

Abstract
Toll-like receptors (TLRs) and RNA helicases (RLHs) are important cell sensors involved in the immunological control of viral infections through production of type I interferon (IFN). The impact of a deficiency in the TRIF and IPS-1 adaptor proteins, respectively, implicated in TLR3 and RLH signaling pathways, was investigated during herpes simplex virus 1 (HSV-1) encephalitis. TRIF−/−, IPS-1−/−, and C57BL/6 wild-type (WT) mice were infected intranasally with 7.5 × 105 PFU of HSV-1. Mice were monitored for neurological signs and survival over 20 days. Groups of mice were sacrificed on days 3, 5, 7, 9, and 11 postinfection for determination of brain viral replication by quantitative PCR (qPCR), plaque assay, and immunohistochemistry and for alpha/beta interferon (IFN-α/β) levels and phosphorylation of interferon regulatory factors 3 and 7 (IRF-3 and -7) in brain homogenates by enzyme-linked immunosorbent assay (ELISA) and Western blotting, respectively. TRIF−/− and IPS-1−/− mice had higher mortality rates than WT mice (P = 0.02 and P = 0.09, respectively). Viral antigens were more disseminated throughout the brain, correlating with a significant increase in brain viral load for TRIF−/− (days 5 to 9) and IPS-1−/− (days 7 and 9) mice compared to results for the WT. IFN-β production was reduced in brain homogenates of TRIF−/− and IPS-1−/− mice on day 5 compared to results for the WT, whereas IFN-α levels were increased on day 7 in TRIF−/− mice. Phosphorylation levels of IRF-3 and IRF-7 were decreased in TRIF−/− and IPS-1−/− mice, respectively. These data suggest that both the TRIF and IPS-1 signaling pathways are important for the control of HSV replication in the brain and survival through IFN-β production.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) is a frequent human pathogen, most commonly associated with orolabial, genital, and ocular infections. Furthermore, HSV-1 is also the most frequent cause of sporadic viral encephalitis in Western countries. Despite antiviral treatment, mortality rates associated with herpes simplex encephalitis (HSE) reach 20 to 30% (1). The cerebral innate immune system is critical to control HSV replication in the brain early after infection, but it may also lead to an exaggerated inflammatory response that could be detrimental to the host.
Mammalian cells sense HSV via distinct pattern recognition receptors (PRRs) to control viral replication (2–4). These include the Toll-like receptors (TLRs) and the RNA helicases (RLHs), the latter comprising both the retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated gene 5 (MDA-5) pathways. Several TLRs, in particular TLR2, -3, and -9, participate in early recognition of HSV components (5–9). HSV particles are first sensed by TLR2, which is expressed on the cell surface and recognizes the virus surface glycoproteins (10). After entry, viral genomic DNA, which contains abundant CpG motifs, is detected by TLR9 (11). Then, replication of the viral genome leads to accumulation of intermediate double-stranded RNAs (dsRNAs), which are sensed by TLR3 (12). Most TLRs use the myeloid differentiation primary response gene 88 (MyD88)-dependent pathway to activate nuclear factor kappa B (NF-κB), resulting in proinflammatory cytokine production (13). In contrast, TLR3 utilizes a MyD88-independent pathway, which activates interferon regulatory factor 3 (IRF-3), resulting in the production of type I interferon (IFN). More precisely, TLR3, which is found in the endosomal compartment, triggers a signal through the Toll/interleukin 1 (IL-1) receptor (TIR) domain-containing adaptor inducing beta IFN (IFN-β) (TRIF) (14). This adaptor protein activates inducible inhibitor of κB kinase 1 (IκK-1) and TANK-binding kinase 1 (TBK-1), leading to the phosphorylation of IRF-3. Once activated, IRF-3 translocates to the nucleus, where it binds to type I IFN promoters to induce their expression.
On the other hand, RIG-I and MDA-5, which are localized in the cytoplasmic compartment, respectively, sense shorter dsRNA or 5′-triphosphate single-stranded RNA (ssRNA) structures and longer dsRNA (15). These sensors recognize HSV dsRNA (16–18) and interact with the IFN-β promoter stimulator 1 (IPS-1) adaptor protein to activate the IRF-3 and IRF-7 pathways (19–21). Phosphorylated IRF-3 and IRF-7 can form homodimers or heterodimers which translocate to the nucleus and differentially modulate the expression of type I IFN gene family members. The initial type I IFN produced acts in both autocrine and paracrine manners through the alpha/beta IFN (IFN-α/β) receptor (IFNAR), which activates components of the Janus kinase-signal transducer and activator of transcription (JAK/STAT) signaling pathway (22). This cascade results in the synthesis of antiviral effectors important to limiting viral spread and to establishing an antiviral state in the infected cells and in neighboring noninfected cells (23).
The relative contributions of the endoplasmic TLR3 and cytoplasmic RIG-I/MDA-5 signaling pathways to the cerebral innate immune response during HSE and their respective involvement in type I IFN production are not well understood. In this study, we investigated the influence of these sensors following intranasal HSV-1 infection of mice deficient for the TRIF and IPS-1 adaptor proteins. The kinetics of viral replication, IFN-α/β production, and activation of downstream effectors (i.e., levels of phosphorylated IRF-3 and IRF-7) were evaluated in brain homogenates by quantitative PCR (qPCR)/plaque assays, enzyme-linked immunosorbent assay (ELISA), and Western blot analysis, respectively. The localization of HSV-1 proteins on thin slices obtained from different brain regions was also assessed by immunohistochemistry. Altogether, our results suggest that the activation of TRIF and IPS-1 signaling pathways is required for the induction of an effective cerebral innate immune response against HSV-1 infection.
MATERIALS AND METHODS
Animals and experimental procedures.
Five- to six-week-old male C57BL/6 wild-type (WT), TRIF−/−, and IPS-1−/− mice were used in this study. C57BL/6 WT mice were purchased from Charles River Canada (St-Constant, Quebec, Canada). TRIF−/− and IPS-1−/− mice (both provided by S. Akira, Osaka University, Osaka, Japan) were generated and maintained in a C57BL/6 background as described previously (14, 24). Animals were housed five per cage and acclimated to standard laboratory conditions. All experimental procedures were approved by the Animal Care Ethics Committee of Laval University.
For survival curve experiments, C57BL/6 WT, TRIF−/−, and IPS-1−/− mice were infected intranasally with HSV-1 (7.5 × 105 PFU of the H25 clinical strain) as described previously (25). HSE-related signs, namely, ruffled fur, ocular swelling, shaking movements, and swollen forehead, as well as weight loss and mortality, were monitored for 20 days. Animals were sacrificed when a ≥20% weight loss or a combination of two other obvious sickness signs were observed.
For determination of the viral DNA load, infectious viral titers, IFN-α/β levels, and phosphorylated IRF-3 and IRF-7 amounts, a subset of 3 to 8 mice per group was sacrificed on days 3, 5, 7, 9, and 11 following infection by intracardiac perfusion with 0.9% cold saline. Brains were then harvested and homogenized in phosphate-buffered saline (PBS) containing protease (cOmplete) and phosphatase (PhosSTOP) inhibitor cocktails (Roche Applied Science, Laval, Quebec, Canada).
For immunohistochemistry studies, 3 mice per group were sacrificed by intracardiac perfusion with 0.9% cold saline followed by a 4% paraformaldehyde solution in 0.1 M borax buffer at pH 9.5 at 4°C. Brains were removed, postfixed in a paraformaldehyde solution for 20 to 24 h, and then placed in a 10% sucrose solution prepared in a 4% paraformaldehyde-borax buffer at 4°C for a maximum of 48 h. Brains were cut in 30-μm coronal sections on dry ice. Sections were then collected in a cold cryoprotectant solution (0.05 M sodium phosphate buffer at pH 7.3 containing 30% ethylene glycol and 20% glycerol) and stored at −20°C.
Viral DNA load and infectious virus titer measurements.
Viral DNA was extracted from brain homogenates using the Magnapure LC total nucleic acid isolation kit (Roche Molecular Systems, Laval, Quebec, Canada) and eluted in 100 μl of elution buffer. Real-time PCR was performed using 5 μl of extracted DNA, and external standards were run in parallel as previously described (26). Titers of virus in brain homogenates were determined by a standard plaque assay. Briefly, monolayers of African green monkey kidney (Vero) cells were infected with serial dilutions of brain homogenates for 90 min in a 5% CO2 humidified incubator at 37°C. The viral suspension was then removed, and cells were incubated with minimal essential medium (MEM) containing 2% fetal bovine serum (FBS) and 0.4% SeaPlaque agarose for 48 h. Cells were fixed and stained, and the number of PFU was determined under an inverted microscope. The limit of detection of the plaque assay is 5 PFU per well.
IFN-α and IFN-β protein levels.
Brain homogenates were centrifuged at 10 000 × g for 10 min at 4°C. Protein levels of IFN-α and IFN-β were measured in the supernatants by ELISA (Verikine, PBL, Piscataway, NJ), following the manufacturer's recommendations.
Phosphorylated IRF-3 and IRF-7 levels.
Western blot analysis was performed with protein samples extracted from brain homogenates. Samples were first homogenized in ice-cold cell lysis buffer (Cell Signaling Technology, Danvers, MA) containing protease (cOmplete) and phosphatase (PhosSTOP) inhibitor cocktails (Roche Applied Science, Laval, Quebec, Canada). Protein concentrations were determined by the bicinchoninic acid (BCA) assay (Pierce, Rockford, IL). Equal amounts of proteins (40 μg) were separated by 10% SDS-PAGE, transferred onto polyvinylidene difluoride (PVDF) membranes, and immunoblotted overnight with selected rabbit anti-phospho-IRF-3 (Ser396) or anti-phospho-IRF-7 (Ser471/472) antibodies (Bioss, Worburn, MA). Membranes were washed in Tris-buffered saline (TBS)–0.1% Tween 20 solution (Thermo Fisher Scientific, Rockford, IL) prior to incubation with a secondary horseradish peroxidase (HRP)-conjugated goat anti-rabbit antibody (Jackson ImmunoResearch, West Grove, PA). HRP activity was revealed by incubation with the LumiGLO chemiluminescent substrate (Gaithersburg, MD) as described in the manufacturer's instructions. Chemiluminescence reactions were visualized and quantitatively analyzed by use of AlphaView software (Alpha Innotech Corp., San Leandro, CA). Relative intensity values were calculated by normalizing each band to its total IRF loading control and expressed as the fold change from results for noninfected control mice.
Immunohistochemistry.
Brain sections (30 μm thick) were washed three times for 15 min (each) in potassium phosphate-buffered saline (KPBS) and incubated for 20 min in KPBS containing 0.4% Triton X-100, 4% goat serum, and 1% bovine serum albumin (BSA) to block nonspecific sites. The sections were then incubated for 2 h with the primary viral antibody (polyclonal rabbit anti-HSV-1/2, diluted 1:750 [Abd Serotec, Kidlington, United Kingdom]) at room temperature. After 3 consecutive washes for 15 min (each) with KPBS, sections were incubated in the dark for 2 h with the fluorochrome-conjugated secondary antibody (Alexa 594-conjugated goat anti-rabbit [Invitrogen, Carlsbad, CA]). Sections were rinsed 3 times for 15 min (each) in KPBS and then incubated with 0.2% 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen, Eugene, OR) at a dilution of 1:3,000 for 5 min and rinsed 3 more times for 15 min (each) in KPBS. Brain sections were mounted on SuperFrost slides (Fisher Scientific, Nepean, Ontario, Canada) and coverslipped with Fluoromount-G (Electron Microscopy Sciences, Hatfield, PA). All images were captured using a Nikon Eclipse 80i microscope (Nikon, Melville, NY) equipped with a digital camera (QImaging, Surrey, British Columbia, Canada).
Silencing of IRF-3 and IRF-7 pathways with small interfering RNAs (siRNAs) in murine embryo fibroblasts.
Murine embryonic fibroblasts (MEFs) were obtained from 13- to 14-day-old C57BL/6 WT mouse embryos digested with 0.5% trypsin-EDTA solution for 30 min at 37°C (27). MEFs were cultured in MEM supplemented with 20% heat-inactivated FBS. Cells were transfected with 1.5 μg of vehicle alone, IRF-3, IRF-7, or both IRF-3 and IRF-7 siRNAs (Silencer select predesigned siRNAs; Ambion, Burlington, Ontario, Canada) using the Lipofectamine RNAiMAX reagent (Invitrogen, Burlington, Ontario, Canada). Forty eight hours posttransfection, MEFs were infected with HSV-1 (strain H25) at a multiplicity of infection (MOI) of 1 or stimulated with polyinosinic:poly(C) (poly I·C; InvivoGen, San Diego, CA) (1 μg/ml, digested or not with RNase III) for 12 h. Cell-free supernatants were then collected and assessed for IFN-β secretion by ELISA.
Statistical analyses.
Differences in group survival rates were compared using a log-rank (Mantel-Cox) test. Viral DNA loads and infectious viral titers were analyzed by a Kruskal-Wallis one-way analysis of variance (ANOVA) with Dunn's posttest. IFN-α/β levels and phosphorylated IRF-3 (p-IRF-3) and p-IRF-7 amounts in brain homogenates, as well as IFN-β levels in MEFs, were analyzed by one-way ANOVA with Tukey's multiple-comparison posttest. All statistical analyses were carried out using the GraphPad Prism software program, version 5.00 (GraphPad Software, San Diego, CA).
RESULTS
TRIF−/− and IPS-1−/− mice are more susceptible than WT mice to HSV-1 infection of the brain.
In order to evaluate the role played by the TRIF and IPS-1 signaling pathways in our model of HSE, the survival rate of C57BL/6 WT mice was first compared with those of TRIF−/− and IPS-1−/− mice after intranasal infection with HSV-1. Mice were infected with an inoculum of 7.5 × 105 PFU and examined daily up to 20 days postinfection (p.i.). All mice started to show sickness signs by days 3 to 8 p.i. Moreover, mice deficient for TRIF and IPS-1 exhibited a swollen forehead, starting on day 5 p.i., which was not seen in C57BL/6 WT mice (Fig. 1A). Both knockout (KO) mice had lower survival rates than the WT group (80% for WT mice, versus 36% for TRIF−/− mice [P = 0.02] [Fig. 1B] and 79% for WT mice, versus 50% for IPS-1−/− mice [P = 0.09] [Fig. 1C]). These results suggest that the TRIF and IPS-1 signaling pathways are important for survival of infected mice and that both adaptor proteins play a role in the mouse cerebral innate immune response against HSV-1 infection.
Fig 1.

Swollen forehead of HSV-1-infected mice on day 7 postinfection (A) and survival rates of TRIF−/− (B) or IPS-1−/− (C) mice compared to those of C57BL/6 WT mice. Ten to fourteen animals per group were infected intranasally with HSV-1 (7.5 × 105 PFU in 20 μl). Mice were carefully examined for HSE-related signs and mortality (≥20% of weight loss and/or neurological signs or mortality were considered endpoints). Group survival rates were compared using a log-rank (Mantel-Cox) test. Survival rates were significantly different between TRIF−/− and C57BL/6 WT mice (P = 0.02) but not between IPS-1−/− and C57BL/6 WT mice (P = 0.09).
TRIF and IPS-1 signaling pathways contribute to control HSV-1 replication in the CNS.
The relative influence of TRIF and IPS-1 signaling pathways in controlling viral replication in the brain was then evaluated in WT, TRIF−/−, and IPS-1−/− mice infected with HSV-1. The viral DNA load was measured in brain homogenates on days 3, 5, 7, 9, and 11 following intranasal infection. As shown in Fig. 2A, there was no difference in the brain viral loads on day 3 p.i. for the different strains of mice. However, the viral load increased significantly in TRIF−/− mice compared to those in WT and IPS-1−/− mice on days 5 and 7 p.i., and it was still significantly higher than that of the WT on day 11 p.i. In IPS-1−/− mice, an increase in the viral load was observed at later time points (i.e., on day 7 and especially on day 9 p.i.). Infectious viral titers were in line with viral DNA load data; in TRIF−/− mice, higher titers of virus were recovered on days 5 to 9 p.i. than for the WT, whereas viral titers significantly increased in brains of IPS-1−/− mice on days 7 and 9 p.i. (Fig. 2B). Overall, our results suggest that both the TRIF and IPS-1 signaling pathways contribute to controlling HSV-1 replication in the central nervous system (CNS), with the former pathway involved at earlier time points after infection. In addition, both deficient mice exhibited a more disseminated HSV-1 infection of the brain as revealed by immunohistochemistry analysis (Fig. 3). Indeed, HSV proteins were restricted to the olfactory bulb in WT mice, whereas their distribution was disseminated to the pons and medulla brain regions in both types of KO mice.
Fig 2.
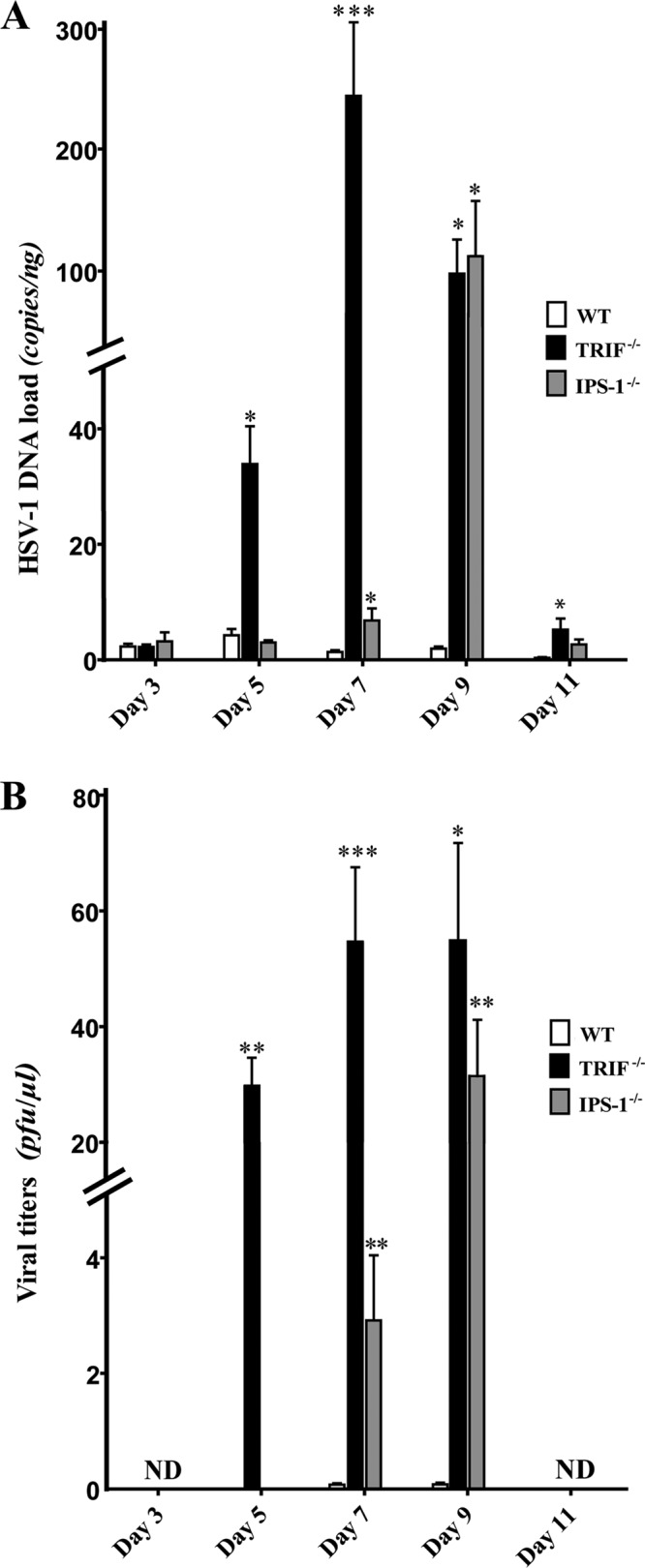
Herpes simplex virus 1 replication in the brains of TRIF−/−, IPS-1−/−, and C57BL/6 WT mice. (A) Viral DNA loads were measured in brain homogenates by qPCR. Results are expressed as copies/ng of extracted DNA. (B) Viral titers were measured in brain homogenates by a standard plaque assay on Vero cells and are reported as PFU/μl of brain homogenate. Statistical analyses were performed using a Kruskal-Wallis one-way ANOVA test with Dunn's posttest. Statistically significant results compared to those for the WT group are indicated as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001. ND, not detected. Data from two independent experiments (of 4 to 6 mice per group) with similar results are combined.
Fig 3.

Representative micrographs illustrating the localization of HSV-1 proteins in different regions of the brains of WT, TRIF−/−, and IPS-1−/− mice (3 mice per group) on day 5 postinfection. Slices (30 μm thick) of brain were processed for immunohistochemistry with a primary polyclonal rabbit anti-HSV-1/2 antibody and a secondary Alexa 594-conjugated goat anti-rabbit antibody (red), followed by staining with DAPI (blue). In WT mice, HSV-1 proteins were restricted to the olfactory bulb and could not be detected in the pons. In TRIF−/− and IPS-1−/− mice, viral antigens were detected in the olfactory bulb and the pons. Magnification, ×20.
TRIF- and IPS-1-deficient mice have delayed production of IFN-β in the CNS.
In order to evaluate the involvement of the innate sensors in type I IFN production during HSE, the levels of IFN-β in brain homogenates of WT, TRIF- and IPS-1-deficient mice were measured at different time points following infection (Fig. 4A). No significant difference in IFN-β production was seen between groups of mice before day 5 p.i. Interestingly, on day 5 p.i., we observed a significant increase in IFN-β production in the WT group compared to levels for noninfected and infected TRIF- and IPS-1-deficient mice. In contrast to declining levels in WT mice on day 7 p.i., both deficient mice subsequently produced significantly higher levels of IFN-β on days 7 and 9 p.i. Overall, these results suggest that a deficiency in the TRIF or IPS-1 signaling pathway is associated with altered production of IFN-β at an early time point that could be critical to controlling HSV-1 infection. Indeed, the mortality observed in KO mice occurred predominantly in the first 6 days after infection.
Fig 4.

Levels of IFN-β (A) or IFN-α (B) in brain homogenates from TRIF−/−, IPS-1−/−, and C57BL/6 WT mice. IFN-α/β levels (expressed in pg/ml) were measured in brain homogenate supernatants by ELISA prior to and on days 3, 5, 7, 9, and 11 following infection. Statistical analyses were performed using a one-way ANOVA test with Tukey's multiple-comparison posttest. Statistically significant results compared to those for the WT group are indicated as follows: *, P < 0.05; ***, P < 0.001. Data from three independent experiments (of 3 to 6 mice per group) with similar results are combined.
The levels of IFN-α in brain homogenates of WT, TRIF−/−, and IPS-1−/− mice were assessed at the same time points described above following infection with HSV-1 (Fig. 4B). No significant difference was observed before day 7 p.i. between the different groups of mice. However, the level of IFN-α measured for TRIF−/− mice was significantly increased over those for WT and IPS-1−/− mice on day 7 p.i. (P < 0.05). A nonsignificant increase of IFN-α was also detected in brain homogenates from TRIF- and IPS-1-deficient mice compared to results for the WT on day 9 p.i. before returning to baseline values on day 11 p.i. These results suggest that the absence of a functional TRIF pathway could be partially compensated by a production of IFN-α possibly mediated through IPS-1 or other cellular sensors that can participate in the recognition of HSV-1.
Expression levels of phosphorylated IRF-3 and IRF-7 in the CNS are increased during HSE.
IRF-3 and IRF-7 are transcription factors essential for the activation of type I IFN genes following triggering of the adaptor proteins TRIF and IPS-1. Thus, the levels of phosphorylated IRF-3 and IRF-7 in brain homogenates obtained on days 3, 5, and 7 p.i. were determined by Western blotting (Fig. 5). As expected, we observed a significant increase (P < 0.05) in both IRF-3 and IRF-7 phosphorylation levels for HSV-1-infected WT mice compared to those for the noninfected group (Fig. 5A and D). The levels of phosphorylated IRF-3 reached a maximum value on day 5 following the infection (P < 0.05), whereas IRF-7 phosphorylation increased steadily with time and peaked on day 7 p.i. (P < 0.01). Conversely, levels of phosphorylated IRF-3 significantly decreased (P < 0.01) for all time points following infection in TRIF−/− mice compared to results for the noninfected group (Fig. 5B), whereas there was a significant increase (P < 0.05) in the phosphorylation of IRF-7 on days 3 and 5 p.i. (Fig. 5E). In IPS-1-deficient mice, there was no change in the phosphorylation levels of IRF-3 following the infection (Fig. 5C), whereas levels of phosphorylated IRF-7 were significantly decreased on days 5 and 7 p.i. (P < 0.01 compared to results for noninfected IPS-1−/− mice) (Fig. 5F). Thus, TRIF and IPS-1 signal mainly through IRF-3 and IRF-7, respectively, in brain of HSV-1-infected mice. These results suggest that the production of type I IFN in TRIF−/− and IPS-1−/− mice, which was higher and sustained compared to that in WT mice at later times, might be due to the activation of other cellular sensors in response to increased viral replication.
Fig 5.

Levels of phosphorylated IRF-3 (A, B, and C) or IRF-7 (D, E, and F) in C57BL/6 WT (A and D), TRIF−/− (B and E), or IPS-1−/− (C and F) mice. Animals were infected intranasally with 7.5 × 105 PFU of HSV-1. Proteins were extracted from brain homogenates prior to and on days 3, 5, and 7 following infection. Left panels show immunoblots for phosphorylated IRF-3 (p-IRF-3) and p-IRF-7 and total IRF-3 and IRF-7 respective loading controls. Relative intensity values were calculated by normalizing each band to its total IRF control. Right panels show the fold increases of normalized p-IRF-3 and p-IRF-7 values relative to those obtained for noninfected (N.I.) mice. Values for each condition represent the means ± standard deviations obtained for 8 different animals. Statistical analyses were performed by one-way ANOVA with Tukey's multiple-comparison posttest. Statistically significant results compared to those for N.I. mice are indicated as follows: *, P < 0.05; **, P < 0.01.
Blocking IRF-3 and IRF-7 pathways decrease IFN-β production in MEFs.
The implication of downstream effectors of TRIF and IPS-1 pathways in the production of IFN-β has been further investigated with WT MEFs silenced for the expression of IRF-3 and IRF-7 with siRNAs. In all cases, positive controls of stimulation were used, i.e., poly(I·C) (recognized mainly by TLR3) and digested polyI·C (recognized mainly by RIG-I). Silencing of the expression of IRF-3 or IRF-7 caused a significant decrease in the production of IFN-β following stimulation of MEFs with poly I·C (digested or not) or with the virus (Fig. 6). Furthermore, silencing of both IRF-3 and IRF-7 expression resulted in nondetectable IFN-β levels regardless of the stimulating agents. These results suggest that both IRF-3 and IRF-7 significantly contributed to the production of IFN-β in response to HSV-1 infection in this ex vivo system.
Fig 6.

Secretion of IFN-β in WT MEFs silenced for IRF-3, IRF-7, or both IRF-3 and IRF-7 expression and infected with HSV-1 or stimulated with poly(I·C). MEFs were transfected with 1.5 μg/ml of vehicle alone (control), IRF-3, IRF-7, or both IRF-3 and IRF-7 siRNAs. At 48 h posttransfection, cells were infected or not with HSV-1 at an MOI of 1 or treated with poly(I·C) or poly(I·C) digested with RNase III (both at 1 μg/ml). Cell culture supernatants were collected 12 h later and assessed for IFN-β production by ELISA. Statistical analyses were performed by a one-way ANOVA with Tukey's multiple-comparison posttest. Statistically significant results compared to those for the respective control MEFs are indicated (***, P < 0.001). ND, not detected.
DISCUSSION
In the present study, we used an experimental model of HSV-1 infection to monitor the development of encephalitis in mice deficient for TRIF and IPS-1 adaptor proteins. IPS-1−/− and especially TRIF−/− mice had increased sickness signs and mortality rates following HSV-1 infection compared to findings for WT mice. These results highlight the crucial role of both signaling pathways in the cerebral innate immune response during HSE. To our knowledge, this report is the first showing the implication of IPS-1 in that context. The production of IFN-β in the brains of deficient mice was impaired at a critical time after infection compared to that of WT mice, and this was associated with a significant increase in the brain viral load and with virus dissemination throughout the brain. These results demonstrate that activation of TRIF and IPS-1 signaling pathways requires a tight coordination to initiate a timely production of IFN-β for effective control of viral replication during HSE.
Protection of the host against viral infections relies on the detection of pathogens followed by efficient activation of the innate immune system through type I IFN production, which is one of the earliest responses to viral invasion (28). Recognition of HSV has previously been proposed to occur in a dual manner through sensing of viral glycoproteins and CpG-rich viral DNA by TLR2 and TLR9, respectively (29). In that respect, TLR2 and especially TLR9 were shown to be important for the control of HSV replication in mouse brain and trigeminal ganglia (30). It was also reported that childhood HSE could be associated with mutations in key components of the TLR3 signaling pathway, including Unc93B (a protein that facilitates translocation of TLR3, -7, -8, and -9 from the endoplasmic reticulum to the endosomes), TRIF, tumor necrosis factor receptor-associated factor 3 (TRAF3) adaptor molecule, and TBK-1 (representing a total of 9 cases among 80 studied children with HSE) (8, 31–33). These inherited deficiencies are responsible for an impaired activation of TLR3 signaling upon recognition of HSV dsRNA, leading to an altered type I IFN production. In addition to endosomal TLR3, the implication of cytoplasmic RIG-I and MDA-5 in the sensing of HSV dsRNA was also found using cell culture models (16–18), but their role during HSE had not yet been investigated. On the one hand, TLR3 signals through TRIF and induces IRF-3 phosphorylation and translocation to the nucleus, which leads to type I IFN production (14). On the other hand, RIG-I and MDA-5 signal through IPS-1 to activate IRF-3 and IRF-7 (19), which act as transcriptional factors to bind IFN promoters (34). However, the relative contributions of these two signaling pathways in type I IFN production during HSE has not yet been elucidated.
The increased susceptibility of IPS-1−/− and especially TRIF−/− mice to HSV-1 infection demonstrated that both adaptor proteins are crucial in the signaling pathways that control viral replication in the CNS and mouse survival. In brain tissue, TLR3 is found in the endosomal compartment of different cell types, such as microglia, astrocytes, oligodendrocytes, and neurons. Moreover, we have recently demonstrated a recruitment of monocytes/macrophages from the blood to the CNS during HSE (35), and these cells also expressed TLR3. Our results suggest that TLR3 is essential for the recognition of HSV-1 in the brain and that viral clearance and survival of mice are linked to appropriate signaling through TRIF. Previous studies have demonstrated that HSV is recognized by IPS-1-dependent RLHs in MEFs and also possibly in macrophages, which accumulate dsRNA in their cytoplasm (12, 16–18). To our knowledge, our results are the first to underscore the role of the IPS-1 signaling pathway during HSE. As with TRIF−/− mice, the brain viral load and mortality rate for IPS-1−/− mice were increased, but these consequences were less important than for TRIF−/− mice. We thus suggest that the IPS-1 signaling pathway contributes to the control of HSV-1 infection of the CNS but to a lesser extent than the TRIF pathway. In both types of deficient mice, the increased mortality rates could be attributed to a more disseminated infection extending to the pons and medulla regions of the brain, which correlated with significantly higher viral loads on days 5, 7, and 9 in TRIF−/− mice and on days 7 and 9 in IPS-1−/− mice than for WT mice. These results suggest that the involvement of the two signaling pathways may be sequential, with TRIF implicated earlier in the course of HSV infection, followed by IPS-1.
The production of IFN-β was increased at an earlier time point (i.e., on day 5) for WT mice than for the deficient mice, and such timely production may be critical to control viral replication in the brain. Indeed, we have previously demonstrated that the intranasal administration of an agonist of TLR3 (poly I·C) to mice before HSV-1 challenge reduced viral replication in the brain, leading to decreased HSE severity and mortality (36). In contrast, poly(I·C) given after the infection exerted no beneficial effect (unpublished data). We thus suggest that the cerebral innate immune response produced during HSE is a double-edged sword, since it is critical to controlling HSV replication in the brain early after infection but if uncontrolled may result in an overzealous inflammatory response that could be detrimental to the host. Thus, these results suggest that the activation of the TRIF and IPS-1 signaling pathways in mouse brain requires close coordination to produce IFN-β in a timely manner to effectively control viral replication. Such cooperation between IPS-1 and TLR9 has already been described for the production of type I IFN in response to HSV-2 infection of macrophages and fibroblasts (17) and to HSV-1 infection of MEFs (16).
In WT mice, levels of phosphorylated IRF-3 and IRF-7, which are important downstream effectors in the IFN-β production cascade, increased over time following HSV-1 infection. Silenced expression of IRF-3 or IRF-7 in WT MEFs confirmed that both downstream effectors participate in IFN-β secretion in response to HSV-1 infection. The delayed production of IFN-β in TRIF- and IPS-1-deficient mice was higher and sustained compared to that in WT mice. Interestingly, TRIF−/− mice also produced significantly larger amounts of IFN-α on day 7 postinfection than WT and IPS-1−/− mice. However, the levels of phosphorylated IRF-3 and/or IRF-7 did not increase on day 7 p.i. but could possibly be higher at later times. Taken together, these data suggest that the production of type I IFN in TRIF−/− and IPS-1−/− mice is not compensated by the functional IPS-1 or TRIF pathway and that other cellular sensors, independent of these signaling pathways, may also be implicated. These sensors may be activated in response to the increased viral replication in the CNS. In this respect, active viral replication correlates with the amount of IFN-β produced over time in the brains of deficient mice. Other signaling pathways involved in IFN-β production following HSV-1 infection have been described, but the data are complex, conflicting, and incomplete. TLR9 has been reported to be involved in early type I IFN responses following HSV infection in vitro and in vivo (6, 11). In contrast, another study has demonstrated that TLR9 knockout mice have type I IFN responses and survival rates comparable to those of WT mice following HSV infection (37). Furthermore, another DNA sensor (IFI16) has also been shown to induce the production of IFN-β following HSV-1 corneal infection, and its expression in epithelial cells is thought to allow for resistance to virus infection (38, 39). Future studies examining the roles of other cellular sensors and their cooperation following HSV-1 infection are thus needed to better characterize the cerebral innate immune response.
We cannot exclude, however, that other inflammatory mediators may also affect the outcome of HSE in both types of deficient mice. In fact, triggering of the IPS-1 adaptor protein leads to NF-κB activation and proinflammatory cytokine production. Therefore, the deficiency in IPS-1 could result in an impaired activation of NF-κB, which in turn may alter the production of innate immune cytokines and affect the control of HSV replication in the CNS. In that regard, production of antigen-specific antibodies is severely decreased in IPS-1-deficient mice, and cytotoxic T cell expansion is also reduced in both TRIF−/− and IPS-1−/− mice (40).
In conclusion, our data suggest that the increased mortality rate observed in TRIF adaptor protein knockout mice is associated with delayed and sustained IFN-β production during HSE. Interestingly, a higher and delayed IFN-α production, possibly through a compensatory mechanism involving IPS-1 or other cellular sensors, was seen in TRIF−/− mice. However, this pathway(s) is not sufficient to control HSV-1 replication in the CNS, and TRIF might thus be crucial in the brain, as found in patients showing deficiencies in the TLR3 pathway (8). Therefore, the TRIF adaptor molecule seems to be more implicated in the innate immune response to HSE than IPS-1. Nevertheless, IPS-1 also plays a pivotal role in the brain immune response against HSV-1 infection. We thus propose that the cerebral innate immune response against HSV-1 infection could be initiated by TLR3 via TRIF and thereafter by RIG-I/MDA-5 via IPS-1 to induce type I IFN gene transcription. Such coordination between TLR and RLH pathways could promote a rapid and effective immune response following HSV-1 infection, suggesting that these receptors could constitute potential therapeutic targets for improving the treatment of HSE. Moreover, our results also suggest the activation of other cellular sensors in response to the increased viral replication in the brains of both types of deficient mice. Studies using double (TRIF × IPS-1) knockout mice should further elucidate the role of those sensors in the cerebral innate immune response to HSV-1 and could confirm the potential implication of other IFN-signaling pathways independent of TRIF and IPS-1.
ACKNOWLEDGMENTS
None of the authors has a conflict of interest.
This study was supported by grants from the Canadian Institutes of Health Research (MOP 97728 to G.B. and MOP 115078 to J.G.). G.B. is the holder of the Canada research chair on emerging viruses and antiviral resistance.
We thank Shizuo Akira, from Osaka University, for kindly providing TRIF−/− and IPS-1−/− mice.
Footnotes
Published ahead of print 17 April 2013
REFERENCES
- 1. Tyler KL. 2004. Update on herpes simplex encephalitis. Rev. Neurol. Dis. 1:169–178 [PubMed] [Google Scholar]
- 2. Melchjorsen J. 2012. Sensing herpes: more than toll. Rev. Med. Virol. 22:106–121 [DOI] [PubMed] [Google Scholar]
- 3. Vandevenne P, Sadzot-Delvaux C, Piette J. 2010. Innate immune response and viral interference strategies developed by human herpesviruses. Biochem. Pharmacol. 80:1955–1972 [DOI] [PubMed] [Google Scholar]
- 4. Paludan SR, Bowie AG, Horan KA, Fitzgerald KA. 2011. Recognition of herpesviruses by the innate immune system. Nat. Rev. Immunol. 11:143–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aravalli RN, Hu S, Rowen TN, Palmquist JM, Lokensgard JR. 2005. Cutting edge: TLR2-mediated proinflammatory cytokine and chemokine production by microglial cells in response to herpes simplex virus. J. Immunol. 175:4189–4193 [DOI] [PubMed] [Google Scholar]
- 6. Krug A, Luker GD, Barchet W, Leib DA, Akira S, Colonna M. 2004. Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood 103:1433–1437 [DOI] [PubMed] [Google Scholar]
- 7. Hochrein H, Schlatter B, O'Keeffe M, Wagner C, Schmitz F, Schiemann M, Bauer S, Suter M, Wagner H. 2004. Herpes simplex virus type-1 induces IFN-alpha production via Toll-like receptor 9-dependent and -independent pathways. Proc. Natl. Acad. Sci. U. S. A. 101:11416–11421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, Segal D, Sancho-Shimizu V, Lorenzo L, Puel A, Picard C, Chapgier A, Plancoulaine S, Titeux M, Cognet C, von Bernuth H, Ku CL, Casrouge A, Zhang XX, Barreiro L, Leonard J, Hamilton C, Lebon P, Heron B, Vallee L, Quintana-Murci L, Hovnanian A, Rozenberg F, Vivier E, Geissmann F, Tardieu M, Abel L, Casanova JL. 2007. TLR3 deficiency in patients with herpes simplex encephalitis. Science 317:1522–1527 [DOI] [PubMed] [Google Scholar]
- 9. Sorensen LN, Reinert LS, Malmgaard L, Bartholdy C, Thomsen AR, Paludan SR. 2008. TLR2 and TLR9 synergistically control herpes simplex virus infection in the brain. J. Immunol. 181:8604–8612 [DOI] [PubMed] [Google Scholar]
- 10. Kurt-Jones EA, Chan M, Zhou S, Wang J, Reed G, Bronson R, Arnold MM, Knipe DM, Finberg RW. 2004. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc. Natl. Acad. Sci. U. S. A. 101:1315–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lund J, Sato A, Akira S, Medzhitov R, Iwasaki A. 2003. Toll-like receptor 9-mediated recognition of herpes simplex virus-2 by plasmacytoid dendritic cells. J. Exp. Med. 198:513–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Weber F, Wagner V, Rasmussen SB, Hartmann R, Paludan SR. 2006. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol. 80:5059–5064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kawai T, Akira S. 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11:373–384 [DOI] [PubMed] [Google Scholar]
- 14. Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. 2003. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 301:640–643 [DOI] [PubMed] [Google Scholar]
- 15. Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, Hiiragi A, Dermody TS, Fujita T, Akira S. 2008. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 205:1601–1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rasmussen SB, Sorensen LN, Malmgaard L, Ank N, Baines JD, Chen ZJ, Paludan SR. 2007. Type I interferon production during herpes simplex virus infection is controlled by cell-type-specific viral recognition through Toll-like receptor 9, the mitochondrial antiviral signaling protein pathway, and novel recognition systems. J. Virol. 81:13315–13324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rasmussen SB, Jensen SB, Nielsen C, Quartin E, Kato H, Chen ZJ, Silverman RH, Akira S, Paludan SR. 2009. Herpes simplex virus infection is sensed by both Toll-like receptors and retinoic acid-inducible gene-like receptors, which synergize to induce type I interferon production. J. Gen. Virol. 90:74–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Melchjorsen J, Rintahaka J, Soby S, Horan KA, Poltajainen A, Ostergaard L, Paludan SR, Matikainen S. 2010. Early innate recognition of herpes simplex virus in human primary macrophages is mediated via the MDA5/MAVS-dependent and MDA5/MAVS/RNA polymerase III-independent pathways. J. Virol. 84:11350–11358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. 2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6:981–988 [DOI] [PubMed] [Google Scholar]
- 20. Seth RB, Sun L, Ea CK, Chen ZJ. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122:669–682 [DOI] [PubMed] [Google Scholar]
- 21. Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. 2005. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 19:727–740 [DOI] [PubMed] [Google Scholar]
- 22. Schindler C, Levy DE, Decker T. 2007. JAK-STAT signaling: from interferons to cytokines. J. Biol. Chem. 282:20059–20063 [DOI] [PubMed] [Google Scholar]
- 23. Sadler AJ, Williams BR. 2008. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 8:559–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kumar H, Kawai T, Kato H, Sato S, Takahashi K, Coban C, Yamamoto M, Uematsu S, Ishii KJ, Takeuchi O, Akira S. 2006. Essential role of IPS-1 in innate immune responses against RNA viruses. J. Exp. Med. 203:1795–1803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sergerie Y, Rivest S, Boivin G. 2007. Tumor necrosis factor-alpha and interleukin-1 beta play a critical role in the resistance against lethal herpes simplex virus encephalitis. J. Infect. Dis. 196:853–860 [DOI] [PubMed] [Google Scholar]
- 26. Boivin G, Goyette N, Sergerie Y, Keays S, Booth T. 2006. Longitudinal evaluation of herpes simplex virus DNA load during episodes of herpes labialis. J. Clin. Virol. 37:248–251 [DOI] [PubMed] [Google Scholar]
- 27. Garfield AS. 2010. Derivation of primary mouse embryonic fibroblast (PMEF) cultures. Methods Mol. Biol. 633:19–27 [DOI] [PubMed] [Google Scholar]
- 28. Stetson DB, Medzhitov R. 2006. Type I interferons in host defense. Immunity 25:373–381 [DOI] [PubMed] [Google Scholar]
- 29. Sato A, Linehan MM, Iwasaki A. 2006. Dual recognition of herpes simplex viruses by TLR2 and TLR9 in dendritic cells. Proc. Natl. Acad. Sci. U. S. A. 103:17343–17348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lima GK, Zolini GP, Mansur DS, Freire Lima BH, Wischhoff U, Astigarraga RG, Dias MF, das Gracas Almeida Silva M, Bela SR, do Valle Antonelli LR, Arantes RM, Gazzinelli RT, Bafica A, Kroon EG, Campos MA. 2010. Toll-like receptor (TLR) 2 and TLR9 expressed in trigeminal ganglia are critical to viral control during herpes simplex virus 1 infection. Am. J. Pathol. 177:2433–2445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Casrouge A, Zhang SY, Eidenschenk C, Jouanguy E, Puel A, Yang K, Alcais A, Picard C, Mahfoufi N, Nicolas N, Lorenzo L, Plancoulaine S, Senechal B, Geissmann F, Tabeta K, Hoebe K, Du X, Miller RL, Heron B, Mignot C, de Villemeur TB, Lebon P, Dulac O, Rozenberg F, Beutler B, Tardieu M, Abel L, Casanova JL. 2006. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science 314:308–312 [DOI] [PubMed] [Google Scholar]
- 32. Perez de Diego R, Sancho-Shimizu V, Lorenzo L, Puel A, Plancoulaine S, Picard C, Herman M, Cardon A, Durandy A, Bustamante J, Vallabhapurapu S, Bravo J, Warnatz K, Chaix Y, Cascarrigny F, Lebon P, Rozenberg F, Karin M, Tardieu M, Al-Muhsen S, Jouanguy E, Zhang SY, Abel L, Casanova JL. 2010. Human TRAF3 adaptor molecule deficiency leads to impaired Toll-like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity 33:400–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sancho-Shimizu V, Perez de Diego R, Lorenzo L, Halwani R, Alangari A, Israelsson E, Fabrega S, Cardon A, Maluenda J, Tatematsu M, Mahvelati F, Herman M, Ciancanelli M, Guo Y, AlSum Z, Alkhamis N, Al-Makadma AS, Ghadiri A, Boucherit S, Plancoulaine S, Picard C, Rozenberg F, Tardieu M, Lebon P, Jouanguy E, Rezaei N, Seya T, Matsumoto M, Chaussabel D, Puel A, Zhang SY, Abel L, Al-Muhsen S, Casanova JL. 2011. Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency. J. Clin. Invest. 121:4889–4902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, Taniguchi T. 2000. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 13:539–548 [DOI] [PubMed] [Google Scholar]
- 35. Boivin N, Menasria R, Gosselin D, Rivest S, Boivin G. 2012. Impact of deficiency in CCR2 and CX3CR1 receptors on monocytes trafficking in herpes simplex virus encephalitis. J. Gen. Virol. 93:1294–1304 [DOI] [PubMed] [Google Scholar]
- 36. Boivin N, Sergerie Y, Rivest S, Boivin G. 2008. Effect of pretreatment with toll-like receptor agonists in a mouse model of herpes simplex virus type 1 encephalitis. J. Infect. Dis. 198:664–672 [DOI] [PubMed] [Google Scholar]
- 37. Wang JP, Bowen GN, Zhou S, Cerny A, Zacharia A, Knipe DM, Finberg RW, Kurt-Jones EA. 2012. Role of specific innate immune responses in herpes simplex virus infection of the central nervous system. J. Virol. 86:2273–2281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Conrady CD, Zheng M, Fitzgerald KA, Liu C, Carr DJ. 2012. Resistance to HSV-1 infection in the epithelium resides with the novel innate sensor, IFI-16. Mucosal Immunol. 5:173–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS, Fitzgerald KA, Paludan SR, Bowie AG. 2010. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 11:997–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kumar H, Koyama S, Ishii KJ, Kawai T, Akira S. 2008. Cutting edge: cooperation of IPS-1- and TRIF-dependent pathways in poly IC-enhanced antibody production and cytotoxic T cell responses. J. Immunol. 180:683–687 [DOI] [PubMed] [Google Scholar]