

Abstract
Hepatitis C Virus (HCV) NS4B protein has many roles in HCV genome replication. Recently, our laboratory (Q. Han, J. Aligo, D. Manna, K. Belton, S. V. Chintapalli, Y. Hong, R. L. Patterson, D. B. van Rossum, and K. V. Konan, J. Virol. 85:6464–6479, 2011) and others (D. M. Jones, A. H. Patel, P. Targett-Adams, and J. McLauchlan, J. Virol. 83:2163–2177, 2009; D. Paul, I. Romero-Brey, J. Gouttenoire, S. Stoitsova, J. Krijnse-Locker, D. Moradpour, and R. Bartenschlager, J. Virol. 85:6963–6976, 2011) have also reported NS4B's function in postreplication steps. Indeed, replacement of the NS4B C-terminal domain (CTD) in the HCV JFH1 (genotype 2a [G2a]) genome with sequences from Con1 (G1b) or H77 (G1a) had a negligible impact on JFH1 genome replication but attenuated virus production. Since NS4B interacts weakly with the HCV genome, we postulated that NS4B regulates the function of host or virus proteins directly involved in HCV production. In this study, we demonstrate that the integrity of the JFH1 NS4B CTD is crucial for efficient JFH1 genome encapsidation. Further, two adaptive mutations (NS4B N216S and NS5A C465S) were identified, and introduction of these mutations into the chimera rescued virus production to various levels, suggesting a genetic interaction between the NS4B and NS5A proteins. Interestingly, cells infected with chimeric viruses displayed a markedly decreased NS5A hyperphosphorylation state (NS5A p58) relative to JFH1, and the adaptive mutations differentially rescued NS5A p58 formation. However, immunofluorescence staining indicated that the decrease in NS5A p58 did not alter NS5A colocalization with the core around lipid droplets (LDs), the site of JFH1 assembly, suggesting that NS5A fails to facilitate the transfer of HCV RNA to the capsid protein on LDs. Alternatively, NS4B's function in HCV genome encapsidation may entail more than its regulation of the NS5A phosphorylation state.
INTRODUCTION
Hepatitis C virus (HCV) infects 2 to 3% of the world population, with ca. 160 to 170 million individuals chronically infected and more than 350,000 deaths annually due to complications from cirrhosis and hepatocellular carcinoma (1, 2). As a result of the error-prone nature of its polymerase (3), HCV is classified into at least 6 genotypes and more than 50 subtypes (4). HCV is an enveloped, positive-sense RNA virus with a 9.6-kb genome flanked by 5′ and 3′ noncoding regions (NCR) and a long open reading frame encoding one polyprotein ∼3,011 amino acids (aa) in length. Processing of the polyprotein by host and viral proteases occurs co- or posttranslationally, giving rise to three structural proteins (the capsid protein core and the envelope glycoproteins E1 and E2), the viroporin protein p7, and six nonstructural (NS) proteins (NS2, -3, -4A, -4B, -5A, and -5B) (5). The p7 and NS2 proteins are involved in HCV assembly (6–8), while NS3 to NS5B are sufficient to promote virus genome replication in vitro (9, 10). Recently, many of the replicase proteins (NS3, NS4B, and NS5A) were also found to play an active role in HCV production (11–15), consistent with the interpretation that the NS proteins have multiple functions in the HCV life cycle.
Recent studies suggest that NS5A physically links the HCV replication complex to the site of HCV assembly on lipid droplets (LDs) or the endoplasmic reticulum (ER) (6, 16). This is possible in part because NS5A is a phosphoprotein that exists in two states, based on its migration distance after SDS-PAGE. Basal phosphorylation (NS5A p56) favors HCV genome replication (17), whereas hyperphosphorylation (NS5A p58) inhibits replication but appears to favor virus production (14, 17). Since NS5A binds HCV RNA (18), it is likely that NS5A keeps the RNA in the replication complex in its p56 form but transfers the RNA to the assembly complex via interaction with core and LDs in its p58 form (13, 19). The molecular mechanism of NS5A phosphorylation is not yet understood, but several host serine/threonine kinases (20–24), as well as virus proteins NS3, NS4A, and NS4B, are involved (25, 26). Since these virus proteins are required for NS5A p58 formation, it is tempting to speculate that these proteins facilitate virus production, in part, by regulating the NS5A phosphorylation state.
NS4B is a 27-kDa polytopic transmembrane protein (27) whose precise membrane topology is still unknown (28–32). The transmembrane region of NS4B is composed of at least four membrane-spanning domains (transmembrane domains [TMDs]) and was recently shown to contain protein-protein interaction motifs crucial for HCV genome replication (33). The N-terminal domain (NTD) of NS4B was reported to have dual-membrane topology (32, 34), whereas the C-terminal domain (CTD) is located on the cytosolic side of the ER membrane. Thus, the CTD is likely engaged in host and viral protein-protein interactions crucial for NS4B function in HCV infection. For example, NS4B expression has been linked in part to the induction of the so-called membrane web structure, or web, the platform upon which HCV genome replication takes place (35–37). Truncations or point mutations in the NS4B CTD have been linked to a disruption of the web and a concomitant drop in HCV replication efficiency, whereas second-site mutations in the NS4B CTD or NS5A were found to rescue the web and virus replication (28, 31, 38). Interestingly, RNA binding motifs in the NS4B CTD were demonstrated to bind to the 3′ NCR of the negative-sense viral RNA and facilitate HCV genome replication (39). Hence, the NS4B CTD may contribute to HCV replication via induction of the membranous web and anchoring the negative-sense viral RNA on this rearranged membrane to optimize replication efficiency. Finally, NS4B interacts with most HCV NS proteins following cotransfection in mammalian cells (28, 40). However, such interactions have not been confirmed during virus infection, nor do we know their significance.
We have recently demonstrated that swapping NS4B CTD sequences between HCV JFH1 (infection results in a relatively high virus titer) (41–43) and HCV Con1 (infection gives rise to a low virus titer) (44) leads to an attenuation of the JFH1 virus titer (33) with a negligible impact on genome replication. This finding suggests that NS4B is a multifunctional protein with distinct roles in both replication and virus production. However, the mechanism whereby NS4B facilitates virus production is unknown. Since NS4B is less likely to directly bind to LDs, the site of JFH1 virus assembly (16), we hypothesized that NS4B regulates the function of host or virus proteins directly involved in JFH1 production. In this study, we demonstrate that the integrity of the JFH1 NS4B CTD is crucial for efficient JFH1 genome encapsidation. Further, we have identified second-site mutations that link HCV NS5A to NS4B's role in virus production. The significance of these findings is discussed.
MATERIALS AND METHODS
Cells.
Huh-7.5 cells, a highly permissive human hepatoma cell line for HCV replication (45), were kindly provided by Apath, LLC (St. Louis, MO). The cells were grown as monolayers in advanced Dulbecco's modified Eagle's medium (advanced DMEM) (Invitrogen, Carlsbad, CA) supplemented with 1.5% fetal bovine serum (FBS), l-glutamine (Invitrogen), 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C in a 5% CO2 incubator.
Antibodies.
Rabbit polyclonal antibody to HCV NS4B was produced by Covance (Denver, PA). Rabbit polyclonal HCV NS5A and NS3 antibodies were kindly provided by Craig Cameron (Penn State, University Park, PA), and mouse monoclonal antibody (9E10) to HCV NS5A (which recognizes NS5A domain III) was kindly provided by Charles Rice (Rockefeller University, New York, NY). Mouse monoclonal antibodies to HCV core and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were obtained from Virostat (Portland, ME) and Fitzgerald (Concord, MA), respectively. Horseradish peroxidase-conjugated secondary antibodies (used in chemiluminescence assays) were obtained from Vector Laboratories (Burlingame, CA). Alexa Fluor 488- and 594-conjugated secondary antibodies (used in immunofluorescence assays) were from Invitrogen.
Construction of plasmids.
pJFH1 and pJFH1/GND vectors carrying wild-type (WT) and replication-defective JFH1 genomes, respectively, were kindly provided by Takaji Wakita (Tokyo Metropolitan Institute for Neuroscience) (42). These vectors were used to generate the various JFH1 chimeras and point mutants, including the full-length genome and the subgenomic luciferase (Luc)-expressing replicons. The plasmid pLuc-JFH1, which contains the T7 promoter sequence fused to nucleotides (nt) 1 to 389 of the JFH-1 consensus sequence, followed by the firefly Luc gene, the encephalomyocarditis virus (EMCV) internal ribosome entry site (IRES), and the nucleotides spanning from the beginning of the NS3 gene to the 3′ nontranslating region (NTR) of JFH1, was constructed according to methods previously described (46, 47). Overlap extension PCR was used to construct the full-length genomes containing the chimeric C-terminal domain of the NS4B gene, where the JFH-1 sequences were replaced with either Con1 (J/Con-C) or H77 (J/H-C) sequences, and was performed according to the methods previously described by Han et al. (33). The various subgenomic Luc replicons were constructed by directly replacing the fragment between the EcoRI site and the SpeI site of their corresponding full-length constructs with EcoRI- and SpeI-cut fragments from pLuc-JFH1. The EcoRI- and SpeI-cut fragment from pLuc-JFH1 contains the HCV 5′ untranslated region (UTR), followed by the Luc reporter, the EMCV IRES, and the beginning of the NS3 sequence (nt 3431 to 4106).
Blunt-end ligation was used to introduce the single point mutations into the JFH-1, J/Con-C, or J/H-C genome, as previously described (33). Since the single mutations were engineered by the same approach, we use the pJ/Con-C N216S (NS4B) vector to illustrate how the constructs were made. First, an F1 PCR product spanning from the NsiI site (in the NS3 gene) to the NS4B N216S region (nt 5281 to 6148) was amplified from the pJ/Con-C vector using the forward primer NsiI-Fwd (5′-CACCCTCACACACCCTGGG-3′) and the phosphorylated reverse primer N216S-Rev (5′-CGAAGCGAACGCTATCAGCC-3′). Next, an F2 PCR product spanning the NS4B N216S–to–RsrII-NS5A region (nt 6149 to 7692) was amplified from the pJ/Con-C vector using the phosphorylated forward primer N216S-Fwd (5′-CGGGGTAGTCACGTCTCCC-3′; the underlined nucleotides correspond to the NS4B N216S mutation) and the reverse primer RsrII-Rev (5′-GTATGACATGGAGCAGCACACG-3′). The F1 and F2 fragments were digested with NsiI and RsrII, respectively, and the resulting fragments were ligated into the NsiI- and RsrII-cut pJFH1 vector.
The multiple point mutations in J/Con-C (or J/H-C) were engineered by using the previous vectors containing single mutations. To construct pJ/Con-C containing p7, NS4B, and NS5A mutations, for instance, the p7 mutation (H31L) was removed from the pJ/Con-C H31L plasmid by digestion with EcoRI (in the 5′ UTR) and NsiI (in NS3) and ligated into an EcoRI- and NsiI-digested pJ/Con-C N216S (NS4B) vector, thereby combining the p7 and NS4B mutations into the resulting vector, pJ/Con-C H31L/N216S. Next, the NS5A mutation (C465S) was removed from the pJ/Con-C C465S plasmid by digestion with RsrII (in NS5A) and EcoRV (in the 3′ UTR) and ligated into an RsrII- and EcoRV-digested pJ/Con-C H31L/N216S vector. The resulting recombinant DNA was sequenced to confirm the presence of the three mutations.
To insert the JFH1 NS3-NS5B sequence into pIRES, the NS3-5B DNA fragment was amplified with the forward primer NS3-Fwd (5′-CG CCG ACG CGT ATG GCT CCC ATC ACT GCT TAT GCC-3′) and the reverse primer NS5B-Rev (5′-CG CCG ACG CGT TTA TCA CCG AGC GGG GAG TAG GAA GAG-3′) with Crimson LongAmp Taq DNA polymerase (New England BioLabs, Ipswich, MA). The PCR product was digested with MluI and ligated into the MluI-cut pIRES vector to generate pIRES-NS3-5BJFH1. The positive clone was further confirmed via sequencing. The construct pIRES-NS3-5BJH/C (which contains an NS4B C-terminal domain swap between strains JFH1 and H77) was obtained in several steps. First, fragment EcoRI-NS3-5BJFH1-XbaI was removed from pIRES-NS3-5BJFH1 by EcoRI and XbaI digestion and ligated with the EcoRI- and XbaI-cut pUC19 vector to generate pUC-NS3-5BJFH1. pUC-NS3-5BJFH1 was cut with NsiI (in NS3) and RsrII (in NS5A), and the resulting fragment was replaced with the chimeric counterpart from the NsiI- and RsrII-cut pJ/H-C vector to generate pUC-NS3-5BJ/H-C. Finally, the pIRES-NS3-5BJ/H-C vector was engineered by subcloning the EcoRI-NS3-5BJ/H-C-XbaI fragment from pUC-NS3-5BJ/H-C into the EcoRI- and XbaI-cut pIRES vector.
In vitro transcription and electroporation of viral RNA into Huh-7.5 cells.
Plasmid DNA constructs containing WT or chimeric genomes or subgenomic replicons were linearized with XbaI and purified using the Cycle Pure Kit (Omega Bio-Tek, Norcross, GA). RNA was synthesized using the T7 RiboMax Express Large Scale RNA Production Systems Kit (Promega, Madison, WI) according to the manufacturer's instructions. The RNA was then isolated using TRIzol-LS (Invitrogen). Prior to electroporation, subconfluent Huh-7.5 cells were trypsinized and resuspended in complete DMEM. The cells were then washed three times with ice-cold phosphate-buffered saline (PBS) and resuspended at a concentration of 1.25 × 107 cells/ml in ice-cold PBS. Briefly, 1 μg of viral RNA or 10 μg of subgenomic replicon (SGR)-Luc RNA was mixed with 2.5 × 106 Huh-7.5 cells in 0.2 ml ice-cold PBS and electroporated with an ElectroSquarePorator (BTX) in a 0.2-mm-gap cuvette. The electroporator was set at 820 V, 99 μs at 1.1-s intervals, and 4 pulses. The actual voltage was around 690 V for each sample. The cells were left to recover for 10 min at room temperature before being diluted in 10 ml of complete medium. The cells were then seeded into a 10-cm dish, with virus samples harvested every 3 days, or into 24-well plates for Luc-expressing replicons and subsequently harvested at 4 h, 2 days (D2), D4, and D6 for Luc assay.
Cell lysate preparation to obtain intracellular virus.
Intracellular-virus samples were generated as previously described (48). Briefly, cells were collected and washed with PBS, followed by resuspension in cell culture medium (the same volume as the supernatant virus samples) and four cycles of flash freezing (in liquid nitrogen) and thawing in a 37°C water bath. The samples were centrifuged at 3,500 rpm for 5 min at 4°C, and virus-containing supernatants were collected.
Intracelluar HCV RNA encapsidation analysis.
Huh-7.5 cells (8 × 106) were electroporated with 10 μg of HCV RNA in 400 μl ice-cold PBS and seeded in a 150-mm dish. For each HCV genome, 3 electroporations were conducted.
At 48 h posttransfection (p.t.), the cells were trypsinized, pooled, and resuspended in 3 ml of complete culture medium. Two hundred microliters of cell suspension each was used for total RNA isolation and core immunoblotting. The remaining cell suspension (2,600 μl) was subjected to four cycles of freeze and thaw, followed by spinning at 2,500 × g for 10 min at 4°C to pellet cellular debris and nuclei. From 2.4 ml of supernatant collected after spinning, 200 μl was used for intracellular-virus titration. The remaining 2.2 ml was diluted with 20 ml of TNE buffer (10 mM Tris, pH 8, 150 mM NaCl, 2 mM EDTA), layered on top of 5 ml of 20% sucrose in TNE buffer, and spun at 31,000 rpm (118,000 × g) for 4 h at 4°C in an SW32 rotor. The supernatant was carefully removed, and the virus pellet was resuspended in 410 μl of PBS. Two hundred microliters of viral-particle suspension was lysed with 4× Laemmli buffer, boiled at 100°C for 10 min, and subjected to SDS-PAGE. The remaining viral-particle suspension was used for viral-RNA extraction with the QIAamp Viral RNA minikit (Qiagen, Valencia, CA).
HCV titration.
Extracellular-supernatant virus titers were determined by endpoint dilution assays as described previously (43, 49). Briefly, Huh-7.5 cells were seeded into 96-well plates at a density of 6 × 103 cells/well. Samples were serially diluted 10-fold in complete growth medium and used to infect the seeded cells. Following 3 days of incubation, the cells were immunostained with NS5A-specific antibody. Positive foci were counted, and the infectivity titer was calculated from the average of the number of foci counted in the last and second-to-last wells of the dilution series that still contained positive foci. The viral titer was expressed as focus-forming units (FFU)/ml.
Quantitative RT-PCR.
Supernatant, intracellular total HCV RNA, and sucrose-purified virus particle (VP) RNA quantitation was conducted according to the method described previously (43) with some modification. Briefly, total intracellular RNA was extracted with TRIzol (Invitrogen) according to the manufacturer's instructions, and 2 μg of total RNA was used as the template for reverse transcription. Supernatant and intracellular-viral-particle RNA was prepared by using the QIAamp Viral RNA minikit (Qiagen). First-strand cDNA was synthesized using random hexamers (Invitrogen) and murine leukemia virus (MuLV) reverse transcriptase (Applied Biosystems) according to the manufacturer's instructions. Quantitative PCR was conducted in triplicate in an iQ 5 Multicolor Real-Time PCR Detection System using 1× iQSYBR Green Supermix (Bio-red) and 1 μM HCV real-time (RT) PCR forward (5′-TCT GCG GAA CCG GTG AGT A-3′) and reverse (5′-TCA GGC AGT ACC ACA AGG C-3′) primers. Standard samples were run in parallel by using serially diluted pJFH1 plasmids with known copy numbers as templates.
Analysis of the revertant virus nucleotide sequences.
Total cellular RNA was extracted from J/Con-C-Ad1 or J/H-C-Ad1 virus-infected cells using the TRIzol-LS reagent (Invitrogen). Five micrograms of total RNA was used for reverse transcription of HCV cDNA with Superscript III (Invitrogen) and a specific primer, 9470R, binding at the HCV 3′ UTR (50). Using a series of overlapping primers, six PCR fragments, which covered the whole HCV genome, were then amplified and sequenced (33, 50).
Luciferase assay.
At the time of Luc assay, the medium was removed from triplicate wells for each construct, and the cells were washed twice in PBS. Fifty microliters of 1× Cell Culture Lysis Reagent (CCLR) buffer (made from a 5× stock; Promega) was added to each well of the 24-well plate, and the plates were gently rocked at room temperature for 15 min to lyse the cells. The lysate was removed from the plate and transferred to a 1.5-ml tube. The supernatant was transferred to a fresh tube after a brief spin at 12,000 × g in a microcentrifuge. Twenty microliters of the lysate was then combined with 20 μl of Luc assay substrate (Promega) and quickly mixed by vortexing prior to enzyme assay in a luminometer.
Immunoblot of HCV and host proteins.
Transfected or infected cells were lysed in RIPA buffer (150 mM NaCl, 50 mM Tris, pH 8.0, 1 mM EDTA, 1% NP-40, 0.1% SDS, 1 mM phenylmethylsulfonyl fluoride, and 2 g/ml leupeptin), and an appropriate amount of protein was resuspended in 4× sample buffer (240 mM Tris, pH 6.8, 4% SDS, 40% glycerol, 4% β-mercaptoethanol, 0.01% bromophenol blue) and boiled for 10 min. The proteins were separated on SDS-PAGE, followed by transfer onto an Immobilon-P membrane (polyvinylidene difluoride [PVDF]; Millipore). After binding with the respective primary antibody and horseradish peroxidase (HRP)-conjugated secondary antibody, proteins were visualized by the enhanced-chemiluminescence (ECL) detection method (Pierce, Rockford, IL).
Indirect immunofluorescence assay.
Transfected cells were seeded onto coverslips in 10-cm dishes or 6-well plates. At 72 h posttransfection, the cells on coverslips were washed with PBS and fixed for 10 min in 4% formaldehyde-PBS. The cells were then permeabilized for 5 min at room temperature in 0.05% Triton X-100–PBS, followed by staining of viral proteins with NS4B or NS5A rabbit polyclonal antibody, core mouse monoclonal antibody, and Alexa Fluor 488- or 594-conjugated secondary antibody. After three washes in PBS, the cells were stained with Deep Red Lipid-Tox neutral lipid stain (Invitrogen; 1:1,000 dilution) for 5 min at room temperature, followed by three more washes in PBS. The cells were mounted on glass slides with Vectashield (Vector Laboratories, Inc., Burlingame, CA) mounting buffer and sealed with nail polish. Immunostained samples were analyzed with a Leica TCS SP5 confocal microscope. Digital images were processed and analyzed with Fiji software.
RESULTS
An NS4B C-terminal-domain swap in HCV JFH1 attenuates virus production, but not genome replication.
We recently reported that the chimeric virus J/Con-C, made by replacing the NS4B CTD of genotype 2a (G2a) strain JFH1 (41–43) with the corresponding part in G1b strain Con1, was defective in virus production but could efficiently replicate its genome (33). This finding is likely the result of NS4B CTD amino acid substitutions between JFH1 and Con1 viruses (Fig. 1A, boldface or underlined). Thus, we predicted that the NS4B CTD swap between JFH1 and the G1a strain H77 (Fig. 1A) would also attenuate JFH1 production, since Con1 and H77 NS4B CTD sequences are quite similar, with the exception of two nonconservative (Q235A and E255S) and three conservative (N254S, D256E, and S258T) amino acid substitutions (Fig. 1A).
Fig 1.

The NS4B protein is involved in HCV production. (A) Alignment of HCV NS4B CTD amino acid sequences from HCV strains JFH1 (G2a), Con1 (G1b), and H77 (G1a). The stars indicate conserved residues. Nonconservative amino acid substitutions between Con1, H77, and JFH1 are in boldface, whereas conservative substitutions are underlined. The dashed boxes indicate residues whose mutation to alanine had no impact on JFH1 virus production (51). The locations of the two alpha helices (helix 1 and helix 2) are also indicated. (B) Schematic of the genomes used to determine the impacts of the NS4B CTD swaps on HCV production. (C) Huh-7.5 cells were electroporated with 1 μg of JFH1 or chimeric RNA. At day 3 or 6 p.t., the cell supernatant was collected, and virus infectivity was measured using the limiting dilution assay (43). The results are representative of at least three independent experiments, each containing triplicate samples. (D) Cells were infected with virus supernatants from the experiment in panel C at an MOI of 0.01, and virus titers were determined at the indicated time points as for panel C. (E) Schematic of the genomes used to measure the impacts of the NS4B CTD swaps on HCV genome replication. (F) Ten micrograms of JFH1 or chimeric replicon RNA (diagrammed in panel E) were electroporated into Huh-7.5 cells, and HCV replication was measured by luciferase reporter activity in cell lysates at 4, 24, 48, and 72 h p.t. The values represent the fold increase in luciferase activity normalized to that at 4 h and to the replication-defective (GND) replicon. The results are representative of at least two independent experiments with triplicate samples. The error bars indicate standard deviations. RLU, relative light units.
We therefore engineered a JFH1 chimera in which the NS4B CTD was replaced with the H77 NS4B CTD to generate the J/H-C virus. The various genomes (JFH1, J/Con-C, and J/H-C) (Fig. 1B) were transcribed in vitro and electroporated into Huh-7.5 cells. At 3 and 6 days p.t., supernatants were collected, and virus titers were determined as focus-forming units/ml (33). As shown in Fig. 1C, cells transfected with the chimeric virus genomes displayed at least a 5-fold (J/Con-C) to 10-fold (J/H-C) decrease in released virus infectivity by day 3 p.t. This defect was even greater (more than 100-fold decrease) when the growth kinetics of the chimeric viruses were analyzed by infecting Huh-7.5 cells at a low multiplicity of infection (MOI) of 0.01 (Fig. 1D). As previously reported (33), the differences in virus titers between Fig. 1C and D can be attributed to the finding that at 48 h postelectroporation, ca. 80% of the cells were positive for NS5A. Therefore, the virus titers from the electroporation results may represent single-step virus growth due to high intracellular levels of virus RNA, whereas for infection at a low MOI (0.01), the difference in virus titers may be the result of multiple cycles of virus infection. Thus, JFH1 produces more infectious virus by 6 days postinfection (p.i.) than J/Con-C and J/H-C viruses, and this difference increases exponentially by 12 days p.i. as more naive cells are infected. Note that virus titers at day 1 and day 2 p.t. (Fig. 1C) were not measured because JFH1 levels at these time points were fairly low (usually less than 2 log units).
To determine whether the attenuation in chimeric virus titers could be explained by a simple decrease in genome replication, Luc-expressing chimeric replicons were engineered (Fig. 1E) and transfected into Huh-7.5 cells, and HCV replication efficiency was monitored in the cell extracts. As shown in Fig. 1F, Luc-J/Con-C and Luc-J/H-C RNAs displayed a delay in genome replication at 48 h p.t., but replication efficiency approached JFH1 levels by 72 h p.t. These data suggest that the NS4B CTD swaps have a marked effect on HCV production but only a minor impact on genome replication.
The NS4B CTD swaps have a negative impact on JFH1 assembly.
Since NS4B CTD swaps did not significantly alter JFH1 genome replication, the data in Fig. 1C and D imply that the NS4B swaps negatively impact postreplication events, such as virus assembly or release. Indeed, two NS4B CTD mutations were recently reported to alter HCV assembly or release (38), but the underlying mechanisms are unknown. Hence, if the mutant viruses, J/Con-C and J/H-C, were defective in assembly, we predicted a marked decrease in cell-associated (intracellular) virus infectivity relative to extracellular (supernatant) virus titers. To test this prediction, Huh-7.5 cells were transfected with the various genomes, and at day 3 p.t., intra- as well as extracellular virus titers were measured as outlined in Fig. 2A. Indeed, Fig. 2B shows that the titers of the extracellular mutant viruses were higher than those of their intracellular counterparts, implying that these viruses were all competent for release. In contrast, the titers of the intracellular mutant viruses were 8- to 20-fold lower than those of JFH1. Additionally, Fig. 2C demonstrates that most of the infectivity in JFH1 and mutant viruses was associated with the extracellular virus. Taken together, these data imply that the mutant viruses with NS4B CTD swaps are defective at the assembly but not the release step. To address the possibility that NS4B affected HCV infectivity in addition to virus assembly, we determined the specific infectivity (virus titer/RNA level) of the crude extracellular virus in the cell culture supernatant. As can be seen in Table 1, the specific infectivity of the mutant viruses was similar to that of their JFH1 counterpart, implying that these viruses were as infectious as JFH1 or that the NS4B chimera did not lead to a putative defect in HCV entry.
Fig 2.
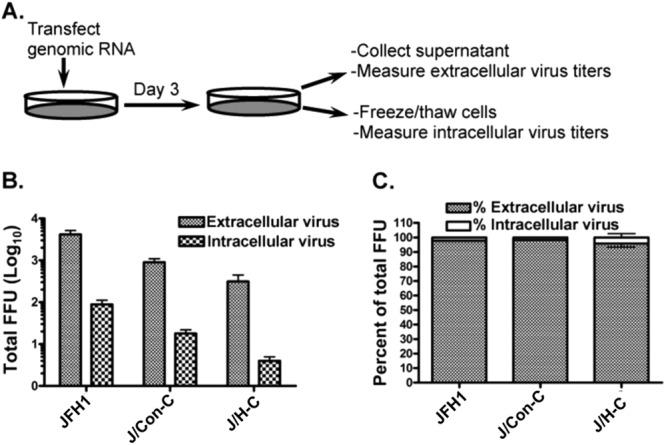
The NS4B chimeric genomes are defective at the virus particle assembly step. (A) Schematic of virus infection and titration. (B and C) One microgram of JFH1 or chimeric viral genomes was electroporated into Huh-7.5 cells. At day 3 p.t., extracellular and intracellular viruses were collected. Virus titers were measured as for Fig. 1C and D and expressed as total FFU (B) or a percentage of the total FFU (C). Virus titers are representative of five independent experiments. The error bars indicate standard deviations.
Table 1.
Specific infectivity associated with released virusa
| Virus | Titer (FFU/ml) | RNA level (copy no./ml) | Specific infectivity (virus titer/RNA level) |
|---|---|---|---|
| JFH1 | 1,900 ± 346 | 3.44 × 105 ± 9.47 × 103 | 5.53 × 10−3 ± 1.00 × 10−3 |
| J/Con-C | 257 ± 15 | 4.57 × 104 ± 5.48 × 103 | 5.63 × 10−3 ± 0.33 × 10−3 |
| J/H-C | 80 ± 26 | 1.46 × 104 ± 1.59 × 103 | 5.46 × 10−3 ± 1.78 × 10−3 |
| JGND | ND | 3.58 × 102 ± 1.70 × 102 | NA |
Data are representative of two independent experiments described in Materials and Methods. ND, not detectable; NA, not applicable.
The revertant viruses can efficiently assemble their genomes.
To further elucidate the mechanism by which the NS4B CTD swaps negatively impact HCV production, transfected cells were passaged every 3 days (Fig. 3A) to obtain revertant viruses. Typically, cells infected with JFH1 virus stopped dividing by day 12 p.t. (Fig. 3B and C) and died thereafter, whereas those containing J/Con-C or J/H-C virus could be passaged for at least 30 days. When the culture supernatant was tested for infectivity, the J/H-C virus titer reached its maximum level at day 54 p.t. (Fig. 3B, arrow), whereas a second round of transfections and passaging was performed for J/Con-C virus to reach JFH1 titer levels at day 30 p.t. (Fig. 3C, arrow). To confirm the phenotypes of the revertant viruses, we infected naive Huh-7.5 cells at an MOI of 0.01 with extracellular virus obtained at day 54 (J/H-C cells) and day 30 (J/Con-C cells, 2nd round) p.t. For controls, cells were infected with day 12 supernatants from JFH1 or parental chimeric viruses (J/Con-C and J/H-C) at low titers. As shown in Fig. 3D, JFH1 virus titers were ca. 10-fold higher than those of the parental chimeric viruses by day 9 p.i. In contrast, infection of naive cells with day 30 and day 54 supernatants led to virus titers that were more than 100-fold higher than those of JFH1 or the parental chimeric viruses at day 6 p.i. These data imply that the day 30 and 54 supernatants contained revertant viruses. J/Con-C-Ad1 and J/H-C-Ad1 refer to the revertant viruses obtained from J/Con-C and J/H-C viruses, respectively.
Fig 3.

Cell culture-adaptive viruses replicate as well as JFH1. (A) Schematic of the strategy used to obtain adaptive viruses. (B and C) One microgram of genomic viral RNA was transfected into Huh-7.5 cells, followed by collection of cell culture supernatant and serial passage of transfected cells every 3 days. The supernatant virus infectivity was measured as for Fig. 1C and D. The arrows indicate peak titer levels for the revertant virus supernatant at day 54 (J/H-C-Ad1) and day 30 (J/Con-C-Ad1), which were used in subsequent studies. (D) Huh-7.5 cells were infected at an MOI of 0.01 with parental and revertant viruses (J/Con-C-Ad1 and J/H-C-Ad1) from panels B and C, and supernatant virus titers were determined at various days postinfection. The titers are representative of two independent experiments. (E) Huh-7.5 cells were infected as for panel D. At day 9 postinfection, extracellular or intracellular virus titers were measured. The virus titers are representative of two independent experiments. The error bars indicate standard deviations.
The finding that the revertant viruses had higher titers than JFH1 led us to test whether these viruses were more competent for assembly or release. Hence, naive cells were infected with the various viruses (from day 9 p.i.) (Fig. 3D), and intracellular, as well as extracellular, virus titers were determined at day 6 p.i. As shown in Fig. 3E, intracellular J/Con-C-Ad1 and J/H-C-Ad1 virus titers were more than 100-fold higher than those of JFH1. These results imply that the revertant viruses had become more efficient at assembling infectious HCV particles than their parental viruses or JFH1. However, these data do not completely rule out the possibility that the revertant viruses had increased replication efficiency compared to JFH1.
The adaptive mutations enhance chimeric virus production rather than replication.
To identify the mutations responsible for the enhanced J/Con-C-Ad1 or J/H-C-Ad1 virus titers, each revertant virus genome was sequenced from a cDNA pool obtained from the infected cells. J/Con-C-Ad1 and J/H-C-Ad1 were found to contain four and two mutations, respectively. Interestingly, two mutations (NS4B N216S and NS5A C465S) were found in both revertant viruses, whereas two other mutations (p7 H31L and NS4B I236T) were detected only in J/Con-C-Ad1 virus. We first tested the possibility that these mutations would increase virus production by stimulating replication (Fig. 4A). This was achieved by measuring the impacts of single or combined mutations on parental luciferase replicons. Figure 4B to E shows that the NS4B and NS5A mutations had negligible impacts on J/Con-C or J/H-C replication efficiency. Most of these mutations resulted in replication kinetics similar to those of the parent chimera: lower replication efficiency at day 2 p.t. but reaching JFH1 levels by day 6 p.t. These data imply that the adaptive mutations did not increase virus titers by stimulating virus genome replication. The impact of the p7 mutation on genome replication was not tested because the replicon contains only the NS3-5B protein-coding region.
Fig 4.

Adaptive mutations do not markedly alter chimeric genome replication efficiency. (A) Steps for measuring HCV replication efficiency. (B to E) Huh-7.5 cells were transfected with 10 μg of JFH1, J/Con-C, or J/H-C replicon RNA with or without the adaptive mutations. At various times posttransfection, the cell extracts were tested for luciferase activity as for Fig. 1F. The impacts of single (B and C) or combined (D and E) mutations on chimeric genome replication were measured. The results are representative of at least two independent experiments with triplicate samples. The error bars indicate standard deviations.
Inasmuch as the adaptive mutations had no significant impact on the chimeric viruses' replication, we postulated that they enhanced postreplication events, in light of the findings shown in Fig. 3E. Thus, single and combined mutations were inserted into the chimeric genomes and tested for their impacts on virus production. First, virus RNAs were transfected into Huh-7.5 cells, and at day 6 p.t., extracellular virus titers were measured. Huh-7.5 cells were then infected at an MOI of 0.01 to rule out differences in virus RNA transfection efficiency (Fig. 5A). As shown in Fig. 5B to D, the adaptive mutations had various impacts on the titers of the J/Con-C and J/H-C viruses. Whereas NS4B I236T had no effect on infectious J/Con-C virus titers (data not shown), p7 H31L, NS4B N216S, and NS5A C465S mutations each resulted in ca. 5-, 50-, and 150-fold increases, respectively, in J/Con-C virus infectivity (Fig. 5B) by day 12 p.i. None of these mutations alone increased J/Con-C titers to JFH1 levels. Further, any combination of two adaptive mutations led to J/Con-C titers similar to those of JFH1, whereas the three mutations led to slightly higher virus titers (Fig. 5C). Nevertheless, the impacts of H31L, C465S, and N216S mutations on J/Con-C infectivity are additive, as these mutations did not result in an exponential increase in J/Con-C titers (Fig. 5C). Similarly, the C465S and N216S single mutations increased J/H-C virus titers but had an additive effect when they were both present in the J/H-C genome (Fig. 5D).
Fig 5.

Adaptive mutations enhance chimeric virus production. (A) Steps for measuring HCV titers. (B to D) Huh-7.5 cells were infected with various virus supernatants at an MOI of 0.01, followed by virus titration at the indicated time points. The titers are representative of two independent assays. The error bars indicate standard deviations.
The adaptive mutations enhance chimeric virus genome encapsidation efficiency.
The likelihood that the chimeric viruses were defective in assembly led us determine the RNA encapsidation efficiencies of these viruses with or without adaptive mutations. We chose to further investigate J/H-C assembly, since this virus was more attenuated than J/Con-C. Hence, JFH1, J/H-C, and the J/H-C-Ad1 genomes with the NS4B N216S and NS5A C465S mutations were electroporated into Huh-7.5 cells. To exclude multiple cycles of infection and therefore different levels of HCV protein expression among the constructs, we collected the virus samples at day 2 p.t. Intracellular virus titers were determined as for Fig. 2 and confirmed our finding that the chimeras are assembly incompetent relative to JFH1 (Fig. 6A). Indeed, the J/H-C-transfected cells produce ca. 6- and 10-fold fewer infectious virus particles than the J/H-C-Ad1 and JFH1 genomes, respectively.
Fig 6.

Adaptive mutations enhance chimeric virus genome encapsidation. Huh-7.5 cells were transfected with various HCV RNA genomes. Forty-eight hours later, the cells were freeze-thawed, intracellular virus particles (VP) were collected from the crude lysate, and the titer was determined (A) or the particles were purified on a 20% sucrose cushion (B to D). (B) A fraction (1/75) of the proteins from total cell lysate and 1/4 of the total viral particles were separated on SDS-PAGE, and core protein levels were detected via immunoblotting with core-specific mouse monoclonal antibody. (C) Core protein levels in panel B were quantified with ImageJ software (left y axis), and the ratio of intracellular viral particle core to total core was determined (right y axis). (D) Total intracellular viral RNA and viral particle-associated RNA were quantified as copy numbers per microgram of total cellular RNA (left y axis). The ratio of viral particle RNA to total intracellular viral RNA was also determined (right y axis). Note that J/H-C-Ad1 refers to J/H-C (N216S plus C465S). The results are representative of three independent experiments. The error bars indicate standard deviations.
For RNA genome encapsidation analysis, transfected cells were homogenized, and HCV particles were pelleted through a 20% sucrose cushion. Total core (virus- and cell-associated) protein levels, as well as pelleted core (virus-associated) protein levels, were determined via immunoblotting. As shown in Fig. 6B and C, comparable levels of total core protein were found in cells transfected with J/H-C, J/H-C-Ad1, and JFH1. Additionally, the pelleted intracellular J/H-C virus particles displayed core protein levels similar to those of J/H-C-Ad1 and JFH1 (Fig. 6B and C). Finally, compared to JFH1 and J/H-C-Ad1 viruses, J/H-C-infected cells displayed similar HCV RNA levels in the total intracellular lysate (Fig. 6D). These findings are all consistent with the interpretation that the chimeric NS4B protein has negligible impact on JFH1 RNA translation or replication. Surprisingly, J/H-C-infected cells displayed significantly less viral RNA associated with pelleted virus particles, resulting in an approximately 5-fold decrease in the ratio of putative VP RNA to total RNA (Fig. 6D). This ratio was 3-fold higher in the revertant virus J/H-C-Ad1 (Fig. 6D). Altogether, these results imply that the chimeric virus J/H-C is defective in HCV RNA encapsidation.
The chimeric viruses display a different NS5A phosphorylation state.
How NS4B protein facilitates HCV particle assembly is currently unknown. In one scenario, NS4B has a direct role in virion production by helping to chaperone the RNA in the HCV assembly complex, but there is no strong evidence that NS4B binds to the positive-sense HCV RNA genome. Alternatively, NS4B may facilitate virion production via interaction with virus and host factors. Indeed, there is evidence that NS4B binds to NS5A (28, 40), a protein involved in HCV assembly (11, 14). Additionally, the NS5A phosphorylation state plays a role in HCV production, with data suggesting that hyperphosphorylated NS5A, or NS5A p58, plays a role in HCV production (14). Koch and Bartenschlager (25) also reported that the integrity of the NS4B CTD plays some role in NS5A p58 formation. Last, but not least, two adaptive mutations in the revertant viruses are located in the C-terminal domains of NS4B and NS5A (NS4B N216S and NS5A C465S, respectively), suggesting perhaps a genetic interaction between NS4B and NS5A via these domains. Thus, we postulated that the HCV NS4B CTD swaps have a negative impact on the JFH1 NS5A hyperphosphorylation state. To test this hypothesis, Huh-7.5 cells were transfected with JFH1, chimeric, or adaptive virus genomes. At day 3 p.t., cell extracts were prepared, followed by immunoblotting (Western blotting [WB]) with NS5A-specific mouse monoclonal antibody. As shown in Fig. 7A, while the NS5A p56 (basal NS5A phosphorylation) and NS4B protein levels were quite similar among the different viruses, the NS5A p58 levels were sharply decreased in cells transfected with the chimeric genomes, especially in cells expressing the J/H-C virus. Quantitative analysis indicated a marked decrease in the NS5A p58/p56 ratio in the chimera relative to JFH1 (Fig. 7B) (P < 0.01). Interestingly, there was only a small increase in the p58/p56 ratio (10%) in the revertant viruses compared to their parents (Fig. 7B) (P > 0.1). Additionally, total NS5A (p56 plus p58), but not the NS3 level, was markedly reduced in the mutant viruses relative to JFH1 (Fig. 7C). These data imply that JFH1 NS4B CTD swaps inhibit NS5A p58 formation and/or NS5A p58 stability.
Fig 7.

The chimeric virus genomes show a decrease in the NS5A hyperphosphorylation state. (A) Ten micrograms of various viral genomes was electroporated into Huh-7.5 cells. At day 3 p.t., cell extracts were prepared and separated on SDS-PAGE, followed by immunoblotting with NS5A-, NS3-, NS4B-, or GAPDH-specific antibody. Note that J/Con-C-Ad1 and J/H-C-Ad1 refer to J/Con-C (N216S plus C465S) and J/H-C (N216S plus C465S), respectively. The results are representative of three independent assays. (B) The ratio of hyperphosphorylated NS5A (p58) to basally phosphorylated NS5A (p56), as in panel A, was quantified using ImageJ software and normalized with the corresponding GAPDH level. **, P < 0.01; *, P > 0.1. The error bars indicate standard deviations. (C) NS3 and NS5A protein levels, as in panel A, were quantified and normalized as in panel B. Note that the results in panels B and C were calculated from three independent experiments. (D) Cells were transfected with a vector expressing HCV NS3-NS5B polyprotein. One hundred fifty micrograms of total protein was separated on SDS-PAGE, followed by immunoblotting with NS4B-, NS3-, or NS5A-specific antibody. (E) Ten micrograms of various viral genomes was electroporated into Huh-7.5 cells, and lysates were processed as in panel A. The results are representative of two independent assays.
To rule out any difference in the NS5A p58/p56 ratio resulting from replication or virus production of the various constructs, we measured NS4B, NS5A, and NS3 levels in cells expressing the HCV polyprotein (NS3-4A, -4B, -5A, and -5B) under the cytomegalovirus (CMV) promoter. As seen in Fig. 7D, NS4B and NS3 levels were similar in cells expressing JFH1 and J/H-C polyproteins, but a significant decrease in the NS5A p58 level was still observed in cells expressing J/H-C polyprotein relative to JFH1. These data indicate that NS4B plays a role in regulating the hyperphosphorylation state of NS5A, regardless of HCV replication.
Since the NS5A p58/p56 ratio of the revertant viruses was 10% higher than that of their parents, we sought to identify the mutation(s) responsible for such an increase. Thus, cells were transfected with various chimeric genomes containing NS4B N216S and NS5A C465S mutations, singly or in combination, and the NS5A p56 and p58 levels were determined. As shown in Fig. 7E, both mutations could lead to some increase in the NS5A p58 level when introduced singly, but the increase was greater when they were combined. Taken together, the data indicate that a defect in NS5A p58 formation might be in part responsible for the low efficiency of chimeric virus RNA encapsidation, but the NS5A p58 requirement was bypassed by the NS4B and NS5A mutations in the revertant viruses.
The chimeric NS4B protein does not significantly alter NS5A association with core and lipid droplets.
LDs are ER-derived intracellular organelles containing esterified lipids surrounded by a single membrane leaflet. Recent studies have indicated a link between LDs and the early steps in HCV assembly (16). Indeed, the presence of core protein on the surfaces of LDs and the recruitment of NS5A to these structures appear to be essential steps in infectious HCV particle assembly (16). Hence, the low efficiency of RNA genome encapsidation, coupled with decreased NS5A p58 levels, led us to predict an alteration in the costaining between NS5A and core proteins around LDs in cells infected with the chimeric J/H-C virus. To test this prediction, Huh-7.5 cells were transfected with various virus genomes, and the subcellular distribution of NS5A and core proteins was determined via confocal microscopy. As previously reported (16), both core and NS5A proteins were present around LDs and displayed significant colocalization in JFH1-infected cells (Fig. 8A, i to v). Surprisingly, a similar subcellular distribution of core and NS5A proteins was observed in cells infected with the chimeric virus J/H-C (Fig. 8A, vi to x) and the adapted virus J/H-C-Ad1 (Fig. 8A, xi to xv). We confirmed these results by determining the ratio of NS5A colocalized with core to total NS5A in 15 infected cells (Fig. 8B).
Fig 8.

The chimeric NS4B protein does not significantly alter NS5A association with core and lipid droplets. Huh-7.5 cells were transfected with various viral genomes. (A) At day 3 p.t., the cells were processed for confocal microscopy with rabbit polyclonal anti-NS5A antibody (i, vi, and xi, green), mouse monoclonal anti-core antibody (ii, vii, and xii, red), and Deep Red LipidTox (i to xv, blue). The boxed areas in iv, ix, and xiv are magnified in v, x, and xv, respectively. (B) For each infection, confocal images were taken of 15 representative cells. The ratio of NS5A colocalized with core pixels to total NS5A pixels was calculated with the JACoP plug-in in Fiji software. Each circle (JFH1), square (J/H-C), or triangle (J/H-C-Ad1) represents one cell.
We also determined the impact of the NS4B CTD swap on putative NS4B association with the core protein or NS5A protein, respectively. NS4B colocalized with NS5A around LDs in JFH1-infected cells, and we observed no marked change in the distribution of these proteins in J/H-C- and J/H-C-Ad1-infected cells (data not shown). These data imply that the NS4B CTD swap did not result in a marked alteration in core, NS5A, or NS4B cellular distribution that could explain the defect in the JFH1 chimeric virus assembly.
DISCUSSION
There is increasing evidence showing that the NS4B protein plays a role in post-HCV replication steps during HCV infection. For example, Jones et al. (51) reported that a mutation in JFH1 NS4B (N216A) had no impact on genome replication but significantly increased the infectious virus load. Further, Paul et al. (38) identified two basic NS4B residues (H217 and H250) that are involved in HCV assembly and release. A concurrent study by Han et al. (33) showed that swapping HCV NS4B CTD sequences between JFH1 and Con1 leads to attenuation of the JFH1 titer. However, the mechanisms underlying the NS4B role in post-HCV genome replication are still unknown.
In this report, we confirmed our previous finding that NS4B protein is involved in HCV production (33), in addition to its broadly recognized function in genome replication. We further revealed that the NS4B CTD swaps lead to a defect in HCV genome encapsidation, consistent with an NS4B role in postreplication events. We hypothesized that second-site mutations in revertant viruses would provide further understanding of the putative NS4B interactions that regulate HCV assembly. Indeed, we successfully generated revertant viruses that assemble their genomes nearly as efficiently as wild-type JFH1. In agreement with their role in virus production, adaptive mutations from the revertant viruses had no impact on HCV genome replication but could rescue the defect in chimeric virus production to almost wild-type JFH1 level. More importantly, the second-site mutations responsible for the revertant phenotype were found in the NS4B and NS5A genes. These data suggest for the first time that genetic interactions between NS4B and NS5A might contribute to HCV production. Finally, taking advantage of the chimeric and revertant viruses, we demonstrate that NS4B facilitates HCV genome encapsidation, in part via regulation of the NS5A phosphorylation state or stability. Thus, these findings identify NS4B as a key regulator of the transition from the HCV replication complex to the virus assembly complex.
In our first attempt to define the role of NS4B in HCV assembly, we used a continuous iodixanol gradient to purify both intracellular and extracellular virus particles with the goal of defining the biophysical properties of the chimeric viruses, as reported previously (12, 48). However, these studies were inconclusive due to the low virus titers of the chimera (or JFH1) and core protein levels in the collected virus fractions (data not shown). Thus, we used sucrose cushion, a less ideal approach, to concentrate and investigate the properties of the intracellular virus particles. From this study, it was apparent that core protein levels in the chimeric virus particles were similar to those in JFH1, although the chimeric virus RNA levels were markedly lower. Although speculative, these findings suggest that the NS4B CTD swap causes the virus to have a significant number of empty virus particles. Alternatively, the intracellular chimeric virus core proteins do not efficiently form a capsid. The use of the high-titer Jc1 virus (41, 52) in future studies will help provide further insight into the biophysical properties of the NS4B chimeric viruses.
The exact location of HCV assembly complexes remains controversial. Miyanari et al. (16) elegantly demonstrated that a crucial step in HCV JFH1 production requires binding of the core protein to LDs, followed by the recruitment of the HCV replication complex, including NS5A, the viral RNA, and the other nonstructural proteins. An equally elegant report demonstrated that the localization of HCV Jc1 core protein to the ER membranes is crucial for efficient HCV Jc1 virus assembly and production (6). Additionally, Boson et al. (6) found that high-titer JFH1, with adaptive mutations, has ER-localized core protein relative to WT JFH1. Therefore, both the ER and LDs appear to be sites for HCV assembly, with the ER compartment favoring higher-titer-producing Jc1 virus while LDs are favored by JFH1. Thus, how NS4B contributes to HCV assembly might depend on the virus strain or where this process takes place. NS4B is an integral membrane protein, with at least four TMDs (29, 33). As such, NS4B's role in JFH1 assembly on LDs could be envisioned in multiple scenarios. For example, NS4B might help to transfer the positive-sense viral RNA from the replication complex to the assembly site. However, the finding that NS4B binds tightly only to the 3′ untranslated region of the HCV negative-sense RNA (Kd, ca. 3 nM) (39) argues against NS4B's role in transporting the viral RNA to the assembly site. Given its multiple TMDs, it is also unlikely that NS4B can bind directly to LDs, the site of JFH1 assembly, unless such interaction occurs via NS4B C-terminal-domain palmitoylation (53). Note that the putative NS4B palmitoylation motif is found in HCV JFH1, Con1, and H77 (the two cysteine residues at the C-terminal end of NS4B [Fig. 1A]) and therefore likely does not account for the low chimeric virus titers in this study. Taking the data together, it is likely that NS4B exerts its influence on HCV assembly via regulation of host or viral factors.
Using a membrane yeast two-hybrid approach, several putative NS4B host partners were identified (Konan laboratory, unpublished data). One such protein is 3-beta-hydroxysteroid-delta(8),delta(7)-isomerase, also known as emopamil binding protein (EBP) (54–56). EBP is an enzyme in the cholesterol biosynthetic pathway. Additionally, HCV NS4B activates sterol regulatory element binding proteins 1 and 2 (SREBP-1 and -2) (57, 58), two proteins involved in the fatty acid and cholesterol biosynthetic pathways, respectively (59, 60). Thus, while NS4B interaction with EBP has yet to be confirmed in mammalian cells, these findings suggest that NS4B facilitates HCV assembly in part by regulating the cholesterol and fatty acid biosynthetic pathways. These pathways in turn control the biogenesis of LDs and other intracellular membrane structures involved in HCV assembly. Although speculative, since the Con1 and H77 strains do not produce high virus titers (44, 61), it is conceivable that these viruses do not alter the cellular lipid metabolism in a way that is favorable to the assembly of highly infectious virus particles. If this is true, then the C-terminal domain of the NS4B protein might be involved in regulating host lipid metabolism.
The evidence in this study suggests that NS4B facilitates HCV assembly via regulation of virus factors. This assertion is supported by the adaptive mutations (p7 H31L, NS4B N216S, and NS5A 465S) in the revertant viruses. Our data suggest that NS4B and NS5A mutations were responsible for most of the increase in the revertant virus titer, whereas the p7 H31L impact was minimal (Fig. 5). Interestingly, while the revertant viruses were at least 10-fold more infectious than JFH1 (Fig. 3), introduction of all the adaptive mutations in the chimeric viruses did result in a slightly more infectious virus than JFH1 (compare Fig. 5 to Fig. 3). Therefore, although the entire genomes from the revertant viruses were sequenced from pooled cDNA, it is conceivable that some mutations that were present in a subpopulation of highly infectious virus were missed during the sequencing reactions. Nevertheless, our study reveals a putative genetic interaction between NS4B and NS5A in the context of JFH1 production. This putative interaction is important, since NS5A (11, 13, 14, 62) is a key factor in the formation of the HCV assembly complex. We have attempted to demonstrate biochemical interaction between NS4B and NS5A in the context of JFH1 infection by performing NS4B pulldown followed by immunoblotting or immunoprecipitation of metabolically labeled cells. However, such experiments have been unsuccessful (data not shown), in part because of the low protein levels in infected cells, the quality of the primary antibody, or both.
Although the idea is controversial, NS5A hyperphosphorylation has been reported to play a role in HCV production (14). Interestingly, the NS4B C-terminal domain was found to facilitate NS5A conversion from its basal phosphorylation (NS5A p56) to the hyperphosphorylation state (NS5A p58) (25). In this study, we demonstrate that the chimeric viruses are defective in NS5A p58 formation. The adapted viruses, which have partially regained the NS5A p58 phenotype, have mutations located in the NS4B and NS5A C-terminal domains, both of which are associated with NS5A p58 formation. Additionally, the chimeric viruses are defective in HCV genome encapsidation. Taken together, these findings suggest that NS5A p58 regulates the transfer of the viral RNA from the site of genome replication to the LD-associated assembly complex. For example, NS5A p58 may bind to but readily release the viral RNA to core protein on LDs, whereas a defect in NS5A p58 could cause NS5A to bind tightly to the RNA, thus inhibiting assembly. The larger number of negative phosphate groups on NS5A p58 could account for this reduced interaction with the negatively charged HCV RNA backbone. This interpretation is supported by the finding that NS5A association with core and lipid droplets is not significantly altered in JFH1- and chimeric virus-infected cells (Fig. 8). How NS4B facilitates NS5A p58 formation is currently unknown. In one scenario, NS4B binds to NS5A, causing NS5A to adopt a conformation favorable for hyperphosphorylation by previously reported kinases (20–23). Conversely, NS4B might bind to and stimulate kinases to hyperphosphorylate NS5A. If this is correct, the failure of the revertant viruses to restore NS5A p58 to its wild-type levels (Fig. 7) may suggest that the adaptive mutations facilitate virus production independently of NS5A p58 production. Alternatively, the absolute level of NS5A, but not NS5A p58, is crucial for HCV production. Consistent with this observation, a closer look at NS5A expression in the chimeric viruses (Fig. 7) indicates a drop in NS5A p58 without a concomitant decrease in the NS5A p56 level. These findings imply that either NS5A p58 is not produced in the chimera at a level similar to that in JFH1 or the p58 form is not as stable in the chimera as it is in JFH1 (Fig. 7C). Thus, we postulate that the NS4B CTD binds to NS5A and inhibits its partial degradation or that the NS4B CTD binds to a putative protease, preventing NS5A degradation. In support of this hypothesis, calcium-dependent calpain protease is known to cleave NS5A (63). Additionally, zinc mesoporphyrin facilitates NS5A downregulation via induction of polyubiquitination and proteasome-mediated degradation (64).
In conclusion, this report has uncovered part of the mechanism by which NS4B facilitates HCV particle production. Future studies will focus on employing genetic, biochemical, and biophysical methods to gain further insights into NS4B's roles in HCV assembly and release.
ACKNOWLEDGMENTS
We are grateful to Takaji Wakita, Charles Rice, Craig Cameron, and Biao He for reagents, suggestions, and critical reading of the manuscript.
This work was supported by grants K22 CA129241 and 1R56AI087769-01A1 from the National Institutes of Health.
Footnotes
Published ahead of print 24 April 2013
REFERENCES
- 1. Averhoff FM, Glass N, Holtzman D. 2012. Global burden of hepatitis C: considerations for healthcare providers in the United States. Clin. Infect. Dis. 55(Suppl. 1):S10–S15 [DOI] [PubMed] [Google Scholar]
- 2. Lavanchy D. 2011. Evolving epidemiology of hepatitis C virus. Clin. Microbiol. Infect. 17:107–115 [DOI] [PubMed] [Google Scholar]
- 3. Bartenschlager R, Lohmann V. 2000. Replication of hepatitis C virus. J. Gen. Virol. 81:1631–1648 [DOI] [PubMed] [Google Scholar]
- 4. Kuiken C, Simmonds P. 2009. Nomenclature and numbering of the hepatitis C virus. Methods Mol. Biol. 510:33–53 [DOI] [PubMed] [Google Scholar]
- 5. Bartenschlager R, Sparacio S. 2007. Hepatitis C virus molecular clones and their replication capacity in vivo and in cell culture. Virus Res. 127:195–207 [DOI] [PubMed] [Google Scholar]
- 6. Boson B, Granio O, Bartenschlager R, Cosset FL. 2011. A concerted action of hepatitis C virus p7 and nonstructural protein 2 regulates core localization at the endoplasmic reticulum and virus assembly. PLoS Pathog. 7:e1002144. 10.1371/journal.ppat.1002144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ma Y, Anantpadma M, Timpe JM, Shanmugam S, Singh SM, Lemon SM, Yi M. 2011. Hepatitis C virus NS2 protein serves as a scaffold for virus assembly by interacting with both structural and nonstructural proteins. J. Virol. 85:86–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yi M, Ma Y, Yates J, Lemon SM. 2009. Trans-complementation of an NS2 defect in a late step in hepatitis C virus (HCV) particle assembly and maturation. PLoS Pathog. 5:e1000403. 10.1371/journal.ppat.1000403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blight KJ, Kolykhalov AA, Rice CM. 2000. Efficient initiation of HCV RNA replication in cell culture. Science 290:1972–1974 [DOI] [PubMed] [Google Scholar]
- 10. Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110–113 [DOI] [PubMed] [Google Scholar]
- 11. Appel N, Zayas M, Miller S, Krijnse-Locker J, Schaller T, Friebe P, Kallis S, Engel U, Bartenschlager R. 2008. Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog. 4:e1000035. 10.1371/journal.ppat.1000035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ma Y, Yates J, Liang Y, Lemon SM, Yi M. 2008. NS3 helicase domains involved in infectious intracellular hepatitis C virus particle assembly. J. Virol. 82:7624–7639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Masaki T, Suzuki R, Murakami K, Aizaki H, Ishii K, Murayama A, Date T, Matsuura Y, Miyamura T, Wakita T, Suzuki T. 2008. Interaction of hepatitis C virus nonstructural protein 5A with core protein is critical for the production of infectious virus particles. J. Virol. 82:7964–7976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tellinghuisen TL, Foss KL, Treadaway J. 2008. Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog. 4:e1000032. 10.1371/journal.ppat.1000032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yi M, Ma Y, Yates J, Lemon SM. 2007. Compensatory mutations in E1, p7, NS2, and NS3 enhance yields of cell culture-infectious intergenotypic chimeric hepatitis C virus. J. Virol. 81:629–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. 2007. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 9:1089–1097 [DOI] [PubMed] [Google Scholar]
- 17. Appel N, Pietschmann T, Bartenschlager R. 2005. Mutational analysis of hepatitis C virus nonstructural protein 5A: potential role of differential phosphorylation in RNA replication and identification of a genetically flexible domain. J. Virol. 79:3187–3194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang L, Hwang J, Sharma SD, Hargittai MR, Chen Y, Arnold JJ, Raney KD, Cameron CE. 2005. Hepatitis C virus nonstructural protein 5A (NS5A) is an RNA-binding protein. J. Biol. Chem. 280:36417–36428 [DOI] [PubMed] [Google Scholar]
- 19. Alvisi G, Madan V, Bartenschlager R. 2011. Hepatitis C virus and host cell lipids: an intimate connection. RNA Biol. 8:258–269 [DOI] [PubMed] [Google Scholar]
- 20. Kim J, Lee D, Choe J. 1999. Hepatitis C virus NS5A protein is phosphorylated by casein kinase II. Biochem. Biophys. Res. Commun. 257:777–781 [DOI] [PubMed] [Google Scholar]
- 21. Quintavalle M, Sambucini S, Di Pietro C, De Francesco R, Neddermann P. 2006. The alpha isoform of protein kinase CKI is responsible for hepatitis C virus NS5A hyperphosphorylation. J. Virol. 80:11305–11312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Quintavalle M, Sambucini S, Summa V, Orsatti L, Talamo F, De Francesco R, Neddermann P. 2007. Hepatitis C virus NS5A is a direct substrate of casein kinase I-alpha, a cellular kinase identified by inhibitor affinity chromatography using specific NS5A hyperphosphorylation inhibitors. J. Biol. Chem. 282:5536–5544 [DOI] [PubMed] [Google Scholar]
- 23. Reed KE, Gorbalenya AE, Rice CM. 1998. The NS5A/NS5 proteins of viruses from three genera of the family flaviviridae are phosphorylated by associated serine/threonine kinases. J. Virol. 72:6199–6206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reed KE, Xu J, Rice CM. 1997. Phosphorylation of the hepatitis C virus NS5A protein in vitro and in vivo: properties of the NS5A-associated kinase. J. Virol. 71:7187–7197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Koch JO, Bartenschlager R. 1999. Modulation of hepatitis C virus NS5A hyperphosphorylation by nonstructural proteins NS3, NS4A, and NS4B. J. Virol. 73:7138–7146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Neddermann P, Clementi A, De Francesco R. 1999. Hyperphosphorylation of the hepatitis C virus NS5A protein requires an active NS3 protease, NS4A, NS4B, and NS5A encoded on the same polyprotein. J. Virol. 73:9984–9991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hugle T, Fehrmann F, Bieck E, Kohara M, Krausslich HG, Rice CM, Blum HE, Moradpour D. 2001. The hepatitis C virus nonstructural protein 4B is an integral endoplasmic reticulum membrane protein. Virology 284:70–81 [DOI] [PubMed] [Google Scholar]
- 28. Aligo J, Jia S, Manna D, Konan KV. 2009. Formation and function of hepatitis C virus replication complexes require residues in the carboxy-terminal domain of NS4B protein. Virology 393:68–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Elazar M, Liu P, Rice CM, Glenn JS. 2004. An N-terminal amphipathic helix in hepatitis C virus (HCV) NS4B mediates membrane association, correct localization of replication complex proteins, and HCV RNA replication. J. Virol. 78:11393–11400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gouttenoire J, Castet V, Montserret R, Arora N, Raussens V, Ruysschaert JM, Diesis E, Blum HE, Penin F, Moradpour D. 2009. Identification of a novel determinant for membrane association in hepatitis C virus nonstructural protein 4B. J. Virol. 83:6257–6268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gouttenoire J, Montserret R, Kennel A, Penin F, Moradpour D. 2009. An amphipathic alpha-helix at the C terminus of hepatitis C virus nonstructural protein 4B mediates membrane association. J. Virol. 83:11378–11384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lundin M, Monne M, Widell A, Von Heijne G, Persson MA. 2003. Topology of the membrane-associated hepatitis C virus protein NS4B. J. Virol. 77:5428–5438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Han Q, Aligo J, Manna D, Belton K, Chintapalli SV, Hong Y, Patterson RL, van Rossum DB, Konan KV. 2011. Conserved GXXXG- and S/T-like motifs in the transmembrane domains of NS4B protein are required for hepatitis C virus replication. J. Virol. 85:6464–6479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lundin M, Lindstrom H, Gronwall C, Persson MA. 2006. Dual topology of the processed hepatitis C virus protein NS4B is influenced by the NS5A protein. J. Gen. Virol. 87:3263–3272 [DOI] [PubMed] [Google Scholar]
- 35. Egger D, Wolk B, Gosert R, Bianchi L, Blum HE, Moradpour D, Bienz K. 2002. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 76:5974–5984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gosert R, Egger D, Lohmann V, Bartenschlager R, Blum HE, Bienz K, Moradpour D. 2003. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J. Virol. 77:5487–5492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Konan KV, Giddings TH, Jr, Ikeda M, Li K, Lemon SM, Kirkegaard K. 2003. Nonstructural protein precursor NS4A/B from hepatitis C virus alters function and ultrastructure of host secretory apparatus. J. Virol. 77:7843–7855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Paul D, Romero-Brey I, Gouttenoire J, Stoitsova S, Krijnse-Locker J, Moradpour D, Bartenschlager R. 2011. NS4B self-interaction through conserved C-terminal elements is required for the establishment of functional hepatitis C virus replication complexes. J. Virol. 85:6963–6976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Einav S, Gerber D, Bryson PD, Sklan EH, Elazar M, Maerkl SJ, Glenn JS, Quake SR. 2008. Discovery of a hepatitis C target and its pharmacological inhibitors by microfluidic affinity analysis. Nat. Biotechnol. 26:1019–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dimitrova M, Imbert I, Kieny MP, Schuster C. 2003. Protein-protein interactions between hepatitis C virus nonstructural proteins. J. Virol. 77:5401–5414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626 [DOI] [PubMed] [Google Scholar]
- 42. Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 102:9294–9299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pietschmann T, Zayas M, Meuleman P, Long G, Appel N, Koutsoudakis G, Kallis S, Leroux-Roels G, Lohmann V, Bartenschlager R. 2009. Production of infectious genotype 1b virus particles in cell culture and impairment by replication enhancing mutations. PLoS Pathog. 5:e1000475. 10.1371/journal.ppat.1000475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 76:13001–13014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kato T, Date T, Miyamoto M, Furusaka A, Tokushige K, Mizokami M, Wakita T. 2003. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology 125:1808–1817 [DOI] [PubMed] [Google Scholar]
- 47. Targett-Adams P, McLauchlan J. 2005. Development and characterization of a transient-replication assay for the genotype 2a hepatitis C virus subgenomic replicon. J. Gen. Virol. 86:3075–3080 [DOI] [PubMed] [Google Scholar]
- 48. Gastaminza P, Kapadia SB, Chisari FV. 2006. Differential biophysical properties of infectious intracellular and secreted hepatitis C virus particles. J. Virol. 80:11074–11081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhong J, Gastaminza P, Chung J, Stamataki Z, Isogawa M, Cheng G, McKeating JA, Chisari FV. 2006. Persistent hepatitis C virus infection in vitro: coevolution of virus and host. J. Virol. 80:11082–11093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Russell RS, Meunier JC, Takikawa S, Faulk K, Engle RE, Bukh J, Purcell RH, Emerson SU. 2008. Advantages of a single-cycle production assay to study cell culture-adaptive mutations of hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 105:4370–4375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jones DM, Patel AH, Targett-Adams P, McLauchlan J. 2009. The hepatitis C virus NS4B protein can trans-complement viral RNA replication and modulates production of infectious virus. J. Virol. 83:2163–2177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pietschmann T, Kaul A, Koutsoudakis G, Shavinskaya A, Kallis S, Steinmann E, Abid K, Negro F, Dreux M, Cosset FL, Bartenschlager R. 2006. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. U. S. A. 103:7408–7413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yu GY, Lee KJ, Gao L, Lai MM. 2006. Palmitoylation and polymerization of hepatitis C virus NS4B protein. J. Virol. 80:6013–6023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Paik YK, Billheimer JT, Magolda RL, Gaylor JL. 1986. Microsomal enzymes of cholesterol biosynthesis from lanosterol. Solubilization and purification of steroid 8-isomerase. J. Biol. Chem. 261:6470–6477 [PubMed] [Google Scholar]
- 55. Paik YK, Trzaskos JM, Shafiee A, Gaylor JL. 1984. Microsomal enzymes of cholesterol biosynthesis from lanosterol. Characterization, solubilization, and partial purification of NADPH-dependent delta 8,14-steroid 14-reductase. J. Biol. Chem. 259:13413–13423 [PubMed] [Google Scholar]
- 56. Silve S, Dupuy PH, Labit-Lebouteiller C, Kaghad M, Chalon P, Rahier A, Taton M, Lupker J, Shire D, Loison G. 1996. Emopamil-binding protein, a mammalian protein that binds a series of structurally diverse neuroprotective agents, exhibits delta8-delta7 sterol isomerase activity in yeast. J. Biol. Chem. 271:22434–22440 [DOI] [PubMed] [Google Scholar]
- 57. Park CY, Jun HJ, Wakita T, Cheong JH, Hwang SB. 2009. Hepatitis C virus nonstructural 4B protein modulates sterol regulatory element-binding protein signaling via the AKT pathway. J. Biol. Chem. 284:9237–9246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Waris G, Felmlee DJ, Negro F, Siddiqui A. 2007. Hepatitis C virus induces proteolytic cleavage of sterol regulatory element binding proteins and stimulates their phosphorylation via oxidative stress. J. Virol. 81:8122–8130 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 59. Horton JD, Goldstein JL, Brown MS. 2002. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109:1125–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jeon TI, Osborne TF. 2012. SREBPs: metabolic integrators in physiology and metabolism. Trends Endocrinol. Metab. 23:65–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yi M, Villanueva RA, Thomas DL, Wakita T, Lemon SM. 2006. Production of infectious genotype 1a hepatitis C virus (Hutchinson strain) in cultured human hepatoma cells. Proc. Natl. Acad. Sci. U. S. A. 103:2310–2315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kim S, Welsch C, Yi M, Lemon SM. 2011. Regulation of the production of infectious genotype 1a hepatitis C virus by NS5A domain III. J. Virol. 85:6645–6656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kalamvoki M, Mavromara P. 2004. Calcium-dependent calpain proteases are implicated in processing of the hepatitis C virus NS5A protein. J. Virol. 78:11865–11878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hou W, Tian Q, Zheng J, Bonkovsky HL. 2010. Zinc mesoporphyrin induces rapid proteasomal degradation of hepatitis C nonstructural 5A protein in human hepatoma cells. Gastroenterology 138:1909–1919 [DOI] [PMC free article] [PubMed] [Google Scholar]