

Abstract
Exposure of the skin to ultraviolet B (UVB) radiation causes oxidative damage to skin, resulting in sunburn, photoaging, and skin cancer. It is generally believed that the skin damage induced by UV irradiation is a consequence of generation of reactive oxygen species (ROS). Recently, there is an increased interest in the use of natural products as chemopreventive agents for non-melanoma skin cancer (NMSC) due to their antioxidants and anti-inflammatory properties. Quercitrin, glycosylated form of quercetin, is the most common flavonoid in nature with antioxidant properties. The present study investigated the possible beneficial effects of quercitrin to inhibit UVB irradiation-induced oxidative damage in vitro and in vivo. Our results showed that quercitrin decreased ROS generation induced by UVB irradiation in JB6 cells. Quercitrin restored catalase expression and GSH/GSSG ratio reduced by UVB exposure, two major antioxidant enzymes, leading to reductions of oxidative DNA damage and apoptosis and protection of the skin from inflammation caused by UVB exposure. The present study demonstrated that quercitrin functions as an antioxidant against UVB irradiation-induced oxidative damage to skin.
Keywords: ultraviolet B, quercitrin, antioxidant, reactive oxygen species, oxidative damage, apoptosis, inflammation
Introduction
Flavonoids, one of the most diverse and widespread groups of natural compounds, are the most common natural phenolics (Shimoi et al., 1996). Daily flavonoid intake (typically present in onion, apple, grape, wine, herbs, and spices) in the human diet is highly variable, with estimations ranging from 23 mg/day to more than 500 mg/day (Felgines et al., 2000; Hertog et al., 1993). Flavonoids are efficient antioxidants that scavenge oxygen radicals and possess anticancer, hypolipidemic, anti-aging, and anti-inflammatory activities (Yang et al., 2012). These biological activities exerted by flavonoids are mainly attributed to their antioxidant properties (Hollman et al., 1995).
Among flavonoids, quercetin is the most common flavonoid in nature and it is present in many fruits and vegetables such as apples, berries, grapes, onions, tea, and tomatoes, as well as many seeds, nuts, barks, and leaves (Kelly, 2011). It is mainly present as its glycosylated forms such as quercitrin (3-rhamnosyl-quercetin) or rutoside (3-rhamnosy-glucosyl quercetin). Quercitrin is a bioflavonoid with antioxidant properties and is better absorbed than other forms of quercetin (Morand et al., 2000). In addition to its chemopreventive activity against a variety of tumors (Granado-Serrano et al. 2006; Nguyen et al., 2004; van Erk et al., 2005), quercitrin also functions as an agent against bacterial infection (Soberon et al., 2007), allergic reaction (Cruz et al., 2008), and H2O2-and UV-induced cell death and apoptosis (Ham, 2012).
Exposure of the skin to ultraviolet B (UVB, 280–320 nm) radiation causes oxidative damage to skin, resulting in sunburn, photoaging, and skin cancer (Fisher et al., 1996). UVB exposure can act directly by inducing inflammation and proliferation in human and animal skin, or indirectly by producing reactive oxygen species (ROS) and by depleting the antioxidants in the skin (Bruner et al., 2000). UVB exposure is the major cause for non-melanoma skin cancer (NMSC), the most common type of human cancer (Yao et al., 2009). It has been demonstrated that most of the mutagenic and carcinogenic properties of sunlight can be attributed to chronic exposure to UVB irradiation (Afaq et al., 2005). Exposure of mammalian cells to UVB causes two major alterations that lead to deleterious outcomes: chemical modifications of DNA and generation of reactive oxygen species (ROS) (Vicentini et al., 2011).
Upon DNA damage by acute UV irradiation, cell cycle arrest will be occurred which allows the repair of damaged DNA. If the DNA damage caused by UV irradiation is too severe and is unable to be repaired, apoptosis pathways are activated to eliminate damaged cells. Long-term and recurrent exposure to UV irradiation causes the gradual deterioration of cutaneous structure and function. It apparently occurs as a result of cumulative DNA damage resulting from recurrent, acute DNA injury, and the effects of chronic inflammation. Those actinic damages could ultimately lead to the development of skin cancers, which is a multistep process involving induction of mutation and escape from immune surveillance.
DNA damage induced by UVB irradiation occurs mainly via oxidative processes and UVB is considered as the most important source of oxidative stress in human skin. Oxidative stress induced by UVB in irradiated cells causes ROS generation by activating certain small molecules such as riboflavin, tryptophan, and porphyrin. These molecules can then activate cellular oxygen (Ikehata and Ono, 2011). Uncontrolled release of ROS is involved in the pathogenesis of a number of human skin disorders by inducing oxidative DNA damage including base damage, single and double-strand breaks, and DNA-protein crosslinking (Black, 2004). ROS attack DNA and produce oxidative base damage such as 8-hydroxyguanine (8-OHdG) and thymine glycol in DNA or can cause strand breaks (Kino and Sugiyama, 2005). ROS also attack cellular nucleotide pools, producing oxidized nucleotides such as 8-hydroxydeoxygunanosine-triphosphate (8-OHdGTP), which can be used as nucleotide precursors for DNA synthesis (Sekiguchi and Tsuzuki, 2002).
Topical application of antioxidants has long been known to protect skin from oxidative damage. A number of natural antioxidant ingredients also have anti-inflammatory properties and can be used in the treatment of conditions associated with oxidative damage such as photoaging and perhaps even skin cancer (Fuchs, 1998; Steenvoorden and van Henegouwen, 1997). Some studies have proven the effectiveness of quercitrin in reducing ROS generation involved in the skin aging process and UVB-irritated inflammation (Casagrande et al., 2006; Vicentini et al., 2011). However, the mechanism of quercitrin, a better absorbed form of quercetin, against UVB-induced oxidative damage has not yet been fully understood. In the present study we examined the protective effect of quercitrin against UVB-induced oxidative DNA damage, apoptosis, and inflammation.
Material and Methods
Chemicals and reagents
Quercitrin and Annexin V/PI were purchased from Sigma-Aldrich (St. Louis, MO). Eagle’s miminal essential medium (EMEM), fetal bovine serum (FBS), gentamicin, and L-glutamine were from Invitrogen (Carlsbad, CA). DeadEnd™ Fluorometric TUNEL system and GSH/GSSG-Glo™ assay were obtained from Promega (Fitchburg, WI). Both 5-(and -6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA) and dihydroethidium (DHE) were obtained from Molecular Probes (Eugene, OR). Antibodies against SOD1, 8-OHdG, and β-actin were obtained from Santa Cruz (Santa Cruz, CA). Antibody against SOD2 was purchased from Millipore (Billerica, MA). Antibodies against catalase, cleaved PARP1, cleaved caspase-3, and γH2AX were purchased from Cell Signaling Tech (Danvers, MA). DNA extraction kit was from Qiagen (Valencia, CA). Single cell gel electrophoresis assay (Comet assay) kit and 8-OHdG ELISA kit were purchased from Trevigen (Gaithersburg, MD).
Cell culture and in vitro UVB exposure
The JB6 P+ mouse epidermal C141 cells (ATCC, CRL-2010) were cultured in monolayer at 37°C, 5% CO2 using MEM containing 5%FBS and 1% antibiotics. UVB Blak-Ray XX-15M UV meter with an output range of 280 to 320 nm was used for in vitro study (Joland, CA). The UVB output is measured with an IL1400A radiometer coupled with the SEL240/UVB-1/TD UVB detector (International Light, Peabody, MA). JB6 cells were seeded in 10-cm dishes. After 80–90% confluence, the cells were pretreated with quercitrin or vehicle (aceton) for 1 h. Then the cells were washed once with phosphate-buffered saline (PBS) before exposed to UVB irradiation in PBS which has no sunscreen effect. Immediately after UV irradiation, PBS was replaced with original media and plates were returned to the incubator for indicated time.
Animal treatment and in vivo UVB exposure
6–8 week old female SKH-1 hairless mice were purchased from Charles River Laboratories (Wilmington, MA) and maintained in animal house facility at the University of Kentucky Health Sciences Center under standard laboratory condition. The UV dosimeter for in vivo study was purchased from Daavlin (Bryan, OH). Mice were exposed to UV irradiation with a distance of 23 cm between the light source and the target skin. The mice were randomly divided in to 6 groups with 12 mice/group. The mouse dorsal skin was topically administrated with either aceton (control group, 200 μl/mouse) or quercitrin (1 mg/mouse in 200 μl aceton) a day before UV exposure to avoid possible sunscreen effect. The Mice were then exposed to 75 mJ/cm2 of UVB for 3 times per week up to 6 weeks. At 1 day and 7 days of post UVB irradiation, the animals were euthanized and dorsal skin tissues were removed for designed experiments.
Apoptosis analysis
Annexin V-fluorescein isothiocyanate (FITC)/Propidium Iodide (PI) double staining was used to measure percentile of apoptosisin JB6 cells according to manufacturer’s protocol (BD Pharmingen). Briefly, JB6 cells were grown in 10-cm dishes with MEM medium. After 80–90% confluence, the cells were pretreated with either quercitrin or aceton followed by UVB exposure. After 24 h, the cells were washed with PBS, digested with 0.25%trypsin/EDTA, and washed with PBS. The cells were re-suspended in 500 μl of 1×binding buffer and added with Annexin V-FITC/PI. After incubation at room temperature for 10min, the cells were measure by flow cytometry. TUNEL assay was used to measure apoptosis for mouse skin. Mouse skin tissue sections were stained using DeadEnd™ Fluorometric TUNEL system according to the manufacturer’s instruction (Promega Corporation).
Immunoblotting analysis
JB6 cells were lysed in RIPA buffer. The protein concentration was measured using Bradford Protein Assay Reagent (Bio-Rad) and 30 μg of sample proteins were separated by SDS-PAGE, and incubated with antibodies. The blots were then reprobed with second antibodies conjugated to horseradish peroxidase. Immunoreactive bands were detected by the enhanced chemiluminescence reagent (Amersham). For mitochondrial fractionation, JB6 cells (50×106) were fractionated by Mitochondrial Fractionation Kit (Active Motif, Carlsbad, CA) according to manufacturer’s instruction. Isolation of nuclear protein was followed by the manufacturer’s protocol (Abcam, Cambridge, MA).
Haematoxylin & Eosin (HE) staining
Isolated mouse skin tissues were fixed at 10% buffered formalin overnight and then immersed in 70% ethanol for storage at room temperature. The tissues were embedded in paraffin. Sections were cut at 5 μm on positively charged slides. The sections were stained with hematoxylin–eosin (HE) solution. All tissue samples were examined and photographed in a blinded fashion. Images were captured using an Olympus BX51 microscopeat ×100 magnification.
Comet assay in vitro and in vivo
Comet assay was carried out for detection of DNA damage according to manufacturer’s protocol. For in vitro assay, JB6 cells were seeded in 12-well plates. The cells were pretreated with or without quercitrin followed by UVB exposure described in “Cell culture” section. After 2 h, the cells were scraped and embedded in low melting agarose on CometSlide™ at 4°C for 10 minutes. The slides were incubated with lysing solution at 4°C for 30 minutes followed by immersion of alkaline unwinding solution for 30 minutes at room temperature. The slides were under electrophoresis at 21 V under neutral conditions. After electrophoresis, the slides were rinsed with distill water and 70% ethanol. Then the slides were stained with SYBR® green for 10 minutes. DNA damage was visualized using Olympus BX5 fluorescence microscope. 50 cells/slide were scored at magnification of x400 and analyzed using CometScore software (TriTek, Sumerduck, VA). Tail length, percent DNA in tail, and tail moment were measured. Percent DNA in the tail is a normalized measurement of total DNA in the cell. Tail moment is a damage measure of combining the amount of DNA in the tail with distance of migration.
For in vivo Comet assay, a small piece of mouse skin tissue was isolated and placed into 1–2 ml of ice cold 1xPBS (Ca2+ and Mg2+ free), 20 mM EDTA. The tissue was minced into very small pieces for 5 minutes using small dissecting scissor. The cell suspension was collected and total cell number was counted. After pelleting by centrifugation and re-suspension at 1×105 cells/ml in ice cold 1x PBS, the cells were embedded in low melting agarose on CometSlide™ at 4°C for 10 minutes. The following procedures were the same as in vitro assay.
8-OHdG detections in vitro and in vivo
For in vitro 8-OHdG detection, JB6 cells were cultured on sterilized cover slips until 80% confluence. The cells were treated with quercitrin and then exposed to UVB as described in the “Cell Culture” section. At the end of treatment, the cells were subjected for DNA extraction using Qiagen Flexigen Kit. A standard ELISA procedure was conducted for quantization of 8-OHdG according to manufacture’s manual. For in vivo 8-OHdG detection, mouse skin sections were deparaffinized and processed for Immunohistochemical staining as described in the “Immunohistochemical staining” below.
Immunohistochemical staining
Mouse skin section were deparaffinized and precessed for immunohistochemical staining according to the Vectastain ABC Kit protocol (Vector Laboratories, Burlingame, CA). Briefly, the sections were incubated with 3% H2O2 in distilled water to block endogenous peroxidase activity. After antigen retrieval, the sections were blocked with normal serum for 20 minutes and then incubated with primary antibody for 1 h. After washing with PBS, the sections were incubated with biotinylated-secondary antibodies for 30 minutes. The sections were then washed with PBS followed by incubation with ABC reagent. The sections were developed in DAB solution until the desired staining intensity was achieved. Finally, the sections were counterstained with hematoxylin.
Cellular hydrogen peroxide (H2O2) and superoxide anion (O2•−) assay
DCFDA and DHE are specific molecular probe for H2O2 and O2•−, respectively. The principle of this assay is that DCFDA diffuses through the cell membrane and is enzymatically hydrolyzed by intracellular esterases to non-fluorescent dichlorofluorescin (DCF). In the presence of H2O2, this compound is rapidly oxidized to highly fluorescent DCF (Bass et al., 1983; LeBel et al., 1992). DHE is oxidized to ethidium that stains nucleus a bright fluorescent red (Ye et al., 1999). The intensity of fluorescence represents the generations of H2O2 and O2•−. JB6 cells were cultured in 12-well plates. Each well contained 3×105 cells. The cells were pretreated with quercitrin prior to UVB exposure. DCFDA and DHE (final concentration, 10μM) were added to the cells and incubated for another 15–20 minutes prior to measurement of fluorescence. After incubation, cells were trypsinized and analyzed by using a Gemini XPS fluorescence microplate spectrofluorometer (Molecular Devices). The fluorescence intensity of DCF was measured at an excitation wavelength of 492 nm and an emission wavelength of 517 nm. The fluorescence intensity of DHE was measured at an excitation wavelength of 535 nm and an emission wavelength of 610 nm.
Electron spin resonance (ESR) assay
ESR measurement was conducted using a Bruker EMX spectrometer (Bruker Instruments, Billerica, MA) and a flat cell assembly. The intensity of the ESR signal is used to measure the amount of hydroxyl radical generation. The spin trapping is a method of choice for detection and identification of hydroxyl radical generation due to its specificity and sensitivity. 5,5-dimethyl-1-pyrroline 1-oxide (DMPO) was used as spin or radical trap. DMPO was charcoal purified and distilled to remove all ESR detectable impurities before use. Hyperfine couplings were measured (to 0.1 G) directly from magnetic field separation using potassium tetraperoxochromate (K3CrO8) and 1,1-diphenyl-2-picrylhydrazyl (DPPH) as reference standard. Reactants were mixed in test tubes to a total final volume of 0.5 ml. The reaction mixture was then transferred to a flat cell for ESR measurement.
GSH/GSSG assay
The ratio of GSH with GSSG in mouse skin tissues was examined according to manufacturer’s instruction (Promega). 10 mg of mouse skin tissue was homogenized in PBS containing 2 mM EDTA and centrifuged at 10,000 g for 15 minutes at 4 °C. 50 μl of supernatant was aliquot to 96-well plate. 50 μl of 2x GSH-GloTM reagent was added and incubated at room temperature for 30 minutes followed by addition of 100 μl of reconstituted luciferin detection reagent. After 15 minutes, the plate was read using luminescence.
Statistical Analysis
The data were expressed as the mean ± standard deviation (SD). Statistical significances of differences among treatment groups were determined by Student’s t-test. A p-value of <0.05 was considered as statistically significant.
Results
Quercitrin reduced UVB-irradiation-induced apoptosis
To investigate the ability of quercitrin against UVB-induced apoptosis, both JB6 cells and SKH-1 mice were used in this study. The results show that UVB irradiation induced apoptosis in a dose-dependent manner after 24 h of UVB irradiation of JB6 cells (Fig. 1A). Pretreatment with 10 μM quercitrin significantly inhibited UVB-induced apoptosis (Fig. 1B). The apoptosis was further inhibited by treatment with higher doses of quercitrin (20 and 40 μM). To verify this in vitro observation, SKH-1 mice were topically administrated with quercitrin one day before UVB irradiation. At 1 day and 7 days of post 6-weeks UVB exposure, the mice were euthanized and mouse skin tissue was isolated for immunohistological staining and immunoblotting. The results shows that 75 mJ/cm2 UVB irradiation induced about 50% of apoptosis in mouse skin epidermis compared to that in control group without UVB irradiation (Figs. 1C and 1D). The apoptosis between 1 day and 7 days post UVB exposure did not display significant difference (Figs. 1C and 1D). Topical administration of quercitrin dramatically ameliorated UVB irradiation-induced apoptosis (Figs. 1C and 1D).
Fig.1.

Effect of quercitrin on apoptosis and apoptotic proteins induced by UVB exposure. A, B, and E, JB6 cells were pretreated with quercitrin (10, 20, and 40 μM) or acetone for 1 h prior UVB exposure. After 24 h, the cells were collected for apoptosis analysis using flow cytometry (A and B) or for immunoblotting assay (E). C and D, 6–8 week old female SKH-1 mice dorsal skin was topically administrated with either aceton (control group) or quercitrin one day before UV exposure. The mice were then exposed to 75 mJ/cm2 of UVB for 3 times per week up to 6 weeks. At 1 day and 7 days, the animals were euthanized and dorsal skin tissues were isolated and subjected for immunofluoresence staining of apoptosis (C and D). The positive cells were counted from a total of 500 cells from 8 random field using Olympus BX51 microscope. The results were expressed as percentage of TUNEL-positive cells (apoptosis index). F, The same as C and D, but mouse dorsal skin tissues were isolated and total protein was extracted for examination of expression levels of cleaved PARP-1 and cleaved caspase-3. * indicates a significant difference compared with control without UVB exposure (p<0.05). # indicates a significant difference compared with 240 mJ/cm2 UVB exposure (p<0.05).
To confirm the involvement of apoptotic proteins, expressions of the two key proteins, cleaved PARP1 (C-PARP1) and cleaved caspase-3 (C-caspase-3) were examined both in JB6 cells and mouse skin tissue. The results shows that in JB6 cells, UVB irradiation caused a dose-dependent increase of C-PARP1 and that pretreatment with quercitrin decreased the activation of C-PARP1 by UVB (Fig. 1E). Similar results were observed on C-caspase-3 expression (Fig. 1E). The results from animal study show that the expressions of both C-PARP1 and C-caspase-3 at 7 days of post UVB exposure were higher than that at 1 day of post UVB exposure (Fig. 1F), indicating that the mouse skin was still struggling from UVB-induced damage due to the continuous stress from UVB exposure (6 weeks, 3 days/week) even though the UV irradiation was terminated. Topical administration of quercitrin on mouse dorsal skin ameliorated the expressions of C-PARP1 and C-caspase-3 induced by UVB in a dose-dependent manner (Fig. 1F), confirming the inhibitory ability of quercitrin on UVB-induced apoptosis.
Quercitrin inhibited NF-κB activation and prevented mouse skin from inflammatory damage induced by UVB irradiation
NF-κB, a transcript factor, is involved in mediation of inflammation. Fig. 2A shows that exposure of UVB to the JB6 cells caused a decrease of cytoplasmic NF-κB expression. Pretreatment with quercitrin reversed the inhibition of NF-κB induced by UVB irradiation. To investigate whether the reduced NF-κB expression in cytoplasm by UVB irradiation is due to translocation of NF-κB to the nuclei, nuclear protein was fractionated from the cells. The results show that the expression of NF-κB in nuclei was increased by UVB irradiation compared to the control (Fig. 2B). Pretreatment with quercitrin reduced the NF-κB activation in nuclei (Fig. 2B). To learn the protective effect of quercitrin against UVB induced skin damage, haematoxylin & eosin (HE) staining was used to examine the alteration of general mouse skin morphology under UVB irradiation and quercitrin administration (Fig. 2C). The results show that in the control mice without UVB irradiation, displayed a normal stratified squamous epithelium with normal keratinization. There was no evidence of inflammatory infiltrates in the superficial dermis. The skin appendages including hair follicles and sebaceous glands were unremarkable. However, the mouse skin euthanized at 1 day of post UVB irradiation showed acute epidermal necrosis with inflammatory infiltrates in the superficial and deep dermis (Fig. 2C-c). The superficial inflammatory infiltrates were perivascularly associated with granulation tissue, indicating that the skin was at least partially under repair. Topical administration of quercitrin did not reduce epidermal necrosis caused by UV irradiation (Fig. 2C-d). However, quercitrin reduced the inflammatory response in the superficial and deep dermis induced by UVB irradiation. Compared Figs. 2C-c, later termination of UVB irradiation (at 7 days, Fig. 2C-e) did not ameliorate skin damage induced by UVB. In contrast, at 7 days of post UVB irradiation, the mouse skin showed significant hyperkeratosis associated with changes consistent with actinic keratosis (thickened epidermis including basal and suprabasal layers of cells with disarray of cell polarity and hyperchromatic nuclei). The mice received quercitrin treatment showed no evidence of hyperkeratosis (Fig. 2C-f). Mild cell polarity changes and hyperchromasia were present in the basal cell layer. However, quercitrin did not alter keratinization (cornification) upon UVB irradiation.
Fig. 2. Quercitrin protects UVB-induced inflammation.
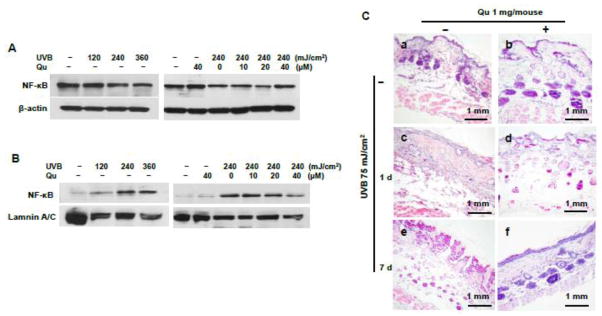
A and B, JB6 cells were pretreated with quercitrin (10, 20, and 40 μM) or acetone for 1 h prior UVB exposure. After 24 h, the cells were collected. Cytoplasmic (A) and nuclear (B) proteins were fractionated and NF-κB expression was examined by immunoblotting. C, Quercitrin administration and UVB irradiation is described in Fig. 1. At 1 day and 7 days, the animals were euthanized and dorsal skin tissues were isolated and fixed in 10% formalin. The mouse skin sections were prepared for Haematoxylin & Eosin (HE) staining. (a), Control without any treatment; (b), Pretreated with 1 mg/mouse of quercitrin, but without UVB exposure; (c) and (e), 1 day and 7 days of post 6-weeks of UVB exposure, respectively, but without pretreatment with quercitrin; (d) and (f), UVB exposure is the same as c and e, but pretreatment with 1 mg/mouse of quercitrin one day before UVB exposure.
Quercitrin reduced oxidative DNA damage induced by UVB irradiation
It has been demonstrated that quercetin possesses protective effect against UV-irradiation induced oxidative stress and inflammation by analyses of activities of myeloperoxidase and proteinase and of GSH level in hairless mice skin (Casagrande et al., 2006). Quercitrin is natural plants-derived antioxidant. We are mainly focusing on the antioxidant capability of quercitrin in the present study. Comet assay was used to monitor DNA single strand break (SSB) in the present study. Our in vitro Comet assay results show that UVB irradiation caused a dose-dependent DNA damage (Figs. 3A and 3C). Percentage of DNA in tail is a normalized measure of the percent of total cell DNA found in tail. Tail moment is defined as the product of the tail length and the fraction of total DNA in the tail. Tail moment is a damage measure combing the amount of DNA in the tail with the distance of migration (severity of damage). The results of Figs. 3A and 3C demonstrate that UVB irradiation increased both amount of DNA in tail and distance of DNA migration. Quercitrin pretreatment at 20 μM and 40 μM doses significantly reduced both percentage of DNA in tail and tail moment elevated by UVB (Figs. 3B and 3D). Similarly, the results from animal study indicate that UVB irradiation induced DNA damage of mouse skin epidermal cells as measured by percentage of DNA in tail and tail moment (Figs. 3E–3H). Topical administration of quercitrin at 1 mg/mouse dramatically reduced the DNA damage as measured by both percentage of DNA in tail and tail moment.
Fig. 3. Quercitrin inhibits DNA damage by UVB exposure.

A–D, JB6 cells were pretreated with quercitrin (10, 20, and 40 μM) or acetone for 1 h prior UVB exposure. After 2 h, the cells were collected for Comet assay. E–H, The animal treatment is the same as that in Fig. 1. At 1 day and 7 days of 6-week treatment, the mice were euthanized and small pieces of skin tissues/mouse was isolated and subjected to Comet assay. A, B, E, and F, Percentage of DNA in tail over that in total. C, D, G, and H, Tail moment. * indicates a significant difference compared with control without UVB exposure (p<0.05). # indicates a significant difference compared with 240 mJ/cm2 (in vitro) or 75 mJ/cm2 (in vivo) of UVB exposure (p<0.05).
It has been well known that UVB irradiation caused oxidative DNA damage. However, the inhibitory capacity of quercitrin on oxidative DNA damage is still unclear. Therefore, the levels of 8-OHdG both in vitro and in vivo were measured (Fig. 4). As shown in Fig. 4A, the production of 8-OHdG was significantly increased in the JB6 cells at 2 h after UVB irradiation (240 and 480 mJ/cm2). Quercitrin pretreatment diminished the 8-OHdG production induced by UVB irradiation (Fig. 4B). The results from immunohistological staining of mouse skin tissue section showed that UVB irradiation caused increase of 8-OHdG (Fig. 4C). 8-OHdG expression was higher at 7 days than at 1 day of post 6-week UVB irradiation. This result is consistent with that of Comet assay. Topical administration of quercitrin resulted in decreased 8-OHdG production (Fig. 4C).
Fig. 4. Quercitrin inhibits 8-OHdG induced by UVB exposure.

A and B, JB6 cells were pretreated with quercitrin (10, 20, and 40 μM) or acetone for 1 h prior UVB exposure. After 2 h, the cells were collected to quantify 8-OHdG production using ELISA analysis. C, The animal treatment is the same as that in Fig. 1. At 1 day and 7 days of 6-week treatment, the mice were euthanized and dorsal skin tissues were isolated and fixed in 10% formalin. The mouse skin sections were used to immunohistological staining with 8-OHdG. Arrows show example of 8-OHdG stained in some cells.
The effects of quercitrin on DNA damage/repair genes induced by UVB irradiation
From previous results we note that quercitrin was able to reduce oxidative damage irradiated by UVB exposure, we would like to investigate ability of quercitrin on those DNA damage/repair genes responsible for the oxidative damage. γH2AX is a marker of DNA double strand breaks (DSBs). Our in vitro results show that UVB irradiation caused a dose-dependent increase of γH2AX at 2 h after UVB exposure, pretreatment with quercitrin reduced γH2AX expression (Fig. 5A). The results from in vivo study show that there was no expression of γH2AX in the epidermal skin cells, the dermal fibroblasts and skin adnexal cells in control mice without any treatment (Fig. 5C). In contrast, γH2AX expressions were significantly increased in epidermal cells, dermal inflammatory cells, and fibroblasts in the mice skin at 1 day of post-UVB exposure. Administration of quercitrin attenuated the γH2AX expressions in the skin cells, dermal inflammatory cells and fibroblasts irradiated by UVB exposure (Fig. 5C). However, no expression of γH2AX was observed in the mice at 7 days of post-UVB exposure (Fig. 5C). XPA, a DNA repair gene, its expression was significantly decreased in the cells exposed by UVB irradiation. Pretreatment of quercitrin restored the reduced XPA expression by UVB in a dose-dependent manner (Fig. 5A). Interestingly, in those mice exposed to UVB for 6 weeks, the expression of XPA was slightly increased at 1 day but not at 7 days of post UVB exposure compared to that in control mice without exposure (Fig. 5B). Administration of quercitrin obviously increased XPA expression in the mice with or without UVB irradiation (Fig. 5B). These results indicate that self-defense of mice skin was activated under the presence of continuous external stress (UVB irradiation in the present study). Once the oxidative stress was terminated, this self-defense was no longer needed. Thus, the expression of XPA was decreased to normal level.
Fig. 5.
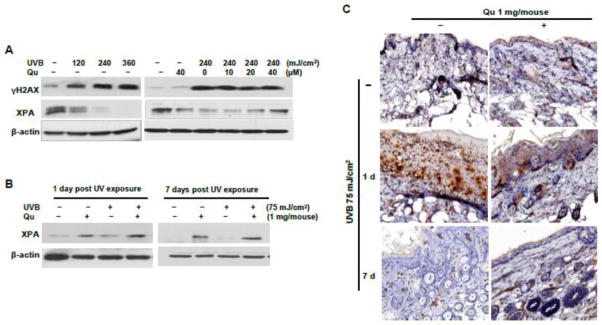
Effect of quercitrin on DNA repair genes induced by UVB exposure. A, JB6 cells were pretreated with quercitrin (10, 20, and 40 μM) or acetone for 1 h prior UVB exposure. After 2 h, the cells were collected for immunostaining analysis. B and C, The animal treatment is the same as that in Fig. 1. At 1 day and 7 days of 6-week treatment, the mice were euthanized and mouse skin tissues were isolated. The protein was lysated for immunoblotting analysis (B). The mouse skin sections were used to immunohistological staining with γH2AX (C).
Quercitrin inhibited ROS generation induced by UVB irradiation
UVB is a potent inducer of various ROS and oxidative stress in difference cell types. Among these ROS, superoxidae radical (O2•−), hydrogen peroxide (H2O2), and hydroxyl radical (•OH) are most important. We analyzed whether UVB-induced ROS generation is responsible for DNA damage and apoptosis and how quercitrin modulated UVB induced ROS generation in JB6 cells. UVB irradiation at 120 mJ/cm2 for 2 h dramatically stimulated O2•− generation indicated by increase of DHE fluorescence fold in Fig. 6A. A dose-dependent increase of O2•− generation was observed when the doses of UVB irradiation increased up to 240 mJ/cm2. Pretreatment with quercitrin at 20 μM and 40 μM significantly decreased UVB-induced O2•− generation (Fig. 6B). Similarly, UVB irradiation increased generations of H2O2 and •OH (Figs. 6C, 6D, and 6E). Quercitrin was able to inhibit both H2O2 and •OH generations.
Fig. 6. Quercitrin inhibits ROS generation induced by UVB exposure.

A, B, C, and D, JB6 cells were pretreated with quercitrin (10, 20, and 40 μM) or acetone for 1 h prior UVB exposure. After 2 h, the cells were subjected for measurement of O2•− (A and B) and H2O2 (C and D). E, 2×106 JB6 cells were collected for measurement of •OH generation. (a) Electron spin resonance (ESR) spectrum recorded 2 minutes from the mixture of JB6 cells and 100 mM 5,5-dimethyl-1-pyrroline 1-oxide (DMPO); (b) the same as (a) but the cells were exposed to 240 mJ/cm2 UVB exposure; (c), The same as (a) the cells were pretreated with 10 μM quercitrin for 1 h before ESR measurement; (d), (e), and (f) the same as (b) but the cells were pretreated with different concentration of quercitrin (10, 20, and 40 μM) 1 h prior to UV exposure. * indicates a significant difference compared with control without UVB exposure (p<0.05). # indicates a significant difference compared with 240 mJ/cm2 UVB exposure (p<0.05).
Quercitrin restored the expressions of antioxidant enzymes reduced by UVB irradiation
So far we discussed capability of quercitrin against UVB-induced ROS generation. Next we investigated the protective ability of quercitrin on UVB-reduced antioxidant enzymes. Catalase and superoxide dismutase (SOD) are important specific scavengers of H2O2 and O2•−, respectively. The expression levels of these antioxidant enzymes were examined in JB6 cells in the present study. SOD1 and SOD2 are two major isoforms of SOD family. SOD1 is located in cytoplasm and SOD2 in mitochondria. The results show that UVB irradiation caused a dose-dependent decrease of both catalase and SOD2 expressions, but not SOD1 (Fig. 7A). To examine the expression level of SOD2 in mitochondria, mitochondrial protein was fractioned and conducted for immunoblotting. Not surprising, the expression of SOD2 in mitochondria was increased upon UVB irradiation (Fig. 7B). Pretreatment with quercitrin restored the expressions of both catalase and SOD2, indicating a potent antioxidant property of quercitrin. To investigate the oxidative redox status after long term UVB irradiation, the expressions of catalase and SOD and the ratio of GSH/GSSG were examined in SKH-1 mice. Similar pattern as those in vitro was observed in catalase expression (Fig. 7C). However, the expressions of SOD1 and SOD2 did not exhibit alignment with that in vitro. Both SOD1 and SOD2 expressions were slightly increased at 1 day but decreased at 7 days of post UVB irradiation. The GSH/GSSH results show that UVB irradiation caused a dramatic depletion of GSH at both 1 day and 7 days (Figs.7D and 7E). Quercitrin restored the GSH depletion back to the control level.
Fig. 7.
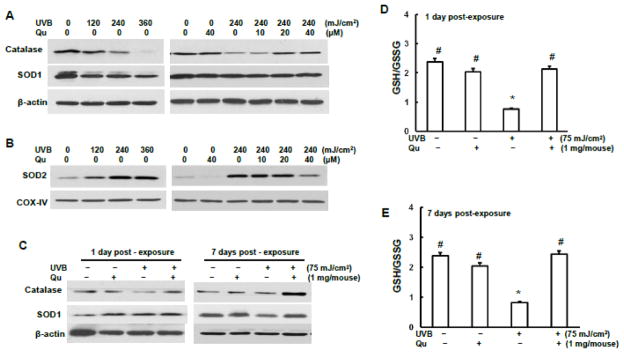
Quercitrin increases antioxidant enzyme expressions reduced by UVB exposure. A, B, and C, JB6 cells were pretreated with quercitrin (10, 20, and 40 μM) or aceton for 1 h followed by UVB exposure. A, The whole cell protein lysate were isolated for immunoblotting to examine the expression levels of catalase, SOD1, and SOD2. B, Mitochondrial protein were fractioned and subjected to immunoblotting to examine SOD2 expression level. C, The animal treatment is the same as Fig. 1. At 1 day and 7 days after 6-week UVB exposure, the mice dorsal skin tissues were isolated and homogenized for GSH/GSSG measurement.* indicates a significant difference compared with control without UVB exposure (p<0.05). # indicates a significant difference compared with 240 mJ/cm2 UVB exposure (p<0.05).
Discussion
Non-melanoma skin cancer (NMSC) is the most commonly diagnosed among all human malignancies and is the cause of 2,000 deaths annually associated with health care costs. New treatment strategies are very much needed to prevent the development of precancerous actinic keratoses and squamous cell tumors caused by prolonged exposure to ultraviolet light. Recently, there is an increase interest in use of natural products as chemopreventive agents for NMSC and other types of cancer due to their anti-oxidant and anti-inflammatory properties in addition to their potential to inhibit numerous signaling pathways involved in cell proliferation, transformation, migration, and survival (Lee et al., 2008; Lin et al., 2008; Xavier et al., 2009). However, the cellular effects of these compounds are often very complicated due to the general lack of specificity for any particular intracellular target, and the end effect on cell fate is often the combination of multiple cellular activities. Various in vitro studies have demonstrated that quercetin is able to block UVB-induced activations of PI-3K, MAPK, c-fos, and AP-1(Olson et al., 2010), the signaling proteins related to inflammation and carcinogenesis. However, the protection of quercitrin, a better absorbed form of quercetin, against UVB irradiation-induced oxidative damage is not well known.
UV radiation causes skin damage, resulting in both pre-cancerous and cancerous skin lesions, and acceleration of skin aging (Casagrande et al., 2006). The alternation of skin pathologies under UV irradiation is a consequence of the generation of ROS. The resulting imbalance between oxidants and antioxidants, in favor to the former, shifts the cellular redox-sensitive cellular signal transduction pathways and gene expression. It has been shown that UVB radiation produced DNA damage directly and indirectly through oxidative stress (Ichihashi et al., 2003). Apoptosis is oxidative responsive process and is tightly bound with DNA damage. UVB irradiation which induces ROS generation and DNA damage is known to evoke apoptosis of the cells (Barzilai and Yamamoto, 2004). Our in vitro and in vivo studies demonstrate that quercitrin blocked UVB-induced oxidative stress and DNA damage, leading to the decrease of apoptosis induced by UVB exposure. While PARP appears to be involved in DNA repair in response to environmental stress (Satoh and Lindahl, 1992), cleavage of PARP (c-PARP) facilitates cellular disassembly and serves as a marker of cells undergoing apoptosis (Oliver et al., 1998). This protein is one of the main cleavage targets of caspase-3 (Nicholson et al., 1995; Tewari et al., 1995). Consistently with the apoptosis analysis, our data from cell and animal studies showed that quercitrin treatment decreased UVB-induced expressions of both c-caspase-3 and c-PARP-1, suggesting important role of these two proteins in reduction of apoptosis by quercitrin.
Oxidative stress plays a significant role in UVB-induced skin carcinogenesis (Agar et al., 2004; Halliday, 2005). Excessive ROS generation causes a range of DNA damages, including DNA strand breaks, DNA-protein cross-links, deletion mutations, and 8-hydroxyl-2-deoxyguanine (8-OHdG)(Kielbassa et al., 1997; Kino and Sugiyama, 2005; Kvam and Tyrrell, 1997). The SSB measured by Comet assay is not only related to oxidation. It might also represent intermediates in the repair process (Collins, 2009). If SSB are not repaired during the G1 phase of the cell cycle, they can generate more hazardous double-strand breaks (DSBs) during the S phase which then can lead to chromosomal aberrations (Wischermann et al., 2008). The generation of the phosphorylated form of histone H2AX (c-H2AX) has been identified as an early event after DSBs formation and is considered the most sensitive assay for their detection (Barnes et al., 2010). As shown in the results of Comet assay in vitro and in vivo, quercitrin attenuated SSB induced by UVB irradiation. Similarly, in the JB6 cells, exposure of UBV in a short term resulted in an increased expression of γH2AX. Pretreatment with quercitrin reduced the UVB-induced γH2AX expression. In the animal study, at 1 day of post 6-week UVB exposure, the expression of γH2AX was significantly increased in the epidermal cells, dermal inflammatory cells, and fibroblasts. Topical administration of quercitrin attenuated the γH2AX expressions in skin cells, dermal inflammatory cells, and fibroblasts induced by UVB. However, the γH2AX expression was not observable at 7 days of post 6-week UVB exposure. That could be explained by the quick recovery of DNA damage from UVB exposure.
8-OHdG is a biomarker of oxidative stress and the major mutagenic form of oxidative DNA damage (Barzilai and Yamamoto, 2004). 8-OHdG lesions can lead to G:C to T:A transversion mutations, and oxidative DNA lesions are detected in many human tumors, including human squamous cell carcinoma (Evans et al., 2004; Valko et al., 2004). The present study hypothesizes that quercitrin functions as an antioxidant against UVB-induced oxidative damage. Our in vitro and in vivo results indicate that quercitrin was able to inhibit 8-OHdG production induced by UVB irradiation. PARP-1 plays multiple roles in cellular response to genotoxic insult. It has been reported that pretreatment of cells with a PARP-1 inhibitor or PARP-1 siRNA significantly increased UVR-induced 8-OHdG lesions, implicating PARP-1 in the repair of oxidative DNA damage (Ding et al., 2009). UVB-induced 8-OHdG formation may coincide with PARP-1 activation. However, in the present animal study, totalPARP-1 expression was not increased upon UVB- irradiation, indicating that PARP-1 is not an important response repair protein at this time point of UVB exposure.
Xeroderma pigmentosum type A (XPA) is known as a scaffold protein without enzymatic activity that shows preferential association to damaged DNA and is indispensable for DNA incision (de Vries et al., 1995; Missura et al., 2001). It functions in a pre-incision step, the recognition of DNA damage (Zhovmer et al., 2010). XPA deficiency is known to decrease antioxidant defenses and to increase susceptibility to UVB-induced skin cancer (de Vries et al., 1995). Our in vitro result shows that acute exposure of UVB caused a sharp decrease of XPA expression, suggesting that the cells were unable to activate the nucleotide-excision repair (NER) which is involved in the removal of a wide spectrum of DNA lesions upon acute external stress. Excitingly, quercitrin is able to restore the reduced expression of XPA by UVB, confirming the dual capabilities of quercitrin against UVB-induced damage by inhibition damage and promotion repair. The results of in vivo study show that XPA expression was slightly increased at 1 day but not 7 days of post UVB irradiation, indicating that immediately after termination of UVB irradiation, the animal’s defense system is evoked under consistent oxidative stress. Once the animals were used to the condition without oxidative stress, they were no longer able to produce self-protection. Administration of quercitrin increased expression of XPA in the mice of both with and without UVB exposure, suggesting that quercitrin boosters the repair ability.
Acute UV skin exposure induces an immediate inflammatory response with erythema and leukocyte infiltration. NF-κB is a redox-sensitive transcription factor that activates multiple target genes involved in the expression of several proinflammatory mediators (Fuchs et al., 2001). Myeloperoxidase (MPO) is a marker of neutrophils presence or inflammation (Bradley et al., 1982; Xia and Zweier, 1997). It has been suggested that UV-induced inflammation is mediated by DNA damage leading to the release of pro-inflammatory cytokines (Widyarini et al., 2001). Quercetin suppressed UVB-irradiation-induced expression of inflammatory cytokines IL-1β, IL-6, IL-8, and tumour necrosis factor (TNF)-α in human keratinocytes (Vicentini et al., 2011). Topical application of quercetin significantly decreased the UVB-induced increase in the MPO in hairless mouse skin (Casagrande et al., 2006). Additionally, quercetin inhibited the carrageenan-induced TNF-α and prostaglandin (PG)E2 production (Morikawa et al., 2003). PGE2 are the major inflammatory mediators of UVB erythema, but play a minor role in UVC and UVA erythema (Podda et al., 1998), suggesting a possible additional anti-inflammatory mechanism for quercetin on UVB-induced skin damage. The results of our immunohistological study indicate that quercitrin reduced the inflammatory response in the superficial and deep dermis and hyperkeratosis induced by UVB irradiation while quercitrin did not display any inhibition on epidermal necrosis caused by UV irradiation. This could be explained that duration of 6 weeks for quercitrin treatment is no longer enough to overcome the accumulative skin damage induced by UVB irradiation. However, our results (data not shown) show that quercitrin significantly reduced mouse squamous skin cancer induced by UVB irradiation after5 months of exposure, indicating the potent antitumor capacity of quercitrin.
It has been reported that UV light is lethal and mutagenic. ROS such as O•− and •OH are responsible for this mutagenesis (Strickland, 1986; Fuchs et al., 1989; Hillebrand et al., 1990; Nishi et al., 1991; Shindo et al., 1993; Evelson et al., 1996). It has been shown that UV light initiates lipid peroxidation in mitochondria and prolongs free radical reactions. Exposure of skin cells to UVB induces an immediate release of labile iron, which can catalyze production of the highly toxic •OH via the Fenton reaction (Kruszewski, 2003). •OH is the primary reactive oxygen species responsible for the formation of lipid radicals in the epidermis following exposure to UV light (Ogura et al., 1991). O2•− induced by UVB irradiation converts to H2O2 either spontaneously (in pH 4.8) or by dismutation catalyzed by superoxide dismutase (SOD) (in neutral and alkaline pH). It has been reported that H2O2 is part of the acute skin response to UVB exposure and can be generated by keratinocytes and infiltrating neutrophils (Wei et al., 2002). In the present study, exposing mouse epidermal JB6 cells to increasing doses, 120–480 mJ/cm2 resulted in a dose-dependent increases of H2O2 and O2•− generation, as well as •OH. Pretreatment with quercitrin reduced the ROS generation induced by UVB irradiation. Quercetin has been shown to scavenge O2•− and •OH radicals to prevent lipid peroxidation (Fuhrman et al., 1995), to inhibit cyclooxygenase and lipoxygenase enzymes (Moroney et al., 1988), and to chelate transition metal ions such as iron (Formica and Regelson, 1995). Our results further show that quercitrin is a potential scavenger of ROS generated by UVB exposure in cellular system.
It has been known for decades that ROS produced in the skin following UV irradiation are key mediators of oxidative damage to the skin, resulting in a rapid depletion of several endogenous skin enzymes and antioxidants such as glutathione reductase, catalase, and superoxide dismutase (SOD), as well as glutathione, tocopherol, and ubiquinone (Liebel et al., 2012). UV-induced oxidative stress is countered in the body by endogenous antioxidants that neutralize the ROS before they can produce oxidative changes in the tissues (Podda and Grundmann-Kollmann, 2001). Catalase and SOD are specific scavengers of H2O2 and O2•−, respectively. Among SOD, three isoforms are present in mammals, SOD1 is located in the cytoplasm, SOD2 in the mitochondria, and SOD3 is extracellular. Mitochondria, the major oxygen-metabolizing organelles of the cell, are also the major source of ROS in the cell, with the O2•− as the initial ROS produced by this organelle (Hoye et al., 2008). O2•− can participate in the production of other radicals, including the reactive nitrogen species (RNS) peroxynitrite (Huie and Padmaja, 1993). SOD2, manganese superoxide dismutase (MnSOD), is the major ROS detoxifying enzyme of cells because of its localization to mitochondria. Altered function or expression of SOD2 can have remarkable consequences on mitochondrial function and the overall health of cells due to oxidative damage to various mitochondria-localized metabolic processes, leading to the development of different diseases (Holley et al., 2011; Miao and St Clair, 2009). These endogenous enzymes are mainly concentrated in the epidermis compared to dermis (Casagrande et al., 2006). The antioxidant systems in the epidermis are more sensitive to UVB irradiation than these in dermis, suggesting that epidermis is the first line of skin defense in response to UVB exposure. It was reported that intraperitonal administration of quercetin increased activities of GSH, SOD, and catalase in rat liver reduced by UVA irradiation (Erden Inal and Kahraman, 2000). In the present study, dose-dependent decreases of both catalase and SOD2 expression were observed, but not SOD1, upon the UVB irradiation. To further investigate the expression level of SOD2 in mitochondria in response to UVB irradiation, the mitochondrial protein was isolated and was found that SOD2 expression in mitochondria was increased, indicating activation of cell defense system against increased O2•− generation. Interestingly, pretreatment with quercitrin not only reduced ROS generation, but also increased catalase expression, suggesting that quercitrin was able to booster the expression of antioxidant enzyme. It maybe noticed that quercitrin failed to increase SOD2 expression in whole cell lysate reduced by UVB irradiation. However, higher concentration of quercitrin treatment (40 μM) do reduced mitochondrial SOD2 expression. Similarly, the results from animal study indicate that catalase is a sensitive target of quercitrin. Both at the 1 day and 7 days of post 6-week UV irradiation, catalase expression was significantly decreased. Topical administration of quercitrin restored the expression of catalase to the control level (1 day of post UVB irradiation) or even higher (7 days of post UVB irradiation). The expressions of both SOD1 and SOD2 were slightly increased at the 1 day of post UVB irradiation. Administration of quercitrin did not affect the expressions of these two antioxidant enzymes. However, expressions of both SOD1 and SOD2 were reduced at 7 days of post UVB irradiation. By counteracting the reduction of UVB, quercitrin restored SOD2 expression to even higher than that in control group, indicating that SOD2, but not SOD1 is the target of quercitrin for decreasing O2•− generation.
The glutathione redox status has been confirmed as an early and sensitive sensor of UVB induced epidermal oxidative stress, suitable for testing the protective antioxidant effect of a substance (Meloni and Nicolay, 2003). The GSH directly scavenges radicals by hydrogen transferring and acts as a cofactor for the enzyme glutathione peroxidase, this enzyme in turn scavenges peroxides and regenerates vitamins E and C (Carini et al., 2000). It has been demonstrated that quercetin protects the GSH depletion induced by buthionine sulfoximine in cultured human skin fibroblast and keratinocytes (Skaper et al., 1997). In agreement, it has been reported that quercetin is able to prevent UVB irradiation induced GSH depletion (Casagrande et al., 2006). The results of our in vivo study show that UVB decreased GSH/GSSG ratio, an indicator of oxidative stress. Quercitrin counteracted the effect of UVB and restored GSH/GSSG ratio to normal level, indicating the protection of quercitrin against UVB-induced oxidative stress.
In conclusion, UVB irradiation caused ROS generations, leading to oxidative DNA damage and therefore apoptosis and quercitrin exhibited protective effects. UVB irradiation-induced oxidative stress decreased expression levels of antioxidant enzymes catalase and SOD1 and GSH/GSSG ratio and activatd mitochondrial SOD. Quercitrin functions as an antioxidant to protect the oxidative damage induced by UVB exposure.
Highlights.
Oxidative stress plays a key role in the mechanism of UV-induced cell and tissue injuries.
Quercitrin decreased ROS generation and restored catalase expression and GSH/GSSG ratio induced by UVB irradiation.
Quercitrin reduced UVB-irradiated oxidative DNA damage, apoptosis, and inflammation.
Quercitrin protects against UV-irradiated skin oxidative damage and inflammation through inhibition of reactive oxygen species generation and increase of antioxidant enzyme levels.
Acknowledgments
This study was supported by NIH 1R21ES019249 to Dr. Z. Zhang and Dr. X. Shi.
Footnotes
Conflict of Interest
This manuscript will not be submitted to any other scientific journal prior to the decision by Toxicology and Applied Pharmacology. Each listed author on the manuscript is aware of and agrees to the contents of the manuscript, including the authorship. None of the listed authors has any financial or other interests that could be of conflict.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Afaq F, Adhami VM, Mukhtar H. Photochemoprevention of ultraviolet b signaling and photocarcinogenesis. Mutat Res. 2005;571:153–173. doi: 10.1016/j.mrfmmm.2004.07.019. [DOI] [PubMed] [Google Scholar]
- Agar NS, Halliday GM, Barnetson RS, Ananthaswamy HN, Wheeler M, Jones AM. The basal layer in human squamous tumors harbors more uva than uvb fingerprint mutations: A role for uva in human skin carcinogenesis. Proc Natl Acad Sci U S A. 2004;101:4954–4959. doi: 10.1073/pnas.0401141101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes L, Dumas M, Juan M, Noblesse E, Tesniere A, Schnebert S, et al. Gammah2ax, an accurate marker that analyzes uv genotoxic effects on human keratinocytes and on human skin. Photochem Photobiol. 2010;86:933–941. doi: 10.1111/j.1751-1097.2010.00744.x. [DOI] [PubMed] [Google Scholar]
- Barzilai A, Yamamoto K. DNA damage responses to oxidative stress. DNA Repair (Amst) 2004;3:1109–1115. doi: 10.1016/j.dnarep.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Bass DA, Parce JW, Dechatelet LR, Szejda P, Seeds MC, Thomas M. Flow cytometric studies of oxidative product formation by neutrophils: A graded response to membrane stimulation. J Immunol. 1983;130:1910–1917. [PubMed] [Google Scholar]
- Black HS. Ros: A step closer to elucidating their role in the etiology of light-induced skin disorders. J Invest Dermatol. 2004;122:xiii–xiv. doi: 10.1111/j.0022-202X.2004.22625.x. [DOI] [PubMed] [Google Scholar]
- Bradley PP, Priebat DA, Christensen RD, Rothstein G. Measurement of cutaneous inflammation: Estimation of neutrophil content with an enzyme marker. J Invest Dermatol. 1982;78:206–209. doi: 10.1111/1523-1747.ep12506462. [DOI] [PubMed] [Google Scholar]
- Bruner SD, Norman DP, Verdine GL. Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature. 2000;403:859–866. doi: 10.1038/35002510. [DOI] [PubMed] [Google Scholar]
- Carini M, Aldini G, Piccone M, Facino RM. Fluorescent probes as markers of oxidative stress in keratinocyte cell lines following uvb exposure. Farmaco. 2000;55:526–534. doi: 10.1016/s0014-827x(00)00037-9. [DOI] [PubMed] [Google Scholar]
- Casagrande R, Georgetti SR, Verri WA, Jr, Dorta DJ, dos Santos AC, Fonseca MJ. Protective effect of topical formulations containing quercetin against uvb-induced oxidative stress in hairless mice. J Photochem Photobiol B. 2006;84:21–27. doi: 10.1016/j.jphotobiol.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Collins AR. Investigating oxidative DNA damage and its repair using the comet assay. Mutat Res. 2009;681:24–32. doi: 10.1016/j.mrrev.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Cruz EA, Da-Silva SA, Muzitano MF, Silva PM, Costa SS, Rossi-Bergmann B. Immunomodulatory pretreatment with kalanchoe pinnata extract and its quercitrin flavonoid effectively protects mice against fatal anaphylactic shock. Int Immunopharmacol. 2008;8:1616–1621. doi: 10.1016/j.intimp.2008.07.006. [DOI] [PubMed] [Google Scholar]
- de Vries A, van Oostrom CT, Hofhuis FM, Dortant PM, Berg RJ, de Gruijl FR, et al. Increased susceptibility to ultraviolet-b and carcinogens of mice lacking the DNA excision repair gene xpa. Nature. 1995;377:169–173. doi: 10.1038/377169a0. [DOI] [PubMed] [Google Scholar]
- Ding W, Liu W, Cooper KL, Qin XJ, de Souza Bergo PL, Hudson LG, et al. Inhibition of poly(adp-ribose) polymerase-1 by arsenite interferes with repair of oxidative DNA damage. J Biol Chem. 2009;284:6809–6817. doi: 10.1074/jbc.M805566200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erden Inal M, Kahraman A. The protective effect of flavonol quercetin against ultraviolet a induced oxidative stress in rats. Toxicology. 2000;154:21–29. doi: 10.1016/s0300-483x(00)00268-7. [DOI] [PubMed] [Google Scholar]
- Evans MD, Dizdaroglu M, Cooke MS. Oxidative DNA damage and disease: Induction, repair and significance. Mutat Res. 2004;567:1–61. doi: 10.1016/j.mrrev.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Felgines C, Texier O, Morand C, Manach C, Scalbert A, Regerat F, et al. Bioavailability of the flavanone naringenin and its glycosides in rats. Am J Physiol Gastrointest Liver Physiol. 2000;279:G1148–1154. doi: 10.1152/ajpgi.2000.279.6.G1148. [DOI] [PubMed] [Google Scholar]
- Fisher GJ, Datta SC, Talwar HS, Wang ZQ, Varani J, Kang S, et al. Molecular basis of sun-induced premature skin ageing and retinoid antagonism. Nature. 1996;379:335–339. doi: 10.1038/379335a0. [DOI] [PubMed] [Google Scholar]
- Formica JV, Regelson W. Review of the biology of quercetin and related bioflavonoids. Food Chem Toxicol. 1995;33:1061–1080. doi: 10.1016/0278-6915(95)00077-1. [DOI] [PubMed] [Google Scholar]
- Fuchs J. Potentials and limitations of the natural antioxidants rrr-alpha-tocopherol, l-ascorbic acid and beta-carotene in cutaneous photoprotection. Free Radic Biol Med. 1998;25:848–873. doi: 10.1016/s0891-5849(98)00161-0. [DOI] [PubMed] [Google Scholar]
- Fuchs J, Zollner TM, Kaufmann R, Podda M. Redox-modulated pathways in inflammatory skin diseases. Free Radic Biol Med. 2001;30:337–353. doi: 10.1016/s0891-5849(00)00482-2. [DOI] [PubMed] [Google Scholar]
- Fuhrman B, Lavy A, Aviram M. Consumption of red wine with meals reduces the susceptibility of human plasma and low-density lipoprotein to lipid peroxidation. Am J Clin Nutr. 1995;61:549–554. doi: 10.1093/ajcn/61.3.549. [DOI] [PubMed] [Google Scholar]
- Granado-Serrano AB, Martin MA, Bravo L, Goya L, Ramos S. Quercetin induces apoptosis via caspase activation, regulation of bcl-2, and inhibition of pi-3-kinase/akt and erk pathways in a human hepatoma cell line (hepg2) J Nutr. 2006;136:2715–2721. doi: 10.1093/jn/136.11.2715. [DOI] [PubMed] [Google Scholar]
- Halliday GM. Inflammation, gene mutation and photoimmunosuppression in response to uvr-induced oxidative damage contributes to photocarcinogenesis. Mutat Res. 2005;571:107–120. doi: 10.1016/j.mrfmmm.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Ham YMY, WJ, Park SY, Song GP, Jung YH, Jeon YJ, Kang SM, Kim KN. Quercitrin protects against oxidative stress-induced injury in lung fibroblast cells via up-regulation of bcl-xl. Journal of Functional Foods. 2012;4:10. [Google Scholar]
- Hertog MG, Hollman PC, Katan MB, Kromhout D. Intake of potentially anticarcinogenic flavonoids and their determinants in adults in the netherlands. Nutr Cancer. 1993;20:21–29. doi: 10.1080/01635589309514267. [DOI] [PubMed] [Google Scholar]
- Holley AK, Bakthavatchalu V, Velez-Roman JM, St Clair DK. Manganese superoxide dismutase: Guardian of the powerhouse. Int J Mol Sci. 2011;12:7114–7162. doi: 10.3390/ijms12107114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollman PC, de Vries JH, van Leeuwen SD, Mengelers MJ, Katan MB. Absorption of dietary quercetin glycosides and quercetin in healthy ileostomy volunteers. Am J Clin Nutr. 1995;62:1276–1282. doi: 10.1093/ajcn/62.6.1276. [DOI] [PubMed] [Google Scholar]
- Hoye AT, Davoren JE, Wipf P, Fink MP, Kagan VE. Targeting mitochondria. Acc Chem Res. 2008;41:87–97. doi: 10.1021/ar700135m. [DOI] [PubMed] [Google Scholar]
- Huie RE, Padmaja S. The reaction of no with superoxide. Free Radic Res Commun. 1993;18:195–199. doi: 10.3109/10715769309145868. [DOI] [PubMed] [Google Scholar]
- Ichihashi M, Ueda M, Budiyanto A, Bito T, Oka M, Fukunaga M, et al. Uv-induced skin damage. Toxicology. 2003;189:21–39. doi: 10.1016/s0300-483x(03)00150-1. [DOI] [PubMed] [Google Scholar]
- Ikehata H, Ono T. The mechanisms of uv mutagenesis. J Radiat Res. 2011;52:115–125. doi: 10.1269/jrr.10175. [DOI] [PubMed] [Google Scholar]
- Kelly GS. Quercetin. Monograph. Altern Med Rev. 2011;16:172–194. [PubMed] [Google Scholar]
- Kielbassa C, Roza L, Epe B. Wavelength dependence of oxidative DNA damage induced by uv and visible light. Carcinogenesis. 1997;18:811–816. doi: 10.1093/carcin/18.4.811. [DOI] [PubMed] [Google Scholar]
- Kino K, Sugiyama H. Uvr-induced g-c to c-g transversions from oxidative DNA damage. Mutat Res. 2005;571:33–42. doi: 10.1016/j.mrfmmm.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Kruszewski M. Labile iron pool: The main determinant of cellular response to oxidative stress. Mutat Res. 2003;531:81–92. doi: 10.1016/j.mrfmmm.2003.08.004. [DOI] [PubMed] [Google Scholar]
- Kvam E, Tyrrell RM. Induction of oxidative DNA base damage in human skin cells by uv and near visible radiation. Carcinogenesis. 1997;18:2379–2384. doi: 10.1093/carcin/18.12.2379. [DOI] [PubMed] [Google Scholar]
- LeBel CP, Ischiropoulos H, Bondy SC. Evaluation of the probe 2′,7′-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem Res Toxicol. 1992;5:227–231. doi: 10.1021/tx00026a012. [DOI] [PubMed] [Google Scholar]
- Lee KW, Kang NJ, Heo YS, Rogozin EA, Pugliese A, Hwang MK, et al. Raf and mek protein kinases are direct molecular targets for the chemopreventive effect of quercetin, a major flavonol in red wine. Cancer Res. 2008;68:946–955. doi: 10.1158/0008-5472.CAN-07-3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebel F, Kaur S, Ruvolo E, Kollias N, Southall MD. Irradiation of skin with visible light induces reactive oxygen species and matrix-degrading enzymes. J Invest Dermatol. 2012;132:1901–1907. doi: 10.1038/jid.2011.476. [DOI] [PubMed] [Google Scholar]
- Lin CW, Hou WC, Shen SC, Juan SH, Ko CH, Wang LM, et al. Quercetin inhibition of tumor invasion via suppressing pkc delta/erk/ap-1-dependent matrix metalloproteinase-9 activation in breast carcinoma cells. Carcinogenesis. 2008;29:1807–1815. doi: 10.1093/carcin/bgn162. [DOI] [PubMed] [Google Scholar]
- Meloni M, Nicolay JF. Dynamic monitoring of glutathione redox status in uv-b irradiated reconstituted epidermis: Effect of antioxidant activity on skin homeostasis. Toxicol In Vitro. 2003;17:609–613. doi: 10.1016/s0887-2333(03)00114-0. [DOI] [PubMed] [Google Scholar]
- Miao L, St Clair DK. Regulation of superoxide dismutase genes: Implications in disease. Free Radic Biol Med. 2009;47:344–356. doi: 10.1016/j.freeradbiomed.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missura M, Buterin T, Hindges R, Hubscher U, Kasparkova J, Brabec V, et al. Double-check probing of DNA bending and unwinding by xpa-rpa: An architectural function in DNA repair. EMBO J. 2001;20:3554–3564. doi: 10.1093/emboj/20.13.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morand C, Manach C, Crespy V, Remesy C. Quercetin 3-o-beta-glucoside is better absorbed than other quercetin forms and is not present in rat plasma. Free Radic Res. 2000;33:667–676. doi: 10.1080/10715760000301181. [DOI] [PubMed] [Google Scholar]
- Morikawa K, Nonaka M, Narahara M, Torii I, Kawaguchi K, Yoshikawa T, et al. Inhibitory effect of quercetin on carrageenan-induced inflammation in rats. Life Sci. 2003;74:709–721. doi: 10.1016/j.lfs.2003.06.036. [DOI] [PubMed] [Google Scholar]
- Moroney MA, Alcaraz MJ, Forder RA, Carey F, Hoult JR. Selectivity of neutrophil 5-lipoxygenase and cyclo-oxygenase inhibition by an anti-inflammatory flavonoid glycoside and related aglycone flavonoids. J Pharm Pharmacol. 1988;40:787–792. doi: 10.1111/j.2042-7158.1988.tb05173.x. [DOI] [PubMed] [Google Scholar]
- Nguyen TT, Tran E, Nguyen TH, Do PT, Huynh TH, Huynh H. The role of activated mek-erk pathway in quercetin-induced growth inhibition and apoptosis in a549 lung cancer cells. Carcinogenesis. 2004;25:647–659. doi: 10.1093/carcin/bgh052. [DOI] [PubMed] [Google Scholar]
- Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, et al. Identification and inhibition of the ice/ced-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- Ogura R, Sugiyama M, Nishi J, Haramaki N. Mechanism of lipid radical formation following exposure of epidermal homogenate to ultraviolet light. J Invest Dermatol. 1991;97:1044–1047. doi: 10.1111/1523-1747.ep12492553. [DOI] [PubMed] [Google Scholar]
- Oliver FJ, de la Rubia G, Rolli V, Ruiz-Ruiz MC, de Murcia G, Murcia JM. Importance of poly(adp-ribose) polymerase and its cleavage in apoptosis. Lesson from an uncleavable mutant. J Biol Chem. 1998;273:33533–33539. doi: 10.1074/jbc.273.50.33533. [DOI] [PubMed] [Google Scholar]
- Olson ER, Melton T, Dickinson SE, Dong Z, Alberts DS, Bowden GT. Quercetin potentiates uvb-induced c-fos expression: Implications for its use as a chemopreventive agent. Cancer Prev Res (Phila) 2010;3:876–884. doi: 10.1158/1940-6207.CAPR-09-0220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podda M, Traber MG, Weber C, Yan LJ, Packer L. Uv-irradiation depletes antioxidants and causes oxidative damage in a model of human skin. Free Radic Biol Med. 1998;24:55–65. doi: 10.1016/s0891-5849(97)00142-1. [DOI] [PubMed] [Google Scholar]
- Podda M, Grundmann-Kollmann M. Low molecular weight antioxidants and their role in skin ageing. Clin Exp Dermatol. 2001;26:578–582. doi: 10.1046/j.1365-2230.2001.00902.x. [DOI] [PubMed] [Google Scholar]
- Satoh MS, Lindahl T. Role of poly(adp-ribose) formation in DNA repair. Nature. 1992;356:356–358. doi: 10.1038/356356a0. [DOI] [PubMed] [Google Scholar]
- Sekiguchi M, Tsuzuki T. Oxidative nucleotide damage: Consequences and prevention. Oncogene. 2002;21:8895–8904. doi: 10.1038/sj.onc.1206023. [DOI] [PubMed] [Google Scholar]
- Shimoi K, Masuda S, Shen B, Furugori M, Kinae N. Radioprotective effects of antioxidative plant flavonoids in mice. Mutat Res. 1996;350:153–161. doi: 10.1016/0027-5107(95)00116-6. [DOI] [PubMed] [Google Scholar]
- Skaper SD, Fabris M, Ferrari V, Dalle Carbonare M, Leon A. Quercetin protects cutaneous tissue-associated cell types including sensory neurons from oxidative stress induced by glutathione depletion: Cooperative effects of ascorbic acid. Free Radic Biol Med. 1997;22:669–678. doi: 10.1016/s0891-5849(96)00383-8. [DOI] [PubMed] [Google Scholar]
- Soberon JR, Sgariglia MA, Sampietro DA, Quiroga EN, Vattuone MA. Antibacterial activity of plant extracts from northwestern argentina. J Appl Microbiol. 2007;102:1450–1461. doi: 10.1111/j.1365-2672.2006.03229.x. [DOI] [PubMed] [Google Scholar]
- Steenvoorden DP, van Henegouwen GM. The use of endogenous antioxidants to improve photoprotection. J Photochem Photobiol B. 1997;41:1–10. doi: 10.1016/s1011-1344(97)00081-x. [DOI] [PubMed] [Google Scholar]
- Tewari M, Quan LT, O’Rourke K, Desnoyers S, Zeng Z, Beidler DR, et al. Yama/cpp32 beta, a mammalian homolog of ced-3, is a crma-inhibitable protease that cleaves the death substrate poly(adp-ribose) polymerase. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- Valko M, Izakovic M, Mazur M, Rhodes CJ, Telser J. Role of oxygen radicals in DNA damage and cancer incidence. Mol Cell Biochem. 2004;266:37–56. doi: 10.1023/b:mcbi.0000049134.69131.89. [DOI] [PubMed] [Google Scholar]
- van Erk MJ, Roepman P, van der Lende TR, Stierum RH, Aarts JM, van Bladeren PJ, et al. Integrated assessment by multiple gene expression analysis of quercetin bioactivity on anticancer-related mechanisms in colon cancer cells in vitro. Eur J Nutr. 2005;44:143–156. doi: 10.1007/s00394-004-0503-1. [DOI] [PubMed] [Google Scholar]
- Vicentini FT, He T, Shao Y, Fonseca MJ, Verri WA, Jr, Fisher GJ, et al. Quercetin inhibits uv irradiation-induced inflammatory cytokine production in primary human keratinocytes by suppressing nf-kappab pathway. J Dermatol Sci. 2011;61:162–168. doi: 10.1016/j.jdermsci.2011.01.002. [DOI] [PubMed] [Google Scholar]
- Wei H, Zhang X, Wang Y, Lebwohl M. Inhibition of ultraviolet light-induced oxidative events in the skin and internal organs of hairless mice by isoflavone genistein. Cancer Lett. 2002;185:21–29. doi: 10.1016/s0304-3835(02)00240-9. [DOI] [PubMed] [Google Scholar]
- Widyarini S, Spinks N, Husband AJ, Reeve VE. Isoflavonoid compounds from red clover (trifolium pratense) protect from inflammation and immune suppression induced by uv radiation. Photochem Photobiol. 2001;74:465–470. doi: 10.1562/0031-8655(2001)074<0465:icfrct>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Wischermann K, Popp S, Moshir S, Scharfetter-Kochanek K, Wlaschek M, de Gruijl F, et al. Uva radiation causes DNA strand breaks, chromosomal aberrations and tumorigenic transformation in hacat skin keratinocytes. Oncogene. 2008;27:4269–4280. doi: 10.1038/onc.2008.70. [DOI] [PubMed] [Google Scholar]
- Xavier CP, Lima CF, Preto A, Seruca R, Fernandes-Ferreira M, Pereira-Wilson C. Luteolin, quercetin and ursolic acid are potent inhibitors of proliferation and inducers of apoptosis in both kras and braf mutated human colorectal cancer cells. Cancer Lett. 2009;281:162–170. doi: 10.1016/j.canlet.2009.02.041. [DOI] [PubMed] [Google Scholar]
- Xia Y, Zweier JL. Measurement of myeloperoxidase in leukocyte-containing tissues. Anal Biochem. 1997;245:93–96. doi: 10.1006/abio.1996.9940. [DOI] [PubMed] [Google Scholar]
- Yang HM, Ham YM, Yoon WJ, Roh SW, Jeon YJ, Oda T, et al. Quercitrin protects against ultraviolet b-induced cell death in vitro and in an in vivo zebrafish model. J Photochem Photobiol B. 2012;114:126–131. doi: 10.1016/j.jphotobiol.2012.05.020. [DOI] [PubMed] [Google Scholar]
- Yao Y, Wolverton JE, Zhang Q, Marathe GK, Al-Hassani M, Konger RL, et al. Ultraviolet b radiation generated platelet-activating factor receptor agonist formation involves egf-r-mediated reactive oxygen species. J Immunol. 2009;182:2842–2848. doi: 10.4049/jimmunol.0802689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Wang S, Leonard SS, Sun Y, Butterworth L, Antonini J, et al. Role of reactive oxygen species and p53 in chromium(vi)-induced apoptosis. J Biol Chem. 1999;274:34974–34980. doi: 10.1074/jbc.274.49.34974. [DOI] [PubMed] [Google Scholar]
- Zhovmer A, Oksenych V, Coin F. Two sides of the same coin: Tfiih complexes in transcription and DNA repair. Scientific World Journal. 2010;10:633–643. doi: 10.1100/tsw.2010.46. [DOI] [PMC free article] [PubMed] [Google Scholar]