

Abstract
Hourly concentrations of ambient fine particle sulfate and carbonaceous aerosols (elemental carbon [EC], organic carbon [OC], and black carbon [BC]) were measured at the Harvard–U.S. Environmental Protection Agency Supersite in Boston, MA, between January 2007 and October 2008. These hourly concentrations were compared with those made using integrated filter-based measurements over 6-day or 24-hr periods. For sulfate, the two measurement methods showed good agreement. Semicontinuous measurements of EC and OC also agreed (but not as well as for sulfate) with those obtained using 24-hr integrated filter-based and optical BC reference methods. During the study period, 24-hr PM2.5 (particulate matter [PM] ≤ 2.5 μm in aerodynamic diameter) concentrations ranged from 1.4 to 37.6 μg/m3, with an average of 9.3 μg/m3. Sulfate as the equivalent of ammonium sulfate accounted for 39.1% of the PM2.5 mass, whereas EC and OC accounted for 4.2 and 35.2%, respectively. Hourly sulfate concentrations showed no distinct diurnal pattern, whereas hourly EC and BC concentrations peaked during the morning rush hour between 7:00 and 9:00 a.m. OC concentrations also exhibited nonpronounced, small peaks during the day, most likely related to traffic, secondary organic aerosol, and local sources, respectively.
INTRODUCTION
Epidemiological studies have reported significant associations between increases in levels of particulate air pollution and excess daily morbidity and mortality from respiratory and cardiovascular causes.1–3 These studies further suggest that observed adverse impacts are due primarily to fine particles (particulate matter [PM] ≤ 2.5 μm in aerodynamic diameter [PM2.5]), with suggestions of different toxicity for different PM2.5 components. Major components of PM2.5 vary by region and by season, but typically include ammonium sulfate ((NH4)2SO4), nitrate (NO3−), and carbonaceous aerosols, elemental carbon (EC) and organic carbon (OC). These PM2.5 components differ substantially in their origins and chemical properties. Sulfate SO42− is a regional, nonvolatile inorganic species produced primarily from the atmospheric oxidation of sulfur dioxide (SO2) emitted from fossil fuel combustion. Like SO42−, EC is also nonvolatile and is emitted primarily from combustion sources, with diesel vehicles being a major source in urban areas. In contrast, OC encompasses many semi-volatile and nonvolatile organic molecules. OC can be of primary origin, such as from combustion processes, residential wood burning, meat cooking, and road dust.4,5 It can also be formed through photochemical processes in the atmosphere as secondary organic aerosol (SOA).6,7
In recent years, several methods have been developed and validated to measure near real-time concentrations of SO42− and carbonaceous particles.8–12 These methods provide the opportunity to better characterize temporal variation in their levels and to improve exposure estimates for these species in health effect assessments. As part of the Program Project of Ambient Particles and Cardiovascular Health, the authors have measured hourly ambient concentrations of PM2.5 and its major components, including semicontinuous measurements of SO42−, EC, OC, and black carbon (BC), in downtown Boston, MA. This paper compares the semicontinuous SO42−, EC, OC, and BC measurements to integrated filter-based measurements and characterizes the diurnal variation of these components by season.
EXPERIMENTAL METHODS
Semicontinuous and integrated filter-based measurements of PM2.5 SO42−, EC, OC, and BC concentrations were made at the Harvard–U.S. Environmental Protection Agency (EPA) Supersite in Boston, MA. The site (42°20′ north latitude, 71°06′ west longitude) is located on the roof of the Countway Library (a six-floor building) of the Harvard Medical School in downtown Boston. This site is located within one block of a four-lane street with truck traffic and with two major highways nearby: Interstate 90 (I-90) is approximately 1.5 km to the north and Interstate 93 (I-93) is approximately 3 km to the south.
SO42− Measurements
Hourly PM2.5 SO42− concentrations were measured using a sulfate particulate analyzer (SPA; Thermo Electron Company, model 5020, Franklin, MA). The SPA consists of converter and analyzer modules. Briefly, particulate SO42− entering the converter module is thermally reduced to gaseous SO2 at the optimal temperature of approximately 1000 °C. The analyzer module uses the pulsed fluorescent technique to continuously measure the SO2 concentration. To minimize potential artifacts, the system also includes an in-line sodium carbonate (Na2CO3)-coated denuder to remove ambient SO2 and other acid gases from the sample flow, as well as a secondary sampling line with an in-line filter to correct for the small amount of background SO2 that passes through the denuder. The SPA measures SO42− concentrations in a 15-min sample cycle (10-min sample mode and 5-min zero mode), yielding four 15-min averages for each hour.
The SPA method differs from a prototype SPA used in previous studies9–10,13: The primary difference between the two methods was the converter oven temperature and configuration. In the prototype, the converter oven, with a stainless steel coil, was set at 800–900 °C, with the first 10 min of every hour used for zero mode (SO2 background). This prototype method reported SO42− concentrations that ranged between 60 and 85% of the filter-based concentrations. In contrast, the SPA converter oven, which contained a stainless steel rod in a quartz converter core, was operated at an optimal temperature of 1000 °C. The heated stainless steel rod is required to reduce particle SO42− to gaseous SO2. Concentrations were measured based on a 15-min sample cycle, with a 10-min sample mode and a 5-min zero mode, yielding four 15-min averages for every hour.
For integrated filter-based measurements, ambient air was drawn through a Harvard impactor (HI) with a 2.5-μm cut point at a flow rate of 4 L/min, which was split into approximately 0.5 L/min used for semicontinuous SO42− measurement (“SPA SO42−”) and the remaining 3.5 L/min drawn through a Teflon filter with a rotameter and a control valve using a diaphragm pump. The filter sample was replaced routinely every 6th day and stored in a refrigerator (~4 °C) until analysis by ion chromatography (IC; Dionex, CX-120, Sunnyvale, CA). Filter-based measurements (IC) were made between January and June 2007 and between December 2007 and October 2008.
Carbonaceous Aerosol Measurements
Measurement of OC and EC
Hourly PM2.5 OC and EC concentrations were measured using the Sunset OC/EC field analyzer (Sunset Laboratory, Inc., model 3, Tigard, OR). The analyzer was programmed to collect aerosol for 47 min at the start of each hour, followed by the analysis of carbonaceous species during the reminder of the hour. Samples were collected at a flow rate of 8 L/min through an inlet equipped with a sharp-cut PM2.5 cyclone (BGI, Waltham, MA) and a carbon-impregnated parallel plate organic denuder (organic denuder), which is designed to remove gaseous organic compounds upstream of the collection filter.
The analytical procedure is based on the modified National Institute for Occupational Safety and Health (NIOSH) method 5040 protocol14 and the thermal-optical transmittance (TOT) method. Additional details on the analytical procedure are available elsewhere.11,12 Briefly, during thermal ramping for the helium (He)-only cycle, some of the OC pyrolyzes, causing the filter to darken (charring), which results in an underestimate of the actual OC. To reduce this error, the analyzer uses a tuned diode laser (660 nm) to monitor the intensity of light transmission while it is being analyzed and to distinguish between the pyrolysis OC and the thermal EC. During analysis using the He/oxygen (O2) mixture, all of the EC is oxidized off and the laser absorbance is reduced to the background level. When the absorbance has decreased to the initial value, called the “split point,”11,12 the method assumes that all of the pyrolyzed OC has been oxidized. The thermal EC concentrations are estimated as the difference of EC minus the pyrolysis OC, whereas the thermal OC is estimated as the sum of the OC and pyrolysis OC.
The Sunset OC/EC field analyzer measures optical EC (“Sunset OptEC”) on the basis of the difference in laser transmission before and after thermal analysis. The optical EC concentration is estimated from a nonlinear fit provided by the manufacturer, derived from rating differences in laser transmission to thermal EC levels over many samples using the modified NIOSH method. However, note that optical EC measurements do not necessarily agree with thermal EC measurements given variation in the optical properties of site- and season-specific particles. Optical OC (“Sunset OptOC”) concentrations were also calculated by subtracting optical EC from total carbon (TC) concentrations determined by thermal analysis.
For integrated filter-based measurement, prebaked quartz filter samples were collected over a 24-hr (12:00 a.m. until 12:00 a.m.) using an HI sampler with a 2.5-μm cut point impactor at a flow rate of 16.7 L/min (with no denuder). The 24-hr filter-based measurements were made every 6th day during two periods: January through July 2007, and December 2007 through August 2008. The sampled filters were stored in a freezer below −20 °C after collection and were analyzed by the modified NIOSH 5040 protocol at the Sunset Laboratory (Tigard, OR). The results are reported as EC (“HI EC”), OC (“HI OC”), and TC (“HI TC”).
BC Measurement
Ambient concentrations of BC, a surrogate measure of EC, were also measured using an Aethalometer (Magee Scientific Company, model AE-16, Berkeley, CA) on the basis of optical transmittance at a single wavelength (λ = 880 nm). The precision of this method was examined using two collocated Aethalometers, serial number (S/N) 280 and S/N 314, during June 26 through August 3, 2007. BC concentrations were determined based on the attenuation of light transmitted through a sampled quartz-fiber filter tape. In contrast to the Sunset OptEC measurement, the attenuation coefficient used was a fixed value of 16.6 m2/g provided by the manufacturer. The method is one of the most commonly utilized and easy-to-use techniques used to measure real-time BC concentrations. Previous studies have shown that the concentrations of BC and EC have been reasonably comparable and well correlated with one another.15–17
Speciation Trends Network
The EPA Speciation Trends Network (STN) site (site ID: 25-025-0042, site name: Dudley Square, Roxbury) is located at a distance of 1.5 km from the Harvard–EPA Supersite. At the STN site, 24-hr integrated PM2.5 filter samples are collected from 12:00 a.m. until 12:00 a.m. following a 1- in 3-day schedule, and then they are shipped to Research Triangle Institute for analysis. For the STN protocol installations, the instrument at this site consists of three sampling modules (Chem-Comb TM cartridges)18 in parallel. Channel 1 contains a Teflon filter that is analyzed gravimetrically for mass and for elements by X-ray fluorescence. Channel 2 contains a denuder-free single quartz filter, which is analyzed for carbonaceous aerosols using the modified NIOSH 5040 method with a temperature profile that was adopted specifically for the STN program.19 Channel 3 includes a Na2CO3-coated honeycomb denuder to remove acid gases, followed by a nylon filter that is analyzed for NO3−, SO42−, ammonium, sodium, and potassium using IC. Comparisons were made between the 24-hr STN measurements of SO42− (“STN SO42−”) and EC (“STN EC”) and the corresponding results (averaged for the same 24-hr intervals) from the semicontinuous concentrations at the measured supersite.
Data Analysis
The differences between two measurements were estimated as relative mean difference (RMD, %). The RMD was calculated as follows:
where the x and y are comparative concentrations and n is total number of samples.
Daily 24-hr PM2.5 mass concentration was measured routinely from 9:00 a.m. until 9:00 a.m. at the site using a custom-made sequential controller with a 2.5-μm cut point HI at a flow rate of 10 L/min. This 24-hr PM2.5 mass concentration was used to estimate mass fractions of each component in this paper. In addition, the 3-hr mixing layer heights were obtained from the National Oceanic and Atmospheric Administration (NOAA) Air Resources Laboratory Web server20 and then analyzed by season. In this paper, winter was defined as December to February, summer as June to August, and transition seasons as March to May and September to November. The hourly concentrations were analyzed for weekdays (Monday through Friday) to investigate the diurnal variation.
RESULTS AND DISCUSSION
Comparison of SO42− Measurement
Several blank experiments were performed using a Teflon filter inlet on the sampling inlet of SPA. For 1-hr averages at a flow rate of 0.5 L/min, the instrumental blank was estimated to be 0.00–0.12 μg/m3 with an average of 0.04 μg/m3. Using 3 times the blank standard deviation, a method detection limit (MDL) of 0.13 μg/m3 was calculated for the semicontinuous measurements.
Hourly SO42− concentrations by the SPA were averaged to compare with the 6-day IC SO42− and the 24-hr STN SO42− concentrations, respectively. Figure 1 shows the association among the different methods. Hourly SO42− concentration was averaged over the collection duration of IC SO42− measurement. The agreement between the SPA SO42− and IC SO42− measurements was excellent, yielding a slope (± standard error) of 1.07 ± 0.01 and an R2 of 0.99 (n = 48). The RMD was 6.9% between the SPA SO42− and IC SO42− concentrations, indicating that the SPA SO42− concentrations were on average 7% higher than corresponding IC measurements. In addition, hourly SO42− concentrations averaged over 24 hr agreed well with corresponding measurements (STN SO42−) at the nearby STN site. Linear regression results in Figure 1 showed a slope of 1.01 ± 0.01 and an R2 of 0.99 (n = 177). The RMD showed a bias in the opposite direction from that for the 6-day measurements, indicating that the STN SO42− measurements were 5% higher than those for the SPA at the supersite.
Figure 1.
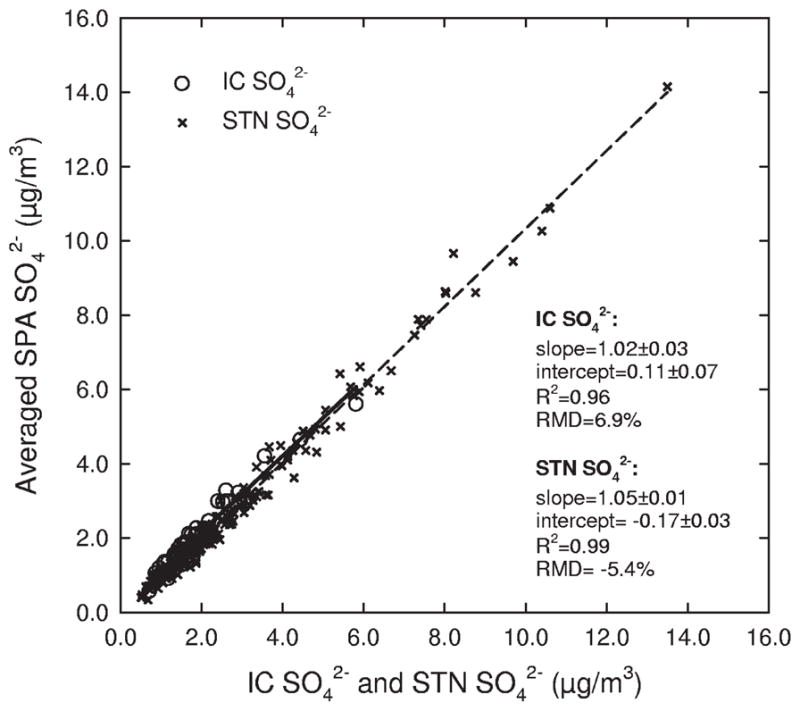
Association among the SO42− concentrations measured using different methods. SPA SO42− is averaged over the collection periods for the filter-based measurements, IC SO42− is the 6-day SO42− concentration, and STN SO42− is the 24-hr SO42− concentration.
Intercomparison of SO42− measurement methods indicated that the SPA method agreed well with filter-based methods, with only small differences between the methods. These small differences are likely because of the SPA sampling cycles, which result in incomplete measurements over the sampling period. As noted earlier, the SPA sampling cycles would result in hourly SO42− concentration on the basis of four sampling cycles or 40 min of total measurement time rather than the full 60 min. Although this impact is likely minor, especially for SO42−, it is possible that it contributed to the observed small difference in the concentrations. In addition, each method has associated measurement error, which would also contribute to the observed differences in the concentrations measured by each method.9 –10,21 The method-specific measurement difference (<10%) in this study was within previous ranges (10–25%) reported from the comparisons of semicontinuous and filter-based measurements. The performance in the study presented here was much better than previous results (60–85%) of studies comparing prototype SPA and filter-based methods, with this improvement likely reflecting improved instrumentation.
Comparison of Carbonaceous Aerosols Measurement
Performance of Sunset OC/EC Analyzer
The organic denuder removes vapor-phase organic compounds by diffusion to carbon-impregnated collection surfaces. This minimizes the artifact OC that results from some of the vapor-phase organic compounds being adsorbed by the quartz-fiber sampling filter. The denuder removal efficiency was determined experimentally. Several experiments were carried out using a Teflon-filter inlet on the sampling line to evaluate the efficiency of the organic denuder. For 1-hr measurements, the OC that passed through the denuder was estimated to be 0.38–1.00 μg/m3 with an average of 0.66 μg/m3. In addition, the instrument blank was obtained by analyzing samples with zero sample volume. The instrument blanks ranged from 0.07 to 0.25 μg carbon, with an average of 0.15 μg carbon. These blank values correspond to atmospheric concentrations of 0.20–0.68 μg carbon/m3 (average of 0.41 μg carbon/m3) for the 1-hr measurement at a flow rate of 8 L/min. At 3 times the blank standard deviation, a MDL of 0.49 μg carbon/m3 was obtained for the semicontinuous measurements.
Internal calibration was accomplished using certified 5% methane automatically balanced with He in a fixed volume loop at the end of each analysis. External calibration was also carried out periodically through the analysis of known sucrose concentrations on a pre-baked filter. Two sucrose spikes, 21.04 and 42.07 μg carbon, were used for external calibration. The concentrations (average ± standard deviation) analyzed by the Sunset semicontinuous analyzer were summarized to be 21.56 ± 0.42 μg carbon (n = 10) and 42.14 ± 1.18 μg carbon (n = 18), respectively. The recoveries of these two sucrose spikes were 1.02 ± 0.02 and 1.00 ± 0.03, respectively.
Semicontinuous and Filter-Based Measurements
Hourly concentrations of EC and OC were averaged over a 24-hr period to compare with the filter-based concentrations. Figure 2 shows the association between semi-continuous and filter-based concentrations. In Figure 2a, a regression of the Sunset ThEC concentrations on HI EC concentrations yielded an R2 of 0.73 (n = 57), indicating relatively good agreement; however, the slope equaled 1.60 ± 0.13, indicating that the semicontinuous method overestimated filter-based EC concentrations (RMD = 20%). As with Sunset ThEC concentration, Sunset OptEC concentrations explained a larger percentage of the variation in filter-based EC concentrations (R2 = 0.80), but with a slope close to unity (0.97 ± 0.06) and an RMD of −0%, consistent with strong agreement between the OptEC and filter-based measurements. Similar results were found when Sunset OptEC concentration was compared with STN EC concentrations (Figure 2b). In addition, the discrepancy between thermal EC and optical EC concentrations was observed when an upgraded model (Sunset Laboratory, Inc., model 4, Tigard, OR) was tested at the supersite during the period of November 23–25, 2009. Thermal EC concentration was on average 27% higher than the corresponding optical EC concentration with the newer model.
Figure 2.

Association among the concentrations of carbonaceous aerosols using different methods. Sunset EC and Sunset OC are the 24-hr averaged concentrations, HI EC and HI OC were measured over a 24-hr period at the supersite, STN EC and STN OC were measured over 24 hr at a nearby STN site, Aeth BC is the 24-hr averaged concentration, Aeth S/N 280 was used for routine measurement, and the Aeth S/N 314 was used for collocating measurement. Regression between (a) HI EC and Sunset ThEC concentrations, (b) STN EC and Sunset OptEC concentrations, (c) HI OC and Sunset OptOC concentrations, (d) HI TC and Sunset TC concentrations, (e) Sunset OptEC and Aeth BC concentrations, and (f) S/N 280 and S/N 314 Aeth BC concentrations.
Several studies have reported a weak agreement between thermal EC and filter-based EC concentrations at low levels.22–24 For example, Saarikoski et al.25 estimated that optical EC concentrations measured by the Sunset OC/EC analyzer were more reliable than that measured by the thermal analysis. The reduced performance of the thermal EC measurement at low levels is likely due to the importance of the split point (e.g., the point at which the transmittance regains its initial value) separating the OC and EC signals, where low particle loadings may increase uncertainties in thermal determination. The reference method for EC relies on 24-hr filter-based EC measurements. Because the optical EC concentrations agree better with the 24-hr filter-based EC concentration, it is more suitable to use the optical EC than to use the thermal EC for the relatively low levels of PM2.5 in Boston. Note that the reason why 24-hr samples are expected to yield more reliable results than hourly samples is that the analytical sensitivity is greater for larger amounts of collected particles. Therefore, subsequent analyses of EC and OC are based on optical EC and optical OC concentrations unless stated otherwise; optical OC concentrations (Sunset OptOC) were estimated by subtracting optical EC concentration (Sunset OptEC) from TC obtained by thermal analysis.
Figure 2c shows the association between 24-hr HI OC concentrations and Sunset OptOC concentrations, with an R2 of 0.59 and a slope of 0.83 ± 0.09 when the semi-continuous measurements were regressed on filter-based measurements. The association was weaker than that for EC presumably because of sampling artifacts. The semi-continuous analyzer but not the filter-based measurement included an organic denuder upstream of the filter, which may result in differences between these two measurements. All OC measurements have been shown to have positive and negative artifacts from the collection of organic gases by filter media and/or volatilization from collected particle-phase OC mass, respectively. The magnitude of these artifacts may vary with sampling conditions, such as method, filter media, sampling velocity, and duration.26 The artifacts can range widely from −80% (e.g., volatilization-induced bias) to +50% (e.g., adsorption-induced bias).27,28 In terms of TC (Figure 2d), the association between two methods was very similar to that for OC concentrations, which was expected given that OC concentrations were dominant contributors to TC concentrations.
In addition, the operating temperatures and analyzing time of the Sunset OC/EC analyzer used for field measurement, the Sunset OC/EC laboratory analyzer used for integrated filter analysis, and the STN program used for the STN data are different, although these methods are both based on the NIOSH TOT protocol. For example, the Sunset OC/EC field analyzer has two temperature steps and analyzing time, whereas the Sunset OC/EC laboratory analyzer uses four steps for carbon analysis. The analytical errors that resulted from different analytical conditions (e.g., method, temperature profiles, and analyzing time) might lead to different concentrations of OC and EC.26,29
EC and BC
Sunset OptEC concentrations are compared with the BC concentrations (“Aeth BC”) by Aethalometer in Figure 2e. For the comparison, the concentrations were averaged for both methods over 24-hr periods to smooth short-term variations. The association yielded a slope of 1.40 ± 0.03 and an R2 of 0.84 (RMD = 29.4%) between two measurements. This indicates that the Aeth BC concentration was well correlated with the Sunset OptEC concentration, but the BC concentration was approximately 29% greater than the OptEC concentration. Similarly, a comparison of the Aeth BC with the HI EC yielded a slope of 1.48 ± 0.13 and an R2 of 0.72 (RMD = 29%). Two Aethalometers were run simultaneously at the supersite during July through August 2007; The S/N 280 was used for routine measurements and the S/N 314 was used for collocated measurements. Using hourly averaged measurements, the regression (Figure 2f) shows a slope of 0.99 ± 0.01 and an R2 of 0.97 (RMD = 6.8%) between two BC concentrations, which indicates an outstanding agreement for this optical BC measurement method.
It is important to note that there was a difference in wavelength for the Aethalometer (λ = 880 nm) and Sunset OC/EC field analyzer (λ = 660 nm). Also, as noted above, these two methods use different algorithms derived from rating differences in laser transmission to EC levels on the basis of different sets of large numbers of samples. Although the Sunset OptEC concentrations were estimated from a nonlinear relation of attenuation coefficients, the Aeth BC concentrations were estimated from a single coefficient of 16.6 m2/g. Because measurements by these two methods are well correlated, it is likely that the overall difference (i.e., RMD = 29%) resulted from the differences in the algorithms used. It has been suggested that the attenuation coefficient needs to be calibrated by sampling site for accurate estimation of EC from optical measurement because of variation in the optical properties of site- and season-specific particles.16,30 The good agreement between the Sunset OptEC concentrations and the HI EC concentrations suggest that the OptEC values are reasonably accurate. Consequently, because the Aethlometer values are consistently higher than the OptEC values, for the particles measured at the Boston supersite, the coefficient used for this site for the Aethelometer should be higher than the default value of 16.6 m2/g. Without this correction, there is a systematic overestimation by the Aeth BC in predicting the ambient EC concentration in the study area.
Temporal Variations of PM2.5, SO42−, OC, EC, and BC
Table 1 shows seasonal statistics of PM2.5, SO42−, OC, EC, and BC concentrations in the Boston urban area during the study period. In the table, SO42− was calculated as the equivalent of (NH4)2SO4 to estimate the composition of PM2.5 mass. As expected, seasonal averages were the highest during summer, followed by winter and then transition season. In the summer, SO42− ((NH4)2SO4 equivalent), OC, EC, and BC concentrations were 5.14, 3.55, 0.43, and 0.71 μg/m3, respectively, accounting for 41.6, 28.7, 3.5, and 5.7% of the PM2.5 concentration, respectively. Overall, SO42− and OC comprised the largest fractions of PM2.5, contributing 39.1 and 35.2%, respectively, of the PM2.5 mass. EC contributed 4.2% to the PM2.5 mass. The remaining 21.5% of PM2.5 mass is likely comprised of ammonium nitrate (NH4NO3), organic material, elements, and particle-bound water.
Table 1.
Seasonal statistics of the PM2.5 components.
| Seasona | PM2.5 | SO42−b | Percentc | OC | Percent | EC | Percent | BC | Percent |
|---|---|---|---|---|---|---|---|---|---|
| Winter | 9.22 ± 4.68d | 3.00 ± 1.61 | 32.5 | 3.31 ± 1.18 | 35.9 | 0.43 ± 0.25 | 4.7 | 0.52 ± 0.34 | 5.6 |
| Summer | 12.35 ± 7.16 | 5.14 ± 3.95 | 41.6 | 3.55 ± 1.24 | 28.7 | 0.43 ± 0.19 | 3.5 | 0.71 ± 0.31 | 5.7 |
| Transition | 7.78 ± 5.22 | 3.05 ± 2.78 | 39.2 | 3.13 ± 1.17 | 40.2 | 0.36 ± 0.20 | 4.6 | 0.54 ± 0.30 | 6.9 |
| Overall | 9.31 ± 6.02 | 3.64 ± 3.11 | 39.1 | 3.28 ± 1.21 | 35.2 | 0.39 ± 0.21 | 4.2 | 0.58 ± 0.32 | 6.2 |
Notes:
Winter (December to February), summer (June to August), transition (March to May and September to November).
Equivalent of (NH4)2SO4.
Weight percent of the PM2.5 concentration.
Average ± standard deviation.
The 3-hr mixing layer heights by season were plotted in Figure 3. As shown in the figure, during the daytime the mixing layer height for summer was the greatest of the seasons, whereas during the morning and nighttime the height was the lowest for summer. As expected, the time of day with the highest observed values was 2:00 p.m. for all seasons. Hourly concentrations of SO42− and carbonaceous aerosols were averaged over the time of day to estimate their diurnal variation. Week-day variations of SO42−, OC, EC, and BC concentrations are plotted in Figure 4. PM2.5 SO42− concentrations did not show a pronounced pattern during the time of day in any season, consistent with the importance of long-range transport for this species.10 SO42− concentrations were slightly lower during the day as compared with during the night, possibly because of diurnal variation of mixing layer height.
Figure 3.

Diurnal variation of mixing layer height by season in Boston.
Figure 4.

Diurnal variations of SO42−, EC, OC, and BC concentrations for weekdays by season in Boston.
Diurnal variations in EC and BC concentrations were similar, as expected, because they measure the same type of pollutant.15,16 In each season, weekday EC and BC concentrations peaked clearly during the morning rush hour (7:00 to 9:00 a.m.). A second, less distinct peak was observed between 6:00 and 10:00 p.m. The evening peaks had nonpronounced pattern. It is more likely due to a higher mixing layer and less traffic density in the evening. In summary, the diurnal pattern in EC and BC concentrations suggests a strong relation to traffic for these components.
In contrast to EC and BC, OC concentration exhibited nonpronounced, small peaks during three time periods: 7:00 to 9:00 a.m., 12:00 to 2:00 p.m., and 8:00 to 9:00 p.m. Although the timing of the second peak varied by season, the observed diurnal pattern in OC levels is consistent with the known sources of urban OC. For example, the morning peak likely reflects the contribution of motor vehicles during rush hour, whereas the evening peaks are consistent with heating appliance use (oil and natural gas), anthropogenic biomass burning from wood stoves and fireplaces, and cooking processes at home and restaurants.4,5 In particular, it is possible that wood stove use for home heating was greater during winter 2007 because of an exceptionally high cost of oil. This increase may explain higher OC concentration in the evening during that winter period. The mid-afternoon peak prevailing in summer presumably reflects the contribution of SOA formed from photochemical oxidation of gas-phase precursors.6,7
CONCLUSIONS
Field validation of semicontinuous measurements of PM2.5 SO42−, OC, EC, and BC was conducted through comparison with filter-based measurements over a period of 22 months at the Harvard–EPA Supersite in Boston. Hourly SO42− concentration measured by the semicontinuous analyzer was in good agreement (high correlations and slopes near unity) with reference method concentrations, indicating that the semicontinuous analyzer provides reliable SO42− concentrations. The results also show that semicontinuous optical EC concentrations agree better with the filter-based EC concentrations, especially for the relatively low PM2.5 levels of Boston. Considering the relatively high uncertainty of carbonaceous measurements, the authors conclude that there is acceptable agreement between semicontinuous measurement and other reference methods. Hourly SO42− concentrations were relatively uniform over the day, which is consistent with the importance of long-range transport of SO42− into Boston. In contrast, hourly EC and BC concentrations had a pronounced pattern, with a peak levels occurring between 7:00 and 9:00 a.m. in the morning rush hour, which suggests a strong relation with traffic. Diurnal variation in OC concentrations also presented with three less distinct, small peaks during the day, likely related to traffic, SOA, and local sources, respectively.
IMPLICATIONS.
Accurate and precise hourly measurements of sulfate and carbonaceous aerosols, two major PM components, are critical to the ability to assess health risks of PM2.5 components in epidemiological studies. This paper compares semicontinuous sulfate, EC, and OC measurement methods to integrated filter-based measurements. Using these comparisons, this paper characterizes the diurnal variations of fine particle sulfate, OC, EC, and BC.
Acknowledgments
This project was supported by the EPA Center for Particle Health Effects at the Harvard School of Public Health (grant R832416) and the National Institute of Environmental Health Sciences (NIEHS) Program Project Grant (5 P01 ES009825-10). However, any opinions, findings, conclusions, or recommendations expressed herein are those of the authors and do not necessarily reflect the views of EPA or the NIEHS. The authors acknowledge the efforts of Mike Wolfson, Erick Vlassidis, and Vasileios Papapostolou for field and laboratory support.
Biography
Choong-Min Kang is a research associate at the Harvard School of Public Health, Exposure, Epidemiology, and Risk Program. Petros Koutrakis is a professor of environmental sciences at the Harvard School of Public Health. Helen H. Suh is an associate professor of environmental chemistry and exposure assessment at the Harvard School of Public Health.
References
- 1.Dockery DW, Pope A, Xu X, Spengler JD, Ware JH, Fay ME, Ferris BG, Speizer FE. An Association between Air Pollution and Mortality in Six U.S. Cities. N Engl J Med. 1993;329:1753–1759. doi: 10.1056/NEJM199312093292401. [DOI] [PubMed] [Google Scholar]
- 2.Pope A, Dockery DW. Acute Health-Effects of PM10 Pollution on Symptomatic and Asymptomatic Children. Am Rev Respir Dis. 1992;145:1123–1128. doi: 10.1164/ajrccm/145.5.1123. [DOI] [PubMed] [Google Scholar]
- 3.Schwartz J, Morris R. Air Pollution and Hospital Admissions for Cardiovascular Disease in Detroit, Michigan. Am J Epidemiol. 1995;142:23–35. doi: 10.1093/oxfordjournals.aje.a117541. [DOI] [PubMed] [Google Scholar]
- 4.Fine PM, Chakrabarti B, Krudysz M, Schauer JJ, Sioutas C. Diurnal Variations of Individual Organic Compound Constituents of Ultrafine and Accumulation Mode Particulate Matter in the Los Angeles Basin. Environ Sci Technol. 2004;38:1296–1304. doi: 10.1021/es0348389. [DOI] [PubMed] [Google Scholar]
- 5.Rogge WF, Hildemann LM, Mazurek MA, Cass GR, Simoneit BR. Sources of Fine Organic Aerosol. 3. Road Dust, Tire Debris, and Organometallic Brake Lining Dust: Roads as Sources and Sinks. Environ Sci Technol. 1993;27:1892–1904. [Google Scholar]
- 6.Pandis SN, Harley RA, Cass GR, Seinfeld JH. Secondary Organic Aerosol Formation and Transport. Atmos Environ. 1992;26:2269–2282. [Google Scholar]
- 7.Strader R, Fred L, Pandis SN. Evaluation of Secondary Organic Aerosol Formation in Winter. Atmos Environ. 1999;33:4849–4863. [Google Scholar]
- 8.Allen GA, Harrison D, Koutrakis P. Proceedings of American Association for Aerosol Research (AAAR)’s 20th Annual Conference. AAAR; Mt. Laurel, NJ: 2001. A New Method for Continuous Measurement of Sulfate in the Ambient Atmosphere. [Google Scholar]
- 9.Drewnick F, Schwab JJ, Hogrefe O, Peters S, Husain L, Diamond D, Weber R, Demerjian KL. Intercomparison and Evaluation of Four Semi-Continuous PM2.5 Sulfate Instruments. Atmos Environ. 2003;37:3335–3350. [Google Scholar]
- 10.Hogrefe O, Schwab JJ, Drewnick F, Lala GG, Peters S, Demerjian KL, Rhoads K, Felton HD, Rattigan OV, Husain L, Dutkiewicz VA. Semicontinuous PM2.5 Sulfate and Nitrate Measurements at an Urban and a Rural Location in New York: PMTACS-NY Summer 2001 and 2002 Campaigns. J Air & Waste Manage Assoc. 2004;54:1040–1060. doi: 10.1080/10473289.2004.10470972. [DOI] [PubMed] [Google Scholar]
- 11.Birch ME, Cary RA. Elemental Carbon-Based Method for Occupational Monitoring of Particulate Diesel Exhaust: Methodology and Exposure Issues. Analyst. 1996;121:1183–1190. doi: 10.1039/an9962101183. [DOI] [PubMed] [Google Scholar]
- 12.Turpin BJ, Cary RA, Huntzicker JJ. An In Situ, Time-Resolved Analyzer for Aerosol Organic and Elemental Carbon. Aerosol Sci Technol. 1990;12:161–171. [Google Scholar]
- 13.Jeong CH, Lee DW, Kim E, Hopke PK. Measurement of Real-Time PM2.5 Mass, Sulfate and Carbonaceous Aerosols at the Multiple Monitoring Sites. Atmos Environ. 2004;38:5247–5256. [Google Scholar]
- 14.Elemental Carbon (Diesel Exhaust): NIOSH Manual of Analytical Methods. 4. National Institute of Occupational Safety and Health; Cincinnati, OH: 1996. [Google Scholar]
- 15.Allen GA, Lawrence J, Koutrakis P. Field Validation of a Semi-Continuous Method for Aerosol Black Carbon (Aethalometer) and Temporal Patterns of Summertime Hourly Black Carbon Measurements in Southwestern PA. Atmos Environ. 1999;33:817–823. [Google Scholar]
- 16.Babich P, Davey M, Allen G, Koutrakis P. Method Comparisons for Particulate Nitrate, Elemental Carbon, and PM2.5 Mass in Seven U.S. Cities. J Air & Waste Mange Assoc. 2000;50:1095–1105. doi: 10.1080/10473289.2000.10464152. [DOI] [PubMed] [Google Scholar]
- 17.Snyder DC, Schauer JJ. An Inter-Comparison of Two Black Carbon Aerosol Instruments and a Semi-Continuous Elemental Carbon Instrument in the Urban Environment. Aerosol Sci Technol. 2007;41:463–474. [Google Scholar]
- 18.Koutrakis P, Sloutas C, Ferguson ST, Wolfson JM. Development and Evaluation of a Glass Honeycomb Denuder/Filter Pack System to Collect Atmospheric Gases and Particles. Environ Sci Technol. 1993;27:2497–2501. [Google Scholar]
- 19.Flanagan JB, Jayanty RK, Rickman EE, Jr, Peterson MR. PM2.5 Speciation Trends Network: Evaluation of Whole-System Uncertainties Using Data from Sites with Collocated Samplers. J Air & Waste Manage Assoc. 2006;56:492–499. doi: 10.1080/10473289.2006.10464516. [DOI] [PubMed] [Google Scholar]
- 20.National Oceanic and Atmospheric Administration. [accessed June 2009];Air Resource Laboratory (ARL) available at http://www.ready.noaa.gov/ready/amet.html.
- 21.Weber R, Orsini D, Duan Y, Baumann K, Kiang CS, Chameides W, Lee YN, Brechtel F, Klotz P, Jongejan P, Brink H, Slania J, Boring CB, Genfa Z, Dasgupta P, Hering S, Stolzenburg M, Dutcher DD, Edgerton E, Hartsell B, Solomon P, Tanner R. Intercomparison of Near Real-Time Monitors of PM2.5 Nitrate and Sulfate at the U.S. Environmental Protection Agency Atlanta Supersite; J Geophys Res. 2003;108 doi: 10.1029/2001JD001220. [DOI] [Google Scholar]
- 22.Arhami M, Kuhn T, Fine PM, Delfino RJ, Sioutas C. Effects of Sampling Artifacts and Operating Parameters on the Performance of a Semicontinuous Particulate Elemental Carbon/Organic Carbon Monitor. Environ Sci Technol. 2006;40:945–954. doi: 10.1021/es0510313. [DOI] [PubMed] [Google Scholar]
- 23.Bae MS, Schauer JJ, DeMinter JT, Tunner JR, Smith D, Cary RA. Validation of a Semi-Continuous Instrument for Elemental Carbon and Organic Carbon Using a Thermal-Optical Method. Atmos Environ. 2004;38:2885–2893. [Google Scholar]
- 24.Venkatachari P, Zhou L, Hopke PK, Schwab JJ, Demerjian KL, Weimer S, Hogrefe O, Felton D, Rattigan O. An Intercomparison of Measurement Methods for Carbonaceous Aerosol in the Ambient Air in New York City. Aerosol Sci Technol. 2006;40:788–795. [Google Scholar]
- 25.Saarikoski S, Timonen H, Saarnio K, Aurela M, Jarvi M, Kenronen P, Kerminen VM, Hillamo R. Sources of Organic Carbon in Fine Particulate Matter in Northern European Urban Air. Atmos Chem Phys. 2008;8:6281–6295. [Google Scholar]
- 26.Hering SV, Appel BR, Cheng W, Salaymeh F, Cadle SH, Mulawa PA, Cahill TA, Eldred RA, Surovik M, Fitz D, Howes JE, Knapp KT, Stockburger L, Turpin BJ, Huntzicker JJ, Zhang X, McMurry PH. Comparison of Sampling Methods for Carbonaceous Aerosols in Ambient Air. Aerosol Sci Technol. 1990;12:200–213. [Google Scholar]
- 27.Eatough DJ, Lewis LJ, Eatough M, Lewis EA. Sampling Artifacts in the Determination of Particulate Sulfate and SO2(g) in the Desert Southwest Using Filter Pack Samplers. Environ Sci Technol. 1995;29:787–791. doi: 10.1021/es00003a029. [DOI] [PubMed] [Google Scholar]
- 28.Turpin BJ, Huntzicker JJ, Hering SV. Investigation of Organic Aerosol Sampling Artifacts in the Los Angeles Basin. Atmos Environ. 1994;28:3061–3071. [Google Scholar]
- 29.Schauer JJ, Mader BT, Deminter JT, Heidermann G, Bae M, Seinfeld JH, Flagan RC, Cary RA, Smith D, Huebert BJ, Bertram T, Howell S, Kline JT, Quinn P, Bates T, Turpin B, Lim HJ, Yu JZ, Yang H, Keywood MD. ACE-Asia Intercomparison of a Thermal-Optical Method for the Determination of Particle-Phase Organic and Elemental Carbon. Environ Sci Technol. 2003;37:993–1001. doi: 10.1021/es020622f. [DOI] [PubMed] [Google Scholar]
- 30.Ballach J, Hitzenberger R, Schultz E, Jaeschke W. Development of an Improved Optical Transmission Technique for Black Carbon (BC) Analysis. Atmos Environ. 2001;35:2089–2100. [Google Scholar]