

Abstract
The important role of the CD8+ T-cell response on HIV control is well established. Moreover, the acute phase of infection represents a proper scenario to delineate the antiviral cellular functions that best correlate with control. Here, multiple functional aspects (specificity, ex vivo viral inhibitory activity [VIA] and polyfunctionality) of the HIV-specific CD8+ T-cell subset arising early after infection, and their association with disease progression markers, were examined. Blood samples from 44 subjects recruited within 6 months from infection (primary HIV infection [PHI] group), 16 chronically infected subjects, 11 elite controllers (EC), and 10 healthy donors were obtained. Results indicated that, although Nef dominated the anti-HIV response during acute/early infection, a higher proportion of early anti-Gag T cells correlated with delayed progression. Polyfunctional HIV-specific CD8+ T cells were detected at early time points but did not associate with virus control. Conversely, higher CD4+ T-cell set points were observed in PHI subjects with higher HIV-specific CD8+ T-cell VIA at baseline. Importantly, VIA levels correlated with the magnitude of the anti-Gag cellular response. The advantage of Gag-specific cells may result from their enhanced ability to mediate lysis of infected cells (evidenced by a higher capacity to degranulate and to mediate VIA) and to simultaneously produce IFN-γ. Finally, Gag immunodominance was associated with elevated plasma levels of interleukin 2 (IL-2) and macrophage inflammatory protein 1β (MIP-1β). All together, this study underscores the importance of CD8+ T-cell specificity in the improved control of disease progression, which was related to the capacity of Gag-specific cells to mediate both lytic and nonlytic antiviral mechanisms at early time points postinfection.
INTRODUCTION
Human immunodeficiency virus (HIV) still represents a major public health concern. Although the instauration of highly active antiretroviral treatment (HAART) had a tremendous impact on the epidemic dynamics, the development of an effective prophylactic vaccine is still a main objective in the HIV-related research field. As HIV is highly diverse among different isolates, evolves continuously under selective pressure, infects immune cells, and encodes proteins with the capacity to modulate immune cell functions, it imposes definite challenges that should be overcome in the race of getting a successful vaccine. However, the description of (i) infected subjects able to control HIV replication over long periods of time to very low levels without therapy (known as long-term nonprogressors [LTNP] and elite controllers [EC]); (ii) uninfected subjects who, despite being highly exposed to the virus, remain seronegative (exposed seronegatives [ESN]); and (iii) the results from the Thai vaccine trial RV-144, which showed 30% efficacy (1), suggests that the objective is reachable. In this line, much of the research work conducted over the past few years was aimed to define the immune correlates of protection, i.e., desirable characteristics that the vaccine-elicited immune response should have in order to contain viral challenge. Within this field, special emphasis has been focused on the HIV-specific CD8+ cytotoxic T lymphocytes (CTLs), which are thought to play a key role in reducing viral replication (2, 3).
The first evidence that specific CD8+ T cells were involved in the control of viral replication was reported in studies conducted in humans and nonhuman primates during the acute phase of infection. After infection, emergence of specific CD8+ T cells correlates with the decline of peak viremia toward set point establishment, which varies from person to person and is a strong predictor of disease progression (4). Also, CTL escape mutants have been described (5, 6), and superior viral control has been attributed to specific human leukocyte antigen (HLA) class I alleles (7, 8). Moreover, recent proof-of-concept vaccine studies in nonhuman primates indicate that vaccine-elicited CD8+ T-cell responses are associated with partial protection from infection and with enhanced control of breakthrough infections (9, 10), reinforcing the notion that specific CD8+ T cells exert a pivotal role in viral control. In-depth analyses of this cellular population, performed in different cohorts and models, suggest that specificity, quality, and phenotype are all determinants of CD8+ T-cell ability to mediate control: specificity in terms of viral targets (11–15); quality in terms of avidity and capacity to mediate viral suppression, proliferate, and secrete a broad spectrum of chemokines and cytokines (16–20); and phenotype in terms of memory sub-subsets and expression of exhaustion markers (21–23).
Cell samples obtained during the acute/early HIV infection constitute invaluable tools to understand the functional features of the HIV-specific CD8+ T cells that best correlate with the lower-set-point/protection-from-progression axis and future control. For sure, these approaches will help dissect the correlates of protection needed to develop an effective prophylactic vaccine. Besides, vaccine-elicited highly suppressive specific CD8+ T cells would help constrain viral replication to very low levels in breakthrough infections occurring in vaccinees, which in turn would contribute to a slower progression of the newly infected person as well as lower transmission risk (24).
We have previously worked with acute phase samples in order to evaluate Nef-specific cross-clade T-cell reactivity in samples from subtype B- and BF-infected subjects (25). In that study, differences in the CD8+ T-cell population functional profile were observed among subjects, finding an association between CD8+ T-cell polyfunctionality and viral control. In the manuscript, we sought to analyze multiple aspects of the HIV-specific CD8+ T-cell compartment (specificity, ex vivo viral inhibitory capacity, and polyfunctionality) arising early after infection in a larger cohort, in comparison with the response found in viremic chronics and elite controllers, with the aim to delineate CD8+ T-cell features that best associate with disease control and that would contribute to rational vaccine design. Briefly, it was found that a higher relative proportion of Gag- than Nef-specific cells, even very early after infection, strongly associated with delayed progression during the first year postinfection, in consonance with Gag immunodominance in EC and chronically infected “viremic controllers.” The advantage of Gag-specific cells may result from their enhanced ability to mediate lysis of infected cells (evidenced by higher capacity to degranulate and to mediate viral inhibition activity in vitro) and simultaneously produce IFN-γ. Also, a direct association between the level of Gag immunodominance and plasma levels of interleukin 2 (IL-2) and macrophage inflammatory protein 1β (MIP-1β) was observed. These data underscore the importance of considering both cell specificity and quality in the design and evaluation of HIV vaccine candidates.
MATERIALS AND METHODS
Study subjects.
A total of 81 subjects participated in this study: 10 healthy HIV-seronegative donors (HD) and 71 HIV-infected patients, of whom 44 were enrolled during acute/early primary HIV infection (PHI), 16 were chronically infected patients (chronics), and 11 were elite controllers (EC) (Table 1; see also Table S1 in the supplemental material). PHI subjects were enrolled by the Grupo Argentino de Seroconversión Study Group under the following inclusion criteria (26): (i) detection of HIV RNA or p24 antigen with a simultaneous negative or indeterminate Western blot assay or (ii) positive Western blot assay with a negative test within the previous 6 months. Chronically infected patients were defined as subjects with established HIV infection older than 3 years, with detectable viral load (VL; >50 HIV RNA copies/ml plasma), and who are HAART naive, while EC were defined as subjects infected for more than 5 years with undetectable VL (<50 HIV RNA copies/ml plasma) who are HAART naïve and have no record of opportunistic infections and/or AIDS-related diseases. The study was reviewed and approved by two institutional review boards (IRB): Comité de Ética Humana, Facultad de Medicina, Universidad de Buenos Aires, and Comité de Bioética, Fundación Huésped (Buenos Aires, Argentina). Both HIV-infected participants and healthy donors provided written informed consent accepting to participate in this study.
Table 1.
Summary of clinical data corresponding to enrolled HIV+ subjects per study group
| Group (no. of subjects) | Viral loada,c |
Viral set pointd (mean log10 ± SD) | CD4+ T-cell count,b,c median no. of cells/μl (IQ) | CD4 set point,d median no. of cells/μl (IQ) | |
|---|---|---|---|---|---|
| Median RNA copies/ml (IQ) | Mean log10 ± SD | ||||
| PHI | |||||
| All (n = 44) | 98,684 (13,161–477,708) | 4.6 ± 1.2 | 4.5 ± 0.8 | 525 (320–678) | 526 (357–656) |
| PHI > 350 (n = 23) | 32,473 (9,532–116,129) | 4.2 ± 1.2 | 4.3 ± 0.6 | 633 (587–786) | 586 (491–680) |
| PHI < 350 (n = 18) | 269,532 (101,394–500,000) | 5.2 ± 1.0 | 5.0 ± 0.8 | 292 (244–375) | 281 (234–327) |
| Chronic (n = 16) | 16,682 (2,136–41,677) | 4.1 ± 0.7 | 455 (164–577) | ||
| Chronic excluding viremic controllers (n = 12) | 22,267 (13,296–44,758) | 4.4 ± 0.5 | 287 (140–544) | ||
| EC (n = 11) | <50 | <1,7 | 595 (562–817) | ||
Versant HIV-1 RNA 3.0 assay, Siemens. Lower and upper detection limits are 50 and 500,000 RNA copies/ml, respectively (1.7 and 5.7log10).
Flow cytometry double platform, FACSCanto, BD Biosciences.
For PHI subjects, data correspond to baseline samples. For chronic and elite controller subjects, data correspond to samples obtained at enrollment.
Set points were not calculated for subjects that initiated HAART during the first year postinfection.
Samples.
Blood samples were collected from study participants at enrollment and centrifuged to separate plasma, which was stored at −80°C until use. Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll-Hypaque density gradient centrifugation (Amersham, Sweden) and cryopreserved for subsequent functional assays. For PHI subjects, additional samples were obtained at 3, 6, 9, and 12 months postinfection. In the case of chronic, EC, and PHI subjects, plasma VL (branched-DNA, Versant HIV-1 RNA 3.0 assay; Siemens Healthcare) and CD4+ T-cell count (flow cytometry double platform, BD FACSCanto; BD Biosciences) were determined.
HLA typing.
Human leukocyte antigen (HLA) class I typing was performed using an in-house protocol consisting of PCR amplification, nucleotide sequencing with nested primers, and Web-based sequence interpretation (R. S. Coloccini and D. C. Monaco, unpublished data). HLA-A exons 2 and 3 were amplified in one amplicon. HLA-B exons 2 and 3 were amplified separately. The amplification of exon 3 was performed by heminested PCR, using 2 different reverse primers. Similarly, HLA-C exons 2 and 3 were amplified separately using a heminested PCR strategy (2 different forward primers) in the case of exon 2. Amplicons were directly sequenced using the BigDye Terminator sequencing kit (Amersham, Sweden) on an automatic sequencer (Applied Biosystems DNA Sequencer 3100). Nucleotide sequences were analyzed and manually adjusted using Sequencher 4.10.1 software (Gene Codes Co.). Sequence interpretation was performed using the NCBI SBT Interpretation software, available online (http://www.ncbi.nlm.nih.gov/gv/mhc/sbt.cgi?cmd=main).
Peptides.
Potential T-cell epitope (PTE) peptide panels corresponding to Nef, Gag, and Env proteins and the cytomegalovirus (CMV), Epstein-Barr virus, and influenza virus (CEF) peptide pool were obtained from the NIH AIDS Reagent Program (27, 28). Lyophilized peptides were dissolved in dimethyl sulfoxide (DMSO) at 40 μg/μl and stored at −20°C.
The PTE peptides are 15 amino acids (aa) in length and contain naturally occurring 9-aa sequences that are potential T-cell determinants embedded in the sequences of circulating HIV-1 strains worldwide. Here, the PTE peptides were grouped in 9 pools: 1× Nef (n = 127 peptides), 3× Gag (corresponding to p17 [n = 97], p24 [n = 128], and p2p7p1p6 [denoted as RG; n = 95]), and 5× Env (Gp120A1 [n = 73, spans HXB2 Env aa positions 1 to 154], Gp120A2 [n = 73, aa 157 to 284], Gp120B [n = 105, aa 287 to 511], Gp41A [n = 114, aa 513 to 689], Gp41B [n = 115, aa 689 to 842]).
ELISPOT assay.
Gamma interferon (IFN-γ)-secreting cells were evaluated using enzyme-linked immunospot (ELISPOT) assays as described previously (25). Briefly, cryopreserved PBMCs were thawed in complete RPMI medium (RPMIc; RPMI 1640 [Gibco BRL], 10% fetal bovine serum [FBS; Gibco BRL], 2 mM l-glutamine [Gibco BRL], 100 U/ml penicillin [Gibco BRL], 100 μg/ml streptomycin [Gibco BRL], 10 mM HEPES [Gibco BRL]) supplemented with 50 U/ml DNase I (Benzonase nuclease; Sigma-Aldrich) and then rested overnight in DNase-free medium at a density of 106 cells/ml. Cell viability was checked by trypan blue exclusion after thawing and overnight rest. Rested PBMCs with >95% viability were plated on sterile 96-well plates (MultiScreen IP plates; Millipore), previously coated with mouse anti-human IFN-γ monoclonal antibody (BD Biosciences) at 105 cells/well, and peptide pools were added at a final concentration of 2 μg/ml of each peptide. Final DMSO concentration was always lower than 0.7%. Negative (peptide-free medium plus 0.5% DMSO), CEF peptide pool (2 μg/ml of each peptide), and phorbol myristate acetate (PMA)-ionomycin (5 ng/ml PMA plus 500 ng/ml ionomycin; Sigma-Aldrich) controls were assayed for each patient. Plates were developed using biotinylated anti-human IFN-γ monoclonal antibody, streptavidin-peroxidase complex, and the 3-amino-9-ethylcarbazole (AEC) substrate reagent set (BD Biosciences). Plates were scanned on an ImmunoSpot reader (Cellular Technology Ltd.). Specific spot count and spot size were calculated using the ImmunoSpot software. Results were expressed as spot-forming units (SFU)/106 PBMCs after subtraction of the negative-control values. Thresholds for positive responses for the test wells were defined as at least 50 SFU/106 PBMCs or as mean SFU greater than three times the mean SFU of the negative-control wells, whichever was higher.
Virus inhibitory activity.
Ex vivo CD8+ T-cell capacity to inhibit viral replication in primary autologous CD4+ T cells was assayed following the protocol published by Sáez-Cirión et al. (29) with minor modifications: bulk CD4+ T cells were isolated from thawed and overnight rested PBMCs by positive selection with anti-CD4 antibody-coated magnetic beads (BD Biosciences) and kept for 3 days in RPMIc supplemented with 1 μg/ml phytohemagglutinin (PHA; Sigma-Aldrich). CD4+ T-cell-depleted PBMCs were then subjected to CD8+ T-cell-positive selection with anti-CD8 antibody-coated magnetic beads (BD Biosciences). Sorted CD8+ cells were cultured for 3 days in RPMIc without PHA.
At day three postisolation, CD4+ and CD8+ cells were washed, counted, and plated in U-bottom 96-well plates at a 1:1 ratio. Cells were then infected with X4-tropic (HIVLAI) or R5-tropic (HIVBAL) HIV-1 laboratory strains at a multiplicity of infection (MOI) of 0.001. In order to improve infection efficiency, plates were first centrifuged at 1,200 × g for 1 h at 22°C (spinoculation), and then adsorption was let to proceed for an extra hour at 37°C in a humidified CO2 incubator. After infection, cells were washed twice and resuspended in RPMIc medium containing IL-2 at a final concentration of 10 U/ml. At day 3 postinfection, the cocultures were fed by replacing one-half of the culture supernatant with fresh medium. At day 7 postinfection, supernatants were removed and p24 antigen was quantified by enzyme-linked immunosorbent assay (ELISA; Vironostika HIV-1 antigen kit; bioMérieux, France). Uninfected cocultures were included as negative controls, and infected CD4+ T cells without added effectors (CD8+ T cells) served as 100% infectivity controls. All conditions were assayed in triplicate. CD8+ T-cell anti-HIV suppressive capacity was calculated as the log10 of the percentage of p24 antigen lost when CD8+ T cells were present in the culture.
Intracellular cytokine staining (ICS).
Production of cytokines and degranulation of HIV-specific cells were measured upon stimulation by flow cytometry following the protocol published before (25) with the following modification: thawed and overnight rested PBMCs were dispensed in U-bottom 96-well plates (5 × 105 cells/well) in duplicate wells. Cell viability was checked before and after overnight rest by trypan blue exclusion (only samples with >95% viability after the overnight rest were used for the assays). Costimulatory antibodies (anti-CD28 and anti-CD49d antibodies, 1 μg/ml; BD Biosciences), monensin (Golgistop, 0.7 μl/ml; BD Biosciences), brefeldin A (10 μg/ml; BD Biosciences), and the corresponding peptide pool (2 μg/ml) were added. An unstimulated (peptide-free medium plus 0.5% DMSO and costimulatory antibodies) and two positive controls (2 μg/ml CEF peptide pool and PMA-ionomycin) were included in each assay. A mixture of anti-CD107A-fluorescein isothiocyanate (FITC) and anti-CD107B-FITC antibodies (BD Biosciences) was added to one of the replicates. Cells were incubated for 6 h at 37°C, washed, and stained for 30 min at 4°C with LIVE/DEAD Fixable NEAR-IR (Invitrogen), in order to exclude dead cells, and with anti-CD3-PerCP and anti-CD8-APC surface antibodies (BD Biosciences). Then, cells were permeabilized and fixed using the Cytofix/Cytoperm kit (BD Biosciences). After the permeabilization/fixation step, one of the replicates was stained using anti-IL-2–phycoerythrin (PE), anti-tumor necrosis factor alpha (TNF-α)-FITC, and anti-IFN-γ–PECy7 antibodies (BD Biosciences), while replicates already containing the anti-CD107A/B mix were stained with anti-IL-2–PE and anti-IFN-γ–PECy7. Cells were then washed and stored until data acquisition in a 2-laser, 6-color BD FACSCanto flow cytometer. Data acquisition and analysis were performed using the BD FACSDiva software. Instrument settings and fluorescence compensation were performed for each day of testing using unstained and single-stained samples. Isotype controls (consisting of stimulated cells stained with conjugated antibodies to surface molecules and isotype controls corresponding to intracellular markers) were performed for each patient in order to accurately set negative populations. First, a plot of forward scatter area (FSC-A) versus height (FSC-H) was constructed to remove doublets. Then, gating was performed on small lymphocytes in a plot of FSC versus side scatter (SSC). At least 80,000 events were acquired in the lymphocyte gate. Dead cells were then excluded on the bases of LIVE/DEAD fluorescence. Subsequently, CD3+ CD8+ cells were gated in a CD3-versus-CD8 dot plot. Following identification of these cells, a gate was made for each function studied (see Fig. 5A). To study 2-function and 3-function positive populations, “derived gate tools” available in the FACSDiva software was used. For this purpose, intersections of two and three gates were created, respectively. Once the percentages of events were determined for each derived gate, the value of triple-positive events was subtracted from those of double-positive events and, in turn, double- and triple-positive events were subtracted from the total events positive for a given function to calculate monofunctional cells. Samples with a nonspecific background higher than 0.05% for any function were retested using a new vial of frozen cells. Data presented correspond to background-subtracted results using the CD28/CD49d-only stimulation. This was performed on a cytokine-subset-by-cytokine-subset basis, i.e., subtracting the result from the CD28/CD49d-only condition for a given cytokine subset to the same subset of a peptide-stimulated condition. Two standard deviations (SDs) above background was set as the threshold for determining positive responses. Values below this threshold were set at 0.
Fig 5.

(A) Gating strategy used for the identification of degranulating and cytokine-secreting HIV-specific T cells. Illustration data were derived from one representative subject, stimulated with an HIV peptide pool. Initial gating was performed on a plot of forward scatter area (FSC-A) versus height (FSC-H) to remove doublets. Then, gating was performed on small lymphocytes in a plot of forward scatter (FSC) versus side scatter (SSC). Dead cells were then excluded on the bases of LIVE/DEAD fluorescence. Subsequently, CD3+CD8+ cells were gated in a CD3-versus-CD8 dot plot. Following identification of these cells, a gate was made for each function studied: degranulation (evidenced as CD107A/B surface expression) and production of IFN-γ, IL-2, and TNF-α. (B) Relative contribution to the total specific CD8+ response made by each function or function combination in PHI (baseline sample), chronic, and EC subjects. Symbols represent the percentage out of the total specific CD8+ T cells expressing the particular combination of functions indicated on the axis, for each subject. Responses shown correspond to background-subtracted results using the CD28/49d control. Values corresponding to bi- and monofunctional cells were corrected by subtracting the values corresponding to triple-positive events and double- and triple-positive events, respectively. Horizontal lines within boxes represent the median values. (C) Summary of functional profile in chronic, EC, and PHI subjects (baseline and year samples in the case of the PHI group). The distinct cellular subsets shown in panel B were grouped by number of functions, so each section of the bars represent, the mean proportion of specific CD8+ T cells expressing one (white), two (gray), or three (black) functions, independently of any particular function, matching the color code used in panel B. (D) Results corresponding to PHI subjects, using either Gag or Nef peptide pools as stimuli. *, P < 0.05; **, P < 0.01; ***, P < 0.005.
For certain cell functions, relative mean fluorescence intensity (rMFI) was calculated as the ratio between MFI corresponding to specific versus total CD8+ T cells for a given channel.
Quantification of plasma soluble factors.
Simultaneous determination of the following 39 cytokines and chemokines was performed in plasma samples from a subset of 28 PHI subjects (at baseline time point only) using Luminex technology (MILLIPLEX MAP Human Cytokine/Chemokine; Millipore): epidermal growth factor (EGF), eotaxin, FGF-2, Flt-3 ligand, fractalkine, granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), GRO, IFN-α2, IFN-γ, IL-1α, IL-1β, IL-1rα, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12 (p40), IL-12 (p70), IL-13, IL-15, IL-17, IP-10, monocyte chemoattractant protein 1 (MCP-1), MCP-3, MDC (CCL22), MIP-1α, MIP-1β, sCD40L, sIL-2Rα, transforming growth factor alpha (TGF-α), TNF-α, TNF-β, VEGF. Samples were processed and analyzed as described by Giavedoni (30).
Data analysis.
For PHI subjects, viral and CD4 set points were calculated as the geometric mean of the determinations obtained between 6 and 12 months after the presumed date of infection. Set points were not calculated for those subjects who started HAART during the first 12 months from infection or if no stable set point was reached during that period.
Statistical analyses were performed using GraphPad Prism 5 (GraphPad Software). All data except log10 VL and breadth of response were analyzed using nonparametric statistics. Two-tailed Wilcoxon and Mann-Whitney tests were used to compare intra- and intergroup variables, respectively. Correlations were determined using Spearman's rank test. All tests were considered significant if the P value obtained was less than 0.05. For correlations between plasma cytokine levels and immune parameters, P values were adjusted for multiple comparisons using a false discovery rate (FDR) procedure, according to the Benjamini & Hochberg method, with R Project software version 2.10.0. Adjusted P values were considered significant when less than 0.1.
RESULTS
Study subject descriptions.
Three groups of HIV-infected subjects were enrolled as follows in order to attempt to dissect immune mechanisms associated with control over disease progression during acute/early HIV infection: 44 subjects recruited during HIV seroconversion and/or within 6 months from presumed date of infection (PHI group), 16 chronically infected subjects (chronics), and 11 subjects defined as elite controllers (EC) according to the criteria defined in Materials and Methods. Detailed descriptions of the HIV-infected participants are shown in Table S1 in the supplemental material. Baseline samples for most PHI subjects were obtained during Fiebig stages V and VI (31). Within this group, median baseline VL was 98,684 RNA copies/ml (25 to 75% interquartile range [IQ], 13,161 to 477,708 copies/ml). As expected, it was statistically higher than chronics' VL (median, 16,682 RNA copies/ml [IQ, 2,136 to 41,677], P = 0.012]) and, of course, EC's VL (Table 1 and Fig. 1). Median baseline CD4+ T-cell count for PHI (525 cells/μl [IQ, 320 to 678]) did not differ significantly from the median CD4+ T-cell count of chronics (455 cells/μl [IQ, 164 to 577]) or EC (595 cells/μl [IQ, 562 to 817]), but a statistically significant difference was found between chronics and EC (P = 0.018) (Fig. 1). For those PHI subjects who remained HAART naive during the first year postinfection, viral and CD4+ T-cell set points were calculated (Table 1 and Fig. 1; see Table S1 in the supplemental material). These parameters did not differ significantly from chronics' VL and CD4+ T-cell count. Of note, the group of chronically infected subjects enrolled for this work is very heterogeneous and includes four subjects that could be classified as what some authors refer to as viremic controllers, i.e., individuals able to spontaneously control viral load below 2,000 RNA copies/ml (32) (C03, C05, C07, C08; Fig. 1; see also Table S1 in the supplemental material). After removing these subjects from the analysis, median VL for the chronic group was 22,267 RNA copies/ml (IQ, 13,296 to 44,758), and median CD4+ T-cell count was 287 cells/μl (IQ, 140 to 544) (Table 1).
Fig 1.
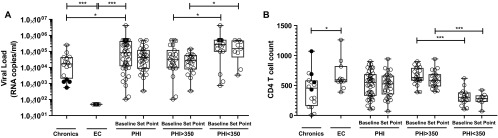
Viral load (A) and CD4+ T-cell count (B) of enrolled HIV+ subjects per study group. For PHI subjects, values corresponding to both baseline and set point are shown. Also, PHI subjects are shown as a whole group (PHI) and split into PHI > 350 and PHI < 350 subgroups whether their CD4+ T-cell count dropped below 350 cells/μl at any time during the first year postinfection or not. Within the chronic group, black dots correspond to viremic controllers (see Table S1 in the supplemental material and the text). Horizontal lines stand for median values. P values were calculated using Mann-Whitney test. VEGF, vascular endothelial growth factor; MDC, macrophage-derived chemokine; GRO, growth-related protein. Asterisks denote different P values: *, P < 0.05; ***, P < 0.005.
PHI subjects were further divided into two subgroups whether their CD4+ T-cell count dropped below 350 cells/μl at any time during the first year postinfection or not (PHI < 350 and PHI > 350). By doing this, we were aimed to differentiate subjects with more rapid or aggressive progression of early infection (PHI < 350 group) and to investigate the association of this pattern with the immune parameters analyzed in this work. The 350-cells/μl endpoint was chosen based on the national and international recommendations for initiation of HAART current by the year 2010, when most of these individuals were already enrolled (26). The PHI < 350 and PHI > 350 groups differed significantly in both baseline and set point VLs and CD4 T-cell counts (Table 1, Fig. 1).
Distribution of HLA alleles in enrolled subjects as a whole reflected the frequency distribution described for Argentina's urban population (www.allelefrequencies.net). To exclude the possibility of enrichment of particular HLA alleles associated with faster (A01, A68, B35) or slower (B27, B51, B57) HIV disease course, the frequency of these alleles was studied per study group. No significant differences in the frequency of A01, A68, or B35 alleles were found among groups, even for the EC group, where these alleles were found in 4 out of 11 subjects (36% versus 41% of chronics and 47% of PHI subjects) (see Table S1 in the supplemental material). On the other hand, “protective alleles” B27, B51, or B57 were present in one subject from the PHI < 350 group, three subjects from the PHI > 350 group, one PHI subject with undetermined progression status, three chronically infected subject, and only three EC. So, the different disease progression rates among groups cannot be explained merely by differences in the genetic background, at least based on the HLA-I locus level.
Screening of HIV-specific T-cell responses by ELISPOT revealed differences between progressive and nonprogressive infection regarding magnitude, preferred targets of specific response, and quality.
To evaluate how the central targets, magnitude, and quality (in terms of breadth and capacity to respond upon stimulation) of the HIV-specific cellular immune response during the acute/early phase of infection impact early disease progression, HIV-specific IFN-γ-secreting cells were screened by ELISPOT in PHI subjects using samples obtained at baseline (defined as the first sample obtained after enrollment according to the criteria defined in Materials and Methods). PTE peptide pools spanning the viral proteins Nef (1 pool), Gag (3 pools), and Env (5 pools) were used as stimuli. Also, these HIV-specific immune responses were screened in chronics and EC for comparison purposes.
(i) Magnitude and targets of HIV-specific cellular immune response in PHI, chronics, and EC.
The median magnitude of the specific immune response (expressed as the number of SFU/106 PBMCs) was higher in EC for all three antigens evaluated than in chronic and PHI subjects. This difference was particularly important in terms of Gag antigen: EC showed a >5-fold-higher median Gag-specific magnitude than chronic and PHI subjects (median Gag-specific magnitude = 3,570, 692, and 145 SFU/106 PBMCs for EC, chronics, and PHI subjects, respectively [EC versus chronics, P = 0.025; EC versus PHI subjects, P = 0.0003]; Fig. 2A). Intragroup analysis showed that anti-Gag magnitude was significantly higher (≈6-fold) in EC than Nef (median, 656 SFU/106 PBMCs; P = 0.005) and Env (median, 590 SFU/106 PBMCs; P = 0.007), while the anti-Nef magnitude in the PHI group (median, 308 SFU/106 PBMCs) tended to be higher than anti-Gag magnitude (median, 145 SFU/106 PBMCs; P = 0.06). Also in the PHI group, both anti-Nef and anti-Gag magnitude were significantly higher than anti-Env magnitude (median, 35 SFU/106 PBMCs; P = 0.015 and P = 0.014, respectively) (Fig. 2A).
Fig 2.

ELISPOT screening of HIV-specific T-cell response magnitude, immunodominant targets, breadth, and functionality in primary HIV infection (PHI, baseline samples), chronic, and elite controller (EC) groups. (A) Magnitude of total anti-Nef, anti-Gag, and anti-Env T-cell responses, expressed as SFU/106 PBMC. (B) Contribution of each antigen relative to the total HIV response expressed as the percentage out of the total sum of the specific response (sum of the magnitude obtained for all Nef, Gag, and Env antigens). (C) Contribution of anti-Nef and anti-Gag responses (relative to the sum of the magnitude obtained for both antigens) on a subject-by-subject basis (each represented by a line) in chronic, EC, and PHI groups. “Viremic chronics” are denoted in the corresponding panel with dashed lines. (D) Gag subunits targeted per study group, expressed as percentage out of the total anti-Gag response. RG = p2p7p1p6. (E) Breadth of the response expressed as the number of peptide pools recognized out of the 9 HIV pools assayed. (F) Mean spot size obtained for both CEF and HIV peptide pools. HIV mean spot sizes represent the average mean spot sizes out of all Nef, Gag, and Env pools for which a positive response was obtained in the ELISPOT assay on a subject-by-subject basis. All data presented in the figure for PHI subjects correspond to baseline samples. PHI > 350 and PHI < 350 stand for subgroups within the PHI group in which subjects were segregated according to whether their CD4+ T-cell count dropped below 350 cells/μl at any time during the first year of infection or not. Horizontal lines within boxes represent the median values. Intra- and intergroup differences were analyzed using Wilcoxon and Mann-Whitney tests, respectively. Asterisks denote different P values: *, P < 0.05; **, P < 0.01; ***, P < 0.005.
The relative contribution of each antigen to the total response, i.e., the proportion of cells specific for each protein relative to the total anti-HIV response, is an important factor to be taken into account when evaluating the association of HIV-specific immune response with viral control (11). Thus, the proportion of specific cells against each HIV antigen in relation to the total response (sum of the T-cell responses against the individual antigens) was analyzed (Fig. 2B). Again, the contribution of the different targets differed among the different study groups: PHI subjects showed a statistically higher proportion of Nef-specific cells (median, 55%) than chronics (median, 17%; P = 0.005) and EC (median, 20%; P = 0.007) and lower proportion of anti-Gag-specific cells (PHI, 32% versus EC, 64% [P = 0.004]; PHI versus chronics, 64% [P > 0.05]). In the same line, intragroup analysis revealed that anti-Gag response (median, 64%) clearly predominated over Nef (20%)- and Env (7.4%)-specific cells in EC (Gag versus Nef, P = 0.005; Gag versus Env, P = 0.007), whereas anti-Nef cells predominated in PHI (median, 55%) over Gag (32%, P = 0.001) and over Env (6%, P < 0.001). The chronic group showed the same immunodominance pattern (Gag > Nef > Env) than EC, although the response dispersion among chronics was much higher, which reflects the heterogeneity of this group. When analyzing separately the chronic subjects referred to as viremic controllers, it could be observed that the median percentage of Gag immunodominance in this chronic subgroup was 86%, considerably higher than that of regular chronics (43%).
When a similar analysis was performed within PHI subgroups, it could be observed that the scenario is clearly different between PHI > 350 and PHI < 350 groups: while no statistically significant difference could be observed between anti-Nef and anti-Gag magnitude or proportion of total response in the PHI > 350 group, anti-Nef cells clearly dominated the anti-HIV response in the PHI < 350 group (median magnitude, 500 SFU/106 PBMCs; median proportion of total response, 62%) over Gag and Env both in terms of magnitude (80 SFU/106 PBMCs [P = 0.0125] and 77 SFU/106 PBMCs [P = 0.001], respectively) and proportion (32% [P = 0.01] and 7% [P = 0.003], respectively) (Fig. 2A and B).
Given the significant role that anti-Nef and anti-Gag responses seem to play in defining the different rates of progression among groups, the contribution of anti-Nef and anti-Gag T-cell responses (relative to the sum of the magnitude obtained for both antigens) was analyzed per individuals included in the different groups (Fig. 2C). Anti-Gag responses clearly dominated over Nef in all EC (P = 0.002). On the opposite sense, anti-Nef responses dominated the anti-HIV response in the group of rapid progressors, PHI < 350 (P = 0.008). In chronics and the PHI > 350 group, an intermediate situation was observed: Gag dominated over Nef only in a subset of individuals, while the rest showed either Nef immunodominance or a balanced situation between both antigens. Among the chronic group, those with clear Gag immunodominance included those previously referred to as “viremic controllers” (C03, C05, C07, and C08).
It must be noted that the immunodominance hierarchy obtained with baseline samples was maintained when using samples obtained 1 year postinfection (data not shown), thus similar tendencies were observed when analysis was performed with the 12-month samples.
Then, a more detailed analysis of the Gag polyprotein subunits targeted by cells from the recruited individuals was performed. In all groups (chronic, EC, and PHI subjects as a whole), the immunodominance pattern (according to medians) was as follows: p24 > p17 > RG (Fig. 2D). Only in EC, the anti-Gag response was focused into p24 protein in a significantly higher proportion over both p17 and RG (P = 0.047 and 0.032, respectively). In chronics and PHI subjects, the proportion of anti-p24 response was significantly higher than the anti-RG response (P = 0.0012 and P = 0.0021, respectively) and also tended to be higher than the anti-p17 response, although the difference was not statistically significant in either group. When PHI subjects were split into PHI > 350 and PHI < 350, it was observed that the PHI > 350 group maintained the immunodominance pattern observed before, whereas a shift to p17 > p24 > RG was observed in the PHI < 350 group.
(ii) Quality (in terms of breadth and intensity of IFN-γ production upon stimulation) of the HIV-specific cell immune response in PHI, chronic, and EC groups.
Afterward, the breadth of the response was calculated for each individual as the number of peptide pools recognized (out of the 9 pools used to screen HIV-specific response) (Fig. 2E). This analysis revealed that EC were able to recognize a higher number of pools than chronics and PHI (P = 0.05 and 0.002, respectively). Even more, EC were able to recognize more Gag-specific pools (n = 3 pools) than chronics and the PHI group (not shown). No statistically significant difference was observed between the PHI < 350 and PHI > 350 groups.
Finally, the mean spot size obtained for both CEF and HIV peptide pools was recorded as the measure of the amount of IFN-γ produced by the individual specific T cells (which in turn correlated with the T-cell functional avidity) (33) (Fig. 2F). No intrasubject difference was observed among the different HIV-specific pools (Nef, Gag, and Env) (data not shown). Hence, for the purpose of this analysis, the mean spot size for all HIV antigens was averaged, taking into account only those pools for which a positive response was obtained in the ELISPOT assay on a subject-by-subject basis. Regarding the control CEF-specific spots, PHI subjects showed the smallest size being statistically different from EC (EC CEF-specific spots were 1.5 times larger than in the PHI group, P = 0.029), which in turn showed the largest CEF-specific spots, even slightly larger than healthy donors (P > 0.05). This difference in the PHI group was driven by PHI < 350 subjects who even differed substantially from their countergroup, PHI > 350 (spots from the PHI > 350 group doubled, on average, those of the PHI < 350 group). A similar scenario was observed for HIV-specific spots but, in this case, differences among groups were stronger. EC showed significantly larger spots than the chronic and PHI groups (1.5× [P = 0.004] and 1.7 × [P = 0.002], respectively). Within the PHI group, a significant difference could be observed between the PHI > 350 and PHI < 350 groups, showing the former group had larger HIV-specific spots (1.87×, P = 0.022). Interestingly, when CEF- versus HIV-specific spot size was analyzed intragroup, the HIV spots were smaller than CEF spots. In fact, the spot size difference between the two antigens reached statistical significance in chronics (P = 0.036), PHI as a whole (P = 0.011), and the PHI < 350 group (P = 0.018). Although the trend was also observed in EC and the PHI > 350 group, the breach between both antigens was less important, especially in EC.
(iii) In acute/early infected subjects (PHI group), baseline relative immunodominance of anti-Gag cellular response correlates with both baseline and set point CD4+ T-cell counts.
In Fig. 2C, it can be observed that the EC and PHI < 350 group (opposed groups in terms of disease progression) showed exactly opposite immunodominance patterns regarding Nef and Gag proteins. Based on this observation, it was hypothesized that in the much heterogenous chronic and PHI > 350 groups, Gag immunodominance correlates with slower disease progression. To test this hypothesis, correlation analyses were performed between the percentages of anti-Gag responses and clinical data of enrolled subjects. It was found that baseline relative Gag immunodominance in PHI subjects (baseline Gag%) significantly correlated with baseline CD4+ T-cell counts (Spearman's r = 0.3836, P = 0.01; Fig. 3A). More interestingly, baseline Gag% also significantly correlated with set point CD4+ T-cell count (Spearman's r = 0.3630, P = 0.04; Fig. 3B), indicating that Gag immunodominance is an important factor shaping disease progression in terms of CD4+ T-cell loss. A similar association between these parameters was found within the chronic group, although the trend did not reach statistical significance, likely due to the smaller sample size (Spearman's r = 0.4842, P = 0.09; Fig. 3C).
Fig 3.
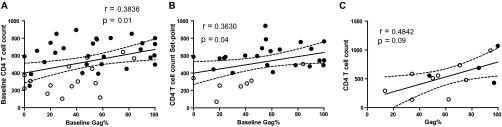
Correlation between the relative baseline immunodominance of anti-Gag response and CD4+ T-cell counts. (A) Baseline CD4+ T-cell count versus baseline percentage of anti-Gag response in PHI subjects. (B) Set point CD4+ T-cell count versus baseline percentage of anti-Gag response in PHI subjects. (C) CD4+ T-cell count versus percentage of anti-Gag response in chronics. In panels A and B, black and white dots denote PHI > 350 and PHI < 350 subjects, respectively. In panel C, black dots correspond to viremic controllers within the chronic group. All r and P values correspond to Spearman's test.
All together, data collected so far demonstrate that although Nef dominates the anti-HIV response in acute/early infection, a higher proportion of early T-cell responses targeting preferentially Gag (in particular p24) and larger spot size (indicative of improved capacity to secrete IFN-γ and higher avidity) were associated with delayed progression during the first year of infection. Also, these same factors were observed in chronically infected elite controllers (EC), indicating that these T-cell features may play an important role in protection from progression. In particular, early Gag immunodominance correlated with higher CD4+ T-cell counts, both at baseline and set point, further denoting it represents a key immune factor involved in delayed progression.
HIV-specific CD8+ T cells elicited during PHI were capable of inhibiting heterologous viral replication in vitro, and the magnitude of this activity was related to Gag immunodominance.
We next aimed to investigate whether HIV-specific CD8+ T cells arising during acute/early HIV infection could suppress the replication of lab-adapted X4 and R5 HIV strains and to which extent, in comparison with chronic infection. It was also our aim to establish a possible relationship between this viral inhibitory activity (VIA) and the viral proteins targeted by the immune response during the early stage of infection. To evaluate this, CD4+ and CD8+ cells were isolated from PBMCs obtained from seven chronic subjects (including viremic controllers C03, C07, and C08), seven EC, and 19 PHI subjects (10 from the PHI > 350 group and 9 from the PHI < 350 group). In PHI subjects, VIA was evaluated at baseline samples as well as samples collected at 1 year postinfection. CD4+ T cells were isolated, activated, infected, and then cultured either alone or in combination with autologous CD8+ T cells at a 1:1 ratio. Viral replication was assayed at day seven postinfection by p24 ELISA. As previously described (16, 19, 20, 34, 35), CD8+ T cells from EC mediated stronger VIA than cells from chronics against both R5 (Fig. 4A) and X4 (data not shown) viruses. Of note, EC equally inhibited both R5 and X4 viruses, indicating that these individuals had a broad VIA. It is worth noting here that the use of heterologous lab-adapted viral strains might lead to a VIA underestimation, and stronger differences among groups may be masked due to this reason (16). As for the PHI group, most subjects (70%) demonstrated to have CD8+ T cells capable of mediating VIA (median, 32%, in p24 reduction, which corresponds to 1.5log10 VIA) against the R5 virus at baseline, indicating that HIV-specific CD8+ T cells able to mediate VIA arise early during infection. Longitudinal analysis using samples obtained at 12 months postinfection indicated that this activity persists over time even beyond set point establishment (Fig. 4A).
Fig 4.

In vitro viral inhibitory activity (VIA) mediated by HIV-specific CD8+ T cells. VIA is expressed as the log10 of the proportion of p24 antigen lost when CD8+ T cells were present in the culture, compared to CD4+ cells alone. (A) Viral inhibitory activity found in chronic, EC, and PHI subjects, against lab-adapted R5 HIV strains; (B) viral inhibitory activity in PHI individuals, reassorted into subjects with Gag immunodominance or Nef immunodominance according to preferred (>50% of total response) viral protein targeted by the immune response at baseline sample in the ELISPOT assay; (C) correlation between the magnitude of Gag-specific response in PHI subjects at baseline sample and VIA; r and P values correspond to Spearman's test; (D) PHI subjects with substantial capacity to mediate VIA (>25%) also showed higher CD4+ T-cell set points. In panels A, B, and D, symbols show the values for each individual subject. Within the chronic group (A), viremic controllers are denoted with black dots. Horizontal lines within boxes represent the median values. Intergroup and intragroup (PHI baseline versus year) differences were analyzed using Mann-Whitney and Wilcoxon tests, respectively; *, P < 0.05.
When the PHI group was split into the PHI > 350 and PHI < 350 groups, no significant differences were observed in VIA either at baseline or 12-month samples. Given our observation obtained from the ELISPOT analysis, where Gag immunodominance during PHI was associated with preservation of the CD4+ T-cell count over the first year postinfection, we decided to reassort PHI individuals into Gag responders or Nef responders according to preferred (>50% of total response) viral protein target. This analysis revealed that CD8+ cells from PHI individuals in which Gag-specific cells dominated the early HIV-specific T-cell response had a stronger capacity to mediate VIA against the R5 virus than Nef responders, both at baseline (P = 0.043) and at the 12-month samples (P = 0.011) (Fig. 4B). This result agrees with the fact that EC consistently show the strongest VIA compared to other HIV+ groups (in this and other studies) and concomitantly show a robust anti-Gag immunodominance of the HIV-specific cellular immune response. In the same line, a statistically significant correlation was found within the PHI group between VIA and the magnitude of Gag-specific SFU (Spearman's r = 0.5885, P = 0.0343; Fig. 4C). Overall, these results suggest that the ability of HIV-specific CD8+ T cells arising early during infection to suppress viral replication in autologous CD4+ T cells might be related, among other factors, to the relative Gag immunodominance out of the total HIV-specific CD8+ T-cell response as well as the absolute number of Gag-specific CD8+ T cells. Also, it was observed that EC and PHI Gag responders more frequently showed VIA against both R5 and X4 viruses, while chronic and PHI Nef responders recognized one, other, or no viruses, suggesting that the former groups have broader activity (data not shown).
Given the consistent association of higher VIA with EC status observed in this and other studies (16, 19, 20, 34, 35), we decided to seek for any association of stronger VIA with clinical parameters related to acute/early infection. No association was observed between VIA magnitude and baseline viral load, viral set point, or baseline CD4+ T-cell count. However, it could be observed that those subjects showing substantial capacity to mediate VIA (>25%) had higher CD4+ T-cell set points (P = 0.02, Fig. 4D), suggesting that the association of Gag immunodominance with delayed progression in terms of CD4+ T-cell count preservation described earlier in the manuscript (Fig. 3) would be related to a higher capacity of Gag-specific cells to mediate VIA.
Low frequency of polyfunctional HIV-specific CD8+ T cells was detected during PHI; it slightly increased over time during the first year postinfection but did not associate with viral or CD4+ T-cell set points.
In order to further characterize HIV-specific CD8+ T cells arising early during infection, the functionality of these cells, in terms of their ability to degranulate (evidenced by CD107A/B mobilization) and to produce IFN-γ, TNF-α, and/or IL-2 upon peptide stimulation, was studied by flow cytometry. For these assays, data obtained in the ELISPOT screening was used as a starting point to define which peptide pools were used as stimuli. So for each subject, only those peptide pools for which positive responses were found by ELISPOT were used as stimuli in the ICS assay. The gating strategy used is illustrated in Fig. 5A. The proportion of cells expressing each of these functions alone or in combination was analyzed, not only in PHI but also in chronic and EC subjects, in order to help distinguish any association with disease progression.
As previously reported by our group (25), HIV-specific CD8+ T cells expressing all the functions analyzed (either alone or in combination) could be measured very early during infection. Among PHI baseline samples, mean ± SD percentages of HIV-specific CD3+ CD8+ cells for each single function were 0.16 ± 0.28 for IFN-γ, 1.13 ± 5.23 for TNF-α, 0.06 ± 0.14 for IL-2, and 0.21 ± 0.4 for CD107. Figure 5B depicts the relative contribution to the total specific CD8+ response made by each function or function combination. No particular function or function combination could be distinguished that differed substantially among PHI, EC, or chronic subjects, except for bifunctional CD107A/B+ IFN-γ+ CD8+ T cells. All EC individuals had detectable bifunctional CD107A/B+ IFN-γ+ CD8+ T cells (mean, 30% ± 11% out of the specific CD8+ T cells), differing, in this sense, significantly from chronic (13% ± 23%; P = 0.007) and PHI individuals (9% ± 14%; P = 0.0004), indicating that this sub-subset of specific CD8+ T cells could be relevant to virus control. Then, the proportion of cells expressing one, two, or three functions was studied among groups independently of any particular function. As expected, the proportion of polyfunctional cells was greater in EC than in chronic and PHI subjects (Fig. 5C). In particular, EC had a significantly higher proportion of trifunctional cells (mean, 8.1% ± 7%) and significantly lower proportion of monofunctional cells (mean, 46% ± 19%) than chronic subjects (mean, 2.7% ± 9% [P = 0.013] and 68% ± 27% [P = 0.033], respectively). As mentioned above, bifunctional and trifunctional cells could be measured very early in PHI, together accounting for 25.5% of the HIV-specific CD8+ pool in these subjects. Moreover, the proportion of these cells seemed to slightly increase over time to the 12-month sample (32.3%), although the difference between the baseline and year samples did not differ significantly. Within the PHI group, no significant differences were observed in any particular function or function combination when individuals were segregated into PHI > 350 and PHI < 350 groups. Similarly, no difference was observed between both groups in the proportion of mono-, bi-, or trifunctional cells. Also, no significant correlation was observed for any function, combination, or proportion either with baseline or set point VL or CD4+ T-cell count (data not shown).
It was then hypothesized that the stronger CD8+ T-cell VIA found in PHI subjects in whom Gag dominated the cellular immune response, as well as the slower progression observed within this subgroup, could be related to higher polyfunctionality of Gag-specific cells. Consequently, the proportion of Gag- and Nef-specific CD8+ T cells expressing one, two, or three functions was compared within the PHI group at baseline and 12-month samples (Fig. 5D). No differences were observed for both antigens, indicating that polyfunctionality was not related to specificity. Moreover, both Gag- and Nef-specific responses showed a nonstatistically significant increase in polyfunctionality over time.
Overall, these results suggest that although a higher proportion of polyfunctional CD8+ T cells is associated to EC status and even when these cells could be measured very early in HIV infection, they would not be associated with better or worse resolution of acute phase. Also, the benefits of Gag-specific cells would not be related to polyfunctionality in the terms analyzed here.
A higher proportion of HIV-specific CD8+ T cells able to degranulate and secrete IFN-γ was associated with improved capacity to suppress viral replication during PHI.
As mentioned before, out of the multiple CD8+ T-cell effector functions studied by flow cytometry, the combined expression of CD107A/B and IFN-γ was significantly associated with EC status (Fig. 5B). Given this, it was reasoned that the particular combination of these two functions (instead of polyfunctionality as a whole) would be associated with improved resolution of acute infection. Thus, associations among percentages of specific CD107A/B+ IFN-γ+ CD8+ T cells and virological and immunological parameters (all corresponding to baseline samples) were analyzed. It was found that, within PHI subjects, those showing substantial CD8+ T-cell VIA (>25%) had higher numbers of CD107A/B+ IFN-γ+ CD8+ T cells than those having weak (<25%) VIA (P = 0.025). Even more, the percentage of CD107A/B+ IFN-γ+ CD8+ T cells in the PHI VIA > 25% group was comparable to that of EC (Fig. 6A and B). Moreover, a significant correlation between the percentage of specific CD107A/B+ IFN-γ+ CD8+ T cells and the CD8 T-cell VIA was found for PHI subjects (Spearman's r = 0.6383, P = 0.01; Fig. 6C).
Fig 6.
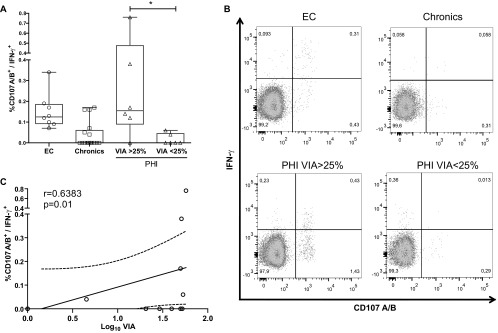
Association between the CD8+ T-cell capacity to suppress HIV replication in vitro and the frequency of HIV-specific T cells able to degranulate and secrete IFN-γ+ upon stimulation. (A) Frequency of HIV-specific CD107A/B+ IFN-γ+ CD8+ T cells in EC, chronic, and PHI subjects (segregated according to CD8+ T-cell VIA magnitude). *, P < 0.05 Mann-Whitney test. (B) Representative IFN-γ+ versus CD107A/B+ dot plots (gated on CD3+CD8+ events) obtained for subjects of each group. (C) Correlation between the frequency of HIV-specific CD107A/B+ IFN-γ+ CD8+ T cells and VIA in PHI subjects at baseline sample. r and P values correspond to Spearman's test.
These observations indicate that the capacity to degranulate and express IFN-γ might play a relevant role in the ability of specific cells to mediate VIA (i.e., to suppress viral replication in vitro), which in turn associated both with the preservation of the CD4+ T-cell subset during acute/early infection and the status of EC. On these bases, the subpopulation of specific CD107A/B+ CD8+ T cells was subjected to a deeper analysis. This analysis revealed that, out of the total CD107A/B+ HIV-specific CD8+ cells, most of them coexpressed IFN-γ and/or IL-2 in the EC cohort, i.e., most degranulating HIV-specific CD8+ T cells from EC were bi- or trifunctional. Of note, all EC had measurable HIV-specific CD107A/B+ IFN-γ+ CD8+ T cells, and around 60% of subjects had measurable HIV-specific CD107A/B+ IL-2+ CD8+ T cells. On the contrary, only a minor portion of chronic and PHI subjects exhibited polyfunctional HIV-specific CD107A/B+ CD8+ T cells. When a similar analysis was performed but considering response specificity, it could be observed that Gag-specific CD107A/B+ CD8+ T cells showed higher levels of polyfunctionality than Nef-specific cells in chronics, even comparable to Gag-specific cells from EC. However, no difference was observed between Nef- and Gag-specific CD8+ T cells in terms of polyfunctionality associated to degranulation capacity in the PHI group, either at baseline or 12-month samples (not shown).
Finally, the relative mean fluorescence intensity (rMFI) of IFN-γ and CD107A/B was studied in specific CD8+ T cells as an indicator of cell function strength (Fig. 7). IFN-γ rMFI analysis (Fig. 7A) produced results that paralleled those obtained after the examination of spot sizes in the ELISPOT assays. CEF-specific cells showed 1.7-fold-higher (1.7×) IFN-γ rMFI than HIV-specific cells in the chronic (P = 0.03) and EC (P > 0.05) groups but not in the PHI group. As for CD107 expression, no difference was observed among groups regarding CEF-specific cells (Fig. 7B). When CD107A/B rMFI was compared intragroup, it was found that Gag-specific cells showed higher CD107A/B rMFI than Nef-specific cells both in chronic (1.7×, P > 0.05) and PHI subjects (1.4×, P = 0.033). Even more, CD107A/B rMFI for Gag-specific cells was comparable to that of CEF-specific cells in all three groups of subjects analyzed. When the same analysis was performed but gated on double-positive CD107A/B+ IFN-γ+ CD8+ T cells, it could be observed that rMFI was higher for each function, as already reported for polyfunctional cells (36), and the same trends shown in Fig. 7 were maintained. Even more, the differences on CD107A/B expression between Nef- versus Gag-specific cells became more pronounced. However, this observation was derived from only a minor subset of patients and a small number of events (especially for chronic and PHI subjects) in order to draw definite statements.
Fig 7.
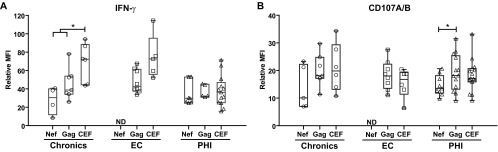
Relative mean fluorescence intensity (rMFI) of IFN-γ (A) and CD107A/B (B) in Nef-, Gag-, and CEF-specific CD8+ T cells, calculated as the ratio between MFI corresponding to specific versus total CD8+ T cells for a given channel, in chronic, EC, and PHI subjects. Symbols show the values for each individual subject. Horizontal lines within boxes represent the median values. *, P < 0.05; ND, not determined.
In summary, EC showed a higher number of degranulating HIV-specific CD8+ cells than chronic and PHI subjects. Even more, in these subjects, this activity was more frequently accompanied by the capacity to secrete cytokines (IFN-γ and/or IL-2), while in the chronic and PHI groups, these cells were mostly monofunctional. Within PHI, a direct correlation between the percentage of HIV-specific CD107A/B+ IFN-γ+ CD8+ T cells and the magnitude of CD8+ T-cell VIA was found. Finally, Gag specificity was associated with a stronger ability to degranulate (evidenced by higher CD107 rMFI), which seems consistent with these cells having a stronger capacity to mediate viral inhibition.
Plasma levels of MIP-1β and IL-2 are associated with Gag immunodominance.
Cytokines play a key role in many infectious diseases, both shaping the immune response mounted against pathogens and contributing to pathogenesis. Several reports have established that plasma cytokine levels measured during acute HIV infection may predict subsequent disease progression (37–39). In this scenario, we sought to analyze the relationship between plasma cytokine levels during acute/early HIV infection and the Gag or Nef immunodominance hierarchy obtained also at PHI baseline samples. This was performed with the aim of determining if there existed an association between both soluble and cellular immune signatures associated with protection from disease progression and following the hypothesis that if two immune parameters are truly determinants of protective immunity, then a direct association between them should be observed. It was found that the percentage of Gag-specific cells out of the total HIV-specific cells (baseline Gag%) significantly correlated in a direct fashion with plasma levels of MIP-1β and IL-2, among PHI subjects at baseline samples (Fig. 8A and B). Although the correlation between plasma MIP-1β and baseline Gag% was not significant after the adjusted analysis, the clear trend indicating a direct association between both parameters is still worth noting. Conversely, the proportion of Nef-specific cells inversely correlated with plasma MIP-1β (Fig. 8C). These findings are of particular importance, since these molecules are produced by CD8+ T cells upon stimulation and the percentage of cells expressing IL-2 and MIP-1β either alone or in combination with other functions were previously associated with virus control (6, 17, 18, 20, 40). Moreover, soluble MIP-1β was also reported to mediate viral inhibition in vitro (41). The fact that there exists an association between important soluble antiviral mediators such as IL-2 and MIP-1β and Gag immunodominance is in line with the evidence indicating that early relative Gag immunodominance is somehow related to the generation of a robust, efficient, and multifaceted antiviral immune response, thus reinforcing the notion that it is one key determinant of protective immunity contributing to slower disease progression.
Fig 8.
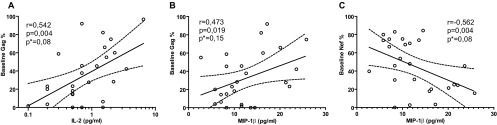
Correlation between anti-Gag and anti-Nef cellular immune response immunodominance hierarchy and plasma cytokine/chemokine levels in PHI subjects at baseline samples. Significant correlations were found between the relative immunodominance of Gag (A and B) and Nef (C) with plasma IL-2 (A) and MIP-1β (B and C) levels. r and P values correspond to Spearman's test. *, P values adjusted by the Benjamini and Hochberg method for false discovery rate (FDR) procedure.
DISCUSSION
Data accumulated over the past years, based on both experimental infections in nonhuman primate models and natural infections in humans, have established that the HIV-specific CD8+ T-cell response plays a critical role on viral control (reviewed in references 2 and 3). This is particularly evident during acute infection, where the up-slope of arising HIV-specific CD8+ T cells is temporarily associated with the decline in the initial peak viremia (4). For this reason, efforts have been made to understand which particular functions and/or phenotypes, out of the global CD8+ T-cell subset, best correlate with control of viral replication (3). Moreover, it is thought that this information will be instrumental for developing and enhancing immunization strategies as well as for defining immune correlates of protection, which are currently lacking, in order to evaluate the performance of vaccine candidates. In line with this, cohort studies addressing the association of particular features of CD8+ T-cell responses arising during acute/early HIV infection with potential markers associated with disease progression are fundamental. To date, most of these studies come from cohorts settled in Europe, North America, or Africa, and scarce information exists on this issue from other settings, such as South America. Here, we report the study of multiple functional aspects of the HIV-specific CD8+ T-cell compartment arising early after infection in a subset of subjects from a well-characterized cohort of Argentinean seroconverters, in comparison to that found in, also local, viremic chronics and elite controllers. Epidemiological, clinical, immunological, and virological characteristics of the patients enrolled in the cohort are described elsewhere (26). Aiming at delineating CD8+ T-cell features that best associate with disease progression in our population, it was found that (i) there exist early differences regarding the immunodominant viral targets of the specific T-cell response between subjects with different rates of progression to disease (in terms of CD4+ T cell loss) during the first year of infection (Fig. 2 and 3), (ii) polyfunctional HIV-specific CD8+ T cells could be detected during PHI but their frequency was not associated with virus control or protection from progression (Fig. 5), (iii) CD8+ T cells capable of mediating viral inhibitory activity (VIA) in vitro could also be detected during PHI (Fig. 4), and (iv) the improved capacity to mediate VIA during PHI was associated with a higher CD4+ T-cell set point and was related to Gag immunodominance and a higher proportion of HIV-specific CD8+ T cells able to degranulate and secrete IFN-γ (Fig. 4 and 6). The main contribution of this study relies on the correlation between the HIV-specific CD8+ T-cell functional properties during acute/early infection and the clinical outcomes during the first year postinfection. On the other hand, to our knowledge, this is the first report to perform an immunological characterization of the T-cell responses in a cohort of acute/early HIV-infected subjects from South America.
Clear differences in the viral proteins that are targeted by the HIV-specific cellular response have been described between the acute/early and chronic phases of infection: while Nef-specific cells dominate the early antiviral response (42), this response afterward broadens toward epitopes contained within other viral proteins, such as Gag, Env, and Vpr (11–15). Many of these studies have also established that Gag-specific CD8+ T-cell responses are associated with low viremia in chronic infection. Other reports indicate that Gag is the immunodominant target in elite controllers (43), both in blood and mucosal tissues (44), providing further support to the important role of Gag-specific cells in restricting viral replication. Possible mechanisms to explain this better ability to control viral replication include ability to kill very recently infected cells (even before viral genome integration to host genome) (45, 46), Gag fitness constraints to escape immune pressure (47, 48), and higher capacity to mediate antiviral activity, which will be discussed in the following paragraphs. In line with these findings, we found that Gag-specific cells (particularly p24-specific cells) dominated the HIV-specific response in EC, both in terms of magnitude and hierarchy (Fig. 2A and B). Also, Gag immunodominance was observed in a subgroup of chronically infected subjects referred to as “viremic controllers.” On the contrary, Nef-specific cells dominated the response in PHI subjects. However, when this group was split into PHI > 350 and PHI < 350 groups, it was observed that in the group with more rapid progression (PHI < 350), Nef clearly dominated the anti-HIV response (Fig. 2A to C). More importantly, significant correlations between baseline Gag immunodominance and both baseline and set point CD4+ T-cell counts were observed in the PHI group (Fig. 3). In line with this result, other authors described a direct correlation between the magnitude of the anti-Nef CD8+ T-cell response and viral load in a cohort of HIV subtype C acutely infected subjects (49). Overall, these results argue in favor of that an earlier and higher contribution of Gag-specific cells to the hierarchy of the total anti-HIV response at very early time points postinfection contributes to a slower disease progression.
The ability of CD8+ T cells to kill virus-infected cells may rely in their specificity, phenotype, and/or functionality and could even be mediated by distinct mechanisms (mediated by soluble factors or cell-to-cell contact). The evaluation of the ex vivo virus inhibitory activity (VIA) results in an overall measurement of the total CD8+ T-cell antiviral potency. Previous reports have used this assay to evaluate VIA mediated by CD8+ T cells obtained from elite controllers, treated and untreated chronically infected individuals (16, 19, 20, 34, 35), as well as human and simian vaccinees (20, 50). In these studies, it was demonstrated that VIA correlates with the magnitude of the Gag-specific CD8+ T-cell response (16, 35), that the expression of “protective” HLA-I alleles is not a required condition (20, 35), and that it is associated with higher frequencies of degranulating (as measured by CD107A/B expression) specific CD8+ T cells, usually accompanied by other functions, such as secretion of IFN-γ and MIP-1β (20, 35, 50). In agreement with this background, we found that CD8+ T cells from EC mediated stronger and broader VIA than those cells from untreated chronic subjects. Additionally, we also demonstrate in this work that VIA can be measured very early after infection in most acutely infected subjects (although to a lower magnitude than EC) and that this activity persists over time (Fig. 4). To our knowledge, there exists only one very recent report studying VIA in acute HIV infection (41). In that work, Freel and collaborators also demonstrate (i) that VIA magnitude correlates with the percentage of HIV-specific CD8+ T cells expressing CD107A, MIP-1β, and IFN-γ and with the secretion of MIP-1α, MIP-1β, IFN-γ, IP-10, and IL-1α after cell stimulation with peptide antigens and (ii) that early escape from CD8+ T-cell-mediated VIA occurs during PHI. In our work, further insights are provided into the role of CD8+ T-cell-mediated VIA during PHI. Aiming at delineating the specificity and functionality of VIA-mediating cells during PHI, it could be established that CD8+ cells from PHI individuals in whom Gag-specific cells dominated the early HIV-specific T-cell response had stronger capacity to mediate VIA than Nef responders, and a statistically significant direct correlation was found within the PHI group between VIA and the magnitude of Gag-specific cellular immune response (Fig. 4). This is in line with results obtained for EC and chronic subjects with broad anti-Gag responses (16, 35) and could provide an additional mechanistic explanation for the pivotal role of Gag-specific cells in delayed disease progression. Although Freel et al. (41) found that Nef-specific cells could mediate VIA to the same extent as Gag-specific cells in acutely infected subjects, no association between these activities and clinical parameters was studied by these authors, as it is the case of the present work. Also, it is important to denote that both studies differ in definition of the specificity of VIA-mediating cells, which may account for the different results obtained. Regarding the functionality of VIA-mediating cells, we found that, within PHI subjects, those showing substantial CD8+ T-cell VIA (>25%) had a higher frequency of CD107A/B+ IFN-γ+ CD8+ T cells (even comparable to EC) than those having weaker VIA. Moreover, a significant correlation between the percentage of specific CD107A/B+ IFN-γ+ CD8+ T cells and the CD8+ T-cell VIA was found for PHI subjects (Fig. 6). In regard to the expression of CD107A/B, this result is in line with previous data obtained for EC, chronic, acute/early infected subjects and simian vaccinees (35, 41, 50). However, in our study, the simultaneous coexpression of both markers (CD107A/B and IFN-γ) is necessary to maintain the association. Other groups reported that MIP-1β secretion is also associated with VIA both in chronic and acute infection (20, 41). Because of technical constrains, we could not include the evaluation of this cytokine in our multicolor flow cytometry assay. However, it is presumable that a similar association would have been found due to the high contribution of MIP-1β to the total response during acute infection and its high rate of coexpression with CD107A/B and IFN-γ (6, 25). What we did observed was a nonsignificant yet border correlation between plasma MIP-1β (see further discussion below) and the proportion of bifunctional degranulating cells, i.e., the proportion of CD107+ IFN-γ+ plus CD107+ IL-2+ HIV-specific CD8+ T cells out of the total CD107+ HIV-specific CD8+ T cells (Spearman's r = 0.4368, P = 0.0699; data not shown), suggesting an association between VIA, degranulation, and expression of MIP-1β. Finally, as it has been documented for chronic infection (35), no association was found between VIA magnitude and viral load. However, we could observe an association between VIA and the CD4+ T-cell set point (Fig. 4D). This result, together with the higher VIA observed in EC (in our and other studies mentioned above) and an observation made in vaccinated rhesus monkeys where strong VIA was related to enhanced virus control in breakthrough infections (50), clearly indicates that CD8+ T-cell-mediated VIA is a desirable feature to be elicited by prophylactic or therapeutic interventions aimed at delaying disease progression.
Early reports comparing the capacity of CD8+ T cells to degranulate and secrete multiple soluble mediators upon stimulation, between individuals with progressive versus long-term nonprogressive HIV infection, have suggested that CD8+ T-cell polyfunctionality would be a functional correlate of virus control (17, 18). However, these studies also raised the question of whether polyfunctionality was not the cause but the consequence of virus control. Results presented here argued in favor of the latter hypothesis: in our PHI cohort, polyfunctional HIV-specific CD8+ T cells could be measured very early during infection (in consonance with our previous observation [25]), and the proportion of these cells seemed to slightly increase over time to the 12-month sample (Fig. 5), as also reported by Ferrari et al. (6). Although significant differences in the proportion of polyfunctional cells were observed in EC versus viremic chronic subjects (as expected, based on previous reports [17, 51]), no significant differences were observed when PHI individuals were segregated as rapid (PHI < 350) or regular (PHI > 350) progressors. Moreover, polyfunctional profiles were similar in PHI individuals with different immunodominant targets, indicating that polyfunctionality was not related to specificity (Fig. 5). These findings shed more light into the notion that, instead of being a marker of antiviral function, T-cell polyfunctionality would be directly affected by viral persistence. In other words, lack of T-cell polyfunctionality would be the consequence of constant antigen stimulation during viremic chronic infection, which ultimately may lead to cell exhaustion and functional impairment, as postulated previously (6, 21). In this sense, CD8+ T-cell phenotypic characterization in terms of exhaustion and memory markers in our cohort is being currently performed. On the other hand, the demonstration that, out of the many CD8+ T-cell function combinations studied, only the frequency of specific bifunctional CD107A/B+ IFN-γ+ CD8+ T cells correlated with the magnitude of CD8+ T-cell VIA (Fig. 6) would indicate that not all the functions exerted by a polyfunctional cell would be equally relevant in mediating antiviral T-cell effector functions.
T-cell functional outcomes upon antigen engagement depend on the strength of the stimulus, which can be directly affected by the level of antigen sensitivity or cell avidity (52, 53). These parameters, in turn, can be indirectly inferred by computing the mean spot size in the IFN-γ ELISPOT assay (33) or the rMFI of a given function evaluated by flow cytometry (52). Here, we found that the analysis of IFN-γ mean spot size and IFN-γ rMFI gave comparable results, together indicating that HIV-specific cells showed less quality in all groups analyzed compared to CEF-specific cells (in EC the difference was not significant) (Fig. 2F and 7). CEF-specific cells were analyzed for comparison since they represent cells that target pathogens that were either cleared (influenza) or controlled (CMV and Epstein-Bar viruses), thus their analysis could provide putative signatures associated with virus control. This result suggests again that constant HIV antigen stimulation may be driving cell exhaustion. Moreover, and regardless of the specificity, cells from PHI subjects showed the lowest quality among all the groups analyzed, suggesting that, during acute/early HIV infection, there exists an overall deterioration of the CD8+ T-cell compartment. In regard to CD107A/B mobilization, it was found that rMFI for Gag-specific cells was higher than for Nef-specific cells and comparable to that of CEF-specific cells in all three groups of subjects analyzed (Fig. 7). A relation among the magnitude of CD107A/B mobilization (evaluated by flow cytometry as in this work), antigen sensitivity, and antiviral activity has previously been established in another setting (52), supporting our association between the stronger ability of Gag-specific cells to degranulate and their stronger capacity to mediate viral inhibition.
A large amount of data indicate that the expression of certain HLA-I molecules is related to HIV control. Part of the evidence came from cohort studies where some alleles were overrepresented in EC and long-term nonprogressors (7, 8), while other studies provided insights into functional performance of the “protective alleles” regarding specificity, proliferation, polyfunctionality, antigen presentation, immune pressure, among other factors (8, 54–61). However, it has also been demonstrated that these protective alleles perform qualitatively different in controllers compared to progressors (60), indicating that a combination of T-cell function and phenotypes should coexist in order to achieve virus control. Conversely, some individuals reach EC status in the absence of protective alleles (as it can be observed in our cohort), indicating that they are not a requirement for virus control. For instance, CD8+ T-cell VIA can be observed both in the presence and absence of protective HLA-I alleles (20, 35). Although in this work a simultaneous evaluation of multiple CD8+ T-cell functions (in different terms such as magnitude, breadth, immunodominant targets, polyfunctionality, and VIA) was performed in HLA-typed individuals with known disease status, both the cohort size and the heterogeneity of alleles and responses preclude us from unequivocally establishing the relationship between CD8+ T-cell function and phenotype and disease outcome. Further studies aimed at developing a model involving these three parameters will be needed.
The cytokine network elicited after HIV/simian immunodeficiency virus (SIV) infection has been studied in order to shed light on the early pathogenic events of the infection and to understand how these events trigger the future course of the disease. Moreover, the level of different plasma cytokines and/or chemokines during acute infection has been proposed as a potential biomarker to predict the rate of disease progression (37–39, 62). However, to our knowledge, no report has established so far the relationship between plasma molecules and the HIV-specific cellular immune response. Here, a correlation between the relative immunodominance of the anti-Gag response and the levels of IL-2 and MIP-1β was found (Fig. 8). As mentioned before, these molecules are produced by CD8+ T cells upon stimulation, and the percentage of cells expressing IL-2 and MIP-1β either alone or in combination with other functions were previously associated with virus control (4, 11, 14, 43, 49). IL-2 is an important molecule for T-cell homeostasis and immune system activation (63). Although different levels of plasma IL-2 during PHI infection have not been previously associated with HIV/AIDS disease outcome, decreased levels of other molecules also involved in T-cell homeostasis and T-cell phenotype determination, such as IL-15 and IL-12, respectively, have been associated with progressive infection in a cohort of chronically infected African-American women (38). On the other hand, it was determined that MIP-1β-producing CD8+ T cells represent an elevated proportion within the HIV-specific CD8+ T-cell response during acute infection and that they exert considerable immune pressure over viral replication (6). At the functional level, it was suggested that the production of MIP-1β is rapidly elicited upon stimulation on the specific cells (52), which may account for an enhanced activity of the MIP-1β-producing CD8+ T cells over cells exerting other functions. Moreover, other authors have found an association between MIP-1β plasma levels and lower VL and higher CD4+ T-cell counts during chronic infection (38). Taken together, all this evidence provides a rationale for the association between the protective role of immunodominant Gag-specific cellular response and plasma levels of IL-2 and MIP-1β. As already mentioned, and although further investigations will be needed to determine the nature of this relationship, these results are in line with the evidence indicating that early relative Gag immunodominance is somehow related to the generation of an effective antiviral immune response leading to slower disease progression.
In summary, multiple aspects of the HIV-specific CD8+ T-cell subset (specificity, ex vivo viral inhibitory capacity, and polyfunctionality) arising early after infection were characterized in a cohort of acute/early infected subjects, in comparison with that found in viremic chronics and elite controllers. Importantly, the association of these functions with disease progression was studied. In the first place, it was found that specificity of cells arising early after the transmission event is critical for immune control: early relative immunodominance of Gag-specific cells was associated with delayed disease progression, in terms of CD4+ T-cell count preservation, during the first year postinfection, in consonance with Gag immunodominance in EC and viremic controllers. Further insights into the functionality of CD8+ T cells from enrolled subjects allowed us to establish that the advantage of Gag-specific CD8+ T cells would rely on their ability to mediate both lysis of infected cells (evidenced by higher capacity to degranulate and to mediate viral inhibition activity in vitro) and soluble inhibition of viral replication by the simultaneous secretion of IFN-γ. Also, higher relative contribution of anti-Gag-specific cells to the total anti-HIV cellular immune response correlated with plasma levels of IL-2 and MIP-1β, molecules that further contribute to viral inhibition.
The identification of phenotypic and functional properties of the CD8+ T-cell subsets associated with viral control is urgently needed to aid in the design and performance evaluation of an effective HIV vaccine. In this scenario, data presented in the manuscript underscore the importance of considering both cell specificity and quality, in terms of both cytotoxic and noncytotoxic mechanisms, to boost preventive and therapeutic anti-HIV immune-based strategies.
Supplementary Material
ACKNOWLEDGMENTS
This research was funded by grants from the Agencia Nacional de Promoción Científica y Tecnológica (ANPCYT; grant numbers 2008/0549 and 2011/1658) to M.M.G. and a grant from Universidad de Buenos Aires (UBACyT 2011-2014 20020100200155) to G.T.
We thank Maria Florencia Quiroga for helpful discussions during experiment design and results interpretation and Sergio Mazzini for assistance during manuscript preparation. We are indebted to all the physicians belonging to the Grupo Argentino de Seroconversión Study Group and their patients for their central contributions to this work.
We have no conflict of interests to declare.
Grupo Argentino de Seroconversión Study Group: Lorena Abusamra, Marcela Acosta, Carolina Acuipil, Viviana Alonso, Liliana Amante, Graciela Ben, M. Belén Bouzas, Ariel Braverman, Mercedes Cabrini, Pedro Cahn, Osvaldo Cando, Cecilia Cánepa, Daniel Cangelosi, Juan Castelli, Mariana Ceriotto, Carina Cesar, María Collins, Fabio Crudo, Darío Dilernia, Andrea Duarte, Gustavo Echenique, María I. Figueroa, Valeria Fink, Claudia Galloso, Palmira Garda, Manuel Gómez Carrillo, Ana Gun, Alejandro Krolewiecki, Natalia Laufer, María E. Lázaro, Alberto Leoni, Eliana Loiza, Patricia Maldonado, Horacio Mingrone, Marcela Ortiz, Patricia Patterson, Héctor Pérez, Norma Porteiro, Daniel Pryluka, Carlos Remondegui, Raúl Román, Horacio Salomón, M. Eugenia Socías, Omar Sued, J. Gonzalo Tomás, Gabriela Turk, Javier Yave, Carlos Zala, Inés Zapiola.
Footnotes
Published ahead of print 24 April 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00865-13.
REFERENCES
- 1. Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, Kaewkungwal J, Chiu J, Paris R, Premsri N, Namwat C, de Souza M, Adams E, Benenson M, Gurunathan S, Tartaglia J, McNeil JG, Francis DP, Stablein D, Birx DL, Chunsuttiwat S, Khamboonruang C, Thongcharoen P, Robb ML, Michael NL, Kunasol P, Kim JH. 2009. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N. Engl. J. Med. 361:2209–2220 [DOI] [PubMed] [Google Scholar]
- 2. Freel SA, Saunders KO, Tomaras GD. 2011. CD8(+)T-cell-mediated control of HIV-1 and SIV infection. Immunol. Res. 49:135–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McDermott AB, Koup RA. 2012. CD8(+) T cells in preventing HIV infection and disease. AIDS 26:1281–1292 [DOI] [PubMed] [Google Scholar]
- 4. Cohen MS, Shaw GM, McMichael AJ, Haynes BF. 2011. Acute HIV-1 infection. N. Engl. J. Med. 364:1943–1954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Borrow P, Lewicki H, Wei X, Horwitz MS, Peffer N, Meyers H, Nelson JA, Gairin JE, Hahn BH, Oldstone MB, Shaw GM. 1997. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat. Med. 3:205–211 [DOI] [PubMed] [Google Scholar]
- 6. Ferrari G, Korber B, Goonetilleke N, Liu MK, Turnbull EL, Salazar-Gonzalez JF, Hawkins N, Self S, Watson S, Betts MR, Gay C, McGhee K, Pellegrino P, Williams I, Tomaras GD, Haynes BF, Gray CM, Borrow P, Roederer M, McMichael AJ, Weinhold KJ. 2011. Relationship between functional profile of HIV-1 specific CD8 T cells and epitope variability with the selection of escape mutants in acute HIV-1 infection. PLoS Pathog. 7:e1001273. 10.1371/journal.ppat.1001273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Altfeld M, Kalife ET, Qi Y, Streeck H, Lichterfeld M, Johnston MN, Burgett N, Swartz ME, Yang A, Alter G, Yu XG, Meier A, Rockstroh JK, Allen TM, Jessen H, Rosenberg ES, Carrington M, Walker BD. 2006. HLA alleles associated with delayed progression to AIDS contribute strongly to the initial CD8(+) T cell response against HIV-1. PLoS Med. 3:e403. 10.1371/journal.pmed.0030403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pereyra F, Jia X, McLaren PJ, Telenti A, de Bakker PI, Walker BD, Ripke S, Brumme CJ, Pulit SL, Carrington M, Kadie CM, Carlson JM, Heckerman D, Graham RR, Plenge RM, Deeks SG, Gianniny L, Crawford G, Sullivan J, Gonzalez E, Davies L, Camargo A, Moore JM, Beattie N, Gupta S, Crenshaw A, Burtt NP, Guiducci C, Gupta N, Gao X, Qi Y, Yuki Y, Piechocka-Trocha A, Cutrell E, Rosenberg R, Moss KL, Lemay P, O'Leary J, Schaefer T, Verma P, Toth I, Block B, Baker B, Rothchild A, Lian J, Proudfoot J, Alvino DM, Vine S, Addo MM, Allen TM, Altfeld M, Henn MR, Le Gall S, Streeck H, Haas DW, Kuritzkes DR, Robbins GK, Shafer RW, Gulick RM, Shikuma CM, Haubrich R, Riddler S, Sax PE, Daar ES, Ribaudo HJ, Agan B, Agarwal S, Ahern RL, Allen BL, Altidor S, Altschuler EL, Ambardar S, Anastos K, Anderson B, Anderson V, Andrady U, Antoniskis D, Bangsberg D, Barbaro D, Barrie W, Bartczak J, Barton S, Basden P, Basgoz N, Bazner S, Bellos NC, Benson AM, Berger J, Bernard NF, Bernard AM, Birch C, Bodner SJ, Bolan RK, Boudreaux ET, Bradley M, Braun JF, Brndjar JE, Brown SJ, Brown K, Brown ST, et al. 2010. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science 330:1551–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hansen SG, Ford JC, Lewis MS, Ventura AB, Hughes CM, Coyne-Johnson L, Whizin N, Oswald K, Shoemaker R, Swanson T, Legasse AW, Chiuchiolo MJ, Parks CL, Axthelm MK, Nelson JA, Jarvis MA, Piatak M, Jr, Lifson JD, Picker LJ. 2011. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 473:523–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Barouch DH, Liu J, Li H, Maxfield LF, Abbink P, Lynch DM, Iampietro MJ, SanMiguel A, Seaman MS, Ferrari G, Forthal DN, Ourmanov I, Hirsch VM, Carville A, Mansfield KG, Stablein D, Pau MG, Schuitemaker H, Sadoff JC, Billings EA, Rao M, Robb ML, Kim JH, Marovich MA, Goudsmit J, Michael NL. 2012. Vaccine protection against acquisition of neutralization-resistant SIV challenges in rhesus monkeys. Nature 482:89–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zuniga R, Lucchetti A, Galvan P, Sanchez S, Sanchez C, Hernandez A, Sanchez H, Frahm N, Linde CH, Hewitt HS, Hildebrand W, Altfeld M, Allen TM, Walker BD, Korber BT, Leitner T, Sanchez J, Brander C. 2006. Relative dominance of Gag p24-specific cytotoxic T lymphocytes is associated with human immunodeficiency virus control. J. Virol. 80:3122–3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Geldmacher C, Currier JR, Herrmann E, Haule A, Kuta E, McCutchan F, Njovu L, Geis S, Hoffmann O, Maboko L, Williamson C, Birx D, Meyerhans A, Cox J, Hoelscher M. 2007. CD8 T-cell recognition of multiple epitopes within specific Gag regions is associated with maintenance of a low steady-state viremia in human immunodeficiency virus type 1-seropositive patients. J. Virol. 81:2440–2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kiepiela P, Ngumbela K, Thobakgale C, Ramduth D, Honeyborne I, Moodley E, Reddy S, de Pierres C, Mncube Z, Mkhwanazi N, Bishop K, van der Stok M, Nair K, Khan N, Crawford H, Payne R, Leslie A, Prado J, Prendergast A, Frater J, McCarthy N, Brander C, Learn GH, Nickle D, Rousseau C, Coovadia H, Mullins JI, Heckerman D, Walker BD, Goulder P. 2007. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat. Med. 13:46–53 [DOI] [PubMed] [Google Scholar]
- 14. Streeck H, Jolin JS, Qi Y, Yassine-Diab B, Johnson RC, Kwon DS, Addo MM, Brumme C, Routy JP, Little S, Jessen HK, Kelleher AD, Hecht FM, Sekaly RP, Rosenberg ES, Walker BD, Carrington M, Altfeld M. 2009. Human immunodeficiency virus type 1-specific CD8+ T-cell responses during primary infection are major determinants of the viral set point and loss of CD4+ T cells. J. Virol. 83:7641–7648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Masemola A, Mashishi T, Khoury G, Mohube P, Mokgotho P, Vardas E, Colvin M, Zijenah L, Katzenstein D, Musonda R, Allen S, Kumwenda N, Taha T, Gray G, McIntyre J, Karim SA, Sheppard HW, Gray CM. 2004. Hierarchical targeting of subtype C human immunodeficiency virus type 1 proteins by CD8+ T cells: correlation with viral load. J. Virol. 78:3233–3243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Saez-Cirion A, Sinet M, Shin SY, Urrutia A, Versmisse P, Lacabaratz C, Boufassa F, Avettand-Fenoel V, Rouzioux C, Delfraissy JF, Barre-Sinoussi F, Lambotte O, Venet A, Pancino G. 2009. Heterogeneity in HIV suppression by CD8 T cells from HIV controllers: association with Gag-specific CD8 T cell responses. J. Immunol. 182:7828–7837 [DOI] [PubMed] [Google Scholar]
- 17. Betts MR, Nason MC, West SM, De Rosa SC, Migueles SA, Abraham J, Lederman MM, Benito JM, Goepfert PA, Connors M, Roederer M, Koup RA. 2006. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood 107:4781–4789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zimmerli SC, Harari A, Cellerai C, Vallelian F, Bart PA, Pantaleo G. 2005. HIV-1-specific IFN-gamma/IL-2-secreting CD8 T cells support CD4-independent proliferation of HIV-1-specific CD8 T cells. Proc. Natl. Acad. Sci. U. S. A. 102:7239–7244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Saez-Cirion A, Lacabaratz C, Lambotte O, Versmisse P, Urrutia A, Boufassa F, Barre-Sinoussi F, Delfraissy JF, Sinet M, Pancino G, Venet A. 2007. HIV controllers exhibit potent CD8 T cell capacity to suppress HIV infection ex vivo and peculiar cytotoxic T lymphocyte activation phenotype. Proc. Natl. Acad. Sci. U. S. A. 104:6776–6781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Freel SA, Lamoreaux L, Chattopadhyay PK, Saunders K, Zarkowsky D, Overman RG, Ochsenbauer C, Edmonds TG, Kappes JC, Cunningham CK, Denny TN, Weinhold KJ, Ferrari G, Haynes BF, Koup RA, Graham BS, Roederer M, Tomaras GD. 2010. Phenotypic and functional profile of HIV-inhibitory CD8 T cells elicited by natural infection and heterologous prime/boost vaccination. J. Virol. 84:4998–5006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Streeck H, Brumme ZL, Anastario M, Cohen KW, Jolin JS, Meier A, Brumme CJ, Rosenberg ES, Alter G, Allen TM, Walker BD, Altfeld M. 2008. Antigen load and viral sequence diversification determine the functional profile of HIV-1-specific CD8+ T cells. PLoS Med. 5:e100. 10.1371/journal.pmed.0050100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Burgers WA, Riou C, Mlotshwa M, Maenetje P, de Assis Rosa D, Brenchley J, Mlisana K, Douek DC, Koup R, Roederer M, de Bruyn G, Karim SA, Williamson C, Gray CM. 2009. Association of HIV-specific and total CD8+ T memory phenotypes in subtype C HIV-1 infection with viral set point. J. Immunol. 182:4751–4761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ganesan A, Chattopadhyay PK, Brodie TM, Qin J, Gu W, Mascola JR, Michael NL, Follmann DA, Roederer M. 2010. Immunologic and virologic events in early HIV infection predict subsequent rate of progression. J. Infect. Dis. 201:272–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cohen MS, Chen YQ, McCauley M, Gamble T, Hosseinipour MC, Kumarasamy N, Hakim JG, Kumwenda J, Grinsztejn B, Pilotto JH, Godbole SV, Mehendale S, Chariyalertsak S, Santos BR, Mayer KH, Hoffman IF, Eshleman SH, Piwowar-Manning E, Wang L, Makhema J, Mills LA, de Bruyn G, Sanne I, Eron J, Gallant J, Havlir D, Swindells S, Ribaudo H, Elharrar V, Burns D, Taha TE, Nielsen-Saines K, Celentano D, Essex M, Fleming TR. 2011. Prevention of HIV-1 infection with early antiretroviral therapy. N. Engl. J. Med. 365:493–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Turk G, Gherardi MM, Laufer N, Saracco M, Luzzi R, Cox JH, Cahn P, Salomon H. 2008. Magnitude, breadth, and functional profile of T-cell responses during human immunodeficiency virus primary infection with B and BF viral variants. J. Virol. 82:2853–2866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Socias ME, Sued O, Laufer N, Lazaro ME, Mingrone H, Pryluka D, Remondegui C, Figueroa MI, Cesar C, Gun A, Turk G, Bouzas MB, Kavasery R, Krolewiecki A, Perez H, Salomon H, Cahn P. 2011. Acute retroviral syndrome and high baseline viral load are predictors of rapid HIV progression among untreated Argentinean seroconverters. J. Int. AIDS Soc. 14:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Currier JR, Kuta EG, Turk E, Earhart LB, Loomis-Price L, Janetzki S, Ferrari G, Birx DL, Cox JH. 2002. A panel of MHC class I restricted viral peptides for use as a quality control for vaccine trial ELISPOT assays. J. Immunol. Methods 260:157–172 [DOI] [PubMed] [Google Scholar]
- 28. Malhotra U, Li F, Nolin J, Allison M, Zhao H, Mullins JI, Self S, McElrath MJ. 2007. Enhanced detection of human immunodeficiency virus type 1 (HIV-1) Nef-specific T cells recognizing multiple variants in early HIV-1 infection. J. Virol. 81:5225–5237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sáez-Cirión A, Shin SY, Versmisse P, Barre-Sinoussi F, Pancino G. 2010. Ex vivo T cell-based HIV suppression assay to evaluate HIV-specific CD8+ T-cell responses. Nat. Protoc. 5:1033–1041 [DOI] [PubMed] [Google Scholar]
- 30. Giavedoni LD. 2005. Simultaneous detection of multiple cytokines and chemokines from nonhuman primates using Luminex technology. J. Immunol. Methods 301:89–101 [DOI] [PubMed] [Google Scholar]
- 31. Fiebig EW, Wright DJ, Rawal BD, Garrett PE, Schumacher RT, Peddada L, Heldebrant C, Smith R, Conrad A, Kleinman SH, Busch MP. 2003. Dynamics of HIV viremia and antibody seroconversion in plasma donors: implications for diagnosis and staging of primary HIV infection. AIDS 17:1871–1879 [DOI] [PubMed] [Google Scholar]
- 32. Okulicz JF, Marconi VC, Landrum ML, Wegner S, Weintrob A, Ganesan A, Hale B, Crum-Cianflone N, Delmar J, Barthel V, Quinnan G, Agan BK, Dolan MJ. 2009. Clinical outcomes of elite controllers, viremic controllers, and long-term nonprogressors in the US Department of Defense HIV natural history study. J. Infect. Dis. 200:1714–1723 [DOI] [PubMed] [Google Scholar]
- 33. Rodriguez AM, Pascutti MF, Maeto C, Falivene J, Holgado MP, Turk G, Gherardi MM. 2012. IL-12 and GM-CSF in DNA/MVA immunizations against HIV-1 CRF12_BF Nef induced T-cell responses with an enhanced magnitude, breadth and quality. PLoS One 7:e37801. 10.1371/journal.pone.0037801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ndhlovu ZM, Proudfoot J, Cesa K, Alvino DM, McMullen A, Vine S, Stampouloglou E, Piechocka-Trocha A, Walker BD, Pereyra F. 2012. Elite controllers with low to absent effector CD8+ T cell responses maintain highly functional, broadly directed central memory responses. J. Virol. 86:6959–6969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Julg B, Williams KL, Reddy S, Bishop K, Qi Y, Carrington M, Goulder PJ, Ndung'u T, Walker BD. 2010. Enhanced anti-HIV functional activity associated with Gag-specific CD8 T-cell responses. J. Virol. 84:5540–5549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Precopio ML, Betts MR, Parrino J, Price DA, Gostick E, Ambrozak DR, Asher TE, Douek DC, Harari A, Pantaleo G, Bailer R, Graham BS, Roederer M, Koup RA. 2007. Immunization with vaccinia virus induces polyfunctional and phenotypically distinctive CD8(+) T cell responses. J. Exp. Med. 204:1405–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Roberts L, Passmore JA, Williamson C, Little F, Bebell LM, Mlisana K, Burgers WA, van Loggerenberg F, Walzl G, Djoba Siawaya JF, Karim QA, Karim SS. 2010. Plasma cytokine levels during acute HIV-1 infection predict HIV disease progression. AIDS 24:819–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Keating SM, Golub ET, Nowicki M, Young M, Anastos K, Crystal H, Cohen MH, Zhang J, Greenblatt RM, Desai S, Wu S, Landay AL, Gange SJ, Norris PJ. 2011. The effect of HIV infection and HAART on inflammatory biomarkers in a population-based cohort of women. AIDS 25:1823–1832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liovat AS, Rey-Cuille MA, Lecuroux C, Jacquelin B, Girault I, Petitjean G, Zitoun Y, Venet A, Barre-Sinoussi F, Lebon P, Meyer L, Sinet M, Muller-Trutwin M. 2012. Acute plasma biomarkers of T cell activation set-point levels and of disease progression in HIV-1 infection. PLoS One 7:e46143. 10.1371/journal.pone.0046143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Saunders KO, Ward-Caviness C, Schutte RJ, Freel SA, Overman RG, Thielman NM, Cunningham CK, Kepler TB, Tomaras GD. 2010. Secretion of MIP-1beta and MIP-1alpha by CD8(+) T-lymphocytes correlates with HIV-1 inhibition independent of coreceptor usage. Cell. Immunol. 266:154–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Freel SA, Picking RA, Ferrari G, Ding H, Ochsenbauer C, Kappes JC, Kirchherr JL, Soderberg KA, Weinhold KJ, Cunningham CK, Denny TN, Crump JA, Cohen MS, McMichael AJ, Haynes BF, Tomaras GD. 2012. Initial HIV-1 antigen-specific CD8+ T cells in acute HIV-1 infection inhibit transmitted/founder virus replication. J. Virol. 86:6835–6846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lichterfeld M, Yu XG, Cohen D, Addo MM, Malenfant J, Perkins B, Pae E, Johnston MN, Strick D, Allen TM, Rosenberg ES, Korber B, Walker BD, Altfeld M. 2004. HIV-1 Nef is preferentially recognized by CD8 T cells in primary HIV-1 infection despite a relatively high degree of genetic diversity. AIDS 18:1383–1392 [DOI] [PubMed] [Google Scholar]
- 43. Ferrando-Martinez S, Casazza JP, Leal M, Machmach K, Munoz-Fernandez MA, Viciana P, Koup RA, Ruiz-Mateos E. 2012. Differential Gag-specific polyfunctional T cell maturation patterns in HIV-1 elite controllers. J. Virol. 86:3667–3674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ferre AL, Lemongello D, Hunt PW, Morris MM, Garcia JC, Pollard RB, Yee HF, Jr, Martin JN, Deeks SG, Shacklett BL. 2010. Immunodominant HIV-specific CD8+ T-cell responses are common to blood and gastrointestinal mucosa, and Gag-specific responses dominate in rectal mucosa of HIV controllers. J. Virol. 84:10354–10365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sacha JB, Chung C, Rakasz EG, Spencer SP, Jonas AK, Bean AT, Lee W, Burwitz BJ, Stephany JJ, Loffredo JT, Allison DB, Adnan S, Hoji A, Wilson NA, Friedrich TC, Lifson JD, Yang OO, Watkins DI. 2007. Gag-specific CD8+ T lymphocytes recognize infected cells before AIDS-virus integration and viral protein expression. J. Immunol. 178:2746–2754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Payne RP, Kloverpris H, Sacha JB, Brumme Z, Brumme C, Buus S, Sims S, Hickling S, Riddell L, Chen F, Luzzi G, Edwards A, Phillips R, Prado JG, Goulder PJ. 2010. Efficacious early antiviral activity of HIV Gag- and Pol-specific HLA-B 2705-restricted CD8+ T cells. J. Virol. 84:10543–10557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Miura T, Brumme ZL, Brockman MA, Rosato P, Sela J, Brumme CJ, Pereyra F, Kaufmann DE, Trocha A, Block BL, Daar ES, Connick E, Jessen H, Kelleher AD, Rosenberg E, Markowitz M, Schafer K, Vaida F, Iwamoto A, Little S, Walker BD. 2010. Impaired replication capacity of acute/early viruses in persons who become HIV controllers. J. Virol. 84:7581–7591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Henn MR, Boutwell CL, Charlebois P, Lennon NJ, Power KA, Macalalad AR, Berlin AM, Malboeuf CM, Ryan EM, Gnerre S, Zody MC, Erlich RL, Green LM, Berical A, Wang Y, Casali M, Streeck H, Bloom AK, Dudek T, Tully D, Newman R, Axten KL, Gladden AD, Battis L, Kemper M, Zeng Q, Shea TP, Gujja S, Zedlack C, Gasser O, Brander C, Hess C, Gunthard HF, Brumme ZL, Brumme CJ, Bazner S, Rychert J, Tinsley JP, Mayer KH, Rosenberg E, Pereyra F, Levin JZ, Young SK, Jessen H, Altfeld M, Birren BW, Walker BD, Allen TM. 2012. Whole genome deep sequencing of HIV-1 reveals the impact of early minor variants upon immune recognition during acute infection. PLoS Pathog. 8:e1002529. 10.1371/journal.ppat.1002529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Radebe M, Nair K, Chonco F, Bishop K, Wright JK, van der Stok M, Bassett IV, Mncube Z, Altfeld M, Walker BD, Ndung'u T. 2011. Limited immunogenicity of HIV CD8+ T-cell epitopes in acute clade C virus infection. J. Infect. Dis. 204:768–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yamamoto T, Johnson MJ, Price DA, Wolinsky DI, Almeida JR, Petrovas C, Nason M, Yeh WW, Shen L, Roederer M, Rao SS, McDermott AB, Lefebvre F, Nabel GJ, Haddad EK, Letvin NL, Douek DC, Koup RA. 2012. Virus inhibition activity of effector memory CD8(+) T cells determines simian immunodeficiency virus load in vaccinated monkeys after vaccine breakthrough infection. J. Virol. 86:5877–5884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Peris-Pertusa A, Lopez M, Rallon NI, Restrepo C, Soriano V, Benito JM. 2010. Evolution of the functional profile of HIV-specific CD8+ T cells in patients with different progression of HIV infection over 4 years. J. Acquir.Immune Defic. Syndr. 55:29–38 [DOI] [PubMed] [Google Scholar]
- 52. Almeida JR, Sauce D, Price DA, Papagno L, Shin SY, Moris A, Larsen M, Pancino G, Douek DC, Autran B, Saez-Cirion A, Appay V. 2009. Antigen sensitivity is a major determinant of CD8+ T-cell polyfunctionality and HIV-suppressive activity. Blood 113:6351–6360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Viola A, Lanzavecchia A. 1996. T cell activation determined by T cell receptor number and tunable thresholds. Science 273:104–106 [DOI] [PubMed] [Google Scholar]
- 54. Borghans JA, Molgaard A, de Boer RJ, Kesmir C. 2007. HLA alleles associated with slow progression to AIDS truly prefer to present HIV-1 p24. PLoS One 2:e920. 10.1371/journal.pone.0000920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Carlson JM, Brumme CJ, Martin E, Listgarten J, Brockman MA, Le AQ, Chui CK, Cotton LA, Knapp DJ, Riddler SA, Haubrich R, Nelson G, Pfeifer N, Deziel CE, Heckerman D, Apps R, Carrington M, Mallal S, Harrigan PR, John M, Brumme ZL. 2012. Correlates of protective cellular immunity revealed by analysis of population-level immune escape pathways in HIV-1. J. Virol. 86:13202–13216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Harari A, Cellerai C, Enders FB, Kostler J, Codarri L, Tapia G, Boyman O, Castro E, Gaudieri S, James I, John M, Wagner R, Mallal S, Pantaleo G. 2007. Skewed association of polyfunctional antigen-specific CD8 T cell populations with HLA-B genotype. Proc. Natl. Acad. Sci. U. S. A. 104:16233–16238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hoof I, Kesmir C, Lund O, Nielsen M. 2008. Humans with chimpanzee-like major histocompatibility complex-specificities control HIV-1 infection. AIDS 22:1299–1303 [DOI] [PubMed] [Google Scholar]
- 58. Horton H, Frank I, Baydo R, Jalbert E, Penn J, Wilson S, McNevin JP, McSweyn MD, Lee D, Huang Y, De Rosa SC, McElrath MJ. 2006. Preservation of T cell proliferation restricted by protective HLA alleles is critical for immune control of HIV-1 infection. J. Immunol. 177:7406–7415 [DOI] [PubMed] [Google Scholar]
- 59. Kloverpris HN, Payne RP, Sacha JB, Rasaiyaah JT, Chen F, Takiguchi M, Yang OO, Towers GJ, Goulder P, Prado JG. 2013. Early antigen presentation of protective HIV-1 KF11Gag and KK10Gag epitopes from incoming viral particles facilitates rapid recognition of infected cells by specific CD8+ T cells. J. Virol. 87:2628–2638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ndhlovu ZM, Chibnik LB, Proudfoot J, Vine S, McMullen A, Cesa K, Porichis F, Jones RB, Alvino DM, Hart MG, Stampouloglou E, Piechocka-Trocha A, Kadie C, Pereyra F, Heckerman D, De Jager PL, Walker BD, Kaufmann DE. 2013. High-dimensional immunomonitoring models of HIV-1-specific CD8 T-cell responses accurately identify subjects achieving spontaneous viral control. Blood 121:801–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Elahi S, Dinges WL, Lejarcegui N, Laing KJ, Collier AC, Koelle DM, McElrath MJ, Horton H. 2011. Protective HIV-specific CD8+ T cells evade Treg cell suppression. Nat. Med. 17:989–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Katsikis PD, Mueller YM, Villinger F. 2011. The cytokine network of acute HIV infection: a promising target for vaccines and therapy to reduce viral set-point? PLoS Pathog. 7:e1002055. 10.1371/journal.ppat.1002055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Boyman O, Sprent J. 2012. The role of interleukin-2 during homeostasis and activation of the immune system. Nat. Rev. Immunol. 12:180–190 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.