

Abstract
Papillomaviruses have complex life cycles that are understood only superficially. Although it is well established that the viral E1 and E2 proteins play key roles in controlling viral transcription and DNA replication, how these factors are regulated is not well understood. Here, we demonstrate that phosphorylation by the protein kinase CK2 controls the biochemical activities of the bovine papillomavirus E1 and E2 proteins by modifying their DNA binding activity. Phosphorylation at multiple sites in the N-terminal domain in E1 results in the loss of sequence-specific DNA binding activity, a feature that is also conserved in human papillomavirus (HPV) E1 proteins. The bovine papillomavirus (BPV) E2 protein, when phosphorylated by CK2 on two specific sites in the hinge, also loses its site-specific DNA binding activity. Mutation of these sites in E2 results in greatly increased levels of latent viral DNA replication, indicating that CK2 phosphorylation of E2 is a negative regulator of viral DNA replication during latent viral replication. In contrast, mutation of the N-terminal phosphorylation sites in E1 has no effect on latent viral DNA replication. We propose that the phosphorylation of the N terminus of E1 plays a role only in vegetative viral DNA replication, and consistent with such a role, caspase 3 cleavage of E1, which has been shown to be necessary for vegetative viral DNA replication, restores the DNA binding activity to phosphorylated E1.
INTRODUCTION
The study of papillomaviruses has resulted in a fair understanding of the overall strategy that these viruses employ to infect their hosts and to generate new virus particles. Papillomaviruses infect the basal layers of the epithelium, where the early viral genes are expressed and the viral DNA is replicated at a low level (1). As the infected cells migrate toward the skin surface and differentiate into keratinocytes, the viral DNA is replicated at high levels, viral capsid proteins are produced, and new virus particles are assembled (1). In contrast to other well-studied viruses, reproduction of the viral life cycle in vitro is difficult but can be achieved with low efficiency (2–4). Consequently, although the general functions of the virus-encoded polypeptides are known, many subtleties, including the consequences of modifications of the viral polypeptides, ranging from alternative splicing to posttranslational modifications, have been difficult to analyze and are poorly understood.
The viral E1 and E2 proteins have been studied biochemically, genetically, and structurally and are among the best-studied polypeptides encoded by the papillomaviruses (5, 6). The E1 protein is a site-specific DNA binding protein that binds to the viral origin of DNA replication (ori) and opens the DNA duplex in preparation for initiation of DNA replication and also serves as the replicative DNA helicase (7–15). The E2 protein is a DNA binding transcription factor that can regulate viral transcription by binding to specific sites in the viral genome (16–21). The E2 protein is also required for initiation of viral DNA replication and binds cooperatively with E1 to the origin of DNA replication, forming an E12E22 complex (22–25). The E1 open reading frame (ORF) encodes at least two different polypeptides. The full-length E1 ORF encodes the viral initiator protein. In addition, the N-terminal domain in E1 can be expressed as a separate polypeptide (M protein) due to alternative splicing. This polypeptide, which has no known function, has been detected in bovine papillomavirus (BPV)-transformed mouse cells and is also likely to exist in other papillomaviruses since these splice sites are highly conserved (26, 27). In full-length E1, the DNA binding domain, the oligomerization domain, and the helicase domain are all well studied and the structures are known (28–30) (Fig. 1A). However, the N-terminal domain has not yielded well-defined functions apart from functions related to nuclear localization (31, 32). In human papillomavirus type 31 (HPV-31) E1, the N terminus has also been shown to interact with a cellular protein, p80 (33). Although the N-terminal domain is less well conserved than the other domains in E1, it still contains features that are conserved within the E1 family, e.g., most E1 proteins contain a highly acidic region in the N-terminal domain.
Fig 1.
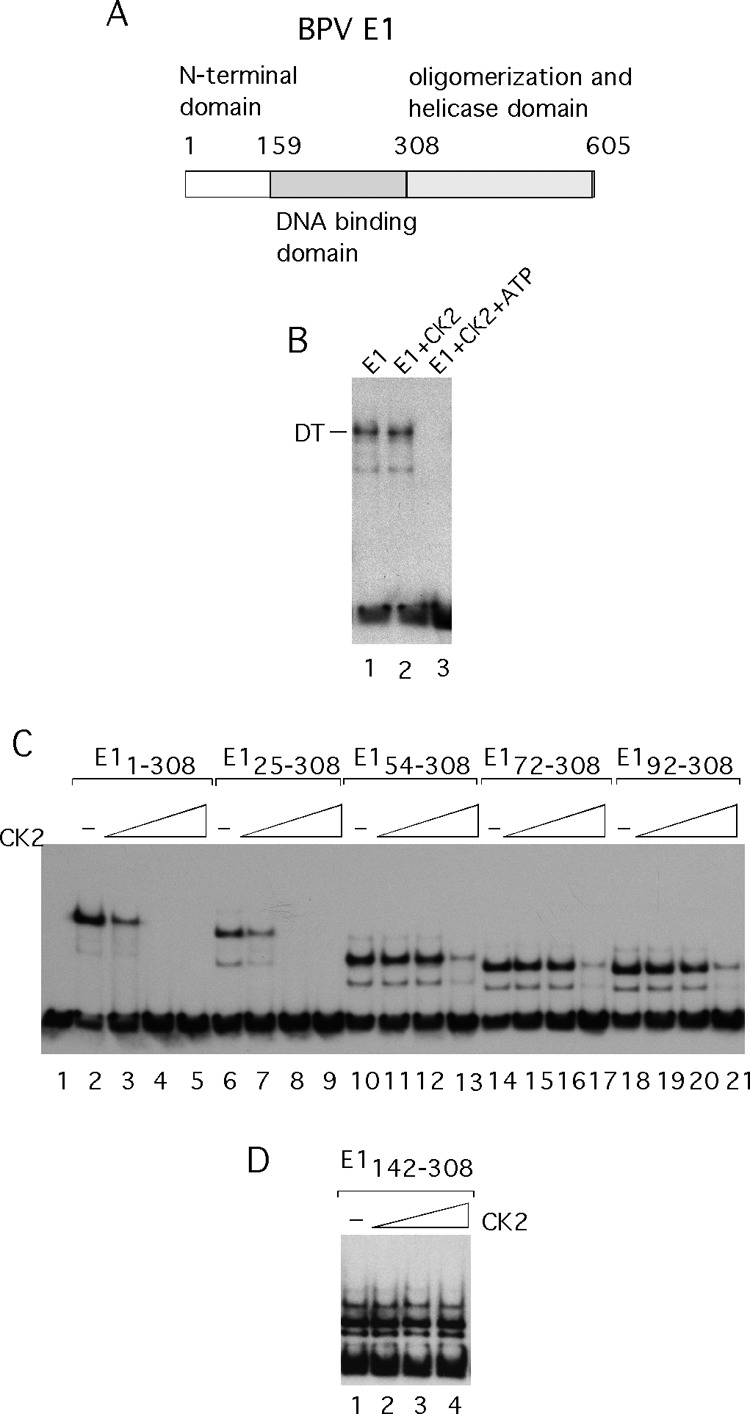
The E1 initiator protein is inactivated for DNA binding by CK2 phosphorylation. (A) A cartoon depicting the E1 polypeptide. (B) The E1 protein was expressed in E. coli, purified, and used in EMSA. E1 was incubated with an 84-bp ori in the presence of 2 mM ADP in the absence (lane 1) or presence (lane 2) of 20 U of the protein kinase CK2 or in the presence of both CK2 and 0.1 mM ATP (lane 3). (C) N-terminal deletions were generated in the N-terminal half of E1 and tested for DNA binding by EMSA. E11–308 (lanes 2 to 5), E125–308 (lanes 6 to 9), E154–308 (lanes 10 to 13), E172–308 (lanes 14 to 17), and E192–308 (lanes 18 to 21) were incubated with ori probe in the absence of CK2 or in the presence of 20, 40, or 80 U of CK2, respectively. (D) E1 DBD (E1142–308) was incubated with ori probe in the absence of CK2 or in the presence of 20, 40, or 80 U of CK2 and used for EMSA.
The E2 protein exists in at least three different versions generated by alternative splicing and alternative transcriptional start sites (5). Two versions, E2C and E8/E2, lack the N-terminal transactivation domain and therefore are likely to serve as competitive repressors of the full-length E2 (18, 21, 34–37). However, the interplay between the different forms of E2 is not well understood, and the functional distinction between the two repressors is not well established (21, 35, 36). The N-terminal activation domain and the C-terminal DNA binding domain have been studied in great detail, including structural studies (38–40). However, the function of the central domain, the “hinge,” is unclear, and this region is believed to be unstructured. Because of the parsimony that generally characterizes viral proteins, the N-terminal domain in E1 and the hinge in E2 likely serve important functions.
A common feature of the N-terminal domain in the E1 protein and the hinge in the E2 protein is that both contain phospho-acceptor sites that are phosphorylated by the protein kinase CK2 (41, 42). Here, we show that phosphorylation on these sites results in a dramatic inactivation of DNA binding of both the E1 and E2 proteins. Phosphorylation of E2 is a negative regulator of the viral replication since mutation of the phosphorylation sites in E2 in the context of the viral genome results in greatly increased viral DNA replication. The CK2 phosphorylation of the N terminus of E1 is a conserved feature, as is the resulting inactivation of E1 DNA binding. This inactivation is unlikely to play a role in the latent stage of the viral life cycle, since mutation of the phosphorylation sites in the N terminus of E1 have no effect in replication and focus assays in mouse C127 cells. More likely, the phosphorylation of E1 plays a role in the vegetative state of the viral life cycle. Consistent with a role in the vegetative stage, the inactivation of E1 can be reversed by cleavage of E1 with the protease caspase 3, an enzyme that is induced and required for the transition from the latent to vegetative stages (43). Although the precise roles that these changes in DNA binding activity play in the viral life cycle are not known, the conservation of these phenomena strongly indicates that they play an important role in how these viruses are propagated.
MATERIALS AND METHODS
Expression and purification of E1 and E2.
Full-length E1 and E1 fragments were expressed in Escherichia coli as N-terminal glutathione S-transferase (GST) fusions and purified by glutathione agarose affinity chromatography. The GST portion was removed by digestion with thrombin, and the material was further purified by ion-exchange chromatography (44). GST fusions that were not intended for thrombin cleavage were further purified by ion-exchange chromatography after the affinity step. Wild-type (wt) E2 and point mutants in E2 were expressed in E. coli without an affinity tag and purified by two rounds of ion-exchange chromatography (44).
EMSA.
Four-percent acrylamide gels (39:1 acrylamide-bis) containing 0.5× Tris-borate-EDTA (TBE) and lacking EDTA were used for all electrophoretic mobility shift assays (EMSAs). E1 was added to 32P-labeled probe (∼2 fmol) in 10 μl binding buffer (BB; 20 mM HEPES [pH 7.5], 70 mM NaCl, 0.7 mg/ml bovine serum albumin [BSA], 0.1% NP-40, 5% glycerol, 5 mM dithiothreitol [DTT], 5 mM MgCl2), and 2 mM ATP or ADP. After incubation at room temperature (∼ 24°C) for 1 h, the samples were loaded and run for 2 h at 9 V/cm (45).
Glycerol gradient sedimentation.
Glycerol gradients (5 ml) were generated in BB. Fifty micrograms of E11–308 was loaded onto a 5 to 30% glycerol gradient, which was run for 23 h at 49,000 rpm in an SW55 rotor. The gradients were fractionated into 150-μl fractions, and the peaks were localized by Bradford assays. The marker protein, carbonic anhydrase (29 kDa), was purchased from Sigma-Aldrich and run in a parallel gradient.
Replication assays.
Replication assays in C127 cells were carried out by electroporation. A total of 2 × 106 cells were trypsinized and resuspended in 250 μl of medium with 10% fetal bovine serum (FBS) containing 50 μg of sonicated carrier DNA and 2 μg of recircularized genomic BPV DNA. The cells were pulsed using a Bio-Rad gene pulser at 210 V and 960 μF and plated on three 100-mm-diameter plates. After 4 and 5 days, low-molecular-weight DNA was prepared and digested with DpnI, linearized with HindIII, and analyzed by agarose gel electrophoresis. The DpnI-resistant replicated DNA was detected by Southern blotting and hybridization using standard procedures (46).
Replication assays in CHO cells were carried out by electroporation as described above, except that the voltage was 230 V. In a standard reaction, 100 ng of ori plasmid (11/12/X), 2 μg of wt or mutant E1 expression vector (46), and 0.5 μg of wt or mutant E2 expression vector (46) were transfected in the absence or presence of 0.5 μg each of the expression vectors for CK2α and CK2β (47, 48). These expression vectors for CK2 were gifts from D. Lichtfield (Addgene plasmids 27086 and 27088). In each transfected sample, 200 ng of the β-galactosidase expression vector pON 260 was included as an internal transfection control (49). Two days after transfection, a sample was taken for determination of β-galactosidase activity as an internal transfection control as described previously (49). For the replication assays, cells were harvested 4 days after transfection and low-molecular-weight DNA was prepared, digested, and analyzed by agarose gel electrophoresis, Southern blotting, and hybridization to an ori probe.
Focus formation assays.
C127 cells were seeded into 6-well plates, and at ∼80% confluence, 100 ng of recircularized BPV genomic DNA was added together with 2 μg of pUC 19 and 3 μl Fugene HD transfection reagent. The following day, the cells from each well were trypsinized and plated onto a 100-mm-diameter plate. The medium was changed every 3 days, and foci were scored after 2 weeks by staining with methylene blue.
Pulldown DNA binding assays.
GST E1 fusion proteins were incubated with glutathione agarose beads in BB for 30 min at room temperature (∼24°C). The beads were washed several times with phosphate-buffered saline (PBS) and then resuspended in BB containing the 32P-labeled probe DNA. After incubation on a rotator at room temperature for 1 h, the beads were washed 4 times with PBS and subjected to phenol chloroform extraction. The samples were analyzed by PAGE and autoradiography.
Caspase 3 was purchased from R&D Inc., and the cleavage conditions were those recommended by the manufacturer. CK2 was purchased from NEB.
Expression of E2 in COS-7 cells.
COS-7 cells were transfected using electroporation. Briefly, 2 × 106 cells were mixed with 200 ng of the E2 expression vector pCGE2-Epi (50), which contains a hemagglutinin (HA) tag, and 50 μg of salmon sperm carrier DNA in 250 μl of Dulbecco's modified Eagle's medium (DMEM) and electroporated at 180 V and 960 μF, and the cells were plated on a 100-mm plate. Three days after transfection, the cells were trypsinized and washed 4 times in PBS with 1% serum. The cell pellet was resuspended in 0.2 ml of a solution containing 10 mM Tris (pH 8.0), 150 mM NaCl, 2 mM EDTA, 0.2% NP-40, and 40% glycerol to generate a cytosolic extract. The nuclei were pelleted by centrifugation, and the supernatant was used for DNA binding assays. A total of 20 μl of extract was incubated with 2 μl of ascites fluid containing the 12CA5 antibody, which recognizes the HA tag present in the overexpressed E2 protein and 20 μl of protein G agarose beads for 1 h at 4°C. The beads were washed 5 times with PBS and subsequently incubated in the presence or absence of lambda phosphatase. The beads were washed to remove the phosphatase and subsequently incubated at room temperature with a mixture of two probes: an ori probe, which contains an E2 binding site (E2 BS), and a larger, nonspecific probe lacking an E2 BS. After multiple washes with PBS containing 0.1% NP-40, the beads were treated with phenol-chloroform, and the DNA present in the aqueous phase was recovered by precipitation and analyzed by gel electrophoresis.
Mass spectrometric analysis.
The mass spectrometric analysis is described in the supplemental materials.
RESULTS
E1 DNA binding is inactivated by CK2 phosphorylation.
We inadvertently discovered that phosphorylation by the protein kinase CK2 affects the biochemical activity of the bovine papillomavirus E1 protein. In the presence of nucleotide, full-length E1 forms a double trimer complex (DT) on the viral origin of DNA replication (ori) (45). This complex, which locally melts the ori, is the precursor for the E1 double hexamer (DH) helicase (12, 51). Incubation of E1 in the presence of CK2 and ATP (Fig. 1B, lane 3) resulted in a complete loss of DNA binding activity, while in the absence of CK2 (lane 1) or with CK2 in the absence of ATP (lane 2), a DT complex was formed, demonstrating that CK2 phosphorylation affects some aspect of E1 DNA binding.
Formation of the E1 DT depends on two DNA binding activities in E1. The E1 DNA binding domain (DBD) binds specifically to four E1 binding sites in the viral ori, and several residues in the E1 helicase domain bind to the DNA flanking the E1 BS, in a sequence-nonspecific manner (7, 30, 52) (Fig. 1A). To determine whether the sequence-specific DNA binding activity present in the E1 DBD was affected by CK2 phosphorylation, we generated an E1 fragment containing the N-terminal 308 residues (E11–308), which includes the E1 DBD (amino acids [aa] 142 to 308) but not the oligomerization and helicase domain. This fragment binds as a dimer to the ori (53). We expressed and purified this fragment and tested it for DNA binding with and without treatment with CK2. E11–308 bound well in the absence of CK2 (Fig. 1C, lane 2); however, the addition of CK2 (lanes 2 to 4) resulted in loss of DNA binding, demonstrating that the defect for DT formation of the full-length E1 is caused by the loss of the specific DNA binding activity present in the E1 DBD. We generated and tested several N-terminal deletions in this fragment. E125–308 (lanes 6 to 9) was almost as sensitive to CK2 as E11–308, while E155–308 (lanes 10 to 13), E172–308 (lanes 14 to 17), and E192–308 (lanes 18 to 21) were much less sensitive. However, at the highest concentrations of kinase, even these polypeptides were inactivated for DNA binding. In contrast, the E1 DBD (E1142–308) was resistant to inactivation even at the highest concentration of CK2 (Fig. 1D, lanes 2 to 4). This result demonstrates that inactivation of the E1 DBD for DNA binding is mediated by phosphorylation events outside the E1 DBD.
We utilized the website ScanSite (54) to predict the presence of CK2 phosphorylation sites in the N-terminal half of E1. The program predicted CK2 sites at residues T20, S26, S43, S48, S94, and S95 (Fig. 2A).
Fig 2.
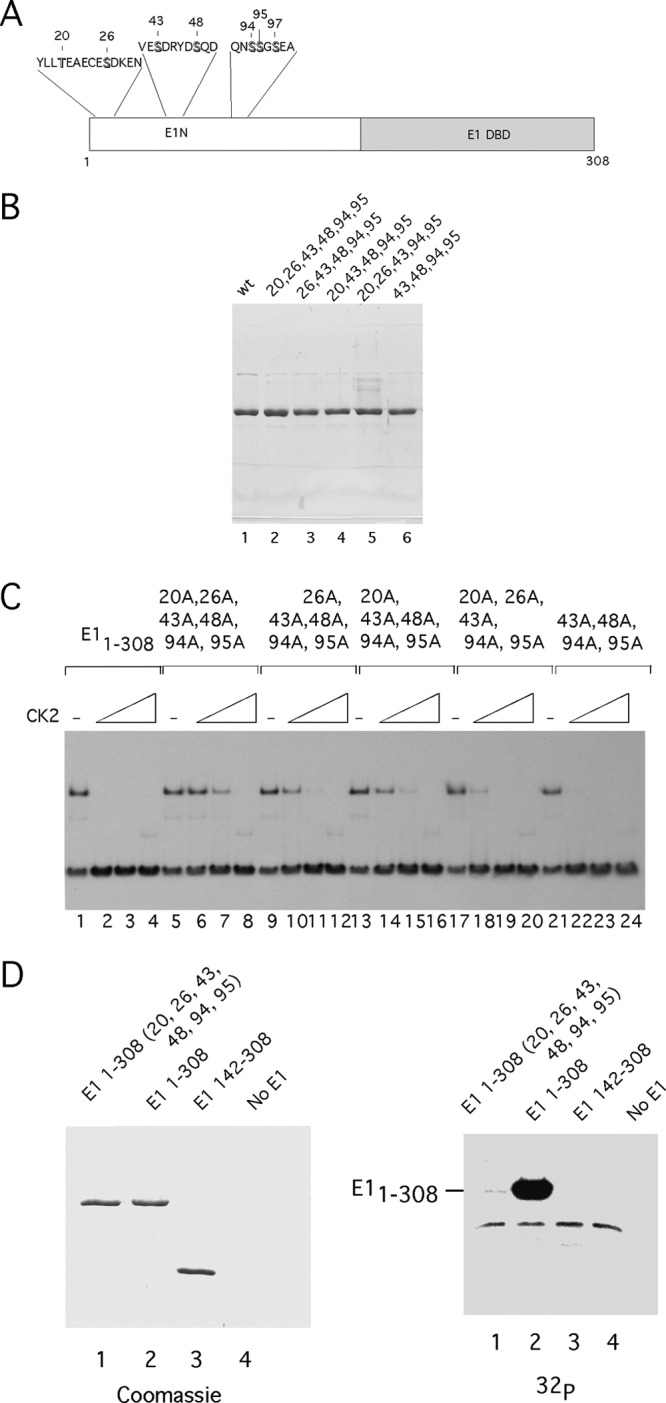
The N-terminal half of E1 contains multiple CK2 phosphorylation sites. (A) A cartoon illustrating the location and sequence of the 7 predicted CK2 phosphorylation sites in the N terminus of E1. (B) Point mutations affecting CK2 phosphorylation sites were generated in E11–308. The resulting proteins were expressed in E. coli, purified, and analyzed by SDS-PAGE. (C) The mutant proteins from panel B were used in EMSA, in the absence of CK2 or in the presence of 20, 40, or 80 U of CK2. (D) E11–308 with alanine substitutions at residues 20, 26, 43, 48, 94, 95 (lane 1), E11–308 (lane 2), E1142–308, or no E1 were incubated with 80 U of CK2 in the presence of γ32P-ATP and loaded onto SDS-PAGE stained with Coomassie brilliant blue. The gel was then dried and subjected to autoradiography (right). The faint band migration below E1 1-308 corresponds to autophosphorylated CK2β.
We mutated these residues and expressed and purified the resulting proteins (Fig. 2B). We then tested the phosphorylation mutants for DNA binding in the absence and presence of CK2 (Fig. 2C). Mutation of 6 residues, 20, 26, 43, 48, 94, and 95, resulted in a large reduction in the sensitivity of E11–308 to CK2 (Fig. 2C, lanes 5 to 8). Interestingly, these mutations did not result in a complete resistance to CK2, however, likely due to some remaining CK2 sites. Different combinations of these phosphorylation site mutations showed different degrees of sensitivity, indicating either that the different sites have different abilities to inactivate DNA binding or that they are phosphorylated at different rates. For example, the protein retaining only the sites at S20 and S26 was inactivated at the same level of kinase as the protein retaining only S48, which was inactivated at the same level of kinase as wt E11–308 (compare lanes 21 to 24 to lanes 17 to 20 and 1 to 4). Thus, no particular phosphorylation site appears to be specifically required for inactivation of DNA binding; however, some sites may be more effective than others (such as S48).
We tested E11–308 with the phosphorylation mutations for CK2 phosphorylation in vitro. We compared E1 DBD, E11–308, and E11–308 with the alanine substitutions at residues 20, 26, 43, 48, 94, and 95 after phosphorylation using γ32P-ATP (Fig. 2D). The mutated E11-308 and E1142-308 (E1 DBD) showed virtually undetectable levels of phosphorylation (>120-fold reduction in the level of phosphorylation compared to E11-308, compare lanes 1 and 2 in Fig. 2D). These results demonstrate that all the significant in vitro CK2 phosphorylation sites are included in the 6 candidate sites.
We subsequently phosphorylated E11-308 in vitro using CK2 and subjected the sample to mass spectrometric analysis after digestion with either trypsin or the nonspecific protease elastase. We could detect phosphorylation on 5 sites, S43, S48, S94, S95, and S97, indicating that a majority of the predicted sites are bona fide CK2 sites in vitro (see the supplemental material).
Inactivation of E1 DNA binding does not involve oligomerization.
Inactivation of the E1 DBD in the context of E11–308 could occur through many possible mechanisms. We wanted to rule out trivial explanations for the loss of DNA binding activity, and we therefore compared the oligomerization status of phosphorylated and unphosphorylated E11–308. We compared the sedimentation properties in a glycerol gradient by mixing E11–308 phosphorylated with γ32P-ATP with unphosphorylated E11–308 and sedimenting the sample through the gradient (Fig. 3A and B). The fractions were analyzed by SDS-PAGE, followed by staining with Coomassie brilliant blue (CBB) and autoradiography. As shown in Fig. 3A, the unphosphorylated E11–308 peaked in fractions 14 and 15. Phosphorylated E11–308 coincided exactly, demonstrating that the two proteins have virtually identical sedimentation properties, consistent with both species sedimenting as monomers (Fig. 3B). This rules out oligomerization or aggregation as a mechanism for inactivation.
Fig 3.
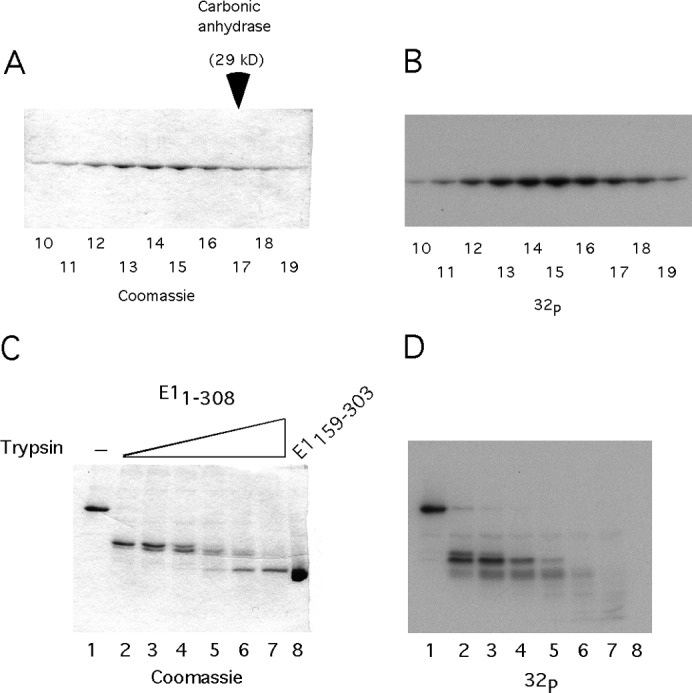
The mechanism of DBD inactivation. (A) A total of 50 ng of E11–308 was phosphorylated in vitro with CK2 and γ32P ATP. The labeled protein was mixed with a 1,000-fold excess of unlabeled E11–308 in the presence of EDTA and analyzed by sedimentation on a 15 to 30% glycerol gradient. The fractions were analyzed by SDS-PAGE, followed by staining with Coomassie and autoradiography. E11–308 peaked in fractions 13 to 15, sedimenting slightly slower than the carbonic anhydrase marker, which peaked in fraction 17. (B) The gel shown in Fig. 3A was dried and subjected to autoradiography. (C) In vitro phosphorylated E11–308 was subjected to partial proteolysis, using 2-fold dilutions of trypsin, followed by SDS-PAGE analysis and staining with Coomassie. (D) The stained gel shown in Fig. 3C was dried and subjected to autoradiography.
It occurred to us that, although we did not observe phosphorylation of the E1 DBD, in the context of the larger E11–308, the DBD might be phosphorylated. To address this question, we devised an experiment where we took advantage of the protease resistance of the E1 DBD, which we have established in our structural studies of the E1 DBD (28). We phosphorylated E11–308 using a trace of γ32P-ATP and then subjected the sample to partial proteolysis with trypsin, using 2-fold increases of trypsin concentration (Fig. 3C, lanes 2 to 7). E11–308 was first reduced to an intermediate band and then eventually reduced to the size of the E1 DBD as detected by CBB staining. When this gel was subjected to autoradiography, we readily observed the presence of radioactive phosphate on E11–308, as well as on the lower-molecular-weight intermediate (Fig. 3D, lanes 1 to 5). However, the 32P signal was absent from the DBD fragment (lanes 6 and 7). This result demonstrates that even in the context of the E11–308, the E1 DBD is not phosphorylated by CK2.
Phosphorylation of two residues in the hinge inactivates E2 DNA binding.
In addition to the viral E1 protein, which is the initiator protein and the replicative DNA helicase, another viral protein is directly involved in initiation of viral DNA replication. The viral transcription factor E2 binds together with E1 to the ori and provides sequence specificity for initiation of DNA replication (24, 25, 55). It is well established that the BPV E2 protein can be phosphorylated by CK2 and that such phosphorylation affects the half-life of E2 (42, 56, 57). However, the activity of E2 in the absence and presence of phosphorylation has not been measured. We used BPV E2 protein purified from E. coli (Fig. 4B) for EMSAs in the absence and presence of CK2 (Fig. 4C). E2 purified from E. coli is highly active for DNA binding, as described previously (44) (Fig. 4C, lane 1). Strikingly, just as for E1, incubation with CK2 resulted in a complete loss of DNA binding activity (lanes 2 and 3). Mutation of two of the known CK2 sites (S298A, S301A) (42) resulted in complete resistance to the kinase (Fig. 4C, lanes 10 to 12), while the individual mutations resulted in an intermediate sensitivity to the kinase (lanes 4 to 9). Mutation of the putative CK2 sites at S290 and S302 did not significantly alter the response to CK2 (data not shown). This result clearly demonstrates that phosphorylation of E2 on two sites present outside the E2 DBD can inactivate the E2 protein for DNA binding. This similarity between the CK2-induced inactivation of E1 and E2 DNA binding is interesting and may indicate similar mechanisms are involved.
Fig 4.
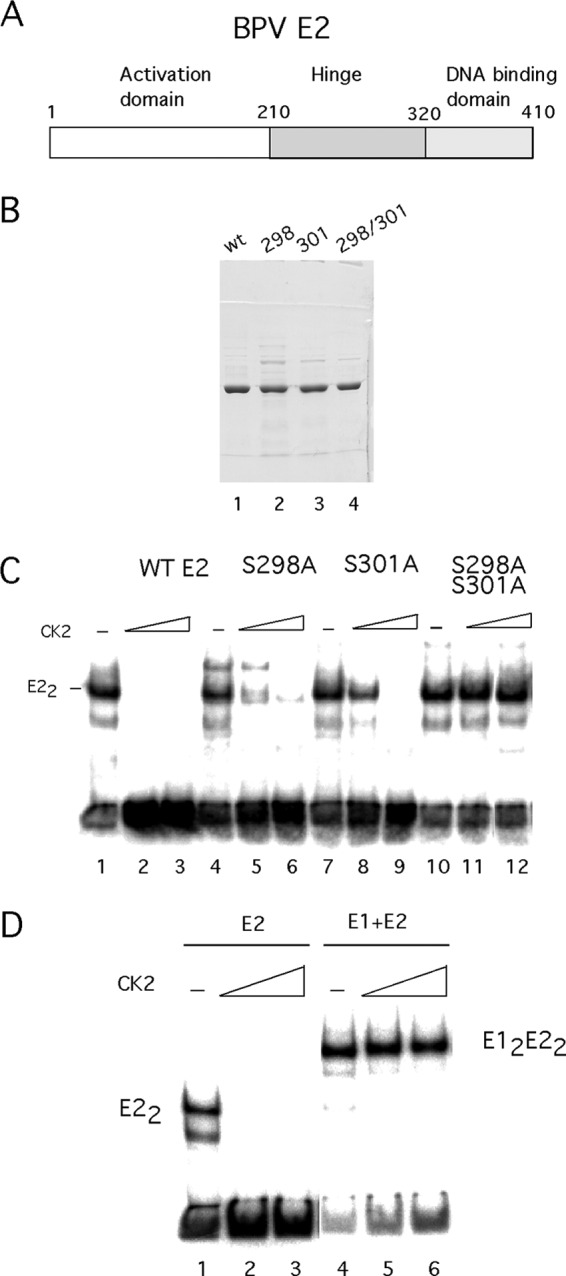
CK2 phosphorylation renders E2 inactive for DNA binding. (A) A cartoon depicting the E2 protein. The N-terminal transactivation domain, the hinge region, and the C-terminal DBD are indicated. (B) Wild-type E2 and the S298A, S301A, and SS298 301AA mutants were expressed in E. coli, purified, and analyzed by SDS-PAGE. (C) EMSA was performed using wt E2 and S298A, S301A, and S298A S301A mutants. E2 was incubated with a probe containing the E2 BS 12 from the BPV genome in the absence of CK2 or in the presence of 20, 40, or 80 U of CK2. (D) E2 was incubated in the absence of CK2 or in the presence of 20, 40, or 80 U of CK2. The phosphorylation reaction was terminated by the addition of EDTA. The phosphorylated E2 was added to the probe either alone or together with E1.
E1 and E2 can bind to their specific sites in DNA individually, but the two proteins can also bind as a cooperative E12E22 complex to the ori, which contains binding sites for both proteins (15, 24, 58). We were interested in addressing whether the phosphorylated E1 and E2 proteins were also inactive for DNA binding in the presence of its binding partner (Fig. 4D). We performed this experiment by first phosphorylating either the E1 or the E2 protein in the presence of ATP and Mg2+ and then adding EDTA to prevent further phosphorylation, followed by the addition of probe and either E1 or E2. Surprisingly, the phosphorylated E2 protein could bind DNA together with the E1 protein, demonstrating that phosphorylated E2 is not irreversibly inactivated (Fig. 4D, lanes 4 to 6). In the converse experiment, phosphorylated E1 failed to bind DNA in the presence of E2, demonstrating that the DNA binding activity of the inactivated E1 protein cannot be restored by the addition of E2 (data not shown). Clearly, the interaction with the E1 protein restores E2 to a conformation that can bind DNA but not vice versa.
Effects of mutations in the CK2 phosphorylation sites in E1 and E2 on DNA replication and morphological transformation.
These results clearly demonstrated that the biochemical activity of both the E1 and E2 proteins could be altered by CK2 phosphorylation. To determine how these phosphorylation events impact the viral life cycle, we generated phosphorylation site mutations in the context of the BPV genome and tested these mutants for DNA replication and morphological transformation in mouse C127 cells, where part of the viral life cycle can be reproduced. The transient replication assays were performed by transfecting circularized viral genomes by electroporation. Low-molecular-weight DNA was isolated 4 and 5 days after transfection, and the DNA was digested with DpnI and linearized with HindIII, run on agarose gels, blotted to nitrocellulose, and probed with a viral genomic probe (Fig. 5A). Strikingly, the phosphorylation mutants in E2 showed a greatly increased level of replication compared to that of the wt. S298A and S301A both showed a 5- to 10-fold increase in the level of DpnI-resistant DNA (lanes 3 and 4 and 7 and 8), while the S298A S301A double mutant (lanes 5 and 6) showed a 10- to 25-fold increase in the level of replicated DNA. Clearly, these mutations generate a significantly higher level of viral DNA replication. In contrast, the phosphorylation site mutations in the N terminus of E1 did not produce a significantly different level of replication than wt E1 (compare lanes 1 and 2 and 9 and 10).
Fig 5.

Effects of E1 and E2 phosphorylation on viral DNA replication. (A) Viral DNA with mutations at S298A and S301A and the S298A S301A double mutant in E2 or with mutations at T20A, S26A, S43A, S48A, S94A, and S95A (All) in E1 was transfected into C127 cells, and time points were taken 4 and 5 days after transfection, harvested by alkaline lysis, and prepared for digestion with DpnI and EcoRI. After Southern transfer, the replicated DpnI-resistant viral DNA was detected by hybridization to a radiolabeled genomic BPV probe. (B) Morphological transformation assays were performed by transfecting the BPV genome into C127 cells using Fugene HD. Cells were fixed and stained with methylene blue after 2 weeks. (C) Transformed foci generated after transfection of C127 cells with the BPV genome containing phosphorylation mutations in E1 and E2 were picked and expanded into cell lines. Genomic DNA was generated and digested with the single cutter BstEII, fractionated by agarose gel electrophoresis, and analyzed by Southern blotting and hybridization with a 32P-labeled BPV probe. Cloned viral DNA was loaded as standards for quantitation. (D) Eight foci each from C127 cells transfected with the wt BPV genome or the S298A phosphorylation mutant were picked and expanded. Low-molecular-weight DNA was prepared from the individual clones and analyzed by Southern blotting and hybridization using a genomic BPV probe. (E) Transient replication assays were performed by transfecting an ori plasmid (11/12/X) together with expression vectors for wt E1 (pCGE1) or E1 with point mutations in the six CK2 phosphorylation sites (T20A, S26A, S43A, S48A, S94A, S95A) in the N-terminal domain (pCGE1 All) and wt E2 (pCGE2) or E2 with the point mutations S298A and S301A (pCGE2 298/301) into CHO cells by electroporation, as indicated. In lanes 5 to 7, expression vectors for CK2α and CK2β were cotransfected with the ori and the E1 and E2 expression vectors. Replicated, DpnI-resistant ori DNA was analyzed by Southern blotting and hybridization with a 32P-labeled probe. The hybridization signal was quantitated, and the relative level of replication in each lane is indicated. (F) Dephosphorylation of E2 expressed in COS-7 cells increases E2's DNA binding activity. An expression vector for E2 carrying an HA tag was transfected into COS-7 cells. Two days after transfection, cytoplasmic extracts were prepared and incubated with the monoclonal antibody 12CA5, which recognizes the HA tag and bound to protein G-Sepharose beads. The beads were divided into three equal parts: one part was treated with 100 U of the protein phosphatase lambda in the presence of Mn2+, one part was treated with 100 U of lambda phosphatase in the absence of Mn2+, and the third sample was left untreated. After being washed, the beads were incubated with two 32P-labeled DNA probes, one of which contained an E2 BS. The bound probes were dissociated from the beads and analyzed by PAGE.
We also performed focus formation assay by transfecting the mutant viral genomes into C127 cells and scoring for morphologically transformed foci 2 weeks after transfection (Fig. 5B). The wt genome and the genome carrying the 6 phosphorylation mutations in the N-terminal domain of E1 (All) showed similar numbers of foci that were also of similar size. The phosphorylation mutants in E2 also showed numbers of foci similar to those of the wt; however, these foci were significantly larger than those generated by the wt genome.
We isolated individual foci from the different transfections and generated cell lines for analysis of the stably replicating viral DNA (Fig. 2C). We generated genomic DNA for two lines for each transfection and compared the levels of viral DNA to those of standards by Southern blotting and hybridization using a genomic BPV probe. We analyzed 2 μg of genomic DNA for each clone. The two clones containing the wt genome (Fig. 5C, lanes 1 and 2) contained approximately 350 pg of viral DNA (compare to the standards in lanes 5 to 7), while the two clones containing the N-terminal phosphorylation mutants (Fig. 5C, All, lanes 3 and 4) showed a slight (∼2-fold) reduction in viral DNA. The three phosphorylation mutations in E2 showed much higher copy numbers. The S298A S301A mutant (Fig. 5C, lanes 12 and 13) had levels of DNA corresponding to ∼3 ng, ∼10-fold higher than wt BPV. The S301A mutant (lanes 10 and 11) showed levels of viral DNA ∼5-fold higher than wt BPV, while the S298A mutant (lanes 8 and 9) had levels of viral DNA 2- to 5-fold higher than wt BPV. These results are consistent with the transient replication assays in panel A, which showed elevated levels of replication for the phosphorylation mutants in E2 but no significant difference between All and wt BPV. We did not detect any indication of integrated viral DNA.
Because one sample from the S298A mutant (lane 8) in panel C showed only a modestly higher level of DNA than the wt samples, we wanted to analyze a larger number of clones to verify that the differences in levels of replicated DNA were representative. We analyzed low-molecular-weight DNA from 8 foci each generated by the wt genome and the S298A mutant. Following linearization and separation on an agarose gel, the samples were transferred to membrane and hybridized with a genomic BPV probe (Fig. 5D). We could observe modest (∼2-fold) variation in the level of viral DNA within each group; however, the difference between the two groups was much greater, ranging from 5- to 20-fold. This result verifies that the S298A mutant replicates to a much higher level than wt BPV.
The fact that the phosphorylation mutations in E2 have a very strong phenotype suggests that in C127 cells, CK2 is active and modulates the activity of the E2 protein. The absence of a significant effect of the phosphorylation mutations in the E1 protein in the replication assays likewise demonstrates that CK2 phosphorylation of E1 does not play an important role in C127 cells either because these sites are not phosphorylated or because phosphorylation of the sites does not affect replication.
To uncouple expression of E1 and E2 and viral DNA replication, we tested viral replication in a different system where we expressed E1 and E2 from expression vectors and monitored replication of a plasmid containing the minimal ori in CHO cells. We transfected pCGE1 and pCGE2 vectors, with and without the phosphorylation mutations (All in E1 and S298A S301A in E2), and the ori plasmid and measured replication 4 days after transfection.
As shown in Fig. 5E, transfection of the wt E1 and E2 expression vectors resulted in robust replication of the ori plasmid (lane 1). Transfection of wt E1 with the S298A S301A E2 mutant resulted in a 2.6-fold increase in the level of replication (lane 2). This is consistent with the results observed for the viral genome, where mutation of the phosphorylation sites in E2 results in a significantly increased replication, presumably because under these conditions higher levels of active E2 are present. When mutant E1 and wt E2 were transfected, we observed replication at the same level as for wt E1 and E2 (compare lanes 1 and 3). Similarly, using the mutant versions of both E1 and E2, we observed a similar level of replication (compare lanes 1 and 4). We wanted to determine whether we could observe an effect of CK2 overexpression. We cotransfected expression vectors for CK2α and CK2β (47, 48) with wt E1 and E2, and we observed an ∼5-fold reduction in replication (compare lanes 1 and 5). Importantly, this effect was not solely related to phosphorylation of E2, since in lane 6, using wt E1 and the S298A S301A E2 mutant, CK2 expression resulted in a 3-fold reduction in replication. When we used the mutant versions of both E1 and E2, we observed no significant reduction in levels of replication (lane 7). These results clearly show that overexpression of CK2 affects replication only when phosphorylation sites are present in E1 and E2, indicating that it is the phosphorylation of E1 and E2 that affects replication. These results support the notion that CK2 can inactivate the E1 and E2 proteins by direct phosphorylation on the phosphorylation sites that we have identified.
It is difficult to demonstrate directly how phosphorylation affects the activity of E1 and E2 in vivo. It is difficult to overexpress E1, and due to the low sequence specificity of full-length E1 for DNA binding, it is also difficult to measure E1 DNA binding activity unless the protein is highly purified, which generally requires recombinant expression. In contrast, E2 can be readily overexpressed, and because of the high sequence specificity of E2, its DNA binding activity can easily be measured even in crude samples. To determine whether we could detect phosphorylation and inactivation of DNA binding in E2, we expressed E2 containing an HA epitope tag in COS-7 cells (50). After 48 h, we generated a cytoplasmic extract which we incubated with protein G-bound monoclonal antibody (12CA5), which recognizes the HA epitope (Fig. 5F). After the beads were extensively washed, we separated them into three equal portions. To one portion we added lambda phosphatase and Mn2+, a required cofactor for lambda phosphatase; to the second sample we added phosphatase only, while the third sample received Mn2+ only. After incubation for 20 min on ice, the beads were then incubated with two DNA probes, one containing the BPV ori, which contains an E2 BS, and an unrelated DNA fragment lacking E2 BS. The beads were then washed, and the bound DNA was recovered by phenol extraction and ethanol precipitation and analyzed by PAGE. As show in Fig. 5F, the E2 BS-containing probe was preferentially recovered in the presence of the 12CA5 antibody, demonstrating that E2 is present and is capable of binding to the E2 BS, establishing the specificity of the assay. In the absence of lambda phosphatase, we recovered the E2 BS probe specifically (lane 3). In the presence of lambda phosphatase but in the absence of added Mn2+, we observed a slight increase in DNA binding, presumably because low levels of Mn2+ are added in the enzyme preparation (lane 4). In the presence of both Mn2+ and lambda phosphatase, we observed an ∼3-fold increase in bound probe (lane 5). These results are consistent with the idea that a significant fraction of the overexpressed E2 protein is inactivated by phosphorylation and can be reactivated by incubation with lambda phosphatase. Similar results were obtained in multiple repeats of this experiment.
DNA binding by E1 from HPV-11 and HPV-31 is also inactivated by CK2 phosphorylation.
We wanted to determine whether E1 from human papillomaviruses could also be phosphorylated by CK2 and, if so, whether such phosphorylation would result in inactivation of DNA binding. Since reliable EMSA has not been developed for HPV E1 proteins, we instead used a pulldown assay to measure DNA binding (Fig. 6). We expressed the N-terminal half of HPV-11 E1 (H11 E12–353), the equivalent of BPV E11–308, as a GST fusion, purified the protein by affinity chromatography and ion-exchange chromatography, rebound the protein to glutathione agarose beads, and performed pulldown experiments either with an ori probe or a nonspecific probe that lacks the ori sequences (Fig. 6A). H11 E12–353 readily recovered the ori probe (lane 5) but not the nonspecific probe (lane 2), demonstrating ori-specific binding. Interestingly, the ori-specific DNA binding was abolished by the incubation with CK2 in the presence of ATP (lane 4), demonstrating that CK2 can inactivate DNA binding also by the HPV-11 E1 DBD.
Fig 6.
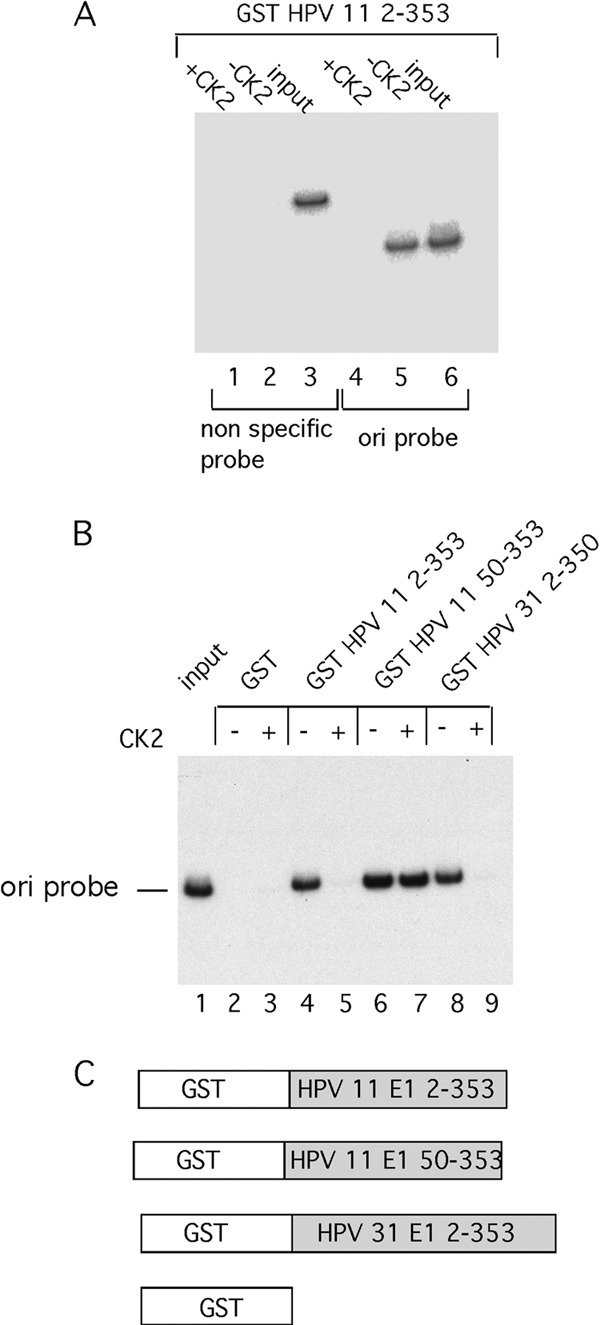
CK2 phosphorylation inactivates DNA binding in HPV-11 and HPV-31 E1. (A) DNA pulldown assays were performed using GST HPV-11 E12–353, a DNA probe representing the BPV ori and a nonspecific probe containing sequences from E1. The assays were performed in the absence or presence of 50 U of CK2 as indicated. (B) Pulldown assays of the BPV ori fragment were performed with the indicated N-terminal fragments of E1 from HPV-11 and HPV-31, in the absence and presence of 50 U of CK2. (C) A cartoon illustrating the GST fusion constructs used in these assays.
We performed the same experiment with HPV-11 E1 lacking the N-terminal 50 residues (H11 E150–353) (Fig. 6B). This protein was not inactivated by CK2 phosphorylation (lane 7), indicating that, indeed, the inactivating phosphorylation sites are present in the N-terminal 50 amino acids. To determine whether the phosphorylation-dependent inactivation of DNA binding could also be detected in other HPV E1 proteins, we tested the N-terminal half of E1 from HPV-31 E1. This protein, HPV-31 E12–332, just as HPV-11 E12–353, was inactivated by CK2 phosphorylation (Fig. 6B, lanes 8 and 9). Similar results were obtained in multiple repetitions of these experiments.
Caspase 3 cleavage restores DNA binding activity to phosphorylated E1.
The failure to observe an effect of the CK2 phosphorylation mutations in the N terminus of E1 in the replication assays in C127 cells was puzzling. We reasoned that maybe the N-terminal phosphorylation could function during the vegetative state of viral DNA replication, which is not reproduced in C127 cells. The transition from the latent to the vegetative stage of the papillomavirus life cycle can be reproduced in a model system developed by Laimins and coworkers (59–61). Viral DNA from HPV-31 is introduced into human foreskin keratinocytes (HFK), which then are induced to differentiate either by the addition of calcium or through anchorage-independent growth. It has been demonstrated that in this system, concomitant with the transition from the latent to vegetative state, the protease caspase 3 is induced (43). Caspase 3 cleaves the HPV-31 E1 protein in the N-terminal domain, and prevention of this cleavage through mutation of the caspase cleavage site impairs the increase in viral DNA content that is associated with the transition from latent to vegetative viral replication (43).
It occurred to us that caspase cleavage of E1 might be one way to reverse the effect of N-terminal phosphorylation of E1. To determine whether caspase 3 could cleave BPV E1, we incubated full-length BPV E1 with recombinant caspase 3 and analyzed the products by SDS-PAGE, followed by staining with CBB. As shown in Fig. 7A, digestion with caspase 3 results in the generation of a very small peptide and a large fragment of ∼65 kDa, consistent with a site close to the N terminus (compare lanes 2 and 3). We next tested the N-terminal halves of BPV E1, HPV-11 E1, and HPV-31 E1 for CK2 phosphorylation and caspase cleavage. BPV E11–308, HPV-11 E12–353, and HPV-31 E12–332 were phosphorylated using CK2 and γ32P-ATP, followed by cleavage by caspase 3 (Fig. 7B and C). Strikingly, in all cases, all the radioactive label was included in the small peptide generated by caspase cleavage, indicating that all the phosphorylation sites are contained in this N-terminal fragment (compare lanes 1 to 6 in Fig. 7B and C). Consistent with the idea that the CK2 phosphorylation sites are located in the first 50 residues in HPV-11, GST HPV-11 E150–355 was not phosphorylated by CK2 (data not shown).
Fig 7.

Cleavage of phosphorylated E1 with caspase 3 removes the phosphorylated residues and restores DNA binding activity to phosphorylated E1. (A) Full-length BPV E1 (10 μg) was cleaved with caspase 3 and run on a 15% SDS-PAGE gel in parallel with uncleaved E1. The position of the small (∼5-kDa) fragment generated by caspase cleavage is indicated by an arrow. (B) The N-terminal E1 fragments from BPV-1 (E11–308), HPV-11 (E12–335), and HPV-31 (E12–332) were phosphorylated in vitro using CK2 and γ32P-ATP. One half of each phosphorylated sample was treated with caspase 3, and the cleaved and uncleaved samples were analyzed by SDS-PAGE and stained with Coomassie. (C) The gel in panel B was dried and subjected to autoradiography. (D) Two concentrations of BPV E11–308 were used for EMSA with an ori probe before or after phosphorylation with CK2. The samples in lanes 1 to 4 were first treated with CK2, and then the samples in lanes 3 and 4 were cleaved with the protease caspase 3 and mixed with the ori probe. (E) Wild-type E11–308 and E11–308 with a D-A mutation at position 55 were phosphorylated with CK2 and γ32P-ATP and then subjected to cleavage with caspase 3, followed by analysis by SDS-PAGE and autoradiography.
The fact that all the phosphorylated residues could be removed by caspase cleavage suggested that the DNA binding activity might be restored by cleaving the phosphorylated protein with caspase 3. To determine whether this was the case, we first phosphorylated BPV E11–308 with CK2. This protein was inactive for DNA binding by EMSA (Fig. 7D, lanes 1 and 2). We next treated the phosphorylated protein with caspase 3 and then tested it for DNA binding using EMSA (Fig. 7D, lanes 3 and 4). Strikingly, the DNA binding activity was restored after cleavage, giving rise to a complex that migrates faster than E11–308 (lanes 5 and 6), consistent with the removal of part of the E1 N terminus. Caspase 3 cleaves sites with the consensus sequence DXXD, and mutational analysis of HPV-31 E1 has shown that a site (DMVD) is present between residues 46 and 49. Alignment of the BPV E1 N terminus shows the presence of a similar site (DFVD) between residues 52 and 55. To determine whether this site corresponded to the bona fide caspase 3 cleavage site, we mutated the aspartic acid at position 55 to an alanine and repeated the experiment shown in Fig. 7C. Phosphorylated E11–308 was cleaved by caspase 3 (Fig. 7E, lanes 1 and 2), while the mutant at residue 55 was not (lanes 3 and 4).
Together, these results show that the phosphorylation-induced inactivation of E1 DNA binding is a feature that is conserved in papillomavirus E1 proteins. The presence of a single caspase site immediately C-terminal to the N-terminal CK2 phosphorylation sites in BPV E1, HPV-11 E1, and HPV-31 E1 is clearly indicative that the reactivation of DNA binding by caspase 3 cleavage is also a conserved feature of E1.
The in vitro phosphorylation experiment, followed by caspase cleavage, demonstrates that all phosphorylation events are N-terminal to the caspase cleavage site at residue 55 (Fig. 7B and C). However, both the mass spectrometry analysis and our experiments with N-terminal deletions and point mutations (Fig. 2) indicate that additional sites that can be phosphorylated are present (e.g., 94/95/97). We do not understand exactly why these results differ, but clearly additional sites are under some conditions available for phosphorylation. The ability to inactivate E192–308 (Fig. 1C) for DNA binding indicates that certain sites in the N-terminal domain may be structurally inaccessible in the intact N-terminal domain but may become accessible upon deletion. It is also possible that the substrate concentration may play a role in whether these additional sites are phosphorylated. The phospho-peptide analysis by mass spectrometry requires much higher concentrations of protein than any of the other assays.
DISCUSSION
Here, we demonstrate that the activities of the papillomavirus E1 and E2 proteins can be dramatically altered as a consequence of phosphorylation by the protein kinase CK2. CK2 phosphorylation results in the inactivation of the sequence-specific DNA binding activity of the E1 protein from BPV-1, as well as of the E1 proteins from HPV-11 and HPV-31. CK2 phosphorylation also inactivates DNA binding by the E2 protein from BPV-1.
The E1 and E2 DBDs are inactivated through phosphorylation events outside the DBDs, and in neither case can phospho-mimetics substitute for the phosphorylation event (data not shown), indicating that these events are highly specific. Another common feature is that the residues phosphorylated by CK2 that result in the inactivation of DNA binding are located in peptides adjacent to the DBDs, in regions that are believed to be unstructured. E2 can be reactivated for DNA binding through the interaction with E1, i.e., phosphorylated E2 can bind DNA in the E12E22 complex (Fig. 4). Phosphorylated E1, however, does not bind DNA together with E2. Inactivation of E1 can be reversed by caspase 3 cleavage at a single specific site in the N terminus, which removes the phosphorylated residues, demonstrating that the inactivation of DNA binding does not involve irreversible changes in the E1 structure (Fig. 7).
Based on our experiments, we can rule out that inactivation is caused by trivial events, such as oligomerization or aggregation (Fig. 3). Instead, we believe two other mechanisms are plausible. The simplest is that the region containing the phosphorylated residues can fold back and interact with the DBD, resulting in blocking of the DNA binding surface. This model does not provide a simple explanation for the fact that numerous phosphorylation sites at different positions can inactivate the E1 DBD but is a satisfactory model for the inactivation of E2. Another possible mechanism is that structural changes are induced by the phosphorylation events and that these structural changes alter the folding of the E1 and E2 DBDs. The reversal of inactivation of E1 DBD by caspase 3 cleavage of the N-terminal tail clearly demonstrates that the inactivation is reversible but does not distinguish between these two models. A well-documented example where protein phosphorylation outside the DNA binding domain regulates DNA binding is the transcription factor Ets-1, which is regulated by phosphorylation by the calmodulin-dependent kinase CaMKII (62, 63). The mechanism utilized in this example is most likely that phosphorylation results in altered folding of the Ets-1 DBD.
The consequences of mutating the CK2 phosphorylation sites in the E2 protein can be clearly observed in replication and transformation assays in C127 cells (Fig. 5). BPV E2 is a positive regulator of viral transcription and viral DNA replication, and the increased copy number generated by mutation of the CK2 phosphorylation sites is consistent with elevated levels of E2 DNA binding activity. The consequences of E2 phosphorylation that we observe are different in several respects from the earlier study where the CK2 sites at 298 and 301 were originally identified (42). First of all, in the previous study, DNA binding was not measured. Second, we observe virtually identical phenotypes of the S298A and the S301A mutations and an aggravated phenotype (resistance to inactivation by phosphorylation) of the S298A S301A double mutant. This demonstrates that phosphorylation at both S298 and S301 affects DNA binding. We currently cannot reconcile this result with the observations by Penrose and McBride, where an effect on viral replication was observed only by S301A and not by S298A. The authors attributed the effect of S301A to an increased half-life of E2 due to failure to degrade E2 by the proteasome and concluded that phosphorylation at S301, but not S298, causes degradation (57). Since we observe the same phenotype with S298A mutation as with S301A mutation, but only the mutation at S301A showed an increased protein half-life, it is unlikely that the phenotype that we observe is caused by an extended protein half-life. It seems possible, however, that degradation is a secondary effect of the inactivation of DNA binding. These questions will have to be examined in more detail to provide clear answers.
The results observed in the experiments where we overexpressed CK2 are interesting in several ways (Fig. 5E). Most importantly, overexpression of CK2 resulted in a significant reduction in viral DNA replication when the inhibitory phosphorylation sites were present on either E1 or E2 but not when the phosphorylation sites in E1 and E2 were mutated, indicating that phosphorylation on either E1 or E2 can indeed suppress viral DNA replication. This result differs in part from the results observed in the context of the viral genome, where mutation of the phosphorylation sites in E1 had no effect on viral replication. There are several possible explanations for this difference. The expression vectors generate very high levels of CK2, which might be required for E1 phosphorylation (64). Alternatively, the viral genome may be able to autoregulate expression of E1 to compensate for E1 inactivated by phosphorylation, a possibility that does not exist when E1 is expressed from an expression vector.
Another surprising result is that the increase in viral replication observed with mutant E2 and wt E1 is not observed when both E1 and E2 are mutated. We know that under our conditions for viral replication, E2 is limiting for replication. It is conceivable that the mutations in E1 result in a protein that is less well expressed or perhaps has a shorter half-life, which might make the level of E1 limiting for replication. Under these conditions, increased E2 activity would not result in increased replication.
The function of E2 phosphorylation.
The greatly elevated level of viral DNA replication for the S298A and S301A E2 phosphorylation mutants indicate that in C127 cells, some fraction of the wt E2 is normally inactivated by phosphorylation, consistent with the data from the transient transfections in COS cells, where treatment with lambda phosphatase results in increased E2 activity (Fig. 5F). Consequently, we believe that CK2 phosphorylation of E2 plays a role in regulating viral DNA replication. The increased level of DNA replication observed with the S298A S301A E2 mutant expressed from an exogenous expression vector is consistent with a direct stimulation of replication by increased levels of active E2 protein (Fig. 5E). In the viral genome, the exact mechanism by which the S298A S301A E2 mutant results in elevated replication is harder to predict. The elevated levels of E2 activity could act directly but likely also result in increased E1 expression. E2 likely also regulates its own expression from the viral genome, although the details of such regulation are not well understood; i.e., how expression of the two repressor forms of E2, which can compete for DNA binding with full-length E2, is regulated from the viral genome is unclear (21, 35, 36). These repressors share the CK2 sites in the C-terminal half of E2 and, if phosphorylated, would be inactive.
BPV-1, in contrast to many other papillomaviruses, contains a large number of high-affinity E2 binding sites concentrated in the E2-dependent enhancer (20, 65). The phosphorylation of E2, which inactivates DNA binding, could be used to downregulate the viral gene expression from this E2 controlled enhancer. An obvious problem with the two separate functions of E2 is how transcription and replication can be regulated separately and how the very-low-affinity E2 binding sites present in the ori can compete for E2 with the very-high-affinity sites in the E2-dependent enhancer (65). The explanation may be that because phosphorylated E2 is active for DNA binding together with E1, it may function to initiate DNA replication, although it would be inactive for transcriptional activation due to the lack of DNA binding in the absence of E1. Consequently, maintaining a low level of phosphorylated E2 at all times would guarantee that some E2 would always be available for replication, even with competition from many strong E2 BS in the E2-dependent enhancer.
The function of CK2 phosphorylation of E1.
The function of CK2 phosphorylation of the N terminus of the E1 protein and, more specifically, the precise function of the inactivation of the E1 DBD remain unclear. However, because this phenomenon is conserved in other E1 proteins, there is little doubt that the phosphorylation and the inactivation of DNA binding play important roles in the viral life cycle. Our failure to detect a clear phenotype in mouse C127 cells indicates that such a function may be related to the vegetative state of the viral life cycle, since that state is not reproduced in these cells.
This possibility led us to test whether caspase 3 cleavage could play a role in the reactivation of phosphorylated E1 protein. Caspase 3 cleavage in the N-terminal domain of HPV-31 E1 has been shown to be important for stimulation of viral DNA replication associated with the switch to vegetative DNA replication (43). An interesting possibility is that the cleavage may be required to relieve the inactivation of DNA binding by simply removing the phosphorylated peptide. A model that would fit with the existing data is that E1 needs to be stockpiled for the vegetative part of the viral life cycle and that a high concentration of active E1 is deleterious for cell proliferation. It is well established that high-level expression of BPV E1 can cause problems for the host cell DNA (66). The inactivation of E1 by phosphorylation would allow stockpiling. In differentiating cells, once the cells are postmitotic, E1 could be reactivated by caspase cleavage and used for vegetative viral DNA replication. Consistent with such a function, the N-terminal domain in E1 appears to be dispensable for viral DNA replication: in a cell-free replication system, BPV E1127–605 is capable of initiating replication in vitro at wt levels, and, similarly, HPV E1 lacking the N-terminal domain has been shown to initiate replication in vitro (S. Schuck and A. Stenlund, unpublished observation) (67). To determine whether this model is correct will require further studies of E1 phosphorylation under conditions where vegetative replication can be reproduced.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by grant RO1 AI072345 to A.S.
Footnotes
Published ahead of print 1 May 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00345-13.
REFERENCES
- 1. Howley PM, Lowy DR. 2001. Papillomaviruses and their replication, vol 2 Lippincott Williams & Wilkins Co., Philadelphia, PA [Google Scholar]
- 2. Dollard SC, Wilson JL, Demeter LM, Bonnez W, Reichman RC, Broker TR, Chow LT. 1992. Production of human papillomavirus and modulation of the infectious program in epithelial raft cultures. OFF. Genes Dev. 6:1131–1142 [DOI] [PubMed] [Google Scholar]
- 3. Frattini MG, Lim HB, Laimins LA. 1996. In vitro synthesis of oncogenic human papillomaviruses requires episomal genomes for differentiation-dependent late expression. Proc. Natl. Acad. Sci. U. S. A. 93:3062–3067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Meyers C, Frattini MG, Hudson JB, Laimins LA. 1992. Biosynthesis of human papillomavirus from a continuous cell line upon epithelial differentiation. Science 257:971–973 [DOI] [PubMed] [Google Scholar]
- 5. McBride AA, Romanczuk H, Howley PM. 1991. The papillomavirus E2 regulatory proteins. J. Biol. Chem. 266:18411–18414 [PubMed] [Google Scholar]
- 6. Wilson VG, West M, Woytek K, Rangasamy D. 2002. Papillomavirus E1 proteins: form, function, and features. Virus Genes 24:275–290 [DOI] [PubMed] [Google Scholar]
- 7. Chen G, Stenlund A. 2001. The E1 initiator recognizes multiple overlapping sites in the papillomavirus origin of DNA replication. J. Virol. 75:292–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gillette TG, Lusky M, Borowiec JA. 1994. Induction of structural changes in the bovine papillomavirus type 1 origin of replication by the viral E1 and E2 proteins. Proc. Natl. Acad. Sci. U. S. A. 91:8846–8850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Leng X, Ludes-Meyers JH, Wilson VG. 1997. Isolation of an amino-terminal region of bovine papillomavirus type 1 E1 protein that retains origin binding and E2 interaction capacity. J. Virol. 71:848–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lin BY, Makhov AM, Griffith JD, Broker TR, Chow LT. 2002. Chaperone proteins abrogate inhibition of the human papillomavirus (HPV) E1 replicative helicase by the HPV E2 protein. Mol. Cell. Biol. 22:6592–6604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sanders CM, Stenlund A. 1998. Recruitment and loading of the E1 initiator protein: an ATP-dependent process catalysed by a transcription factor. EMBO J. 17:7044–7055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schuck S, Stenlund A. Mechanistic analysis of local ori melting and helicase assembly by the papillomavirus E1 protein. Mol. Cell 43:776–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sedman J, Stenlund A. 1998. The papillomavirus E1 protein forms a DNA-dependent hexameric complex with ATPase and DNA helicase activities. J. Virol. 72:6893–6897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Titolo S, Pelletier A, Sauve F, Brault K, Wardrop E, White PW, Amin A, Cordingley MG, Archambault J. 1999. Role of the ATP-binding domain of the human papillomavirus type 11 E1 helicase in E2-dependent binding to the origin. J. Virol. 73:5282–5293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang L, Mohr I, Fouts E, Lim DA, Nohaile M, Botchan M. 1993. The E1 protein of bovine papilloma virus 1 is an ATP-dependent DNA helicase. Proc. Natl. Acad. Sci. U. S. A. 90:5086–5090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Androphy EJ, Lowy DR, Schiller JT. 1987. Bovine papillomavirus E2 trans-activating gene product binds to specific sites in papillomavirus DNA. Nature 325:70–73 [DOI] [PubMed] [Google Scholar]
- 17. Hawley-Nelson P, Androphy EJ, Lowy DR, Schiller JT. 1988. The specific DNA recognition sequence of the bovine papillomavirus E2 protein is an E2-dependent enhancer. EMBO J. 7:525–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hirochika H, Hirochika R, Broker TR, Chow LT. 1988. Functional mapping of the human papillomavirus type 11 transcriptional enhancer and its interaction with the trans-acting E2 proteins. Genes Dev. 2:54–67 [DOI] [PubMed] [Google Scholar]
- 19. Hou SY, Wu SY, Chiang CM. 2002. Transcriptional activity among high and low risk human papillomavirus E2 proteins correlates with E2 DNA binding. J. Biol. Chem. 277:45619–45629 [DOI] [PubMed] [Google Scholar]
- 20. Spalholz BA, Yang YC, Howley PM. 1985. Transactivation of a bovine papilloma virus transcriptional regulatory element by the E2 gene product. Cell 42:183–191 [DOI] [PubMed] [Google Scholar]
- 21. Stubenrauch F, Hummel M, Iftner T, Laimins LA. 2000. The E8E2C protein, a negative regulator of viral transcription and replication, is required for extrachromosomal maintenance of human papillomavirus type 31 in keratinocytes. J. Virol. 74:1178–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen G, Stenlund A. 2002. Sequential and ordered assembly of E1 initiator complexes on the papillomavirus origin of DNA replication generates progressive structural changes related to melting. Mol. Cell. Biol. 22:7712–7720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lusky M, Hurwitz J, Seo YS. 1994. The bovine papillomavirus E2 protein modulates the assembly of but is not stably maintained in a replication-competent multimeric E1-replication origin complex. Proc. Natl. Acad. Sci. U. S. A. 91:8895–8899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sedman J, Stenlund A. 1995. Co-operative interaction between the initiator E1 and the transcriptional activator E2 is required for replicator specific DNA replication of bovine papillomavirus in vivo and in vitro. EMBO J. 14:6218–6228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang L, Li R, Mohr IJ, Clark R, Botchan MR. 1991. Activation of BPV-1 replication in vitro by the transcription factor E2. Nature 353:628–632 [DOI] [PubMed] [Google Scholar]
- 26. Hubert WG, Lambert PF. 1993. The 23-kilodalton E1 phosphoprotein of bovine papillomavirus type 1 is nonessential for stable plasmid replication in murine C127 cells. J. Virol. 67:2932–2937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Thorner L, Bucay N, Choe J, Botchan M. 1988. The product of the bovine papillomavirus type 1 modulator gene (M) is a phosphoprotein. J. Virol. 62:2474–2482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Enemark EJ, Chen G, Vaughn DE, Stenlund A, Joshua-Tor L. 2000. Crystal structure of the DNA binding domain of the replication initiation protein E1 from papillomavirus. Mol. Cell 6:149–158 [PubMed] [Google Scholar]
- 29. Enemark EJ, Joshua-Tor L. 2006. Mechanism of DNA translocation in a replicative hexameric helicase. Nature 442:270–275 [DOI] [PubMed] [Google Scholar]
- 30. Enemark EJ, Stenlund A, Joshua-Tor L. 2002. Crystal structures of two intermediates in the assembly of the papillomavirus replication initiation complex. EMBO J. 21:1487–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Deng W, Lin BY, Jin G, Wheeler CG, Ma T, Harper JW, Broker TR, Chow LT. 2004. Cyclin/CDK regulates the nucleocytoplasmic localization of the human papillomavirus E1 DNA helicase. J. Virol. 78:13954–13965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lentz MR, Pak D, Mohr I, Botchan MR. 1993. The E1 replication protein of bovine papillomavirus type 1 contains an extended nuclear localization signal that includes a p34cdc2 phosphorylation site. J. Virol. 67:1414–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cote-Martin A, Moody C, Fradet-Turcotte A, D'Abramo CM, Lehoux M, Joubert S, Poirier GG, Coulombe B, Laimins LA, Archambault J. 2008. Human papillomavirus E1 helicase interacts with the WD repeat protein p80 to promote maintenance of the viral genome in keratinocytes. J. Virol. 82:1271–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Choe J, Vaillancourt P, Stenlund A, Botchan M. 1989. Bovine papillomavirus type 1 encodes two forms of a transcriptional repressor: structural and functional analysis of new viral cDNAs. J. Virol. 63:1743–1755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hubbert NL, Schiller JT, Lowy DR, Androphy EJ. 1988. Bovine papilloma virus-transformed cells contain multiple E2 proteins. Proc. Natl. Acad. Sci. U. S. A. 85:5864–5868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lambert PF, Monk BC, Howley PM. 1990. Phenotypic analysis of bovine papillomavirus type 1 E2 repressor mutants. J. Virol. 64:950–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stubenrauch F, Zobel T, Iftner T. 2001. The E8 domain confers a novel long-distance transcriptional repression activity on the E8E2C protein of high-risk human papillomavirus type 31. J. Virol. 75:4139–4149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Antson AA, Burns JE, Moroz OV, Scott DJ, Sanders CM, Bronstein IB, Dodson GG, Wilson KS, Maitland NJ. 2000. Structure of the intact transactivation domain of the human papillomavirus E2 protein. Nature 403:805–809 [DOI] [PubMed] [Google Scholar]
- 39. Harris SF, Botchan MR. 1999. Crystal structure of the human papillomavirus type 18 E2 activation domain. Science 284:1673–1677 [DOI] [PubMed] [Google Scholar]
- 40. Hegde RS, Grossman SR, Laimins LA, Sigler PB. 1992. Crystal structure at 1.7 A of the bovine papillomavirus-1 E2 DNA-binding domain bound to its DNA target. Nature 359:505–512 [DOI] [PubMed] [Google Scholar]
- 41. Lentz MR, Stevens SM, Jr, Raynes J, Elkhoury N. 2006. A phosphorylation map of the bovine papillomavirus E1 helicase. Virol. J. 3:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McBride AA, Howley PM. 1991. Bovine papillomavirus with a mutation in the E2 serine 301 phosphorylation site replicates at a high copy number. J. Virol. 65:6528–6534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Moody CA, Fradet-Turcotte A, Archambault J, Laimins LA. 2007. Human papillomaviruses activate caspases upon epithelial differentiation to induce viral genome amplification. Proc. Natl. Acad. Sci. U. S. A. 104:19541–19546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sedman T, Sedman J, Stenlund A. 1997. Binding of the E1 and E2 proteins to the origin of replication of bovine papillomavirus. J. Virol. 71:2887–2896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schuck S, Stenlund A. 2005. Assembly of a double hexameric helicase. Mol. Cell 20:377–389 [DOI] [PubMed] [Google Scholar]
- 46. Ustav M, Stenlund A. 1991. Transient replication of BPV-1 requires two viral polypeptides encoded by the E1 and E2 open reading frames. EMBO J. 10:449–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Graham KC, Litchfield DW. 2000. The regulatory beta subunit of protein kinase CK2 mediates formation of tetrameric CK2 complexes. J. Biol. Chem. 275:5003–5010 [DOI] [PubMed] [Google Scholar]
- 48. Vilk G, Saulnier RB, St Pierre R, Litchfield DW. 1999. Inducible expression of protein kinase CK2 in mammalian cells. Evidence for functional specialization of CK2 isoforms. J. Biol. Chem. 274:14406–14414 [DOI] [PubMed] [Google Scholar]
- 49. Spaete RR, Mocarski ES. 1985. Regulation of cytomegalovirus gene expression: alpha and beta promoters are trans activated by viral functions in permissive human fibroblasts. J. Virol. 56:135–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ustav E, Ustav M, Szymanski P, Stenlund A. 1993. The bovine papillomavirus origin of replication requires a binding site for the E2 transcriptional activator. Proc. Natl. Acad. Sci. U. S. A. 90:898–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu X, Schuck S, Stenlund A. 2007. Adjacent residues in the E1 initiator beta-hairpin define different roles of the beta-hairpin in Ori melting, helicase loading, and helicase activity. Mol. Cell 25:825–837 [DOI] [PubMed] [Google Scholar]
- 52. Liu X, Schuck S, Stenlund A. Structure-based mutational analysis of the bovine papillomavirus E1 helicase domain identifies residues involved in the nonspecific DNA binding activity required for double trimer formation. J. Virol. 84:4264–4276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen G, Stenlund A. 1998. Characterization of the DNA-binding domain of the bovine papillomavirus replication initiator E1. J. Virol. 72:2567–2576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Obenauer JC, Cantley LC, Yaffe MB. 2003. Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 31:3635–3641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Stenlund A. 2003. E1 initiator DNA binding specificity is unmasked by selective inhibition of non-specific DNA binding. EMBO J. 22:954–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Penrose KJ, Garcia-Alai M, de Prat-Gay G, McBride AA. 2004. Casein kinase II phosphorylation-induced conformational switch triggers degradation of the papillomavirus E2 protein. J. Biol. Chem. 279:22430–22439 [DOI] [PubMed] [Google Scholar]
- 57. Penrose KJ, McBride AA. 2000. Proteasome-mediated degradation of the papillomavirus E2-TA protein is regulated by phosphorylation and can modulate viral genome copy number. J. Virol. 74:6031–6038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lusky M, Hurwitz J, Seo YS. 1993. Cooperative assembly of the bovine papilloma virus E1 and E2 proteins on the replication origin requires an intact E2 binding site. J. Biol. Chem. 268:15795–15803 [PubMed] [Google Scholar]
- 59. Fehrmann F, Klumpp DJ, Laimins LA. 2003. Human papillomavirus type 31 E5 protein supports cell cycle progression and activates late viral functions upon epithelial differentiation. J. Virol. 77:2819–2831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Fehrmann F, Laimins LA. 2005. Human papillomavirus type 31 life cycle: methods for study using tissue culture models. Methods Mol. Biol. 292:317–330 [DOI] [PubMed] [Google Scholar]
- 61. Hebner CM, Wilson R, Rader J, Bidder M, Laimins LA. 2006. Human papillomaviruses target the double-stranded RNA protein kinase pathway. J. Gen. Virol. 87:3183–3193 [DOI] [PubMed] [Google Scholar]
- 62. Cowley DO, Graves BJ. 2000. Phosphorylation represses Ets-1 DNA binding by reinforcing autoinhibition. Genes Dev. 14:366–376 [PMC free article] [PubMed] [Google Scholar]
- 63. Pufall MA, Lee GM, Nelson ML, Kang HS, Velyvis A, Kay LE, McIntosh LP, Graves BJ. 2005. Variable control of Ets-1 DNA binding by multiple phosphates in an unstructured region. Science 309:142–145 [DOI] [PubMed] [Google Scholar]
- 64. Soufi A, Noy P, Buckle M, Sawasdichai A, Gaston K, Jayaraman PS. 2009. CK2 phosphorylation of the PRH/Hex homeodomain functions as a reversible switch for DNA binding. Nucleic Acids Res. 37:3288–3300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Li R, Knight J, Bream G, Stenlund A, Botchan M. 1989. Specific recognition nucleotides and their DNA context determine the affinity of E2 protein for 17 binding sites in the BPV-1 genome. Genes Dev. 3:510–526 [DOI] [PubMed] [Google Scholar]
- 66. Mannik A, Runkorg K, Jaanson N, Ustav M, Ustav E. 2002. Induction of the bovine papillomavirus origin “onion skin”-type DNA replication at high E1 protein concentrations in vivo. J. Virol. 76:5835–5845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Amin AA, Titolo S, Pelletier A, Fink D, Cordingley MG, Archambault J. 2000. Identification of domains of the HPV-11 E1 protein required for DNA replication in vitro. Virology 272:137–150 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.