

Abstract
Alphaviruses are small enveloped RNA viruses that include important emerging human pathogens, such as chikungunya virus (CHIKV). These viruses infect cells via a low-pH-triggered membrane fusion reaction, making this step a potential target for antiviral therapies. The E1 fusion protein inserts into the target membrane, trimerizes, and refolds to a hairpin-like conformation in which the combination of E1 domain III (DIII) and the stem region (DIII-stem) pack against a core trimer composed of E1 domains I and II (DI/II). Addition of exogenous DIII proteins from Semliki Forest virus (SFV) has been shown to inhibit E1 hairpin formation and SFV fusion and infection. Here we produced and characterized DIII and DI/II proteins from CHIKV and SFV. Unlike SFV DIII, both core trimer binding and fusion inhibition by CHIKV DIII required the stem region. CHIKV DIII-stem and SFV DIII-stem showed efficient cross-inhibition of SFV, Sindbis virus, and CHIKV infections. We developed a fluorescence anisotropy-based assay for the binding of SFV DIII-stem to the core trimer and used it to demonstrate the relatively high affinity of this interaction (Kd [dissociation constant], ∼85 nM) and the importance of the stem region. Together, our results support the conserved nature of the key contacts of DIII-stem in the alphavirus E1 homotrimer and describe a sensitive and quantitative in vitro assay for this step in fusion protein refolding.
INTRODUCTION
The Alphavirus genus contains about 40 recognized species of small enveloped plus-sense RNA viruses (1, 2). Most alphaviruses are transmitted by mosquito vectors and can infect a variety of mammalian, avian, and other hosts. Alphaviruses can produce fever, arthritis, and encephalitis in humans and include potential bioterrorism agents and emerging infectious disease threats, such as Venezuelan, Eastern, and Western equine encephalitis viruses and chikungunya virus (CHIKV). Recent outbreaks caused millions of CHIKV infections in south India and on La Reunion and other islands in the Indian Ocean (3, 4). Although past epidemics of CHIKV have been confined to Africa and Asia, increased air travel has produced increased transmission in Europe and the Western hemisphere (4–6). A mutation enhancing the ability of CHIKV to infect and be transmitted by the Aedes albopictus mosquito has also increased the risk for infection due to the continuing global spread of this vector (7–9). There are currently no approved vaccines or antiviral therapies for CHIKV infection.
Alphaviruses infect cells by binding to cell surface receptors, internalization by endocytosis, and low-pH-triggered fusion of the viral and endosomal membranes (reviewed in references 1 and 10). Similarly, CHIKV infection has been shown to require endocytic uptake and low pH (11). Fusion and infection by alphaviruses, such as Semliki Forest virus (SFV) and Sindbis virus (SINV), are promoted by cholesterol in the host cell membrane (12–16), and studies of CHIKV suggest that its infection is also promoted by cholesterol (11, 17).
The alphavirus membrane is covered by a highly organized protein lattice composed of trimers of heterodimers of the E2 and E1 viral envelope proteins (1, 18). E2 mediates receptor binding, and E1 is the membrane fusion protein (reviewed in references 1 and 10). The ectodomain of E1 is composed of three domains: the central domain I (DI), domain II (DII) containing the hydrophobic fusion loop, and domain IIII (DIII), an Ig-like domain that is connected to the transmembrane (TM) domain via a stem region of about 28 residues (19–22). During fusion, E1 inserts into the target membrane via the fusion loop, forms a core trimer composed of domains I and II, and refolds to a hairpin-like conformation in which DIII and the stem (DIII-stem) pack against the central core trimer (23). This refolding reaction moves the TM domain and the fusion loop to the same side of the trimer, bringing the viral and target membranes together and driving membrane fusion. Recombinant SFV DIII proteins can bind to the core trimer, preventing refolding to the hairpin and thereby inhibiting SFV fusion and infection (24, 25). The dengue virus (DENV) membrane fusion protein is structurally similar to alphavirus E1 (e.g., see references 26 and 27), and its refolding and fusion activity are analogously inhibited by DENV DIII proteins (24, 28) or by stem peptides (29–32).
We have previously described an in vitro system that reproduces the protein-protein interactions during trimerization of SFV E1 (33). Truncated E1 proteins containing domains I and II (E1 DI/II) were produced in S2 Drosophila cells. When treated at low pH in the presence of target membranes, such DI/II proteins form a stable core trimer that can bind recombinant DIII proteins. DIII binding is specific, occurs at either low or neutral pH, and is stabilized by the stem region. Because of its importance in driving the membrane fusion reaction, the interaction of DIII-stem with the core trimer represents a potential target for inhibitory drugs.
Here we have addressed the significance of our reconstituted SFV system to infection by medically important alphaviruses. We report the production of recombinant CHIKV DIII and DI/II proteins and the analysis of CHIKV DIII's inhibition of membrane fusion and infection. We found differences in the requirements for expression and in vitro refolding of SFV and CHIKV DIII proteins. We also found that the first nine amino acids of the stem region promoted inhibition by CHIKV DIII protein. Despite these differences between SFV and CHIKV, our results showed efficient cross-inhibition by DIII proteins from both viruses. We developed a fluorescence anisotropy-based assay to monitor the binding of SFV DIII with the core trimer. This assay demonstrated the increase in affinity conferred by the E1 stem region and provides a candidate screen for small molecule inhibitors of E1 refolding and fusion.
MATERIALS AND METHODS
Cells and viruses.
BHK-21 cells were cultured at 37°C in complete medium (Dulbecco's modified Eagle's medium containing 5% fetal calf serum, 10% tryptose phosphate broth, 100 U penicillin/ml, and 100 μg streptomycin/ml). Cholesterol-containing C6/36 mosquito cells and cholesterol-depleted C6/36 cells were cultured at 28°C as previously described (13). Wild-type (WT) SFV was a well-characterized plaque-purified stock (34). The chikungunya virus strain used was the CHIK vaccine strain 181/clone 25 (181/25), kindly provided by Robert B. Tesh (University of Texas Medical Branch, Galveston, TX). Sindbis virus (SINV) was derived from the Toto1101 infectious clone (35).
Fusion infection assay.
Virus fusion with the plasma membrane was quantitated using a previously described assay (24). In brief, dilutions of CHIKV, SFV, or SINV stocks were bound to BHK-21 cells on ice for 90 min and treated at 37°C for 1 min with medium buffered to the indicated pH. Cells were then cultured overnight at 28°C in medium containing 20 mM NH4Cl to prevent secondary infection. Cells infected due to virus fusion with cell membrane were quantitated by immunofluorescence (24). SFV- and CHIKV-infected cells were detected with a polyclonal antibody to the SFV E1 and E2 proteins (24), and SINV-infected cells were detected with a mixture of the monoclonal antibodies (MAbs) R6 and R2 against SINV E1 and E2 proteins, respectively, obtained from hybridomas kindly provided by William Klimstra (36). Fusion and infection were similarly assayed on control and cholesterol-depleted C6/36 cells, using 1 min of treatment at the indicated pH at 28°C.
Bacterial protein expression constructs.
DNA sequences of CHIKV E1 DIII with or without the stem region or of CHIKV E1 DI/II were amplified by PCR from the plasmid p353T-EIC, kindly donated by Felix Rey and Karine Moncoq (Pasteur Institute). CHIKV DIII sequences were subcloned into the bacterial expression vector pET-14b (Novogen) to express DIII proteins with an added N-terminal methionine and with or without an N-terminal 6×His tag, consisting of 36 extra residues. The plasmids expressing SFV E1 DIII proteins and dengue virus DV2 DIIIH1 protein were previously described (24). The residue numbers in the CHIKV and SFV DIII proteins were 291 to 383 (DIII) or 291 to 412 (DIIIS). SFV E1 DI/II was amplified from the previously reported plasmid pMTA-SFV E1DI/II (33). The CHIKV and SFV E1 DI/II sequences were cloned into the bacterial expression vector pET 21a(+) (Novagen). All sequences were confirmed by DNA sequencing.
Protein production in bacteria.
Plasmids were transformed in BL21(DE3) Rosetta cells (Stratagene). DIII proteins were expressed, refolded, and purified as previously described (24, 33). CHIKV and SFV E1 DI/II proteins were expressed and refolded using a modified version of a published protocol (37). In brief, bacterial cultures were induced with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for 3 h, bacterial pellets were treated to obtain inclusion bodies (24, 33), and proteins were refolded in vitro by a fast dilution method in a 1-liter oxidative refolding buffer (100 mM, Tris-HCl, 0.4 M nondetergent sulfobetaine 201 [NDSB-201], 5 mM reduced glutathione, 0.5 mM oxidized glutathione [pH 8.0]) (37). The refolded proteins were concentrated and purified by fast protein liquid chromatography on a Superdex S200 column (GE Healthcare). The protein concentration was determined by Bradford assay or by absorption at 205 nM (38).
Protein production in S2 Drosophila cells.
C-terminally truncated forms of SFV or CHIKV E1 proteins were obtained by inducible expression in Drosophila S2 cells as previously described (33). The SFV E1 DI/II protein includes domains I and II and 13 residues of the DI-DIII linker region and ends at E1 P294, followed by two Strep-tags separated by a flexible linker. Our previous studies showed that the DI-DIII linker was essential to form the stable core trimer (39). The analogous CHIKV E1 DI/II protein was constructed by DNA amplification of the plasmid p353T-EIC mentioned above. Inducible S2 cells expressing these constructs were prepared by cotransfection with the pT371 puromycin selection vector. Proteins were expressed by growth of the cells in Drosophila serum-free medium (HyClone, Logan, UT) in the presence of 750 μM copper sulfate at 27°C for 8 days. Strep-tagged proteins were purified from the culture medium by affinity chromatography using a Strep-Tactin column (IBA, Lifesciences, Goettingen, Germany).
Fluorescence-based thermostability analysis.
Protein thermostability was analyzed using an assay that detects the increase in fluorescence of SYPRO orange upon binding to exposed hydrophobic residues (Invitrogen, Carlsbad, CA). Proteins were diluted to 50 μM in 150 mM NaCl–100 mM HEPES (pH 7.5), and 40-μl samples were mixed with 1 μl of a 1:200 dilution of SYPRO orange dye stock. Replicate 20-μl aliquots were added to a standard 96-well PCR plate and covered with optical sealing film. Fluorescence was monitored versus temperature using a real-time PCR device (Bio-Rad iCycler iQ5 PCR thermal cycler; Bio-Rad Hercules, CA). The companion software was used to analyze the data and calculate the midpoint of the protein unfolding transition (Tm).
Liposome preparation.
Liposomes were prepared in TAN buffer (20 mM triethanolamine [TEA] [pH 8.0], 130 mM NaCl) by freeze-thawing and extrusion through 2 stacked 400-nm polycarbonate filters as previously described, stored at 4°C under N2, and used within 2 weeks of preparation (33). Complete liposomes were composed of a 1:1:1:3 molar ratio of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine, bovine brain sphingomyelin (Avanti Polar Lipids, Alabaster, AL), and cholesterol (Steraloids, Inc., Wilton, NH), plus trace amounts of [3H]cholesterol (Amersham, Arlington Heights, IL). Cholesterol-minus liposomes maintained the 1:1:1 molar ratio of phospholipids without cholesterol.
Protein-liposome interaction.
SFV or CHIKV E1 DI/II proteins were mixed with liposomes at a final concentration of 50 μg protein/ml and 1 mM lipid in MES (morpholineethanesulfonic acid) buffer (20 mM MES [pH 8.0], 130 mM NaCl) or TAN buffer. DIII proteins were added as indicated, using a molar ratio of 1:1 with E1 DI/II. Samples were preincubated at 25°C for 5 min, adjusted to pH 5.75 by addition of precalibrated amounts of 0.3 M MES, incubated at 25°C for 30 min, and adjusted to pH 8.0 by addition of a precalibrated volume of 0.3 M TEA. Samples were then adjusted to a final concentration of 20% sucrose, loaded on top of a 300-μl 60% sucrose cushion in a TLS55 ultracentrifuge tube, and overlaid with 1.2 ml 15% sucrose and 200 μl 5% sucrose, all wt/wt in TAN buffer (pH 8.0). Gradients were centrifuged for 3 h at 54,000 rpm at 4°C in a TLS55 rotor and fractionated into seven 300-μl fractions. Fractions were pooled into the 3 top and 4 bottom fractions or into the 2 top, 2 middle, and 3 bottom fractions. The [3H]cholesterol marker was quantitated by scintillation counting; >70% of the marker was routinely recovered in the top 3 fractions. Aliquots of the pooled fractions were precipitated with 10% trichloroacetic acid and analyzed by SDS-PAGE on 11% acrylamide gels. Proteins were detected by Western blotting using domain-specific MAb E1-3 against SFV DI/II and MAb E1-2 against SFV DIII (33), a polyclonal antibody against SFV E1 and E2 proteins that also detects CHIKV E1 DI/II protein (33), and a MAb against the His tag (Lifetein LLC, South Plainfield, NJ) to detect His-tagged CHIKV DIII proteins, followed by Alexa Fluor 680-conjugated secondary antibodies (Molecular Probes, Grand Island, NY). Western blots were quantitated using an Odyssey infrared imaging system and Odyssey InCell Western software (LI-COR Biosciences, Lincoln, NE). The Prism program (Graphpad Software) was used to calculate P values by two-tailed Student's t test with Bonferroni's correction for multiple comparisons.
FP assay of DIII-core trimer interaction.
To produce the core trimer in bulk, 350 μg/ml of bacterially expressed SFV E1 DI/II protein was mixed with 1.5 mM cholesterol-containing liposomes, treated at pH 5.75 for 30 min, and adjusted to pH 8.0. An aliquot was taken to determine the efficiency of trimer formation using flotation in sucrose gradients or trypsin resistance (33). The mixture was then adjusted to a final concentration of 1.5% octyl-glucopyranoside (OG) (Sigma-Aldrich, St. Louis, MO) and 1 M NaCl, and solubilized for 1 h at room temperature. A mock reaction without DI/II protein was prepared in the same way, and used for trimer dilutions in fluorescence polarization (FP) studies as described below. SFV DIIIS and DIII proteins were labeled with an amino-reactive form of fluorescein (N-hydroxysuccinimide-fluorescein), purified by a desalting column, and eluted in TAN buffer, and the degree of labeling was determined, all as described by the manufacturer (Thermo Scientific, Waltham, MA). The labeling ratio for each DIII preparation was 2 to 3 mol of dye per mol of protein.
The labeled DIII was then used to establish an FP assay for binding of DIII to the core trimer. Serial 1:2 dilutions of labeled DIII proteins were analyzed for fluorescence and FP with core trimer targets; a concentration of 100 nM DIII proved optimal for FP measurements. The assay was thus performed using 100 nM DIII and serial 1:2 dilutions of trimer starting at 5 μM. Duplicate aliquots of the samples were loaded into 384-well plates and incubated at room temperature for 1 h. FP was then measured in a Tecan Infinite 200 PRO M1000 plate reader. The Kd (dissociation constant) value was estimated as the concentration of core trimer protein producing a half-maximal increase in fluorescence polarization and was calculated by nonlinear regression analysis of binding curves using the Prism program.
RESULTS
Characterization of CHIKV fusion.
The CHIKV vaccine strain (181/25) was derived from the 15561 Southeast Asian human isolate by passage in MRC-5 cells (40). To develop this virus as a system to study inhibition by recombinant DIII, we first evaluated its membrane fusion requirements. The pH dependence of virus-membrane fusion was determined for CHIKV, SFV, and SINV by following the fusion of virus with the plasma membrane of BHK-21 cells (24) (Fig. 1). Virus was prebound to cells on ice and then pulsed with media of various pHs for 1 min at 37°C to trigger the fusion of virus with the cell membrane. As previously described, under these conditions, SFV showed a pH threshold of ∼pH 6.0 and maximal fusion at ∼pH 5.75 to 5.5, while SINV Toto1101 had a pH threshold of ∼pH 5.5 and maximal fusion at ∼pH 5.0 (34). Similar to SFV, CHIKV had a pH threshold of ∼pH 6.0, but maximal fusion occurred after treatment at pH 5.25, and unlike SFV, CHIKV did not show significant inactivation of fusion activity at lower pH. Fusion of this strain of CHIKV was also strongly promoted by target membrane cholesterol, similar to SFV and SINV (Table 1) (13, 14).
Fig 1.

Comparison of the pH dependence of CHIKV, SFV, and SINV membrane fusion. Serial dilutions of CHIKV, SFV, or SINV were prebound to BHK-21 cells on ice. Virus-plasma membrane fusion was then triggered by treatment at the indicated pH for 1 min at 37°C, and the cells infected due to low-pH-induced fusion were quantitated by immunofluorescence. Results were expressed as the percentage of maximal fusion for each virus. Shown are means and standard deviations from 3 independent experiments.
Table 1.
Effect of cholesterol on alphavirus fusion
| Virus | Viral titera |
Ratiob | |
|---|---|---|---|
| +cholesterol | −cholesterol | ||
| SFV | 9 × 107 | 8 × 102 | 1 × 105 |
| CHIKV | 9.7 × 107 | 2 × 102 | 5 × 105 |
| SINV | 4 × 106 | 9 × 102 | 4 × 103 |
Viral titers from fusion infection assays performed at pH 5.5 (SFV and CHIKV) or pH 5.0 (SINV).
Ratio of titers on cells ± cholesterol.
Production of CHIKV DIII proteins.
We then wished to test if CHIK virus fusion could be inhibited by exogenous DIII and to compare autologous and heterologous inhibition by CHIKV versus SFV DIII proteins. Recombinant CHIKV DIII proteins were expressed in Escherichia coli, refolded in vitro, and purified by gel filtration as previously described for SFV DIII proteins (24). We tested CHIKV DIII constructs with or without a His tag (H) at the amino terminus and with or without the C-terminal stem region (S). Unlike the comparable SFV DIII proteins, CHIKV DIII proteins without the His tag were neither efficiently expressed nor accumulated in inclusion bodies (data not shown). In contrast, His-tagged CHIKV DIII proteins with or without the stem were expressed and purified at levels comparable to those of SFV. SDS-PAGE analysis of these CHIKV DIII proteins demonstrated the presence of disulfide bonds (data not shown). As a test for correct folding and protein stability, we performed thermal denaturation assays using SYPRO orange to detect the exposure of hydrophobic regions. As a control, we tested His-tagged and untagged SFV proteins with or without the stem, which were extensively characterized in our previous work and shown to contain the correct disulfide bonding pattern and structure as DIII in SFV E1 (24) (Protein Data Bank [PDB] accession no. 2V33). The CHIKV HDIII and HDIIIS proteins showed low SYPRO orange binding below 70°C, a sharp rise in fluorescence of the probe at higher temperatures, and a single Tm of ∼81 to 82°C (Fig. 2). All of the SFV DIII proteins behaved similarly but showed lower melting temperatures ranging from ∼64 to 70°C (Fig. 2). Together, our results indicated that recombinant CHIKV HDIII and HDIIIS were correctly folded and highly stable, in keeping with the known immunoglobulin-like fold and 3 disulfide bonds of alphavirus E1 DIII.
Fig 2.

Thermal stability of SFV and CHIKV DIII proteins. Shown are differential fluorescence scanning denaturation assays of the indicated SFV and CHIKV DIII proteins at neutral pH, plotted as a function of temperature. A.U., arbitrary units. An average of 8 samples for each protein were used.
Inhibition of membrane fusion by CHIKV DIII proteins.
We then compared the inhibitory activities of CHIKV HDIII and HDIIIS proteins with those of the analogous SFV proteins. The proteins were tested by inclusion during a 1-min low-pH treatment of CHIKV, SFV, or SINV prebound to BHK cells. In agreement with our previous studies (24), SFV HDIII efficiently inhibited membrane fusion and infection by SFV and SINV, and we observed that it also showed strong cross-inhibition of CHIKV fusion (Fig. 3A). In contrast, CHIKV HDIII protein showed no inhibition of CHIKV, SFV, or SINV fusion (Fig. 3A), and this lack of activity was observed even at high concentrations (up to 50 μM) of the CHIKV protein (data not shown). As previously described (24), the SFV stem region produced an increase in the efficiency of inhibition, with the SFV HDIIIS protein completely inhibiting SFV, SINV, and CHIKV fusion at a concentration of ∼0.5 μM (Fig. 3B). The CHIKV HDIIIS protein very efficiently inhibited CHIKV fusion, with complete inhibition at 0.5 μM (Fig. 3B). CHIKV HDIIIS also inhibited SFV and SINV fusion, although complete inhibition of these viruses required concentrations of ∼2 μM (Fig. 3B). Thus, CHIKV HDIIIS showed both homologous and cross-inhibition of alphavirus fusion. Unlike SFV, however, the CHIKV stem region was required for inhibition.
Fig 3.

Cross-species inhibition of virus membrane fusion by CHIKV and SFV DIII proteins. Serial dilutions of CHIKV, SFV, or SINV were prebound to BHK cells on ice, and virus fusion was triggered by treatment at 37°C for 1 min at the optimal pH value determined for each virus in Fig. 1 (SFV/CHIKV, pH 5. 5; SINV, pH 5.0). The indicated concentrations of exogenous CHIK or SFV DIII proteins without (A) or with (B) the stem region were included during the 1-min low-pH treatment. Results are expressed as the percentage of fusion in the absence of any added DIII protein. Shown are means and standard deviations from 3 independent experiments.
Requirement for CHIKV stem region.
We hypothesized that the difference in inhibition by SFV HDIII versus CHIK HDIII was due to a difference in the binding affinity of these proteins for their target, the E1 core trimer intermediate formed during virus fusion (24, 25). The core trimer can be produced in vitro by low-pH treatment of purified DI/II proteins in the presence of cholesterol-containing liposomes (33). The membrane-inserted core trimer binds exogenous DIII, and the complex can be floated with the liposomes on sucrose gradients (33). We used this system to assay DIII binding to SFV or CHIKV DI/II core trimers. As predicted by our fusion results, the SFV HDIII and HDIIIS proteins interacted with the CHIKV core trimer (Fig. 4A) or the SFV core trimer (33; data not shown). In contrast, CHIKV HDIII did not cofloat with either the CHIKV core trimer (Fig. 4B, first two lanes) or the SFV core trimer (data not shown), in keeping with its lack of inhibition of fusion.
Fig 4.
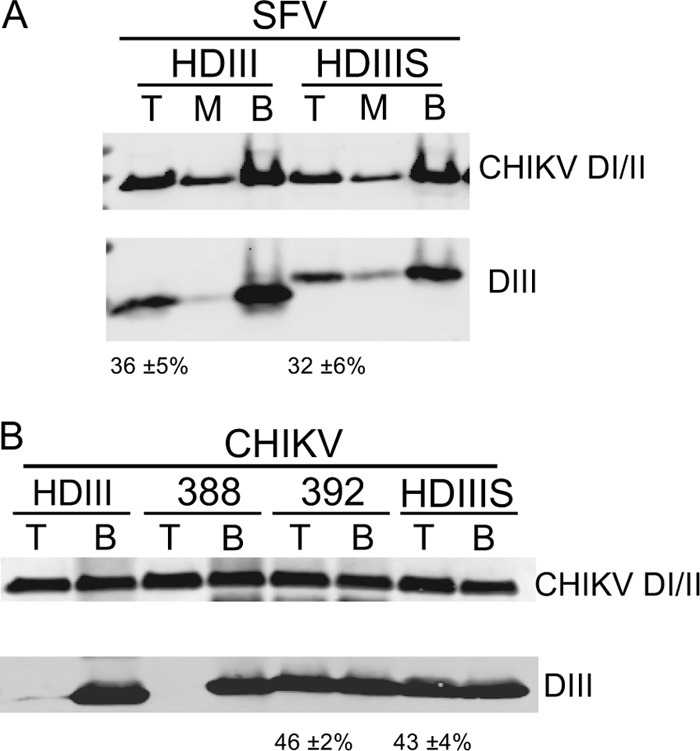
Binding of CHIKV and SFV DIII proteins to membrane-inserted CHIKV DI/II core trimers. CHIKV DI/II protein (50 μg/ml) was mixed with liposomes and a 1:1 molar ratio of the indicated SFV (A) or CHIKV (B) DIII proteins, with truncations and nomenclature as shown in Fig. 5A. The samples were treated at pH 5.75 at room temperature for 30 min, adjusted to pH 8.0, and separated on sucrose flotation gradients. Gradients were fractionated and pooled into the top (T), middle (M), and bottom (B) fractions in panel A or into the top (T) and bottom (B) fractions in panel B. Pooled fractions were acid precipitated and analyzed by SDS-PAGE and Western blotting to detect E1 DI/II and DIII as indicated. Both panels are representative examples of 3 independent experiments, with the percentage of DIII protein in the top fraction (average ± standard deviation) listed below each panel.
The E1 stem region is ∼28 residues long, extending from the end of DIII at P384 to K412 just prior to the membrane anchor (Fig. 5A). To define the extension of the stem required to block fusion, we produced CHIKV HDIIIS proteins truncated after position 388, 392, or 397, as well as HDIII and HDIIIS, which end at P384 and K412, respectively (Fig. 5A). We tested 10 μM concentrations of these proteins for inhibition of CHIKV or SFV fusion with BHK cells, using the assay described in Fig. 3. CHIKV HDIII proteins that extend to at least residue 392 inhibited membrane fusion by both viruses, while a version truncated at residue 388 showed little inhibition (Fig. 5B). Binding studies of these DIII proteins with the CHIKV core trimer demonstrated that DIII protein truncated at residue 392 cofloated with the core trimer, while DIII truncated at residue 388 did not (Fig. 4B). Thus, the stem residues 389NYPA392 (Fig. 5A) and/or this stem length were important in promoting the binding of CHIKV HDIII protein to the core trimer and the inhibition of virus fusion.
Fig 5.
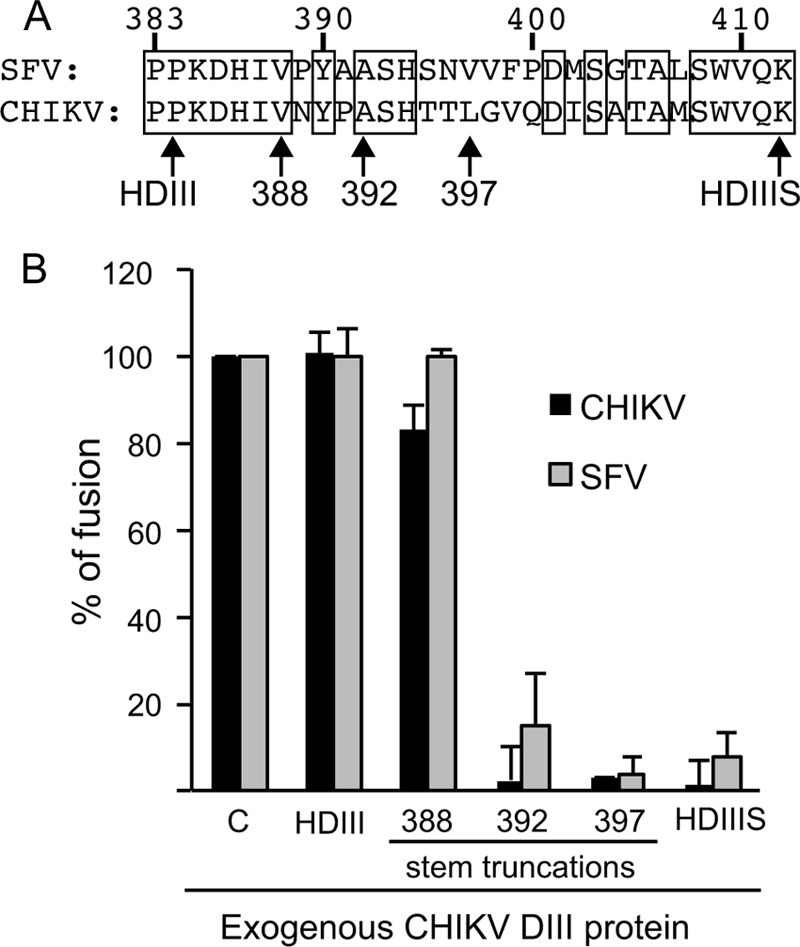
Definition of the stem requirement for inhibition of virus fusion by CHIKV DIII proteins. (A) Alignment of the SFV and CHIKV stem sequences, with the E1 residue numbers indicated above. Below the sequences, “HDIII” indicates the C-terminal boundary of DIII, and the numbers indicate C-terminal residues of the DIII proteins used in panel B. HDIIIS contains the complete stem and is truncated after residue 412 as shown. (B) Inhibition of CHIKV and SFV fusion by CHIKV DIII proteins was monitored by the fusion assay described in Fig. 1. Cells with bound virus were treated at pH 5.5 in the presence of 10 μM the indicated CHIKV DIII protein or the control dengue virus E HDIIIS protein (C). Data were calculated as the percentage of fusion compared to that of the control. Shown are means and standard deviations from 3 independent experiments. Significant differences compared to control samples (P ≤ 0.001) were observed for SFV and CHIKV with 392, 397, and HDIIIS constructs and for CHIKV with the 388 construct.
Binding assay for core trimer and DIII proteins.
To quantitatively analyze the affinity of the DIII-core trimer interaction, we developed an assay based on the change in fluorescence anisotropy upon binding of fluorescently labeled DIII protein to the DI/II core trimer. SFV or CHIKV DIII proteins were labeled with an amine-reactive form of fluorescein (details in Methods). While SFV DIII and DIIIS proteins were successfully labeled, the CHIKV DIII proteins precipitated under a variety of dye conjugation conditions. We also noted that the CHIKV DI/II core trimer was not stable in octylglucoside. We therefore used the SFV proteins for further studies. The labeled SFV DIIIS and DIII proteins showed comparable coflotation with the SFV DI/II core trimer as unlabeled DIII proteins (data not shown). We also established a bacterial expression and in vitro refolding system for the SFV E1 DI/II protein (details in Materials and Methods). Similar to the protein produced in Drosophila S2 cells, the SFV DI/II protein produced in bacteria formed stable core trimers in the presence of liposomes at low pH, but not at neutral pH or in the absence of target membranes. These core trimers bound DIII proteins at neutral pH, again similar to the S2 core trimers (data not shown).
Fluorescence polarization (FP) measurements were then performed to characterize the binding of labeled SFV DIII proteins to SFV E1 DI/II trimers. Dilutions of DI/II trimers or the DI/II monomer were mixed with DIII proteins and incubated for 1 h at neutral pH, and the FP was quantitated (Fig. 6A). The FP of SFV DIIIS showed a reproducible increase in the presence of DI/II trimers, while the DI/II monomer did not change the DIIIS FP. Comparable changes in FP were obtained using the S2 cell-expressed core trimer as the target for DIIIS binding (data not shown). The Kd for binding of SFV DIIIS with the DI/II core trimer was calculated as 85 ± 5 nM (Fig. 6A). Binding of labeled DIII without the stem did not reach saturation even at the maximal concentration of trimer used (Fig. 6A), and analysis using the Prism program estimated a Kd ∼100 times higher than that of DIIIS. These values are similar to those previously determined for DIII and DIIIS inhibition of virus fusion (24). The binding of DIIIS to the DI/II core trimer was efficiently competed by excess unlabeled DIIIS, confirming the specificity of the FP assay (Fig. 6B).
Fig 6.

Fluorescence polarization assay of interaction of SFV DIII proteins and solubilized SFV core trimer. (A) Binding assay based on fluorescence polarization. Serial dilutions of the solubilized SFV E1 DI/II trimer or monomer were mixed with 100 nM fluorescein-labeled DIIIS or DIII and incubated at room temperature for 1 h. Fluorescence polarization (FP) was measured and expressed as a function of SFV E1 DI/II protein concentration (molarity [M]). FP (mP) measurements were fitted in each case to a sigmoidal curve (solid lines) and used to derive the Kd value and 95% confidential interval (CI) for DIIIS plus trimer. (B) Competition by unlabeled SFV DIIIS. Serial dilutions of unlabeled SFV DIIIS (as indicated on the x axis) were mixed with 5 μM SFV E1 DI/II trimer; 100 nM fluorescein-labeled DIIIS was added, and FP was measured as in Fig. 6A. The values shown by the graphs and the Kd values are the averages and standard deviations from 3 independent experiments with duplicate samples in each.
DISCUSSION
Here we produced a series of CHIKV DIII proteins and analyzed their activity against virus-membrane fusion. The first nine residues of the stem region promoted the inhibition of virus fusion by CHIKV DIII proteins. Similarly, these stem residues were required for stable binding of CHIKV DIII proteins to the CHIKV E1 DI/II core trimer. Cross-inhibition and comparative binding studies strongly supported the conservation of the DIII-stem–core trimer interface between the SFV, SINV, and CHIKV fusion proteins. We then developed a fluorescence anisotropy-based assay for the binding of SFV DIII proteins to the core trimer. This assay defined the high affinity of the DIII-stem–core trimer interaction and confirmed the stabilizing effect of the stem region.
SFV and CHIKV E1 proteins.
Our initial characterization experiments confirmed that CHIKV fusion was both low pH and cholesterol dependent. However, even though E1's sequence and structure are strongly conserved among the SFV, SINV, and CHIKV E1 proteins (20–22), we observed several differences in the properties of DIII proteins from CHIKV versus SFV. The CHIKV DIII proteins required the N-terminal His tag for successful expression. We have not tested alternative sequence tags for their effects. Once refolded in vitro, thermal denaturation tests showed that CHIKV HDIII and HDIIIS have a higher melting temperature (+ ∼10 to 18°C) than the analogous SFV proteins, suggesting higher stability. Unlike SFV DIII proteins, labeling of the CHIKV DIII proteins with amine-reactive fluorescein caused their aggregation under all conditions we tested, making them poor candidates for fluorescence anisotropy assays of DIII-core trimer interaction. In contrast, E1 DI/II proteins from both CHIKV and SFV were efficiently expressed and purified from S2 cells and showed similar activities in formation of core trimers that bound DIII. We also successfully produced SFV E1 DI/II by bacterial expression and refolding. Although in this system DI/II is not glycosylated, it was comparable to the Drosophila-expressed protein in formation of stable core trimers that efficiently bound DIII. This system avoids the higher cost of eukaryotic cell expression, and we anticipate that the successful bacterial production of active alphavirus DI/II and DIII will aid in large-scale production of these proteins as needed for structural studies and small molecule screens.
Role of the stem region.
The structure of the SFV E1 postfusion trimer shows that DIII is positioned within the groove formed by the DI/II regions of two E1 molecules (23). The inhibition of the rapid virus fusion reaction by exogenous DIII proteins (24, 25) and the formation of a stable complex between exogenous DIII protein and the core trimer (24, 33) suggest that the affinity of this interaction is high. DIII proteins inhibit across alphavirus species (24; this work), and the CHIKV and SINV DIII-stem proteins are 49% and 45% identical to SFV DIII-stem, respectively. Here we observed a prominent difference between the SFV and CHIKV DIII proteins in their requirements for the stem region. Unlike SFV, the CHIKV DIII required the first 9 residues of the stem in order for the protein to inhibit fusion and stably bind the core trimer. Similar CHIKV DIII melting temperatures were observed with or without the stem, so this requirement was not due to effects on overall domain folding or stability.
The length of the alphavirus E1 stem region is conserved. A minimal length of the SFV E1 stem is required in order to allow DIII foldback, hairpin formation, and fusion (25, 41). The presence of the stem causes increased stability of the SFV DIII-DI/II core trimer complex (33). However, SFV mutagenesis studies showed that a specific stem sequence is not essential for fusion (41), and tests of several isolated SFV stem peptides showed that they did not inhibit fusion (41). These results suggested that DIII-core trimer interactions are important in promoting the hairpin. However, clearly the previous data and the results of this study also suggest a role for the stem region, probably occurring after the initial foldback of the DI-DIII linker and DIII (25, 39).
Using the SFV E1 homotrimer structure, we analyzed the residues in DIII-stem for interactions with the core trimer (Table 2 and Fig. 7). (Note that in the structure the stem ends at residue 391.) Initial analysis used the PISA (Protein Interfaces, Surface and Assemblies Service) program at the European Bioinformatics Institute (42) and a modified SFV E1 trimer PDB file containing one DI/II and the DIII from the same E1 protein or the adjacent E1 protein. All DIII residues containing surface area buried by contacts with DI/II were further analyzed with the programs CSU (Contact of Structural Units) (43) and PyMOL (44). Residues within 4 Å of each other were compared with the corresponding residues in CHIKV E1. Predicted interactions between SFV DIII-stem residues and the core trimer are listed in Table 2 and detailed in Fig. 7, and can occur between DIII and DI/II of the same E1 chain or the adjacent E1 chain.
Table 2.
Predicted interactions between SFV DIII-stem and the SFV E1 core trimer
| Structure | Residue(s) at which core trimer interactsa |
|---|---|
| Side chain | |
| DIII | |
| Asp311 | Thr41 (hydrogen bond) |
| His331 | Asn149 (hydrogen bond) |
| His333b | Asp145 (salt bridge) |
| Stem | |
| His386 | Glu246 (salt bridge) |
| Tyr390 | Pro237 (hydrophobic) |
| Main chain | |
| DIII | |
| Ser309 | Leu44 |
| Gln340b | Phe257, Gly258 |
| Stem | |
| Lys384 | Arg206 |
| His386 | Asp212 |
The type of predicted interaction is shown in parentheses. Note that for the results shown for the side chain, all interactions are with the side chain of the core trimer residue, except for Thr41, which interacts with the main chain. Note that for the results shown for the main chain, Leu44, Phe257, and Gly258 interact via the main chain of the core trimer, and Arg206 and Asp212 interact via their side chains.
Residues that differ between SFV and CHIKV DIII-stem (Met333 and Arg340 in CHIKV). Note that for His333, the predicted core trimer-interacting partner also differs between SFV (Asp145) and CHIKV (Thr145).
Fig 7.
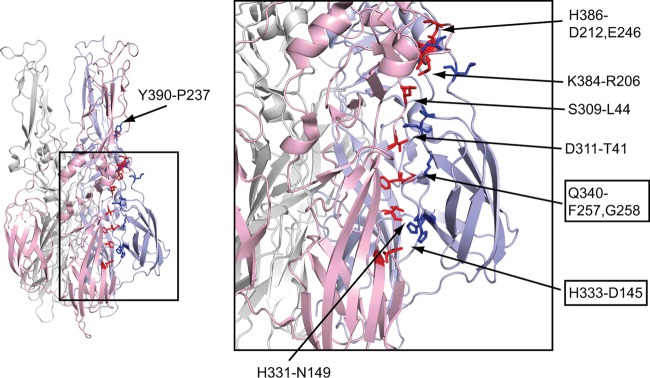
E1 trimer structure and predicted interactions between SFV DIII-stem and SFV E1 core trimer. The structure of the postfusion trimer of the SFV E1 ectodomain (PDB accession no. 1RER) is on the left, with the fusion loops oriented toward the top of the page. The three chains are colored in light pink, light blue, and light gray. The residues of DIII and the stem involved in the predicted interactions listed in Table 2 are highlighted as stick structures in blue. The corresponding interacting residues of DI/II are highlighted in red. The locations of residues Y390 and P237 are indicated on the left panel. The region inside the square is zoomed in the right panel to show the details. The location of each pair is indicated by arrows pointing between the residues, with the title indicating the DIII-stem residue followed by the residue(s) on DI/II. The residues on DIII (and their pairs on DI/II) that differ between CHIKV and SFV are boxed. Note that the residue pairs Q340-F257/G258, H386-E246, and Y390-P237 are located in the same chain. This figure was prepared with the program PyMOL (44).
We then compared these interactions with the residues at the same positions in CHIKV E1. The interacting residues are highly conserved between the sequences of SFV and CHIKV E1, in keeping with the cross-inhibition observed with DIII proteins from these two viruses. While a number of the predicted interactions occur via the main chain, both DIII and stem show several side-chain interactions with the core trimer. Most of these residues are conserved between SFV and CHIKV, with the exception of H333 on SFV DIII. In the case of CHIKV, the interaction of DIII H333 with DI D145 is replaced by an M333-T145 pair. Although prior studies with SFV showed that alanine substitution of H333 did not detectably affect virus growth or fusion (45), it is possible that these sequence differences contribute to the difference in CHIKV DIII interaction. The stem truncation experiments in Fig. 5 implicate stem residues 389NYPA392 in stabilizing CHIKV DIII binding. The SFV homotrimer structure predicts interaction of Y390 with P237 in the ij loop of DII. This Y390-P237 pair is conserved between SFV and CHIKV and is relatively conserved among alphaviruses. Although Y390 is not essential for SFV fusion (41), it may be important in stabilizing the binding of CHIKV DIII protein, perhaps by compensating for the loss of the H333-D145 interaction. It is also possible that in the context of CHIKV E1, other residues of the stem promote binding and inhibition by exogenous DIII.
Significance of cross-inhibition of membrane fusion in alphaviruses.
Subtle differences in fusion protein sequence or structure could result in a significant difference in the requirements for the fusion process. This is an important issue to consider in designing strategies for inhibitors of medically important alphaviruses, such as CHIKV. Since DIII inhibition of virus fusion and infection has been mainly developed and studied using the SFV model, we wanted to validate this approach for CHIKV. We here confirmed cross-inhibition of SFV and CHIKV fusion by SFV and CHIKV DIII proteins. CHIKV HDIIIS inhibition of membrane fusion was optimal for CHIKV, while somewhat higher concentrations were required to inhibit SFV and SINV. In contrast, SFV HDIIIS protein inhibited the fusion of all three viruses with comparable high efficiencies. In flotation experiments, the SFV and CHIKV HDIIIS proteins bound to the core trimer of SFV or CHIKV to a similar extent. The measured high affinity of SFV DIIIS for the DI/II core trimer (85 nM) from our fluorescence assay is in good agreement with the previously observed 50% inhibitory concentration (IC50) of ∼100 nM for this protein in virus fusion assays (24). Our results suggest that the sites of contact for SFV HDIII or HDIIIS with the core trimer are present during its interaction with CHIKV core trimer and result in cross-inhibition. Thus, the assay we describe for the in vitro interaction of SFV DIIIS with the core trimer has potential for use as a screen for broad-specificity inhibitors of alphavirus membrane fusion.
ACKNOWLEDGMENTS
We thank all the members of our lab for helpful discussions and experimental suggestions. We thank Anuja Ogirala and Youqing Xiang for excellent technical assistance, Rafael Toro and Steve Almo for help with SYPRO orange experiments, Chenyang Zhan and Rotem Rubinstein for assistance in structural analysis, and Evripidis Gavathiotis and Denis Reyna for help with the FP measurements.
This work was supported by grants to M.K. from the National Institute of Allergy and Infectious Diseases (R01-AI075647) and the Northeast Biodefense Center (U54-AI057158-Lipkin) and by Cancer Center Core Support Grant NIH/NCI P30-CA13330. W.F. was supported by the Training Program in Cellular and Molecular Biology and Genetics, T32 GM007491.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health.
Footnotes
Published ahead of print 1 May 2013
REFERENCES
- 1. Kuhn RJ. 2007. Togaviridae: the viruses and their replication, p 1001–1022 In Knipe DM, Howley PM. (ed), Fields virology, 5th ed, vol 1 Lippincott, Williams and Wilkins, Philadelphia, PA [Google Scholar]
- 2. Strauss JH, Strauss EG. 1994. The alphaviruses: gene expression, replication, and evolution. Microbiol. Rev. 58:491–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schwartz O, Albert ML. 2010. Biology and pathogenesis of chikungunya virus. Nat. Rev. Microbiol. 8:491–500 [DOI] [PubMed] [Google Scholar]
- 4. Enserink M. 2007. Infectious diseases. Chikungunya: no longer a Third World disease. Science 318:1860–1861 [DOI] [PubMed] [Google Scholar]
- 5. Reiskind MH, Pesko K, Westbrook CJ, Mores CN. 2008. Susceptibility of Florida mosquitoes to infection with chikungunya virus. Am. J. Trop. Med. Hyg. 78:422–425 [PMC free article] [PubMed] [Google Scholar]
- 6. Rezza G, Nicoletti L, Angelini R, Romi R, Finarelli AC, Panning M, Cordioli P, Fortuna C, Boros S, Magurano F, Silvi G, Angelini P, Dottori M, Ciufolini MG, Majori GC, Cassone A. 2007. Infection with chikungunya virus in Italy: an outbreak in a temperate region. Lancet 370:1840–1846 [DOI] [PubMed] [Google Scholar]
- 7. Vazeille M, Moutailler S, Coudrier D, Rousseaux C, Khun H, Huerre M, Thiria J, Dehecq JS, Fontenille D, Schuffenecker I, Despres P, Failloux AB. 2007. Two chikungunya isolates from the outbreak of La Reunion (Indian Ocean) exhibit different patterns of infection in the mosquito, Aedes albopictus. PLoS One 2:e1168. 10.1371/journal.pone.0001168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tsetsarkin KA, Vanlandingham DL, McGee CE, Higgs S. 2007. A single mutation in chikungunya virus affects vector specificity and epidemic potential. PLoS Pathog. 3:e201. 10.1371/journal.ppat.0030201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schuffenecker I, Iteman I, Michault A, Murri S, Frangeul L, Vaney MC, Lavenir R, Pardigon N, Reynes JM, Pettinelli F, Biscornet L, Diancourt L, Michel S, Duquerroy S, Guigon G, Frenkiel MP, Brehin AC, Cubito N, Despres P, Kunst F, Rey FA, Zeller H, Brisse S. 2006. Genome microevolution of chikungunya viruses causing the Indian Ocean outbreak. PLoS Med. 3:e263. 10.1371/journal.pmed.0030263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kielian M, Chanel-Vos C, Liao M. 2010. Alphavirus entry and membrane fusion. Viruses 2:796–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bernard E, Solignat M, Gay B, Chazal N, Higgs S, Devaux C, Briant L. 2010. Endocytosis of chikungunya virus into mammalian cells: role of clathrin and early endosomal compartments. PLoS One 5:e11479. 10.1371/journal.pone.0011479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Phalen T. 1993. Analysis of the Semliki Forest virus requirement for cholesterol during membrane fusion and infection. Ph.D. thesis Albert Einstein College of Medicine, Bronx, NY [Google Scholar]
- 13. Vashishtha M, Phalen T, Marquardt MT, Ryu JS, Ng AC, Kielian M. 1998. A single point mutation controls the cholesterol dependence of Semliki Forest virus entry and exit. J. Cell Biol. 140:91–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lu YE, Cassese T, Kielian M. 1999. The cholesterol requirement for Sindbis virus entry and exit and characterization of a spike protein region involved in cholesterol dependence. J. Virol. 73:4272–4278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marquardt MT, Kielian M. 1996. Cholesterol-depleted cells that are relatively permissive for Semliki Forest virus infection. Virology 224:198–205 [DOI] [PubMed] [Google Scholar]
- 16. Marquardt MT, Phalen T, Kielian M. 1993. Cholesterol is required in the exit pathway of Semliki Forest virus. J. Cell Biol. 123:57–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tsetsarkin KA, McGee CE, Higgs S. 2011. Chikungunya virus adaptation to Aedes albopictus mosquitoes does not correlate with acquisition of cholesterol dependence or decreased pH threshold for fusion reaction. Virol. J. 8:376. 10.1186/1743-422X-8-376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Paredes AM, Brown DT, Rothnagel R, Chiu W, Schoepp RJ, Johnston RE, Prasad BVV. 1993. Three-dimensional structure of a membrane-containing virus. Proc. Natl. Acad. Sci. U. S. A. 90:9095–9099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lescar J, Roussel A, Wien MW, Navaza J, Fuller SD, Wengler G, Rey FA. 2001. The fusion glycoprotein shell of Semliki Forest virus: an icosahedral assembly primed for fusogenic activation at endosomal pH. Cell 105:137–148 [DOI] [PubMed] [Google Scholar]
- 20. Roussel A, Lescar J, Vaney M-C, Wengler G, Wengler G, Rey FA. 2006. Structure and interactions at the viral surface of the envelope protein E1 of Semliki Forest virus. Structure 14:75–86 [DOI] [PubMed] [Google Scholar]
- 21. Li L, Jose J, Xiang Y, Kuhn RJ, Rossmann MG. 2010. Structural changes of envelope proteins during alphavirus fusion. Nature 468:705–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Voss JE, Vaney MC, Duquerroy S, Vonrhein C, Girard-Blanc C, Crublet E, Thompson A, Bricogne G, Rey FA. 2010. Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature 468:709–712 [DOI] [PubMed] [Google Scholar]
- 23. Gibbons DL, Vaney M-C, Roussel A, Vigouroux A, Reilly B, Lepault J, Kielian M, Rey FA. 2004. Conformational change and protein-protein interactions of the fusion protein of Semliki Forest virus. Nature 427:320–325 [DOI] [PubMed] [Google Scholar]
- 24. Liao M, Kielian M. 2005. Domain III from class II fusion proteins functions as a dominant-negative inhibitor of virus-membrane fusion. J. Cell Biol. 171:111–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Roman-Sosa G, Kielian M. 2011. The interaction of alphavirus E1 protein with exogenous domain III defines stages in virus-membrane fusion. J. Virol. 85:12271–12279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Modis Y, Ogata S, Clements D, Harrison SC. 2003. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 100:6986–6991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Modis Y, Ogata S, Clements D, Harrison SC. 2004. Structure of the dengue virus envelope protein after membrane fusion. Nature 427:313–319 [DOI] [PubMed] [Google Scholar]
- 28. Liao M, Sanchez-San Martin C, Zheng A, Kielian M. 2010. In vitro reconstitution reveals key intermediate states of trimer formation by the dengue virus membrane fusion protein. J. Virol. 84:5730–5740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schmidt AG, Yang PL, Harrison SC. 2010. Peptide inhibitors of dengue-virus entry target a late-stage fusion intermediate. PLoS Pathog. 6:e1000851. 10.1371/journal.ppat.1000851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schmidt AG, Yang PL, Harrison SC. 2010. Peptide inhibitors of flavivirus entry derived from the E protein stem. J. Virol. 84:12549–12554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hrobowski YM, Garry RF, Michael SF. 2005. Peptide inhibitors of dengue virus and West Nile virus infectivity. Virol. J. 2:49. 10.1186/1743-422X-2-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pierson TC, Kielian M. 2013. Flaviviruses: braking the entering. Curr. Opin. Virol. 3:3–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sanchez-San Martin C, Sosa H, Kielian M. 2008. A stable prefusion intermediate of the alphavirus fusion protein reveals critical features of class II membrane fusion. Cell Host Microbe 4:600–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Glomb-Reinmund S, Kielian M. 1998. The role of low pH and disulfide shuffling in the entry and fusion of Semliki Forest virus and Sindbis virus. Virology 248:372–381 [DOI] [PubMed] [Google Scholar]
- 35. Rice CM, Levis R, Strauss JH, Huang HV. 1987. Production of infectious RNA transcripts from Sindbis virus cDNA clones: mapping of lethal mutations, rescue of a temperature-sensitive marker, and in vitro mutagenesis to generate defined mutants. J. Virol. 61:3809–3838l9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Meyer WJ, Gidwitz S, Ayers VK, Schoepp RJ, Johnston RE. 1992. Conformational alteration of Sindbis virion glycoproteins induced by heat, reducing agents, or low pH. J. Virol. 66:3504–3513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Luca VC, AbiMansour J, Nelson CA, Fremont DH. 2012. Crystal structure of the Japanese encephalitis virus envelope protein. J. Virol. 86:2337–2346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Scopes RK. 1974. Measurement of protein by spectrophotometry at 205 nm. Anal. Biochem. 59:277–282 [DOI] [PubMed] [Google Scholar]
- 39. Zheng Y, Sanchez-San Martin C, Qin ZL, Kielian M. 2011. The domain I-domain III linker plays an important role in the fusogenic conformational change of the alphavirus membrane fusion protein. J. Virol. 85:6334–6342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Levitt NH, Ramsburg HH, Hasty SE, Repik PM, Cole FE, Jr, Lupton HW. 1986. Development of an attenuated strain of chikungunya virus for use in vaccine production. Vaccine 4:157–162 [DOI] [PubMed] [Google Scholar]
- 41. Liao M, Kielian M. 2006. Functions of the stem region of the Semliki Forest virus fusion protein during virus fusion and assembly. J. Virol. 80:11362–11369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Krissinel E, Henrick K. 2007. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372:774–797 [DOI] [PubMed] [Google Scholar]
- 43. Sobolev V, Sorokine A, Prilusky J, Abola EE, Edelman M. 1999. Automated analysis of interatomic contacts in proteins. Bioinformatics 15:327–332 [DOI] [PubMed] [Google Scholar]
- 44. DeLano WL. 2002. The PyMOL user's manual. DeLano Scientific, San Carlos, CA [Google Scholar]
- 45. Qin ZL, Zheng Y, Kielian M. 2009. Role of conserved histidine residues in the low-pH dependence of the Semliki Forest virus fusion protein. J. Virol. 83:4670–4677 [DOI] [PMC free article] [PubMed] [Google Scholar]