate that Tr/E appear to be important in the host's defense against HSV-2, and these molecules could represent a vulnerable target of the viral evasion mechanisms that should be taken into consideration when designing protective measures against STIs.
In this study, E appeared ∼7 times more potent against HSV-2 than its precursor, Tr. Interestingly, we reported a similar observation of E being more potent (∼100 times) than Tr against HIV-1 (49). These findings are also in line with the earlier-mentioned report on a divergent anti-HSV-2 effect of defensins (24). It is unclear at the moment why E was more potent than Tr against HSV-2 infection. However, our data likely reflect the result of elafin's greater ability to inhibit viral binding/entry and replication, to enter the nucleus, as well as to attenuate NF-κB phosphorylation/nuclear translocation and to modulate gene and protein expression of proinflammatory mediators.
Here, we found that IL-8 and TNF-α, as well as NF-κB nuclear translocation, were significantly diminished following exposure to each Ad/Tr and rTr/E, indicating that Tr/E can moderate HSV-2-induced antiviral immune-inflammatory responses that are also associated with decreased viral replication. We speculate, and our data would support this notion, that the observed moderation of inflammatory responses could be a result of Tr/E acting not only at the entry level, by reducing the number of attached virions, but also at the postentry level, or intracellularly, perhaps directly (DNA binding) or indirectly (upstream) targeting NF-κB transcriptional activity, all of which remain to be elucidated. The potential intracellular mechanism would be supported by earlier reports showing Tr/E-mediated reduction in LPS-induced AP-1 and NF-κB by targeting the ubiquitin-proteosome pathway (63) and upregulating IκBα (66). Moreover, the study demonstrating that SLPI can bind to DNA and compete for NF-κB binding sites, thus preventing NF-κB activation (74), and our recent finding of the dependence of anti-HIV-1 and immunomodulatory activity of Tr/E on its intranuclear localization (31) would also support the proposed intracellular mode of anti-HSV-2 action of Tr/E. Collectively, these observations may support DNA binding by Tr/E as one plausible mechanism of Tr/E antiviral activity that would explain both our reduced viral titers as well as anti-inflammatory mediators, which might be worth focusing on in future investigations.
In support of the argument that antimicrobial factors can act at multiple levels, α-defensin 1 was shown, in the absence of serum, to act directly on HIV-1 but, in the presence of serum, to inhibit HIV-1 replication by interfering with PKC phosphorylation in primary CD4+ T cells (75). Interestingly, the latter report is similar to an earlier study by McMichael et al. showing that, in serum-containing conditions, Tr/E had an inhibitory effect on LPS-induced release of TNF-α (36). However, the same study reported that in serum-free conditions and at higher concentrations, Tr/E had a stimulatory activity on TNF-α release.
Recently, we demonstrated that in HEC-1A cells, following adenoviral augmentation of Tr/E and in response to poly(I·C), the secretion of inflammatory factors and the expression of innate viral sensors RIG-I and MDA5 (35) were reduced. We also showed that genital ECs, pretreated with recombinant Tr and E proteins, secreted reduced levels of proinflammatory IL-8 as well as demonstrated attenuated mRNA expression of innate viral sensors TLR3 and RIG-I in response to R5-tropic HIV-1 (31). In contrast, no changes in expression levels of PRRs were found in this study following HSV-2 challenge (data not shown), suggesting that reduced levels of proinflammatory factors and NF-κB nuclear translocation observed here cannot be accounted for by the modulatory effect of Tr/E on PRR expression. These findings could also indicate that Tr/E could modulate immune responses toward various pathogens through differential mechanisms.
IRF3 has been shown to mediate antiviral protection through the activation of ISGs that may or may not depend on the induction of IFN-β (76, 77). Thus, it is likely that increased antiviral protection observed in Tr/E-treated and HSV-2-exposed cells was due in part to increased IRF3 nuclear translocation and consequentially augmented IFN-β induction. However, other IRFs, including IRF7 and IRF9, could also contribute, since IRF3 was shown to act in homo- and heterodimers (78). Overall, this is the first report of Tr/E-mediated increased IFN-β secretion and IRF3 nuclear translocation in the context of a viral infection that is also linked with increased antiviral protection of genital ECs against HSV-2 challenge. These observations clearly indicate a potential interaction between Tr/E and the IRF3 pathway, perhaps by acting through the upstream regulators of IRF3 induction. Taking into account that viruses, including HSV-2 and HIV-1, specifically target and disrupt functional activity of IRF3 as part of their evasion strategy (73, 79), such interaction might be of importance in future applications of antiviral properties of Tr/E.
A protective role of innate and adaptive immune responses mediated by NK cells, CD4+ and CD8+ cells, IL-15, and IFN-β (12, 80) has been well characterized and established in murine models of HSV-2 genital infection. However, a beneficial role of restricted antiviral and inflammatory responses was also recently highlighted (16, 81). In vivo experiments using a murine transgenic model of lethal HSV-2 intravaginal infection showed that Etg mice, expressing human Tr, had reduced viral replication in the CNS, which could be attributed to a better containment and less efficient viral translocation/spread from the local target organ, GT, into the CNS. It is unclear at the moment why viral clearance was not affected in the GT of Etg mice, as would be expected based on our in vitro data. However, this lack of antiviral protection in the GT could be related to the experimental conditions of high HSV-2 inoculum and a progesterone-driven system. Arguably, these conditions could be dampening specifically antiviral activity of Tr but sparing its immunomodulatory effects, as would be supported by decreased levels of TNF-α in GT and CNS in the Etg group. These findings could imply that Tr-mediated protection is perhaps conditional and can be overwhelmed by a higher viral load or other parameters of the infection, which was also seen in our siRNA experiments.
It is also unclear why viral translocation into the CNS was initially delayed or reduced in Etg animals at day 2, despite similar viral load in the GT. However, the sustained lower HSV-2 replication in the CNS of Etg mice is likely related to lower TNF-α levels in the GT and CNS, thus suggesting that immunomodulatory effects might be less conducive to viral systemic dissemination and might be better for a disease outcome. Our results are in line with studies demonstrating that mice lacking RNase L or with neutralized TNF-α had a better disease outcome and reduced mortality with genital HSV-2 infection (16, 81), hence confirming the beneficial role of Tr-mediated lower inflammation and viral translocation in genital HSV-2 infection. However, that only a trend of increased survival and a modest improvement in disease presentation were observed in Etg mice is a limiting factor in interpreting the significance of our observations and their extrapolations on human studies.
Although this is not the first study using Etg animals against viral challenge (37, 82), it is the first report pertaining to HSV-2 infection. Nevertheless, our study shares key observations with earlier publications. Despite the differential experimental designs and suggested antiviral mechanisms of Tr, our findings of increased antiviral protection and reduced mortality are in agreement with Zaidi et al. (acute model of viral myocarditis) (82) and Roghanian et al. (a model of adenoviral lung challenge assessing adaptive immunity) (37). That beneficial effects of Tr/E were observed in such distinct models attests to the existence of differential mechanisms of Tr/E antiviral protection that could be attributed to the pleiotropic nature of the molecules as well as the target organ specificity. Taken together, our results from in vivo studies provide evidence indicating the existence of a protective and contextual anti-HSV-2 effect of Tr/E.
In conclusion, this is the first study showing a Tr/E anti-HSV-2 effect in vitro and in vivo. This study presented novel evidence of multifactorial and complex activities of Tr/E that targeted virus-cell interaction and attachment/entry as well as mounting of antiviral and inflammatory cellular responses to HSV-2. These observations importantly complement and expand previously shown antibacterial and anti-HIV-1 activities of Tr/E and may translate into a broader use of Tr/E as microbicides in the female genital mucosa.
ACKNOWLEDGMENTS
We thank Ficherova for kindly providing End1/E6E7 cells for our experiments. We appreciate the expert and valuable technical support of J. Newton and A. Patrick.
This work was supported by a grant as part of the Comprehensive T Cell Vaccine Immune Monitoring Consortium (CTC-VIMC), a key component of the Collaboration for AIDS Vaccine Development (CAVD) funded by the Bill & Melinda Gates Foundation. A.G.D. was supported by a Studentship Award from the Ontario HIV Treatment Network (OHTN) and K.L.R. by a Career Scientist Award from the OHTN.
Footnotes
Published ahead of print 1 May 2013
REFERENCES
- 1. Chen CY, Ballard RC, Beck-Sague CM, Dangor Y, Radebe F, Schmid S, Weiss JB, Tshabalala V, Fehler G, Htun Y, Morse SA. 2000. Human immunodeficiency virus infection and genital ulcer disease in South Africa: the herpetic connection. Sex. Transm. Dis. 27:21–29 [DOI] [PubMed] [Google Scholar]
- 2. Xu F, Sternberg MR, Kottiri BJ, McQuillan GM, Lee FK, Nahmias AJ, Berman SM, Markowitz LE. 2006. Trends in herpes simplex virus type 1 and type 2 seroprevalence in the United States. JAMA 296:964–973 [DOI] [PubMed] [Google Scholar]
- 3. Koelle DM, Wald A. 2000. Herpes simplex virus: the importance of asymptomatic shedding. J. Antimicrob. Chemother. 45(Suppl T3):1–8 [DOI] [PubMed] [Google Scholar]
- 4. Freeman EE, Weiss HA, Glynn JR, Cross PL, Whitworth JA, Hayes RJ. 2006. Herpes simplex virus 2 infection increases HIV acquisition in men and women: systematic review and meta-analysis of longitudinal studies. AIDS 20:73–83 [DOI] [PubMed] [Google Scholar]
- 5. Corey L. 2007. Synergistic copathogens—HIV-1 and HSV-2. N. Engl. J. Med. 356:854–856 [DOI] [PubMed] [Google Scholar]
- 6. Ouedraogo A, Nagot N, Vergne L, Konate I, Weiss HA, Defer MC, Foulongne V, Sanon A, Andonaba JB, Segondy M, Mayaud P, Van de Perre P. 2006. Impact of suppressive herpes therapy on genital HIV-1 RNA among women taking antiretroviral therapy: a randomized controlled trial. AIDS 20:2305–2313 [DOI] [PubMed] [Google Scholar]
- 7. Rasmussen SB, Jensen SB, Nielsen C, Quartin E, Kato H, Chen ZJ, Silverman RH, Akira S, Paludan SR. 2009. Herpes simplex virus infection is sensed by both Toll-like receptors and retinoic acid-inducible gene-like receptors, which synergize to induce type I interferon production. J. Gen. Virol. 90:74–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yu M, Levine SJ. 2011. Toll-like receptor, RIG-I-like receptors and the NLRP3 inflammasome: key modulators of innate immune responses to double-stranded RNA viruses. Cytokine Growth Factor Rev. 22:63–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li H, Li X, Wei Y, Tan Y, Liu X, Wu X. 2009. HSV-2 induces TLRs and NF-kappaB-dependent cytokines in cervical epithelial cells. Biochem. Biophys. Res. Commun. 379:686–690 [DOI] [PubMed] [Google Scholar]
- 10. Mogensen TH, Paludan SR. 2005. Reading the viral signature by Toll-like receptors and other pattern recognition receptors. J. Mol. Med. (Berl.) 83:180–192 [DOI] [PubMed] [Google Scholar]
- 11. Kawai T, Akira S. 2006. Innate immune recognition of viral infection. Nat. Immunol. 7:131–137 [DOI] [PubMed] [Google Scholar]
- 12. Gill N, Deacon PM, Lichty B, Mossman KL, Ashkar AA. 2006. Induction of innate immunity against herpes simplex virus type 2 infection via local delivery of Toll-like receptor ligands correlates with beta interferon production. J. Virol. 80:9943–9950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nazli A, Yao XD, Smieja M, Rosenthal KL, Ashkar AA, Kaushic C. 2009. Differential induction of innate anti-viral responses by TLR ligands against herpes simplex virus, type 2, infection in primary genital epithelium of women. Antiviral Res. 81:103–112 [DOI] [PubMed] [Google Scholar]
- 14. Bauer CM, Dewitte-Orr SJ, Hornby KR, Zavitz CC, Lichty BD, Stampfli MR, Mossman KL. 2008. Cigarette smoke suppresses type I interferon-mediated antiviral immunity in lung fibroblast and epithelial cells. J. Interferon Cytokine Res. 28:167–179 [DOI] [PubMed] [Google Scholar]
- 15. Li Y, Li C, Xue P, Zhong B, Mao AP, Ran Y, Chen H, Wang YY, Yang F, Shu HB. 2009. ISG56 is a negative-feedback regulator of virus-triggered signaling and cellular antiviral response. Proc. Natl. Acad. Sci. U. S. A. 106:7945–7950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thapa M, Carr DJ. 2008. Herpes simplex virus type 2-induced mortality following genital infection is blocked by anti-tumor necrosis factor alpha antibody in CXCL10-deficient mice. J. Virol. 82:10295–10301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Thapa M, Carr DJ. 2008. Chemokines and chemokine receptors critical to host resistance following genital herpes simplex virus type 2 (HSV-2) infection. Open Immunol. J. 1:33–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kurt-Jones EA, Belko J, Yu C, Newburger PE, Wang J, Chan M, Knipe DM, Finberg RW. 2005. The role of Toll-like receptors in herpes simplex infection in neonates. J. Infect. Dis. 191:746–748 [DOI] [PubMed] [Google Scholar]
- 19. Kurt-Jones EA, Chan M, Zhou S, Wang J, Reed G, Bronson R, Arnold MM, Knipe DM, Finberg RW. 2004. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc. Natl. Acad. Sci. U. S. A. 101:1315–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cunningham AL, Diefenbach RJ, Miranda-Saksena M, Bosnjak L, Kim M, Jones C, Douglas MW. 2006. The cycle of human herpes simplex virus infection: virus transport and immune control. J. Infect. Dis. 194(Suppl 1):S11–S18 [DOI] [PubMed] [Google Scholar]
- 21. Rebbapragada A, Wachihi C, Pettengell C, Sunderji S, Huibner S, Jaoko W, Ball B, Fowke K, Mazzulli T, Plummer FA, Kaul R. 2007. Negative mucosal synergy between herpes simplex type 2 and HIV in the female genital tract. AIDS 21:589–598 [DOI] [PubMed] [Google Scholar]
- 22. Ghosh M, Shen Z, Fahey JV, Cu-Uvin S, Mayer K, Wira CR. 2010. Trappin-2/elafin: a novel innate anti-human immunodeficiency virus-1 molecule of the human female reproductive tract. Immunology 129:207–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Iqbal SM, Ball TB, Levinson P, Maranan L, Jaoko W, Wachihi C, Pak BJ, Podust VN, Broliden K, Hirbod T, Kaul R, Plummer FA. 2009. Elevated elafin/trappin-2 in the female genital tract is associated with protection against HIV acquisition. AIDS 23:1669–1677 [DOI] [PubMed] [Google Scholar]
- 24. Hazrati E, Galen B, Lu W, Wang W, Ouyang Y, Keller MJ, Lehrer RI, Herold BC. 2006. Human alpha- and beta-defensins block multiple steps in herpes simplex virus infection. J. Immunol. 177:8658–8666 [DOI] [PubMed] [Google Scholar]
- 25. MasCasullo V, Fam E, Keller MJ, Herold BC. 2005. Role of mucosal immunity in preventing genital herpes infection. Viral Immunol. 18:595–606 [DOI] [PubMed] [Google Scholar]
- 26. Venkataraman N, Cole AL, Svoboda P, Pohl J, Cole AM. 2005. Cationic polypeptides are required for anti-HIV-1 activity of human vaginal fluid. J. Immunol. 175:7560–7567 [DOI] [PubMed] [Google Scholar]
- 27. Tsunemi M, Matsuura Y, Sakakibara S, Katsube Y. 1996. Crystal structure of an elastase-specific inhibitor elafin complexed with porcine pancreatic elastase determined at 1.9 A resolution. Biochemistry 35:11570–11576 [DOI] [PubMed] [Google Scholar]
- 28. Schalkwijk J, Wiedow O, Hirose S. 1999. The trappin gene family: proteins defined by an N-terminal transglutaminase substrate domain and a C-terminal four-disulphide core. Biochem. J. 340(Part 3):569–577 [PMC free article] [PubMed] [Google Scholar]
- 29. Nara K, Ito S, Ito T, Suzuki Y, Ghoneim MA, Tachibana S, Hirose S. 1994. Elastase inhibitor elafin is a new type of proteinase inhibitor which has transglutaminase-mediated anchoring sequence termed “cementoin”.J. Biochem. 115:441–448 [DOI] [PubMed] [Google Scholar]
- 30. Drannik AG, Henrick BM, Rosenthal KL. 2011. War and peace between WAP and HIV: role of SLPI, trappin-2, elafin and ps20 in susceptibility to HIV infection. Biochem. Soc. Trans. 39:1427–1432 [DOI] [PubMed] [Google Scholar]
- 31. Drannik AG, Nag K, Yao XD, Henrick BM, Ball TB, Plummer FA, Wachihi C, Kimani J, Rosenthal KL. 2012. Anti-HIV-1 activity of elafin depends on its nuclear localization and altered innate immune activation in female genital epithelial cells. PLoS One 7:e52738. 10.1371/journal.pone.0052738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Baranger K, Zani ML, Chandenier J, Dallet-Choisy S, Moreau T. 2008. The antibacterial and antifungal properties of trappin-2 (pre-elafin) do not depend on its protease inhibitory function. FEBS J. 275:2008–2020 [DOI] [PubMed] [Google Scholar]
- 33. Guyot N, Zani ML, Maurel MC, Dallet-Choisy S, Moreau T. 2005. Elafin and its precursor trappin-2 still inhibit neutrophil serine proteinases when they are covalently bound to extracellular matrix proteins by tissue transglutaminase. Biochemistry 44:15610–15618 [DOI] [PubMed] [Google Scholar]
- 34. Simpson AJ, Maxwell AI, Govan JR, Haslett C, Sallenave JM. 1999. Elafin (elastase-specific inhibitor) has anti-microbial activity against Gram-positive and Gram-negative respiratory pathogens. FEBS Lett. 452:309–313 [DOI] [PubMed] [Google Scholar]
- 35. Drannik AG, Nag K, Yao XD, Henrick BM, Sallenave JM, Rosenthal KL. 2012. Trappin-2/elafin modulate innate immune responses of human endometrial epithelial cells to polyI:C. PLoS One 7:e35866. 10.1371/journal.pone.0035866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McMichael JW, Roghanian A, Jiang L, Ramage R, Sallenave JM. 2005. The antimicrobial antiproteinase elafin binds to lipopolysaccharide and modulates macrophage responses. Am. J. Respir. Cell Mol. Biol. 32:443–452 [DOI] [PubMed] [Google Scholar]
- 37. Roghanian A, Williams SE, Sheldrake TA, Brown TI, Oberheim K, Xing Z, Howie SE, Sallenave JM. 2006. The antimicrobial/elastase inhibitor elafin regulates lung dendritic cells and adaptive immunity. Am. J. Respir. Cell Mol. Biol. 34:634–642 [DOI] [PubMed] [Google Scholar]
- 38. King AE, Critchley HO, Sallenave JM, Kelly RW. 2003. Elafin in human endometrium: an antiprotease and antimicrobial molecule expressed during menstruation. J. Clin. Endocrinol. Metab. 88:4426–4431 [DOI] [PubMed] [Google Scholar]
- 39. King AE, Paltoo A, Kelly RW, Sallenave JM, Bocking AD, Challis JR. 2007. Expression of natural antimicrobials by human placenta and fetal membranes. Placenta 28:161–169 [DOI] [PubMed] [Google Scholar]
- 40. Pfundt R, van Ruissen F, van Vlijmen-Willems IM, Alkemade HA, Zeeuwen PL, Jap PH, Dijkman H, Fransen J, Croes H, van Erp PE, Schalkwijk J. 1996. Constitutive and inducible expression of SKALP/elafin provides anti-elastase defense in human epithelia. J. Clin. Invest. 98:1389–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pfundt R, Wingens M, Bergers M, Zweers M, Frenken M, Schalkwijk J. 2000. TNF-alpha and serum induce SKALP/elafin gene expression in human keratinocytes by a p38 MAP kinase-dependent pathway. Arch. Dermatol. Res. 292:180–187 [DOI] [PubMed] [Google Scholar]
- 42. Sallenave JM, Marsden MD, Ryle AP. 1992. Isolation of elafin and elastase-specific inhibitor (ESI) from bronchial secretions. Evidence of sequence homology and immunological cross-reactivity. Biol. Chem. Hoppe Seyler 373:27–33 [DOI] [PubMed] [Google Scholar]
- 43. Schalkwijk J, de Roo C, de Jongh GJ. 1991. Skin-derived antileukoproteinase (SKALP), an elastase inhibitor from human keratinocytes. Purification and biochemical properties. Biochim. Biophys. Acta 1096:148–154 [DOI] [PubMed] [Google Scholar]
- 44. Wiedow O, Schroder JM, Gregory H, Young JA, Christophers E. 1990. Elafin: an elastase-specific inhibitor of human skin. Purification, characterization, and complete amino acid sequence. J. Biol. Chem. 265:14791–14795 [PubMed] [Google Scholar]
- 45. Fisman DN, Lipsitch M, Hook EW, III, Goldie SJ. 2002. Projection of the future dimensions and costs of the genital herpes simplex type 2 epidemic in the United States. Sex. Transm. Dis. 29:608–622 [DOI] [PubMed] [Google Scholar]
- 46. Simpson AJ, Cunningham GA, Porteous DJ, Haslett C, Sallenave JM. 2001. Regulation of adenovirus-mediated elafin transgene expression by bacterial lipopolysaccharide. Hum. Gene Ther. 12:1395–1406 [DOI] [PubMed] [Google Scholar]
- 47. Fakioglu E, Wilson SS, Mesquita PM, Hazrati E, Cheshenko N, Blaho JA, Herold BC. 2008. Herpes simplex virus downregulates secretory leukocyte protease inhibitor: a novel immune evasion mechanism. J. Virol. 82:9337–9344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sallenave JM, Shulmann J, Crossley J, Jordana M, Gauldie J. 1994. Regulation of secretory leukocyte proteinase inhibitor (SLPI) and elastase-specific inhibitor (ESI/elafin) in human airway epithelial cells by cytokines and neutrophilic enzymes. Am. J. Respir. Cell Mol. Biol. 11:733–741 [DOI] [PubMed] [Google Scholar]
- 49. Drannik AG, Nag K, Yao XD, Henrick BM, Jain S, Ball TB, Plummer FA, Wachihi C, Kimani J, Rosenthal KL. 2012. Anti-HIV-1 activity of elafin is more potent than its precursor's, trappin-2, in genital epithelial cells. J. Virol. 86:4599–4610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fichorova RN, Rheinwald JG, Anderson DJ. 1997. Generation of papillomavirus-immortalized cell lines from normal human ectocervical, endocervical, and vaginal epithelium that maintain expression of tissue-specific differentiation proteins. Biol. Reprod. 57:847–855 [DOI] [PubMed] [Google Scholar]
- 51. Bett AJ, Haddara W, Prevec L, Graham FL. 1994. An efficient and flexible system for construction of adenovirus vectors with insertions or deletions in early regions 1 and 3. Proc. Natl. Acad. Sci. U. S. A. 91:8802–8806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sallenave JM, Xing Z, Graham F, Gauldie J. 1997. In vivo adenovirus-mediated expression of human pre-elafin, a potent neutrophil elastase inhibitor. Chest 111:128S–129S [DOI] [PubMed] [Google Scholar]
- 53. Sallenave JM, Xing Z, Simpson AJ, Graham FL, Gauldie J. 1998. Adenovirus-mediated expression of an elastase-specific inhibitor (elafin): a comparison of different promoters. Gene Ther. 5:352–360 [DOI] [PubMed] [Google Scholar]
- 54. Lester RT, Yao XD, Ball TB, McKinnon LR, Kaul R, Wachihi C, Jaoko W, Plummer FA, Rosenthal KL. 2008. Toll-like receptor expression and responsiveness are increased in viraemic HIV-1 infection. AIDS 22:685–694 [DOI] [PubMed] [Google Scholar]
- 55. Sallenave JM, Cunningham GA, James RM, McLachlan G, Haslett C. 2003. Regulation of pulmonary and systemic bacterial lipopolysaccharide responses in transgenic mice expressing human elafin. Infect. Immun. 71:3766–3774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Drannik AG, Pouladi MA, Robbins CS, Goncharova SI, Kianpour S, Stampfli MR. 2004. Impact of cigarette smoke on clearance and inflammation after Pseudomonas aeruginosa infection. Am. J. Respir. Crit. Care Med. 170:1164–1171 [DOI] [PubMed] [Google Scholar]
- 57. Spear PG. 2004. Herpes simplex virus: receptors and ligands for cell entry. Cell. Microbiol. 6:401–410 [DOI] [PubMed] [Google Scholar]
- 58. Murphy JA, Duerst RJ, Smith TJ, Morrison LA. 2003. Herpes simplex virus type 2 virion host shutoff protein regulates alpha/beta interferon but not adaptive immune responses during primary infection in vivo. J. Virol. 77:9337–9345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Guo J, Peters KL, Sen GC. 2000. Induction of the human protein P56 by interferon, double-stranded RNA, or virus infection. Virology 267:209–219 [DOI] [PubMed] [Google Scholar]
- 60. Mossman KL, Macgregor PF, Rozmus JJ, Goryachev AB, Edwards AM, Smiley JR. 2001. Herpes simplex virus triggers and then disarms a host antiviral response. J. Virol. 75:750–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cook WJ, Kramer MF, Walker RM, Burwell TJ, Holman HA, Coen DM, Knipe DM. 2004. Persistent expression of chemokine and chemokine receptor RNAs at primary and latent sites of herpes simplex virus 1 infection. Virol. J. 1:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Corey L, Adams HG, Brown ZA, Holmes KK. 1983. Genital herpes simplex virus infections: clinical manifestations, course, and complications. Ann. Intern. Med. 98:958–972 [DOI] [PubMed] [Google Scholar]
- 63. Butler MW, Robertson I, Greene CM, O'Neill SJ, Taggart CC, McElvaney NG. 2006. Elafin prevents lipopolysaccharide-induced AP-1 and NF-kappaB activation via an effect on the ubiquitin-proteasome pathway. J. Biol. Chem. 281:34730–34735 [DOI] [PubMed] [Google Scholar]
- 64. McMichael JW, Maxwell AI, Hayashi K, Taylor K, Wallace WA, Govan JR, Dorin JR, Sallenave JM. 2005. Antimicrobial activity of murine lung cells against Staphylococcus aureus is increased in vitro and in vivo after elafin gene transfer. Infect. Immun. 73:3609–3617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5:730–737 [DOI] [PubMed] [Google Scholar]
- 66. Henriksen PA, Hitt M, Xing Z, Wang J, Haslett C, Riemersma RA, Webb DJ, Kotelevtsev YV, Sallenave JM. 2004. Adenoviral gene delivery of elafin and secretory leukocyte protease inhibitor attenuates NF-kappa B-dependent inflammatory responses of human endothelial cells and macrophages to atherogenic stimuli. J. Immunol. 172:4535–4544 [DOI] [PubMed] [Google Scholar]
- 67. Nahmias AJ, Lee FK, Beckman-Nahmias S. 1990. Sero-epidemiological and -sociological patterns of herpes simplex virus infection in the world. Scand. J. Infect. Dis. Suppl. 69:19–36 [PubMed] [Google Scholar]
- 68. Koelle DM, Corey L. 2008. Herpes simplex: insights on pathogenesis and possible vaccines. Annu. Rev. Med. 59:381–395 [DOI] [PubMed] [Google Scholar]
- 69. Saidi H, Magri G, Nasreddine N, Requena M, Belec L. 2007. R5- and X4-HIV-1 use differentially the endometrial epithelial cells HEC-1A to ensure their own spread: implication for mechanisms of sexual transmission. Virology 358:55–68 [DOI] [PubMed] [Google Scholar]
- 70. Ma G, Greenwell-Wild T, Lei K, Jin W, Swisher J, Hardegen N, Wild KT, Wahl S. 2004. Secretory leukocyte protease inhibitor binds to annexin II, a cofactor for macrophage HIV-1 infection. J. Exp. Med. 200:1337–1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Duffy CL, Phillips SL, Klingelhutz AJ. 2003. Microarray analysis identifies differentiation-associated genes regulated by human papillomavirus type 16 E6. Virology 314:196–205 [DOI] [PubMed] [Google Scholar]
- 72. Duerst RJ, Morrison LA. 2004. Herpes simplex virus 2 virion host shutoff protein interferes with type I interferon production and responsiveness. Virology 322:158–167 [DOI] [PubMed] [Google Scholar]
- 73. Yao XD, Rosenthal KL. 2011. Herpes simplex virus type 2 virion host shutoff protein suppresses innate dsRNA antiviral pathways in human vaginal epithelial cells. J. Gen. Virol. 92:1981–1993 [DOI] [PubMed] [Google Scholar]
- 74. Taggart CC, Cryan SA, Weldon S, Gibbons A, Greene CM, Kelly E, Low TB, O'Neill JS, McElvaney NG. 2005. Secretory leucoprotease inhibitor binds to NF-kappaB binding sites in monocytes and inhibits p65 binding. J. Exp. Med. 202:1659–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Chang TL, Vargas J, Jr, DelPortillo A, Klotman ME. 2005. Dual role of alpha-defensin-1 in anti-HIV-1 innate immunity. J. Clin. Invest. 115:765–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Samuel CE. 2001. Antiviral actions of interferons. Clin. Microbiol. Rev. 14:778–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Yoneyama M, Fujita T. 2010. Recognition of viral nucleic acids in innate immunity. Rev. Med. Virol. 20:4–22 [DOI] [PubMed] [Google Scholar]
- 78. Hiscott J. 2007. Triggering the innate antiviral response through IRF-3 activation. J. Biol. Chem. 282:15325–15329 [DOI] [PubMed] [Google Scholar]
- 79. Doehle BP, McNevin HFJP, McElrath MJ, Gale M., Jr 2009. Human immunodeficiency virus type 1 mediates global disruption of innate antiviral signaling and immune defenses within infected cells. J. Virol. 83:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Chan T, Barra NG, Lee AJ, Ashkar AA. 2011. Innate and adaptive immunity against herpes simplex virus type 2 in the genital mucosa. J. Reprod. Immunol. 88:210–218 [DOI] [PubMed] [Google Scholar]
- 81. Duerst RJ, Morrison LA. 2007. Herpes simplex virus type 2-mediated disease is reduced in mice lacking RNase L. Virology 360:322–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zaidi SH, Hui CC, Cheah AY, You XM, Husain M, Rabinovitch M. 1999. Targeted overexpression of elafin protects mice against cardiac dysfunction and mortality following viral myocarditis. J. Clin. Invest. 103:1211–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]