

SUMMARY
The extensive physiological influence of transmission through the CB2 cannabinoid receptor makes this G protein-coupled receptor (GPCR) a promising therapeutic target for treating neuropathic pain, inflammation, and immune disorders. However, there is little direct structural information pertaining to either GPCR or CB2-receptor ligand recognition and activation. The present work helps characterize experimentally the ligand-binding interactions of the human CB2 (hCB2) receptor. This study illustrates how our overall experimental approach, “ligand-assisted protein structure” (LAPS), affords direct determination of the requirements for ligand binding to the hCB2 receptor and discrimination among the binding motifs for ligands that activate therapeutically relevant GPCRs.
INTRODUCTION
Some 30% of marketed drugs are small-molecule ligands of G protein coupled receptors (GPCRs), the most prevalent integral membrane proteins (Lagerström and Schiöth, 2008). Two GPCRs, the principal cannabinoid (CB) receptors CB1 and CB2, are critical components of the endogenous CB (endocannabinoid) signaling system (Vemuri et al., 2008). Activation of CB receptors elicits the dissociation of Gαi proteins and a consequent decrease in intracellular adenylyl cyclase activity (Rhee et al., 1998). CB1 or CB2 receptor transmission can also stimulate mitogen-activated protein kinase (Bouaboula et al., 1996), and the CB1 receptor directly modulates L-, N-, Q-, and P-type calcium channels (Gebremedhin et al., 1999; Mackie et al., 1995; Pan et al., 1996). The wide-ranging physiological and regulatory effects of endocannabinoid signaling place great interest on targeting CB1 and CB2 receptors for therapeutic gain. Structurally diverse CB1- or CB2-receptor ligands can modulate endocannabinoid signaling (Raitio et al., 2005; Vemuri et al., 2008). The virtual absence of CB2 receptors in the central nervous system limits the potential of selective CB2-receptor agonists/antagonists to elicit (CB1-mediated) psychoactive effects (Malan et al., 2003) and lends particular appeal to activating ligands selective for the CB2 receptor as potential anti-inflammatory, anti-nociceptive, and neuroprotective drugs (Marriott and Huffmann, 2008). CB1 and CB2 receptors share only 44% overall identity at the level of their amino acid residues, increasing to 68% shared identity within their transmembrane domains (Munro et al., 1993). Despite the limited homology between these two CB-receptor subtypes, receptor discrimination represents an ongoing concern in the design and therapeutic application of CB-receptor ligands (Marriott and Huffmann, 2008; Vemuri et al., 2008). Nonetheless, the selectivity displayed by at least some cannabinergic compounds for either the CB1 or CB2 receptor (Jagerovic et al., 2008; Marriott and Huffmann, 2008) suggests that ligand-induced activation of each CB receptor subtype might occur through a distinct ligand-binding motif.
Largely because of inherent difficulties in isolating the requisite quantities of purified GPCRs for analysis by X-ray crystallography and nuclear magnetic resonance spectroscopy, there is a paucity of direct, experimentally derived data on GPCR structure and ligand recognition. Notwithstanding descriptions of the crystal structures of bovine rhodopsin (Rho) (Palczewski et al., 2000), an engineered, human β2-adrenergic receptor (Cherezov et al., 2007), the ligand-free opsin receptor (Park et al., 2008), and the β1-adrenergic receptor-cyanopindolol complex (Warne et al., 2008), the conformational plasticity of GPCRs and their incompletely understood activation dynamics further complicate GPCR structural elucidation. According to the predominant, extended ternary-complex model, GPCRs exist in an equilibrium between inactive (R) and activated (R*) states. Depending on how they affect the interstate equilibrium, ligands are classified as agonists, neutral antagonists, or inverse agonists, (De-Lean et al., 1980). However, this paradigm inadequately explains the complex behavior of GPCRs, which likely exist in multiple conformational states between R and R* (Lagerström and Schiöth, 2008; Samama et al., 1993). The limited direct information on GPCR ligand-interaction sites mandates further experimentation to establish more precisely the ligand-binding and pharmacophore requirements of these CB receptors and refine CB1- and CB2-receptor structural models. The three-dimensional Rho structure has been used to formulate CB-receptor homology models, which remain speculative because of the attendant extrapolations used (Poso and Huffman, 2008; Reggio, 2006). Likewise, although profiling of cannabinergic ligand binding to CB1- and CB2-receptor mutants has helped identify amino acid residues influencing ligand recognition (Picone et al., 2005; Tao et al., 1999), such studies cannot offer direct demonstration of discrete receptor-ligand interactions at the aminoacid level (Peracchi, 2001; Admiraal et al., 2001).
Work in this laboratory has sought to define the structural aspects of ligand recognition by endocannabinoid-system enzymes and CB receptors by utilizing a direct experimental approach we have termed “ligand-assisted protein structure” (LAPS) (Zvonok et al., 2008). This approach exploits the ability of CB receptors to recognize several chemical classes of ligands, including prototypic tricyclic CBs such as the phytocannabinoid (−)-Δ9-tetrahydrocannabinol (Δ9-THC); nonclassical synthetic bicyclic terpenoids (e.g., CP55940); the aminoalkylindole WIN55212-2; the endocannabinoid anandamide (AEA); and the biarylpyrazoles SR141716A (CB1-receptor antagonist) and SR144528 (CB2-receptor antagonist) (Palmer et al., 2002; Rhee and Kim, 2002) (Figure 1A). Among the many unique cannabinergic compounds we have generated are ligands with exceptionally high affinity and selectivity for either the CB1 or CB2 receptor (Charalambous et al., 1992; Guo et al., 1994; Morse et al., 1995; Picone et al., 2002). Some have been rationally designed to incorporate pharmacophores that react irreversibly in a chemically selective manner with CB-receptor amino acid residues at or near the receptor’s ligand-binding site. This feature enables the ligands to be used as cannabinoid-receptor affinity probes that, in conjunction with site-directed mutagenesis, are integrated into the LAPS paradigm for characterizing CB-receptor-binding domains. One such compound, (−)-7′-isothiocyanato-11-hydroxy-1′,1′-di-methylheptylhexahydrocannabinol (AM-841), is a classical CB analog with a 7-isothiocyanate (NCS) moiety at the terminus of its C-3 alkyl side chain (Figure 1A). We have previously implicated cysteine C6.47(355) as the site of covalent attachment of the AM-841 NCS group to the CB1 receptor, leading to receptor activation (Picone et al., 2005).
Figure 1. Chemical Structures of Cannabi-nergic Ligands and Effect of AM-841 on WT and Mutant hCB2 Receptor Ligand Binding.

(A) Chemical structures of representative cannabi-nergic ligands used in this study.
(B and C) Preincubation with AM-841 eliminates [3H]-CP55940 binding to the WT hCB2 receptor and the hCB2 C7.38(284)S and hCB2 C7.42(288)S mutant receptors, but not to the hCB2 C6.47(257)A or hCB2 C6.47(257)S mutant receptors. Membranes prepared from HEK293 cells expressing either the WT or a mutant hCB2 receptor were preincubated with 9 nM (six-fold the Ki) AM-841 for 1hr at 30°C and then extensively washed to remove unbound, noncovalently associated ligand. The washed membranes were subjected to a saturation binding assay using [3H]-CP55940 as the radioligand. (B) Saturation-binding curves using [3H]-CP55940 for WT and mutant hCB2 receptors preincubated with AM-841 as described above and “control” membranes processed in parallel, but without prior exposure to AM-841. Data represent the means ± SEM of at least 2 independent experiments performed in duplicate. (C) Comparison of the difference in Bmax values of each hCB2 receptor with or without preincubation with AM-841. Data shown represent the means ± SEM of at least 2 independent experiments performed in duplicate.
Receptor amino acid residues are numbered herein using the Ballesteros and Weinstein (1995) scheme. Accordingly, the most highly conserved residue across GPCRs within a given family in each TMH is assigned a locant of 50. This number is preceded by the TMH number and followed in parentheses by the sequence number. All other residues in a TMH are numbered relative to this residue. For example: The most highly conserved residue in TMH6 of the hCB2 receptor is P6.50(260). The residue that immediately precedes it is designated F6.49(259).
The present study examines experimentally the ligand-binding motif of the hCB2 receptor. We identify C6.47(257) as the cysteine residue in transmembrane helix (TMH) 6 of the human CB2 (hCB2) receptor homologous to C6.47(355) in the CB1 receptor with which the NCS moiety of AM-841 interacts. Direct evidence is presented that C6.47(257) is an essential residue within the hCB2 receptor-binding pocket. We demonstrate that AM-841 elicits a sustained activation of the hCB2 receptor with significantly greater potency than it displays at the hCB1 receptor. C6.47(257) is part of the highly conserved CWXP motif, a putative molecular hinge essential for ligand recognition by family A, group I GPCRs implicated in their activation (Lagerström and Schiöth, 2008; Reggio, 2006). Experimental evidence is also advanced that the alkyl tail of AM-841 is oriented within a binding pocket in TMH6 of the hCB2 receptor via a ligand-binding motif quite distinct from that of the CB1 receptor. Additionally, we have targeted a highly conserved (Jensen et al., 2001; Shi et al., 2002) lysine residue, K3.28(109), in TMH3 of the hCB2 receptor, which has been implicated by mutational analysis in the recognition of several cannabinergic compounds by the hCB1 receptor (Song and Bonner, 1996). The present work directly shows that this conserved residue appears to play little role in ligand-hCB2 receptor interaction. The aggregate data illustrate how our LAPS approach, by integrating the complementary strengths of affinity labeling with covalent probes and site-directed mutagenesis, enables direct discrimination between the ligand-binding motifs of the CB1 and CB2 receptors. These data help satisfy the need for structural detail on ligand recognition by the hCB2 receptor and, perhaps, GPCRs in general while helping inform the development of selective hCB2-receptor ligands as potential drugs.
RESULTS
Characterization of Heterologously Expressed Wild-Type and Mutant hCB2 Receptors
Stably transfected polyclonal human embryonic kidney (HEK) 293 cell lines were generated that express either the nonmutated wild-type (WT) hCB2 receptor or hCB2 receptors with amino acid substitutions at C6.47(257). Cell lines expressing the hCB1 or hCB2 receptor mutated to replace lysine K3.28(192) or K3.28(109), respectively, with alanine were also generated. Receptor Kd and Bmax values for individual cell lines were then determined in saturation-binding assays performed on isolated membranes with either [3H]-CP55940 or [3H]-WIN55212-2 as radioligand. The WT hCB2 receptor and the hCB2 C6.47(257)A and hCB2 C6.47(257)S mutants demonstrated characteristic Kd values of 0.67 nM, 1.23 nM, and 0.85 nM, respectively, with [3H]-CP55940 (Table 1), whereas no specific binding was observed with C6.47(257)K, C6.47(257)I, C6.47(257)L, C6.47(257)D or C6.47(257)Y mutant hCB2 receptors (data not shown). WT and hCB2 C6.47(257)A and hCB2 C6.47(257)S mutant receptors displayed comparable affinity for [3H]-WIN55212-2 (Table 1). No specific radioligand binding to membranes from nontransfected HEK293 cells was observed (data not shown), ruling out gross, nonspecific ligand-membrane interactions.
Table 1.
Ligand-Binding Parameters for WT and C6.47(257) Mutant hCB2 Receptors
| [3H]-CP55940
|
[3H]-WIN55212-2
|
|||
|---|---|---|---|---|
| Kd (nM) | Bmax (pmol/mg) | Kd (nM) | Bmax (pmol/mg) | |
| WT hCB2 | 0.67 (0.51–0.83) | 1.19 (1.01–1.37) | 3.54 (2.27–4.80) | 2.96 (2.66–3.26) |
|
| ||||
| hCB2 C6.47(257)A | 1.23 (0.73–1.78) | 0.96 (0.84–1.08) | 2.89 (1.80–3.99) | 3.39 (3.05–3.73) |
|
| ||||
| hCB2 C6.47(257)S | 0.85 (0.48–1.22) | 1.07 (0.95–1.18) | 3.10 (0.92–5.29) | 2.82 (2.28–3.36) |
Saturation-binding assays were performed using membranes from stably transfected HEK293 cells and [3H]-CP55940 and [3H]-WIN55212-2 as the radioligands. The Kd and Bmax values shown are the means of at least 3 independent experiments performed in triplicate; 95% confidence intervals are given in parentheses.
Ligand Binding Affinities for WT hCB2 and Mutant Receptors
Binding affinities for the WT hCB2 receptor and C6.47(257) hCB2 receptor mutants were defined using ligands representative of different cannabinergic classes in competitive binding assays with [3H]-CP55940 and [3H]-WIN55212-2. The covalent ligand AM-841, a structurally optimized and functionalized classical CB agonist, was employed along with its noncovalent congeners AM-4056 and AM-4043 (Deng et al., 2005; Picone et al., 2005; Shen et al., 2006), in which the NCS group of AM-841 is substituted by H or Br, respectively. We also included the classical plant-derived CB Δ9-THC, the aminoalkylindole analog AM-2233, the endocannabinoid AEA, and the CB2-receptor selective biarylpyrazole SR144528 (Figure 1A).
The binding affinities of the hCB2 C6.47(257)S mutant receptor for the classical CB analogs AM-841, AM-4043, and AM-4056 in competition with radioligand [3H]-CP55940 were comparable to those of the WT hCB2 receptor (Table 2A). Conversely, the ability of these three ligands to displace CP55940 from the hCB2 C6.47(257)A mutant was enhanced by at least 2-fold. Consistent with the saturation-binding data (Table 1), no specific binding of AM-841 by the C6.47(257)K, C6.47(257)I, C6.47(257)L, C6.47(257)D, and C6.47(257)Y hCB2 receptor mutants was observed in a competitive binding assay with [3H]-CP55940 radioligand (data not shown). The phytocannabi-noid Δ9-THC, which contains a 5-carbon alkyl chain as compared to the longer 3-dimethylheptyl chain in AM-841 (Figure 1A), evidenced a comparably lower affinity for the WT and each mutant hCB2 receptor.
Table 2.
Binding Affinities for hCB2 WT and C6.47(257) Mutant Receptors and the hCB1 K3.28(192)A Mutant Receptor Using Either [3H]-CP55940 or [3H]-WIN55212-2 as the Radioligand
| (A) [3H]-CP55940 Ki (nM) | ||||
|---|---|---|---|---|
| AM-841 | AM-4043 | AM-4056 | Δ9-THC | |
| WT hCB2 | 1.51 (1.17–1.93) | 2.64 (2.15–3.23) | 2.14 (1.72–2.66) | 50.08 (36.4–68.9) |
| hCB2 C6.47(257)S | 1.68 (1.08–2.62) | 1.51 (1.20–1.89) | 1.52 (1.08–2.13) | 38.72 (24.73–60.62) |
| hCB2 C6.47(257)A | 0.71 (0.40–1.26) | 0.88 (0.64–1.22) | 1.03 (0.65–1.63) | 34.10 (18.53–62.75) |
| hCB2 K3.28(109)A | 2.31 (1.56–2.83) | 1.14 (0.81–1.59) | ||
| hCB1 K3.28(192)A | No Specific Binding | No Specific Binding | ||
| B | (B) [3H]-WIN55212-2 Ki (nM) | |||||
|---|---|---|---|---|---|---|
| Δ9-THC AM-841 | AM-4043 | AM-4056 | AEA | AM-2233 | SR144528 | |
| WT hCB2 | 17.1 (13.0–22.5) 4.5 (3.8–5.3) | 2.4 (2.0–2.9) | 2.3 (1.7–3.0) | 208.7 (163–267) | 9.2 (7.3–11.6) | 32.4 (25.0–42.1) |
| hCB2 C6.47(257) A | 2.9 (1.1 – 7.9) 0.7 (0.6–1.0) | 0.8 (0.6–1.0) | 0.5 (0.31–0.8) | 211.0 (176–252) | 0.5 (0.38–0.6) | 14.3 (10.6–19.2) |
| hCB2 C6.47(257)S | 17.6 (11.0–28.2) 1.5 (1.3–1.8) | 1.4 (1.1–1.8) | 1.2 (0.9–1.6) | 365.8 (307–435) | 0.8 (0.6–0.9) | 14.0 (11.4–17.2) |
Competitive binding assays were performed, and binding affinities were determined, using membrane preparations from stably transfected HEK293 cells. Ki values are the means of at least 3 independent experiments performed in triplicate, with 95% confidence intervals shown in parentheses.
Δ9-THC displaced [3H]-WIN55212-2 and showed a 5-fold increase in affinity for the hCB2 C6.47(257)A mutant over the WT hCB2 receptor (Table 2B). However, complete [3H]-WIN55212-2 dissociation by Δ9-THC was not observed in either the WT or mutant receptors. Compared to the WT hCB2 receptor, hCB2 C6.47(257)A and hCB2 C6.47(257)S mutant receptors showed the greatest (over 11-fold) incremental increases in affinity for AM-2233, a potent aminoakylindole cannabinergic ligand developed by us and related to WIN55212-2 (Deng et al., 2005). The two mutant hCB2 receptors also evidenced some 3–6-fold greater affinity than the WT hCB2 receptor for AM-841. These data establish that the hCB2 receptor recognizes and binds AM-841, necessary prerequisites for experiments detailed below with this ligand. Because binding of AM-841 to the WT hCB2 receptor is irreversible, reported affinities of this receptor for AM-841 should be regarded as “apparent Ki” values (Picone et al., 2005).
To determine ligand-binding specificity and, ultimately, any differences in recognition site and activation between CB1 and CB2 receptors, the amino acid at position K3.28, a highly conserved site across family A, group 1 GPCRs (Jensen et al., 2001; Shi et al., 2002), was modified. The hCB2 K3.28(109)A mutant receptor evidenced Ki values similar to those for WT hCB2 receptor for our experimental compounds AM-841 and AM-4056 using [3H]-CP55940 as the radioligand (Table 2A). Conversely, no specific binding of AM-841 or AM-4056 was observed by the hCB1 K3.28(192)A mutant receptor in a competition binding assay with [3H]-CP55940 as the radioligand (data not shown). In addition, no saturable binding by the hCB1 K3.28(192)A mutant receptor was observed using [3H]-CP55940 or [3H]-SR141716A (data not shown). Mutational evidence (Tao et al., 1999) has implicated K3.28(192) in the binding of various cannabinergic agonists by the hCB1 receptor. The lack of effect of mutating this highly conserved position on ligand recognition by the hCB2 receptor suggests that the hCB2 receptor has a unique binding architecture with respect to that of the hCB1 receptor.
Covalent hCB2 Receptor Labeling
AM-841 covalently binds with high affinity to the WT hCB2 receptor, whereas its binding is prohibited by the amino acid substitutions in hCB2 C6.47(257)A and hCB2 C6.47(257)S (vide supra; Table 2). After a 1 hr preincubation of membranes with 9 nM AM-841 (~6-fold the receptor’s apparent Ki for AM-841; Table 2A) at 30°C followed by extensive buffer wash-out to remove unbound ligand, the WT hCB2 receptor displayed a significantly lower Bmax for [3H]-CP55940 binding, compared with “control” membranes without prior AM-841 exposure (0.22 versus 1.05 pmol/mg, respectively) (Figure 1B). Membranes preincubated only with buffer prior to washing and incubation with [3H]-CP55940 demonstrated ~80% irreversible AM-841 binding (data not shown). The hCB2 C6.47(257)K, C6.47(257)I, C6.47(257)L, C6.47(257)D, and C6.47(257)Y mutant receptors were not evaluated, since they evidenced no specific binding of [3H]-CP55940 in saturation binding assays (data not shown).
To explore the potential involvement of other residues in the covalent attachment of AM-841 to the WT hCB2 receptor, we also developed the hCB2 C7.38(284)S and hCB2 C7.42(288)S mutants (Figure 2). Our prior hCB2 receptor modeling would predict that these two cysteine residues, located within the upper half of TMH7, are candidates for a covalent nucleophilic reaction with AM-841’s NCS moiety (Tao et al., 1999; Zhang et al., 2005). In [3H]-CP55940 displacement experiments, the WT hCB2 receptor and the hCB2 C7.38(284)S and hCB2 C7.42(288)S mutant receptors exhibited an ~80% decrease in [3H]-CP55940 binding after AM-841 pretreatment. In marked contrast, the hCB2 C6.47(257)A and hCB2 C6.47(257)S mutant receptors displayed no distinct decrease in [3H]-CP55940 Bmax after AM-841 pretreatment (Figure 1B). These results, summarized in Figure 1C, demonstrate that mutating the cysteine residues in TMH7 to serine did not affect AM-841’s covalent binding to the hCB2 receptor. However, mutation of C6.47(257) abrogated the irreversible interaction between the hCB2 receptor and AM-841. Additionally, preincubation of the WT hCB2 receptor with either AM-4043 or AM-4056 (two nonelectrophilic congeners of AM-841 unable to bind covalently to the hCB2 receptor) did not affect its [3H]-CP55940 Bmax in a subsequent saturation-binding assay (data not shown). Taken together, these results provide strong and complementary evidence that C6.47(257) is the site of covalent attachment of the NCS moiety of AM-841 to the hCB2 receptor.
Figure 2. Schematic Representation of the hCB2 Receptor.

Amino acids subjected to mutation in this study, C6.47(257), C7.38(284), and C7.42(288), are circled in bold. Alignment of the V6.43/I6.46 groove and the CWXP motif in the CB1 and CB2 receptors is highlighted.
Functional Characterization of hCB2 WT and Mutant Receptors
Since the hCB2 receptor is negatively coupled to adenylyl cyclase (Rhee et al., 1998), inhibition of forskolin-stimulated cAMP production was used to index WT and mutant hCB2 receptor function. Both AM-841 and its structural analog, AM-4056, acted as agonists for the WT hCB2 receptor, inhibiting forskolin-stimulated cellular cAMP formation in a concentration-dependent manner, as did the prototypic agonist, WIN55212-2 (Figure 3). Notably, AM-841 displayed a strikingly lower IC50 (0.079 nM) at the WT hCB2 receptor, compared with AM-4056 (3.27 nM) and WIN55212-2 (12.92 nM) (Table 3). No significant difference was observed between the respective IC50 values for AM-841 and AM-4056 inhibition of cAMP production by hCB2 C6.47(257)A-expressing or hCB2 C6.47(257)S-expressing cells (Table 3).
Figure 3. Concentration-Dependent Inhibition of Forskolin-Stimulated cAMP Accumulation in HEK293 Cells Expressing hCB2 WT or Mutant Receptors by Various Agonists.
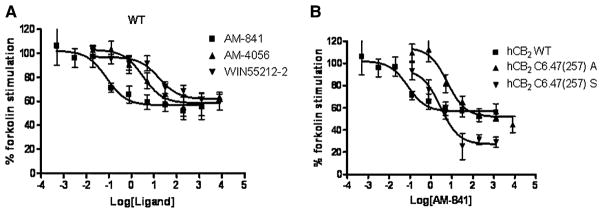
(A) Comparison among AM-841, AM-4056, and WIN55212-2 to inhibit forskolin-stimulated cAMP accumulation in the HEK293 cells expressing WT hCB2 receptor.
(B) Comparison of the ability of AM-841 to compete with forskolin-stimulated cAMP accumulation in HEK293 cells expressing either the WT hCB2 receptor or the hCB2 C6.47(257)A or C6.47(257)S mutant receptor.
Table 3.
Inhibition of Forskolin-Stimulated cAMP Accumulation in Cells Expressing the WT or a C6.47(257) Mutant hCB2 Receptor by AM-841, AM-4056, and WIN55212-2
| AM-841 (nM) | AM-4056 (nM) | WIN55212-2 (nM) | |
|---|---|---|---|
| WT hCB2 | 0.079 (0.024–0.267) | 3.27 (0.73–14.59) | 12.92 (3.87–43.15) |
| hCB2 C6.47(257)A | 6.01 (2.09–17.25) | 2.62 (0.75–9.14) | |
| hCB2 C6.47(257)S | 2.88 (0.88–9.42) | 8.82 (3.94–19.78) |
cAMP assays were performed using cells from stably transfected HEK293 WT and mutant lines. Data are the mean IC50 values from at least 3 independent experiments performed in triplicate with 95% confidence intervals shown in parentheses.
Molecular Modeling of hCB2 R*-Ligand Complexes
Protein cysteine residues are most likely to react with NCS-containing molecules such as AM-841 (Tahtaoui et al., 2003). This finding and the data presented above on AM-841 covalent binding to WT and mutant hCB2 receptors offer sound justification for interactive docking studies aimed at modeling AM-841 binding interactions with the hCB2 receptor. Our previously advanced model of the hCB2 R* receptor (Zhang et al., 2005) implicated each of the five cysteine residues in the TMH domains of the hCB2 receptor at the level of the ligand-binding pocket—C1.39(40), C2.59(89), C6.47(257), C7.38(284) and C7.42(288) —as having the potential to form a covalent bond with AM-841. C1.39(40) is disposed facing into the binding pocket between TMH2 and TMH7, but M7.40(286) sterically blocks this residue. C2.59(89) is located in the TMH2-3 interface and is accessible to the thiol-directed agent (2-aminoethyl)methane thiosulfonate hydrobromide (MTSEA). C7.38(284) is extracellular to C7.42(288) by one helix turn. In our model, C7.38(284) and C7.42(288) are located at the TMH6–7 interface, with C7.42(288) comparatively more accessible to the ligand-binding pocket. C6.47(257) is one turn below the level of C7.42(288) and fairly deep within the binding pocket. C6.47(257) changes its orientation upon activation of the β2-adrenergic receptor, becoming accessible within the binding pocket only when that receptor is R* (Javitch et al., 1997). According to our prior modeling of the hCB2 receptor (Zhang et al., 2005), this cysteine residue faces lipid in R and is within the TMH6–7 interface in R*.
To identify possible sites of interaction between AM-841 and the hB2 receptor, the covalent bond between the NCS-functionalized tail of AM-841 and each accessible candidate cysteine residue—C2.59(89), C6.47(257), C7.38(284), and C7.42(288) —was first formed. Potential binding sites were screened using the criterion from hCB2-receptor mutational studies that S7.39(285) is an interaction site for classical cannabinoids (Rhee and Kim, 2002). It was found that only when AM-841 was covalently attached to C6.47(257) could it hydrogen-bond with S7.39(285). In this case, the carbocyclic CH2OH substituent at the γ position of AM-841 was the hydrogen-bonding partner with S7.39(285) (d = 2.62 Å; O – H- -O angle = 175°). With this as an anchoring interaction, the model was probed using interactive computer graphics for additional sites that could hydrogen-bond with the pyran oxygen or with the phenolic hydroxyl of AM-841. No interaction site for the pyran oxygen was identified in the hCB2 receptor. However, we found that the phenolic hydroxyl of AM-841 could hydrogen-bond with S6.58(268) (d = 2.61 Å; O – H- -O angle = 176°) and still maintain its interaction with S7.39(285) and covalently link to C6.47(257).
The hCB2-receptor R* binding site for AM-841 modeled here is the energy-minimized complex depicted in Figure 4. Most notably, the orientation of AM-841 in the binding pocket of the hCB2 receptor is quite different in a number of salient aspects from its orientation in the binding pocket of the CB1-receptor (Picone et al., 2005). In our hCB2 receptor model, the tricyclic ring of AM-841 is oriented nearly perpendicularly to the TMH helices. Conversely, its orientation in the hCB1 receptor is parallel to the TMHs (see Figure 9 in Picone et al., 2005). Figure 4 also illustrates the formation of a salt bridge involving K3.28(109). Contrary to the documented importance of K3.28 to classical, nonclassical, and endogenous cannabinoid binding by the CB1 receptor (Chin et al., 1998; Hurst et al., 2002; Song and Bonner, 1996), mutational analysis suggests that K3.28 is not essential for ligand binding to the hCB2 receptor (Tao et al., 1999 and present study). Extracellular loop (EC) 3 in the hCB2 receptor (TTLSDQVKK), when compared to its paralog in the hCB1 receptor (GKMNKLIKT), exhibits one important amino acid difference, the negatively charged D275 residue centrally located in EC3. We have hypothesized that D275 forms a salt bridge with K3.28(109) in the hCB2 receptor, rendering K3.28(109) less available for ligand interaction. This assumption was used to select an EC3 conformation in the modeling studies reported here. In the model illustrated (Figure 4), K3.28(109) is involved in a salt bridge with D275 of EC3 (d = 2.55 Å; N – H- -O angle = 171°) and in a hydrogen bond with N2.63(93) (d = 2.66 Å; N – H- -N angle = 168°). D275 also forms a hydrogen bond with S274 in EC3 (d = 2.67 Å; O – H- -O angle = 170°) and with S2.60(90) (d = 2.63 Å; O – H- -O angle = 160°), a residue that is accessible from within the binding pocket in the hCB2 receptor due to the helix distortion produced by S2.54(84) (see Experimental Procedures and Zhang et al., 2005).
Figure 4. Illustration of the CB2 R*/AM-841 Binding Site from Modeling Studies.
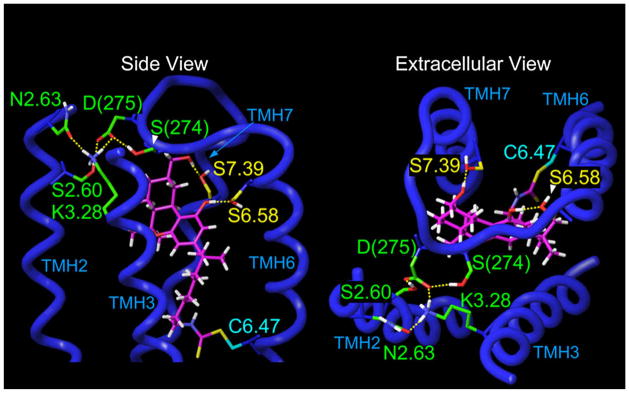
TMHs 1, 4, and 5 have been omitted from this view for simplicity. In the energy-minimized CB2 R*/AM-841complex in which AM-841 is covalently attached to C6.47(257), the carbocyclic ring CH2OH of AM-841 hydrogen-bonds with S7.39(285) (d = 2.62 Å; O – H- -O angle = 175°), while the phenolic hydroxyl of AM-841 hydrogen-bonds with S6.58(268) (d = 2.61 Å; O – H- -O angle = 176°). Also illustrated here is the formation of a salt bridge between D275 in EC3 and K3.28(109), a residue that is crucial for classical CB binding to the CB1 receptor (Song and Bonner, 1996), but which is not important for binding to the CB2 receptor. In the final, energy-minimized complex illustrated here, K3.28(109) is involved in a salt bridge with D275 of the EC3 loop (d = 2.55 Å; N – H- -O angle = 171°) and in a hydrogen bond with N2.63(93) (d = 2.66 Å; N – H- -N angle = 168°). D275 also forms a hydrogen bond with S274 in EC3 (d = 2.67 Å; O – H- -O angle = 170°) and with S2.60(90) (d = 2.63 Å; O – H- -O angle = 160°), a residue that is accessible from within the binding pocket in the CB2 receptor due to the helix distortion produced by S2.54(84) (Experimental Procedures and Zhang et al., 2005).
DISCUSSION
The present report has employed the LAPS approach using a high-affinity cannabinergic compound, AM-841, as covalent probe for direct experimental characterization of the ligand-binding architecture of the hCB2 receptor. AM-841 is one of several novel cannabinergic ligands we have rationally designed to interact irreversibly and covalently with specific amino acid residues at or immediately adjacent to the CB1- and/or CB2-receptor-binding pocket (Charalambous et al., 1992; Guo et al., 1994; Morse et al., 1995; Picone et al., 2005). AM-841’s reactive NCS group does not interfere with its binding to the hCB2 receptor, since competitive-binding data herein show that the affinity of AM-841 for the hCB2 receptor is comparable to that of two AM-841 analogs (AM-4043 and AM-4056) lacking the NCS moiety. Because of their highly nucleophilic thiol group, cysteines are the most likely amino acids to participate in a nucleophilic addition reaction with the AM-841 NCS moiety (Tahtaoui et al., 2003). Although the amino group of lysine is capable of interacting with an NCS group, under our experimental conditions a lysine amino group is a significantly weaker nucleophile than a cysteine thiol. Furthermore, the only lysine located in the putative binding pocket of the hCB2 receptor is in TMH3, a region unlikely to associate with the alkyl tail of nonclassical or classical cannabinoids (Tian et al., 2005).
Although in the binding experiments reported we utilized membranes from HEK cells overexpressing hCB2 receptors, it is possible that a non-CB2-binding site is responsible for the ~2–3-fold greater mean hCB1-receptor Bmax values between [3H]-WIN55212-2 over [3H]-CP55940 (Table 1). Putative binding site(s) for WIN55212-2 distinct from CB1 and CB2 receptors have been proposed and largely remain to be characterized fully (Dhawan et al., 2006; Fride et al., 2003; Monory et al., 2002). We detected no specific radioligand binding to membranes from nontransfected HEK cells, however. Alternatively, the mean Bmax differences we observe between the two radioligands employed appear to be within an acceptable range for quantitative CB-receptor assays among replicate cell cultures/membrane preparations. Mean cannabinoid-receptor Bmax values over a 2–3-fold range using a single radioligand have been considered “comparable” or “similar” (Shire et al., 1996; Tao and Abood, 1998; Tao et al., 1999).
Prior modeling predicted that C6.47 is the cysteine residue in both the hCB1 and hCB2 receptors closest to the terminal position of the alkyl tail of classical and nonclassical CBs (Picone et al., 2005; Raitio et al., 2005). In the present study, two alternate cysteine residues, C7.38(284) and 7.42(288), located in TMH7 and in moderate proximity to C6.47(257) (Zhang et al., 2005) were also probed in order to rule out the possibility that they may be alternate sites of interaction with AM-841. C6.47(257) was mutated to seven different amino acids representing a wide diversity of side-chain reactivities and steric constraints: C6.47(257)A, S, K, L, I, Y, and D. Our data show that the hCB2 C6.47(257)A and hCB2 C6.47(257)S mutant receptors bind with similar Ki values as WT hCB2 receptor using AM-841 as displacing ligand and CP55940 or WIN-55212-2 as radioligand. These mutant receptors did not bind AM-841 covalently. Importantly, neither C7.38(284) nor C7.42(288) covalently interacted with AM-841. These aggregate data unequivocally demonstrate that C6.47(257) is the site of covalent attachment of AM-841 to the hCB2 receptor. Covalent AM-841 binding was shown to activate the hCB2 receptor (i.e., inhibit forskolin-stimulated cAMP production) with exceptional potency as compared to either AM-841’s effect on the hCB1 receptor or AM-4056’s activity as hCB2-receptor agonist. This enhanced potency is observed only with the WT hCB2 receptor, but not with the two binding-competent hCB2 C6.47(257)A and hCB2 C6.47(257)S mutant receptors. The molecular basis for the exceptional agonist potency of AM-841 following its covalent interaction with the hCB2 receptor’s C6.47(257) residue is currently being explored.
Misfolding leading to improper interacellular protein translocation/sequestration is a particular problem with mutated GPCRs, including CB receptors (Shire et al., 1996; Tao et al., 1999). Consequently, the lack of specific ligand binding we have observed by some mutant hCB2 receptors could potentially reflect functional receptors improperly integrated into/disposed within the cell membrane, obviating ligand binding; dysfunctional receptors properly integrated into the cell membrane; and/or abnormal receptor translocation/compartmentalization such that receptors do not reach the cell membrane. In any event, the result is the same,—that is, an abortive receptor is unable to bind ligand at the physiologically relevant membrane site, which would translate operationally into the observed lack of specific ligand binding to select mutant hCB2 receptors in our membrane preparations.
The ligand-binding properties of the individual mutants offer insight into the binding motif of classical cannabinoids to the hCB2 receptor. Our data show that, compared with the WT hCB2 receptor, the mutation C6.47(257)A maintains or enhances the binding affinities of the hCB2 receptor for the cannabinergic ligands examined in this study. Likewise, the hCB2 C6.47(257)S mutant and WT receptors also exhibit comparable affinities for the majority of ligands tested. The basis for the higher affinity of hCB2 C6.47(257)S versus WT receptor for the aminoalkylin-dole AM-2233 (Table 2B) is under investigation. The aggregate binding data for AM-841, [3H]-CP55940, and [3H]-WIN55212-2 suggest that there is partial convergence of hCB2-receptor-binding site for these three ligands, which appear to occupy a similar, though not necessarily identical, space. For example, the data in Table 2 indicate that C6.47(257) plays a rather permissive role in both Δ9-THC and [3H]-CP55940 binding by the hCB2 receptor, since C6.47(257) mutation to either serine or alanine did not alter Δ9-THC’s ability to displace [3H]-CP55940, whereas C6.47 may be functionally more relevant to [3H]-WIN55212-2 binding when challenged by competing ligand, since C6.47(257) mutation to alanine markedly facilitated [3H]-WIN55212-2, but not [3H]-CP55940, displacement by Δ9-THC and most other ligands tested. Because cysteine and serine are very conservative, isosteric analogs of one another bearing polar, uncharged side-chains, whereas alanine has a nonpolar, smaller aliphatic (i.e., methyl) side-chain, these data suggest that a combination of amino-acid polarity and side-chain bulk at hCB2-receptor C6.47(257) is particularly influential on [3H]-WIN55212-2 binding and, hence, would represent a consideration in the design and targeting of ligands to the hCB2 receptor for therapeutic purposes.
From the structural and functional evidence presented in this study, we propose that the hCB1 and hCB2 receptors have different binding motifs with respect to cannabinergic ligands such as AM-841. Our previous computational docking models for hCB receptors (Picone et al., 2005; Zhang et al., 2005) as elaborated by the modeling presented herein support this conclusion. The centrality of position C6.47 is conserved in the CB1 and CB2 receptors (as with most other members of family A, group 1 GPCRs). In contrast, as demonstrated with ligands AM-841 and AM-4056, the overall binding template for each principal cannabinoid receptor is unique with respect to the highly conserved residue K3.28A.
It is tempting to relate our observations to GPCRs other than the hCB2 receptor. The cysteine residue C6.47(285) in the human β2-adrenergic receptor homologous to hCB2 C6.47(257) becomes accessible to the binding pocket only when the receptor is in R*, proline P6.50(288) initiating the rotational movement of C6.47(285) leading to activation (Javitch et al., 1997). We hypothesize that a similar conformational change also occurs upon activation of the WT hCB2 receptor such that its C6.47(257) residue becomes accessible within the binding pocket only in R*. Agonists appear to bind to the CB2 R* receptor, but not the R (Leff, 1995; Samama et al., 1993). According to our published hCB2 receptor model (Zhang et al., 2005), C6.47(285) faces lipid in R and is located in the TMH6-7 interface in R*. The cAMP functional data herein show that AM-841 and its noncovalent analog, AM-4056, both act as hCB2-receptor agonists. Thus, it is likely that AM-841 binds favorably to hCB2 R* receptor, wherein the C6.47(257) residue is accessible.
Of the five cysteine residues modeled in the ligand-binding pocket of the hCB2 receptor, C6.47(257) is located most deeply within the pocket, near the center of the lipid bilayer (Zhang et al., 2005). On the assumption that the NCS functional group of AM-841 reacts with the first cysteine residue with which it comes into contact in the hCB2 receptor’s ligand-binding pocket, identification of C6.47(257) as the site for covalent attachment of AM-841 to the hCB2 receptor suggests that the tail of AM-841 enters the receptor’s binding pocket at great depth. We therefore hypothesize that AM-841, a highly lipophilic molecule, enters the hCB2-receptor-binding pocket from the surrounding membrane domain and not from the aqueous extra-cellular milieu, the lipid bilayer helping direct AM-841 to the hCB2 receptor’s binding pocket. This view is supported by results from small-angle X-ray diffraction and differential scanning calorimetry studies of classical cannabinoids in model membranes. We have previously shown that Δ8-THC intercalates between contiguous acyl chains in the membrane lipid bilayer, Δ8-THC’s phenol group located near the phospholipid head-groups and its alkyl tail deeper within the bilayer and oriented parallel to the fatty-acyl chains (Mavromoustakos et al., 1991). The terminal iodo group of 5′-I-Me-Δ8-THC resides in a region extending ≈5 Å from the center of the membrane bilayer (Mavromoustakos et al., 1995). Another lipophilic CB2-receptor ligand structurally related to the endocannabinoid anandamide orients its terminal carbon tail in a similar fashion within the bilayer, positioned for a productive interaction with C6.47 (Tian et al., 2005).
More generally, the current study extends prior demonstration (Picone et al., 2002 and 2005; Zvonok et al., 2008) that functionalized, covalent molecular probes are valuable experimental tools for obtaining direct information on functionally relevant interactions between proteins and small-molecule ligands. As afforded by our LAPS paradigm, characterization of the hCB2 receptor’s ligand-recognition site and its similarities to and differences from that of the hCB1 receptor should help inform the rational design of future generations of selective hCB2-receptor ligands having potential therapeutic utility.
SIGNIFICANCE
Pharmacological modulation of endocannabinoid signaling by altering transmission through the two main CB receptors, CB1 and CB2, holds far-reaching therapeutic promise. The differential tissue distributions of CB1 and CB2 receptors, their distinct physiological and regulatory roles, and the potential psychobehavioral side-effects of centrally acting CB1-receptor ligands have placed great emphasis on targeting novel, high-affinity ligands that bind selectively to one or the other CB receptor subtype. In particular, selective CB2-receptor agonists are increasingly being sought as potential therapeutics for neurodegenerative, inflammatory, and immunological diseases. Thorough understanding of the respective ligand-binding interactions of the CB1 and CB2 receptors, the structural features of receptor activation, and the pharmacophore requirements at each receptor’s binding domain is complicated by the lack of direct experimental characterization of these (and virtually all other) GPCRs. This information is essential to the optimal design and exploitation of CB1- and CB2-selective ligands for therapeutic gain. The present work has utilized a chemically selective, covalent affinity probe (AM-841) and site-directed mutational analysis to characterize experimentally the binding domain of the hCB2 receptor. Our data demonstrate the importance of TMH6 C6.47(257) in ligand recognition by and activation of the hCB2 receptor, a role similar to that of the CB1 receptor’s C6.47(355). In marked contrast, another highly conserved GPCR residue, K3.28(109), was shown to play little role in ligand-hCB2 receptor interaction, although it is crucial to the recognition of cannabinergic ligands by the CB1 receptor. The direct experimental evidence provided supports general conclusion that CB1 and CB2 receptors have distinct ligand-binding motifs. Our data invite extrapolation from other GPCRs as to the importance of C6.47(257) in the conformational changes leading to hCB2-receptor activation. By characterizing the hCB2 receptor’s binding motif and how it differs with respect to that of the hCB1 receptor, the present study helps inform the rational design of selective hCB2-receptor (activating) ligands with therapeutic potential. The data also add to the paucity of experimentally-derived general structural detail for GPCRs, which constitute a prime class of drug targets.
EXPERIMENTAL PROCEDURES
Materials
Chemicals and reagents were obtained from Sigma (St. Louis, MO) at highest purity/grade unless otherwise noted. Δ9-THC, AM-841, AM-2233, AM-4043, and AM-4056 were synthesized at the Center for Drug Discovery, Northeastern University (Boston, MA). CP55940, [3H]-CP55940, AEA, and SR144528 were supplied by the National Institute on Drug Abuse (Bethesda, MD). [3H]-WIN55212-2 was purchased from PerkinElmer (Wellesley, MA). pcDNA 3.1+ was purchased from Invitrogen (Carlsbad, CA). Oligonucleotide primers were synthesized by Integrated DNA Technologies (Coralville, IA).
Site-Directed Mutagenesis, Cell Culture, Transfection, and Transgene Integrity
The partial-length cDNA encoding the translated region of the hCB2 receptor was provided by Sean Munro (MRC Laboratory of Molecular Biology, Cambridge, UK). The hCB2-receptor translated region was sequenced for integrity and subcloned into pcDNA3.1+ by polymerase chain reaction (PCR) using primers spanning the native start and stop codons. An appropriate Kozak initiation site was part of the endogenous sequence. Site-directed mutagenesis of pcDNA 3.1+-hCB2 was performed with the QuickChange™ Site-Directed Mutagenesis system (Stratagene, La Jolla, CA). Primers were annealed and extended using 18 cycles with an Eppendorf Mastercycler (Westbury, NY) and Pfu DNA polymerase (Stratagene). Primers used to make the following mutations in hCB2, C6.47(257)A, C6.47(257)S, C6.47(257)K, C6.47(257)I, C6.47(257)L, C6.47(257)D, C6.47(257)Y, C7.38(284)S and C7.42(288)S, and K3.28(109)A, were as follows (in the order listed; targeted codons are in bold and underlined): Forward 5′-GCT GTG CTC CTC ATC GCC TGG TTC CCA GTG CTG-3′, Reverse 5′-CAG CAC TGG GAA CCA GGC GAT GAG GAG CAC AGC-3′; Forward 5′-GCT GTG CTC CTC ATC AGC TGG TTC CCA GTG CTG-3′, Reverse 5′-CAG CAC TGG GAA CCA GCT GAT GAG GAG CAC AGC-3′; Forward 5′-G GCT GTG CTC CTC ATC AAG TGG TTC CCA GTG CTG G-3′, Reverse 5′-C CAG CAC TGG GAA CCA CTT GAT GAG GAG CAC AGC C-3′; Forward 5′-G GCT GTG CTC CTC ATC ATC TGG TTC CCA GTG CTG G-3′, Reverse 5′-C CAG CAC TGG GAA CCA GAT GAT GAG GAG CAC AGC C-3′; Forward 5′-GTG CTC CTC ATC CTT TGG TTC CCA GTG-3′, Reverse 5′-CAC TGG GAA CCA AAG GAT GAG GAG CAC-3′; Forward 5′-GTG CTC CTC ATC GAT TGG TTC CCA GTG-3′, Reverse 5′-CAC TGG GAA CCA ATC GAT GAG GAG CAC-3′; Forward 5′-GTG CTC CTC ATC TAT TGG TTC CCA GTG-3′, Reverse 5′-CAC TGG GAA CCA ATA GAT GAG GAG CAC-3′; Forward 5′-GCC TTT GCT TTC TCC TCC ATG CTG TG- 3′, Reverse 5′-CA CAG CAT GGA GGA GAA AGC AAA GGC- 3′; Forward 5′-GC TCC ATG CTG TCC CTC ATC AAC TCC-3′, Reverse 5′-GGA GTT GAT GAG GGA CAG CAT GGA GC-3′. Forward 5′-GCT GTC TTC CTG CTG GCC ATT GGC AGC GTG ACT ATG-3′, Reverse- 5′-CAT AGT CAC GCT GCC AAT GGC CAG CAG GAA GAC AGC-3′. Primers designed for the generation of the hCB1-receptor mutant line K3.28A(192) were as follows: Forward 5′-CGC AAC GTG TTT CTG TTC GCC CTG GGT GGG GTC ACG GCC TCC-3′, Reverse 5′-GGA GGC CGT GAC CCC ACC CAG GGC GAA CAG AAA CAC GTT GCG-3′. DpnI-treated DNA was transformed into One Shot Top10 competent Escherichia coli cells (Invitrogen). Plasmid DNA was isolated using the QIAGEN Midi-Prep Kit (Valencia, CA). Plasmid DNA sequencing confirmed that only the desired mutations had been effected. All other DNA manipulations were performed as described elsewhere (Ausubel et al., 2006).
HEK293 cells (American Type Culture Collection, Manassas, VA) and HEK293-derived cell lines were cultured using Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum, 1% penicillin-streptomycin, 4.5 g/l glucose, and 2 mM glutamine (Tao et al., 1999). HEK293 cells were transfected with verified mutagenic plasmid DNA utilizing Lipofectamine 2000 with an appropriate amount of linearized plasmid DNA harboring the transgene cassette according to the vendor’s (Invitrogen) technical manual. Typically, 3–5 independent transfections were performed in parallel and duplicated over a 3-day period to maximize cell line integrity. Cultures were selected with the appropriate antibiotics (600 μg/ml G418, as determined by performing a standard kill curve) over a 10-day period, passed to adherent culture flasks, grown to a cell volume sufficient to perform preliminary saturation binding assays, and harvested in PBS with centrifugation and repeated washing. This method allowed data regarding the efficacy of the receptor transgene to be collected within 15 days from design to analysis. Cells were cryopreserved after characterization under liquid nitrogen.
We routinely tested for the presence of all introduced transgenes by PCR amplification using primers corresponding to the parental vector sequence flanking the receptor-encoding cDNA (5′ and 3′) matched with internal primers corresponding to the receptor sequence. By this method, we could generate amplified fragments of the transgene cassette and avoid endogenous hCB2 receptor amplification. Corresponding transgene cassettes were sequence-confirmed for integrity of the incorporated encoding sequence and respective mutation. The results of these analyses ensured that the phenotype correlated with the genotype in all cases.
Cell Membrane Preparations and Radioligand Binding Assays
Cells were disrupted by cavitation, and the membrane fraction was obtained by ultracentrifugation, as detailed elsewhere (Xu et al., 2005). Saturation-binding assays were performed in a 96-well format. Membrane pellets were resuspended in 25 mM Tris base/5 mM MgCl2/1 mM ethylenediaminetetraacetic acid (EDTA) (TME) containing 0.1% (w/v) BSA (TME-BSA). Membrane, equivalent to 25 μg protein (DC Protein Assay System; BioRad, Hercules, CA), was added to each assay well. Radioligands ([3H]-CP55940 or [3H]-WIN55212-2) were diluted in TME-BSA to yield final assay concentrations from an order of magnitude below to an order of magnitude above each predicted Kd. Nonspecific binding was assayed in the presence of 5 μM respective unlabeled ligand. The assay was performed at 30°C for 1 hr with gentle agitation. After incubation, samples were transferred to Unifilter GF/B filter plates, and unbound ligand was removed using a Packard Filtermate-96 Cell Harvester (Perkin Elmer Packard, Shelton, CT). Filter plates were washed four times with ice-cold wash buffer (50 mM Tris-base and 5 mM MgCl2 containing 0.5% BSA [pH 7.4]). Bound radioactivity was quantified with a Packard TopCount Scintillation Counter. Nonspecific binding was subtracted from total bound radioactivity to calculate specific radioligand binding (as pmol/mg protein). Kd values are presented as means with 95% confidence intervals from at least 3 independent experiments performed in triplicate (n = 3 or greater). Bmax and Kd values were calculated by nonlinear regression using GraphPad Prism 3.03 (GraphPad Software, San Diego, CA) on a Windows platform; one-site binding analysis equation Y = Bmax*X/(Kd + X). Specific binding in all positive saturable binding assays was typically between 60%–90% of total binding.
Competition binding assays were performed in a 96-well format, as modified from Lan et al. (1999). Membrane pellets were resuspended in TME-BSA, and membrane (equivalent to 25 μg protein) was added to each assay well. The radioligands [3H]-CP55940 and [3H]-WIN55212-2 were brought to a final concentration of 0.76 nM or 0.91 nM, respectively, in a total volume of 200 μl TME-BSA. Concentrations of the displacing experimental ligands were determined using the IGOR Pro (Lake Oswego, OR) software by inputting an IC50 estimated from the literature or from prior structure-activity studies and a log-range of 4. Binding incubation and filtration were performed as described above. IC50 values were calculated by nonlinear regression using GraphPad Prism software, and Ki values were determined for each ligand. Ki values are presented as means with 95% confidence intervals from at least 3 independent experiments performed in triplicate.
Affinity-labeling assays were performed using membranes prepared as described above. Five milliliters of 0.8 mg/ml membrane protein was preincu-bated with 9.0 nM AM-841 (a concentration of AM-841 previously shown for each covalent ligand studied to inhibit [3H]-CP55940 binding to the WT hCB2 receptor by at least 80% across a range of [3H]-CP55940 concentrations). Membranes were allowed to equilibrate with 9.0 nM AM-841 for 1 hr at 30°C with agitation and were then sedimented at 27 × g, 30°C, and washed 3 times in TME containing 1% (w/v) BSA at 30°C. The membranes were allowed to equilibrate at 30°C in buffer for 15 min between each wash. Two final washes were then performed with BSA-free TME. Saturation-binding assays were then performed with the washed membranes and [3H]-CP55940 as radioligand. The resultant data were analyzed as described above for at least two independent experiments performed in duplicate.
cAMP Assay
cAMP was quantified as described elsewhere (Tao et al., 1999), with minor modification. HEK293 cells expressing the hCB2 receptor were grown to 70% confluency under selection, as described above. Cells were harvested by centrifugation at 500 × g for 5 min and resuspended in DMEM containing phosphodiesterase inhibitors (0.1 mM RO-20-1724 [Calbiochem, La Jolla, CA] and 1 mM IBMX), 20 mM HEPES (pH 7.3), and 0.1% (w/v) BSA to a final concentration of 1 × 106 cells/ml and incubated at 30°C for 30 min. Cells (1 × 106 per assay) were then incubated with 5 μM forskolin (Fisher Scientific, Pittsburgh, PA) and the appropriate test ligand at concentrations ranging from 0.01 to 10,000 nM for 5 min. Basal cAMP levels were determined from cells incubated in the absence of forskolin and ligand. cAMP levels were also determined in control cells incubated with forskolin alone. Reactions were stopped by boiling for 5 min and then immediately lysing the cells by rapid freeze-thaw. Lysates were centrifuged at 12,000 × g for 5 min to pellet cellular debris. cAMP was determined in the supernatants with a radiochemical competitive-binding assay (Diagnostic Products Corporation, Los Angeles, CA). Each cAMP determination was made at least three independent times, each in triplicate. IC50 values for inhibition of net forskolin-induced cAMP production (above basal) were determined by nonlinear regression (GraphPad Prism).
Molecular Modeling of the hCB2 Receptor
A TMH model of the hCB2 R receptor was created with the 2.8 Å crystal structure of bovine Rho (Palczewski et al., 2000) as the starting point. The sequences of the hCB2 receptor (Munro et al., 1993) and bovine Rho were aligned using the same highly conserved residues as guides that had been employed to generate our initial CB1-receptor model (Bramblett et al., 1995). Helix ends for Rho and the hCB2 receptor were considered analogous (Palczewski et al., 2000). Changes from the general Rho structure necessitated by sequence differences were then imposed. For example, the absence of helix-kinking proline residues in TMH1 and TMH5 necessitated modeling these as normal alpha helices. We recently showed that the conformation of TMH2 diverges between Rho and the hCB2 receptor and changes with time due to the absence of the Rho GGXTT motif in TMH2 of the hCB2 receptor. It was found that in the hCB2 receptor, S2.54(84) in a g- (+60o) χ1 can form an intrahelical hydrogen bond that induces a deviation from normal α-helicity by causing both a bend and a change in wobble angle and face shift in TMH2 of the hCB2 receptor. The result, a decrease in the number of residues per turn above S2.54(84), tightens the helix and alters the placement of residue C2.59(89) from one facing lipid in Rho to a position in the TMH2-3 interface, allowing it to react with the substituted cysteine accessibility method reagent MTSEA (Zhang et al., 2005).
A TMH model of the hCB2 R* receptor was created by modifying our Rho-based hCB2 R model (Zhang et al., 2005). R* model construction was guided by the biophysical literature on the R to R* transition in Rho and the β-2-adrenergic and muscarinic M3 receptors. The literature indicates that: a salt bridge between R3.50 and E/D6.30 at its intracellular end stabilizes the R receptor (Ballesteros and Weinstein, 1995); a conformational change in TMH6 occurs upon activation (Farrens et al., 1996; Jensen et al., 2001) mediated by the straightening of the CWXP flexible hinge region of TMH6 (Jensen et al., 2001); and rotation of TMH3 (Lin and Sakmar, 1996) and TMH6 (Javitch et al., 1997; Lin and Sakmar, 1996) occurs upon activation. The above experimental findings were used to create the hCB2 R* model in the current study. Specifically, the R* hCB2 TMH bundle was modeled from the R model of the hCB2 receptor by rotating TMH3 so that residue 3.41(122) moves into the less hydrophobic environment of the TMH3-4 interface (Lin and Sakmar, 1996). This was accomplished by a 20° counterclockwise (extracellular view) rotation of TMH3 from its orientation in the inactive (R) bundle. A conformer of TMH6 with a reduced bend (Barnett-Norris et al., 2002) was incorporated into the bundle. TMH6 was also rotated (counterclockwise from extracellular view) so that C6.47(257) became accessible from inside the binding site crevice (Javitch et al., 1997).
Loop segments were added in stages to the resultant R* TMH bundle model using MODELER v8.0 (Sali and Blundell, 1993). In each case, 1000 loop conformations were generated. The objective function of MODELER was used to rank conformations. The results were screened for the lowest energy loop that did not produce steric interference with the ligand-binding pocket. In all cases, this loop came from the first 50 low energy structures.
The EC3 conformation was identified by adding T(272) thru K(278) to the model and then performing a conformational search of this range of residues in MODELER. On the basis of the hypothesis of a salt bridge between K3.28 and D(275) (see Discussion), a single constraint was applied during the search: the C gamma atom of D(275) was required to be within 4.0 Å of the NZ atom of K3.28(109). One thousand loops were generated, and a structure, salt-bridged with K3.28(109) but not overlapping sterically with the ligand binding pocket, was incorporated into the computation.
EC1 was subsequently generated by adding F(97) thru K(103) to the model and then performing a conformational search of this range of residues in MODELER in the presence of EC3. The initial conformation for EC2 was taken from our Biased Scaled Collective Variable in Monte Carlo study of the EC2 (Barnett-Norris et al., 2003; Hassan et al., 2002). This loop has an internal C4.66(174)-C179 disulfide bridge, one which has been suggested to be present in the hCB2 receptor from mutagenesis studies (Gouldson et al., 2000). To allow the EC2 to adjust in the presence of EC1 and EC3, MODELER was used to vary S(180) to N(188). The conformation of the smaller internal loop (C4.66(174)-C179) was not varied. Intracellular loops (ICs) were generated using the same protocol, with IC3 added first.
Ligand Conformations and Docking Positions
Because AM-841 is a CB2-receptor agonist, it was docked in the CB2 R* model. The binding-site conformation and anchoring interactions within the receptor used for AM-841 were based on published computational and experimental structure-activity studies. AM-841 was docked in the global minimum energy conformation of its tricyclic hexahydrocannabinol ring system (Reggio et al., 1993). Cysteine residues are the most likely candidates for reaction with isothiocyanates (Tahtaoui et al., 2003), providing the rationale for interactive docking studies to elucidate binding interactions for AM-841. The hCB2 receptor has five cysteine residues in its TMH domains at the level of the ligand-binding pocket: C1.39(40), C2.59(89), C6.47(257), C7.38(284), and C7.42(288). C1.39(40) orients into the binding pocket between TMH2 and TMH7, but M7.40(286) sterically blocks this residue. C2.59(89) is located at the TMH2-3 interface and is accessible to MTSEA (Zhang et al., 2005). C7.38(284) is by one turn extracellular to C7.42(288). C6.47(257) is one turn below the level of C7.42(288) and fairly deep within the binding pocket. Upon activation of the β-2-adrenergic receptor, C6.47 becomes accessible within the binding pocket (Javitch et al., 1997). According to our prior hCB2 receptor model (Zhang et al., 2005), C6.47 faces lipid in R and is located in the TMH6–7 interface in R*. C7.38(284) and C7.42(288) are also located in the TMH6–7 interface, with C7.42(288) more accessible to the ligand-binding pocket.
Rhee (2002) has reported that the classical cannabinoid, HU-243, loses affinity by over 10-fold in a S7.39A mutant, suggesting that S7.39 is important for classical cannabinoid binding at the CB2 receptor. For this reason, S7.39(285) was used as the primary interaction site for AM-841 in our hCB2 R* model. To identify possible sites of interaction of AM-841 in the hCB2 receptor, a covalent bond between the NCS-functionalized tail of AM-841 and each accessible candidate cysteine residue—C2.59(89), C6.47(257), C7.38(284), and C7.42(288)—was first formed. Interactive computer graphics revealed that only when AM-841 was covalently attached to C6.47(257) could this ligand hydrogen-bond with S7.39(285). In this case, the CH2OH moiety of the carbocyclic-ring of AM-841 was hydrogen-bonded with S7.39(285) to establish an anchoring interaction. Further computer modeling demonstrated that the phenolic hydroxyl of AM-841 could hydrogen-bond with S6.58(268) while maintaining its interaction with S7.39(285) and covalent link with C6.47(257). No interaction site for the pyran oxygen was identified in the hCB2 receptor.
Energy Minimization
The OPLS_2005 all-atom force field in Macromodel 9.1 (Schrödinger LLC, NY, USA) was used to minimize the energy of the full hCB2-receptor bundle-AM-841 complex. An 8.0 Å extended nonbonded cutoff (updated every 10 steps), 20.0 Å electrostatic cutoff, and 4.0 Å hydrogen-bond cutoff were used in each stage of the calculation. All residues except D2.50(80), K3.28(109), and D(275) were neutralized during this initial minimization. C alpha atom restraints (100 kcal/mol) for all C alpha atoms were applied, and the full bundle was energy minimized until an energy gradient of 0.1 kcal/mol was reached. The C alpha atom restraints were then reduced in steps to 50 kcal/mol, 10 kcal/mol, and 0 kcal/mol (no restraints) until an energy gradient of 0.1 kcal/mol was achieved at each step. To allow the loops to adjust in their proper environment, atoms of the TMH regions were frozen, and the bundle was re-minimized in water solvent to 0.1 kcal/mol gradient with loop residues fully charged.
Acknowledgments
We are indebted to Robert Picone and Pusheng Fan for assistance in generating preliminary cannabinoid-receptor binding data. This work has been supported by grants DA03934 and DA000489 (to P.R.) and DA9158 and DA3801 (to A.M.) from the National Institutes of Health, National Institute on Drug Abuse.
References
- Admiraal SJ, Meyer P, Schneider B, Deville-Bonne D, Janin J, Herschlag D. Chemical rescue of phosphoryl transfer in a cavity mutant: a cautionary tale for site-directed mutagenesis. Biochemistry. 2001;40:403–413. doi: 10.1021/bi002472w. [DOI] [PubMed] [Google Scholar]
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struh K. Current Protocols in Molecular Biology. New Jersey: J. Wiley and Sons; 2006. [Google Scholar]
- Ballesteros JA, Weinstein H. Integrated Methods for the Construction of Three Dimensional Models and Computational Probing of Structure Function Relations in G Protein-Coupled Receptors. In: Conn PM, Sealfon SM, editors. In Methods in Neuroscience. Vol. 25. San Diego: Academic Press; 1995. pp. 366–428. [Google Scholar]
- Barnett-Norris J, Hurst DP, Buehner K, Ballesteros JA, Guarnieri F, Reggio PH. Agonist alkyl tail interaction with cannabinoid CB1 receptor V6.43/I6.46 groove induces a helix 6 active conformation. Int J Quantum Chem. 2002;88:76–86. [Google Scholar]
- Barnett-Norris J, Hurst DP, Reggio PH. The influence of cannabinoid receptor second extracellular loop conformation on the binding of CP55940. In 2003 Symposium on the Cannabinoids; Cornwall, Ontario: International Cannabinoid Research Society; 2003. p. 79. [Google Scholar]
- Bouaboula M, Poinot-Chazel C, Marchand J, Canat X, Bourrie B, Rinaldi-Carmona M, Calandra B, Le-Fur G, Casellas P. Signaling pathway associated with stimulation of CB2 peripheral cannabinoid receptor: involvement of both mitogen-activated protein kinase and induction of Krox-24 expression. Eur J Biochem. 1996;237:704–711. doi: 10.1111/j.1432-1033.1996.0704p.x. [DOI] [PubMed] [Google Scholar]
- Bramblett RD, Panu AM, Ballesteros JA, Reggio PH. Construction of a 3D model of the cannabinoid CB1 receptor: determination of helix ends and helix orientation. Life Sci. 1995;56:1971–1982. doi: 10.1016/0024-3205(95)00178-9. [DOI] [PubMed] [Google Scholar]
- Charalambous A, Yan G, Houston DB, Howlett AC, Compton DR, Martin BR, Makriyannis A. 5′Azido-delta-8-THC: a novel photo-affinity label for the cannabinoid receptor. J Med Chem. 1992;35:3076–3079. doi: 10.1021/jm00094a023. [DOI] [PubMed] [Google Scholar]
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin CN, Lucas-Lenard J, Abadji V, Kendall DA. Ligand binding and modulation of cyclic AMP levels depend on the chemical nature of residue 192 of the human cannabinoid receptor 1. J Neurochem. 1998;70:366–373. doi: 10.1046/j.1471-4159.1998.70010366.x. [DOI] [PubMed] [Google Scholar]
- De-Lean A, Stadel JM, Lefkowitz RJ. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled β-adrenergic receptor. J Biol Chem. 1980;255:7108–7117. [PubMed] [Google Scholar]
- Deng H, Gifford AN, Zvonok AM, Cui G, Li X, Fan P, Deschamps JR, Flippen-Anderson JL, Gatley SJ, Makriyannis A. Potent cannabinergic indole analogues as radioiodinatable brain imaging agents for the CB1 cannabinoid receptor. J Med Chem. 2005;48:6386–6392. doi: 10.1021/jm050135l. [DOI] [PubMed] [Google Scholar]
- Dhawan J, Deng H, Gatley SJ, Makriyannis A, Akinfeleye T, Bruneus M, Dimaio AA, Gifford AN. Evaluation of the in vivo receptor occupancy for the behavioral effects of cannabinoids using a radiolabeled cannabinoid receptor agonist, R-[125/131I]AM2233. Synapse. 2006;60:93–101. doi: 10.1002/syn.20277. [DOI] [PubMed] [Google Scholar]
- Farrens DL, Altenbach C, Yang K, Hubbell WL, Khorana HG. Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science. 1996;274:768–770. doi: 10.1126/science.274.5288.768. [DOI] [PubMed] [Google Scholar]
- Fride E, Foox A, Rosenberg E, Faigenboim M, Cohen V, Barda L, Blau H, Mechoulam R. Milk intake and survival in newborn cannabinoid CB1 receptor knockout mice: evidence for a “CB3” receptor. Eur J Pharmacol. 2003;461:27–34. doi: 10.1016/s0014-2999(03)01295-0. [DOI] [PubMed] [Google Scholar]
- Gebremedhin D, Lange AR, Campbell WB, Hillard CJ, Harder DR. Cannabinoid CB1 receptor of cat cerebral arterial muscle functions to inhibit L-type Ca2+ channel current. Am J Physiol. 1999;276:H2085–H2093. doi: 10.1152/ajpheart.1999.276.6.H2085. [DOI] [PubMed] [Google Scholar]
- Gouldson P, Calandra B, Legoux P, Kerneis A, Rinaldi-Carmona M, Barth F, Le Fur G, Ferrara P, Shire D. Mutational analysis and molecular modeling of the antagonist SR 144528 binding site on the human cannabinoid CB(2) receptor. Eur J Pharmacol. 2000;401:17–25. doi: 10.1016/s0014-2999(00)00439-8. [DOI] [PubMed] [Google Scholar]
- Guo Y, Abadji V, Morse KL, Fournier DJ, Li X, Makriyannis A. (−)-11-Hydroxy-7′-isothiocyanato-1′,1′-dimethylheptyl-delta-8-THC: a novel, high-affinity irreversible probe for the cannabinoid receptor in brain. J Med Chem. 1994;37:3867–3870. doi: 10.1021/jm00049a002. [DOI] [PubMed] [Google Scholar]
- Hassan SA, Mehler EL, Weinstein H. Structure calculation of protein segments connecting domains with defined secondary structure: A simulated annealing Monte Carlo combined with biased scaled collective variables technique. In: Gan HH, editor. Computational Methods for Macromolecules: Challenges and Applications. Vol. 24. New York: Springer-Verlag; 2002. pp. 197–231. [Google Scholar]
- Hurst DP, Lynch DL, Barnett-Norris J, Hyatt SM, Seltzman HH, Zhong M, Song ZH, Nie J, Lewis D, Reggio PH. N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyraxole-3-carboxamide (SR141716A) interaction with LYS 3.28(192) is crucial for its inverse agonism at the cannabinoid CB1 receptor. Mol Pharmacol. 2002;62:1274–1287. doi: 10.1124/mol.62.6.1274. [DOI] [PubMed] [Google Scholar]
- Jagerovic N, Fernandez-Fernandez C, Goya P. CB1 cannabinoid antagonists: structure-activity relationships and potential therapeutic applications. Curr Top Med Chem. 2008;8:205–230. doi: 10.2174/156802608783498050. [DOI] [PubMed] [Google Scholar]
- Javitch JA, Fu D, Liapakis G, Chen J. Constitutive activation of the β2 adrenergic receptor alters the orientation of its sixth membrane-spanning segment. J Biol Chem. 1997;272:18546–18549. doi: 10.1074/jbc.272.30.18546. [DOI] [PubMed] [Google Scholar]
- Jensen AD, Guarnieri F, Rasmussen SG, Asmar F, Ballesteros JA, Gether U. Agonist-induced conformational changes at the cytoplasmic side of transmembrane segment 6 in the β2-adrenergic receptor mapped by site-selective fluorescent labeling. J Biol Chem. 2001;276:9279–9290. doi: 10.1074/jbc.M004871200. [DOI] [PubMed] [Google Scholar]
- Lan R, Liu Q, Fan P, Lin S, Fernendo SR, McCallion D, Pertwee R, Makriyannis A. Structure-activity relationships of pyrazole derivatives as cannabinoid receptor antagonists. J Med Chem. 1999;42:769–776. doi: 10.1021/jm980363y. [DOI] [PubMed] [Google Scholar]
- Lagerström MC, Schiöth HB. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov. 2008;7:339–357. doi: 10.1038/nrd2518. [DOI] [PubMed] [Google Scholar]
- Leff P. The two-state model of receptor activation. Trends Pharmacol Sci. 1995;16:89–97. doi: 10.1016/s0165-6147(00)88989-0. [DOI] [PubMed] [Google Scholar]
- Lin SW, Sakmar TP. Specific tryptophan UV-absorbance changes are probes of the transition of rhodopsin to its active state. Biochemistry. 1996;35:11149–11159. doi: 10.1021/bi960858u. [DOI] [PubMed] [Google Scholar]
- Mackie K, Lai Y, Westenbroek R, Mitchell R. Cannabinoids activate an inwardly rectifying potassium conductance and inhibit Q-type calcium currents in AtT20 cells transfected with rat brain cannabinoid receptor. J Neurosci. 1995;15:6552–6561. doi: 10.1523/JNEUROSCI.15-10-06552.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malan TP, Jr, Ibrahim MM, Lai J, Vanderah TW, Makriyannis A, Porreca F. CB2 cannabinoid receptor agonists: pain relief without psychoactive effects? Curr Opin Pharmacol. 2003;3:62–67. doi: 10.1016/s1471-4892(02)00004-8. [DOI] [PubMed] [Google Scholar]
- Marriott KS, Huffmann JW. Recent advances in the development of selective ligands for the cannabinoid CB(2) receptor. Curr Top Med Chem. 2008;8:187–204. doi: 10.2174/156802608783498014. [DOI] [PubMed] [Google Scholar]
- Mavromoustakos T, Yang DP, Broderick W, Fournier D, Makriyannis A. Small angle X-ray diffraction studies on the topography of cannabinoids in synaptic plasma membranes. Pharmacol Biochem Behav. 1991;40:547–552. doi: 10.1016/0091-3057(91)90361-5. [DOI] [PubMed] [Google Scholar]
- Mavromoustakos T, Yang DP, Makriyannis A. Small angle X-ray diffraction and differential scanning calorimetric studies on O-methyl(−)-delta-8-tetrahydrocannabinol and its 5′ iodinated derivative in membrane bilayers. Biochim Biophys Acta. 1995;1237:183–188. doi: 10.1016/0005-2736(95)00101-8. [DOI] [PubMed] [Google Scholar]
- Monory K, Tzavara ET, Lexime J, Ledent C, Parmentier M, Borsodi A, Hanoune J. Novel, not adenyl cyclase-coupled cannabinoid binding site in cerebellum of mice. Biochem Biophys Res Commun. 2002;292:231–235. doi: 10.1006/bbrc.2002.6635. [DOI] [PubMed] [Google Scholar]
- Morse KL, Fournier DJ, Li X, Grzybowska J, Makriyannis A. A novel electrophilic high affinity irrreversible probe for the cannabinoid receptor. Life Sci. 1995;56:1957–1962. doi: 10.1016/0024-3205(95)00176-7. [DOI] [PubMed] [Google Scholar]
- Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, et al. Crystal structure of rhodopsin: a G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- Palmer SL, Thakur GA, Makriyannis A. Cannabinegic ligands. Chem Phys Lipids. 2002;121:3–19. doi: 10.1016/s0009-3084(02)00143-3. [DOI] [PubMed] [Google Scholar]
- Pan X, Ikeda SR, Lewis DL. Rat brain cannabinoid receptor modulates N-type Ca2+ channels in a neuronal expression system. Mol Pharmacol. 1996;49:707–714. [PubMed] [Google Scholar]
- Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP. Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature. 2008;454:183–187. doi: 10.1038/nature07063. [DOI] [PubMed] [Google Scholar]
- Peracchi A. Enzyme catalysis: removing chemically “essential” residues by site-directed mutagenesis. Trends Biochem Sci. 2001;26:497–503. doi: 10.1016/s0968-0004(01)01911-9. [DOI] [PubMed] [Google Scholar]
- Picone RP, Fournier DJ, Makriyannis A. Ligand based structural studies of the CB1 cannabinoid receptor. J Pept Res. 2002;60:348–356. doi: 10.1034/j.1399-3011.2002.21069.x. [DOI] [PubMed] [Google Scholar]
- Picone RP, Khanolkar AD, Xu W, Ayotte LA, Thakur GA, Hurst DP, Abood ME, Reggio PH, Fournier DJ, Makriyannis A. (−)-7′-Isothiocyanato-11-hydroxy-1′,1′-dimethylheptylhexahydrocannabinol (AM841), a high-affinity electrophilic ligand, interacts covalently with a cysteine in helix six and activates the CB1 cannabinoid receptor. Mol Pharmacol. 2005;68:1623–1635. doi: 10.1124/mol.105.014407. [DOI] [PubMed] [Google Scholar]
- Poso A, Huffman JW. Targeting the cannabinoid CB2 receptor: modeling and structural determinants of CB2 selective ligands. Br J Pharmacol. 2008;153:335–346. doi: 10.1038/sj.bjp.0707567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raitio KH, Salo OM, Nevalainen T, Poso A, Jarvinen T. Targeting the cannabinoid CB2 receptor: mutations, modeling and development of CB2 selective ligands. Curr Med Chem. 2005;12:1217–1237. doi: 10.2174/0929867053764617. [DOI] [PubMed] [Google Scholar]
- Reggio PH. Computational methods in drug design: modeling G protein-coupled receptor monomers, dimers, and oligomers. AAPS J. 2006;8:E322–E336. doi: 10.1007/BF02854903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reggio PH, Panu AM, Miles S. Characterization of a region of steric interference at the cannabinoid receptor using the active analog approach. J Med Chem. 1993;36:1761–1771. doi: 10.1021/jm00064a010. [DOI] [PubMed] [Google Scholar]
- Rhee MH. Functional role of serine residues of transmembrane dopamin VII in signal transduction of CB2 cannabinoid receptor. J Vet Sci. 2002;3:185–191. [PubMed] [Google Scholar]
- Rhee MH, Kim SK. SR144528 as inverse agonist of CB2 cannabinoid receptor. J Vet Sci. 2002;3:179–184. [PubMed] [Google Scholar]
- Rhee MH, Bayewitch M, Avidor-Reiss T, Levy R, Vogel Z. Cannabinoid receptor activation differentially regulates the various adenylyl cyclase isoenzymes. J Neurochem. 1998;71:1525–1534. doi: 10.1046/j.1471-4159.1998.71041525.x. [DOI] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modeling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Samama P, Cotecchia S, Costa T, Lefkowitz RJ. A mutation-induced activated state of the β2-adrenergic receptor. Extending the ternary complex model. J Biol Chem. 1993;268:4625–4636. [PubMed] [Google Scholar]
- Shen CP, Xiao JC, Armstrong H, Hagmann W, Fong TM. F200A substitution in the third transmembrane helix of human cannabinoid CB1 receptor converts AM2233 from receptor agonist to inverse agonist. Eur J Pharmacol. 2006;531:41–46. doi: 10.1016/j.ejphar.2005.12.026. [DOI] [PubMed] [Google Scholar]
- Shi L, Liapakis G, Xu R, Guarnieri F, Ballesteros JA, Javitch JA. β2 adrenergic receptor activation. Modulation of the proline kink in transmembrane 6 by a rotamer toggle switch. J Biol Chem. 2002;277:40989–40996. doi: 10.1074/jbc.M206801200. [DOI] [PubMed] [Google Scholar]
- Shire D, Calandra B, Delpech M, Dumont X, Kaghad M, Le Fur G, Caput D, Ferrara P. Structural features of the central cannabinoid CB1 receptor involved in binding of the specific CB1 antagonist SR 141716A. J Biol Chem. 1996;271:6941–6946. doi: 10.1074/jbc.271.12.6941. [DOI] [PubMed] [Google Scholar]
- Song ZH, Bonner TI. A lysine residue of the cannabinoid receptor is critical for receptor recognition by several agonists but not WIN55212-2. Mol Pharmacol. 1996;49:891–896. [PubMed] [Google Scholar]
- Tahtaoui C, Balestre MN, Klotz P, Rognan D, Barberis C, Mouillac B, Hibert M. Identification of the binding sites of the SR49059 nonpeptide antagonist into the V1a vasopressin receptor using sulfhydryl-reactive ligands and cysteine mutants as chemical sensors. J Biol Chem. 2003;278:40010–40019. doi: 10.1074/jbc.M301128200. [DOI] [PubMed] [Google Scholar]
- Tao Q, Abood ME. Mutation of a highly conserved aspartate residue in the second transmembrane domain of the cannabinoid receptors, CB1 and CB2, disrupts G-protein coupling. J Pharmacol Exp Ther. 1998;285:651–658. [PubMed] [Google Scholar]
- Tao Q, McAllister SD, Andreassi J, Nowell KW, Cabral GA, Hurst DP, Bachtel K, Ekman MC, Reggio PH, Abood ME. Role of a conserved lysine residue in the peripheral cannabinoid receptor (CB2): evidence for subtype specificity. Mol Pharmacol. 1999;55:605–613. [PubMed] [Google Scholar]
- Tian X, Guo J, Yao F, Yang DP, Makriyannis A. The conformation, location, and dynamic properties of the endocannabinoid ligand anandamide in a membrane bilayer. J Biol Chem. 2005;280:29788–29795. doi: 10.1074/jbc.M502925200. [DOI] [PubMed] [Google Scholar]
- Vemuri VK, Janero DR, Makriyannis A. Pharmacotherapeutic targeting of the endocannabinoid signaling system: drugs for obesity and the metabolic syndrome. Physiol Behav. 2008;93:671–686. doi: 10.1016/j.physbeh.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Handerson R, Leslie AGW, Tate CG, Schertler GFX. Structure of a β1-adrenergic G-protein-coupled receptor. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Filppula S, Mercier R, Yaddanapudi S, Pavlopoulos S, Cai J, Pierce WM, Makriyannis A. Purification and mass spectroscopic analysis of human CB1 cannabinoid recpetor functionality expressed using the baculovirus system. J Pept Res. 2005;66:138–150. doi: 10.1111/j.1399-3011.2005.00283.x. [DOI] [PubMed] [Google Scholar]
- Zhang R, Hurst DP, Barnett-Norris J, Reggio PH, Song ZH. Cysteine 2.59(89) in the second transmembrane domain of human CB2 receptor is accessible within the ligand binding crevice: evidence for possible CB2 deviation from a rhodopsin template. Mol Pharmacol. 2005;68:69–83. doi: 10.1124/mol.104.007823. [DOI] [PubMed] [Google Scholar]
- Zvonok N, Pandarinathan L, Williams J, Johnston M, Karageorgos I, Janero DR, Krishnan SC, Makriyannis A. Covalent inhibitors of human monoacylglycerol lipase: ligand-assisted characterization of the catalytic site by mass spectrometry and mutational analysis. Chem Biol. 2008;15:854–862. doi: 10.1016/j.chembiol.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]