

Abstract
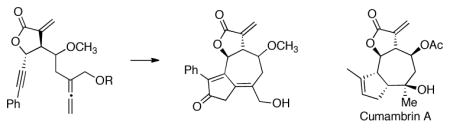
Syntheses of two 6,12-guaianolide analogs are reported within. The scope of the tandem allylboration/lactonization chemistry is expanded to provide a functionalized alleneyne-containing α-methylene butyrolactone that undergoes a Rh(I)-catalyzed cyclocarbonylation reaction to afford a 5-7-5 ring system. The resulting cycloadducts bear a structural resemblance to other NF-κB inhibitors such as cumambrin A and indeed were shown to inhibit NF-κB signaling and cancer cell growth.
Guaianolides are the most abundant group of sesquiterpene lactones (SLs), possessing a privileged natural product status and a wide range of biological activities.1 Yet there is only one guaianolide, arglabin, available as a marketed drug, constituting one of only twenty-four natural products approved for therapeutic use between 1974 and 2006. 2, 3 Reasons for the slow realization of their therapeutic potential include poor bioavailability due to high plasma protein interactions, poor toxicological profiles, and hydrophobicity. 4 Moreover, the biological activity of these compounds is attributed to covalent bonding to the α,β-unsaturated carbonyl groups, the same functionality responsible for their toxicity.5 Despite potential toxicities, three of the top-ten drugs in the US, and one third of all enzyme targets for which there is an FDA approved inhibitor, operate by a covalent mechanism of action.6 These proven biomedical applications, combined with the finding that irreversible binding may be an important factor against drug resistance, have led to a reinvestment of the pharmaceutical community in covalent drugs.6,7
Natural products, such as guaianolides can serve as excellent leads for drug development, but molecular complexity can pose formidable synthetic challenges.8 To date, most synthetic approaches towards 6,12-guaianolides can be characterized as target-oriented synthesis (TOS) strategies that have not been explored for analog preparation of these highly oxygenated skeletons;9 the synthesis of thapsigargin (2) being one exception (Figure 1).10 Oxidation level [O] constitutes one measure of molecular complexity which can be directly correlated with synthetic accessibility when performing a TOS.11 For example, the synthetic steps required to prepare arglabin (8) and chinensiolide (7) where [O] = 4, were fewer than twenty. In contrast, more than forty steps were required to complete the synthesis of thapsigargin (2).10 Given the highly oxidized nature of 6,12-guaianolides, a synthetic approach employing the principles of redox economy would greatly alleviate the synthetic challenges associated with the class of compounds.11
Figure 1.

Examples of highly oxidized 6,12-guaianolides
Described within is an eleven-step synthesis of two guaianolide analogs with oxidation levels equivalent to thapsigargin and eupatochinilide VI; concise syntheses that were realized by limiting the number of redox adjustments in the synthetic sequence. We have previously demonstrated the advantages of early-stage incorporation of an α-methylene butyrolactone on the Rh(I)-catalyzed allenic Pauson-Khand reaction (APKR).12 This study expands on the scope of the APKR by incorporating additional functionality into the alleneyne precursor 10. Furthermore, bioactivity studies provides support for the preparation of non-naturally occuring guaianolide analogs such as 11 (Scheme 1).13
Scheme 1.
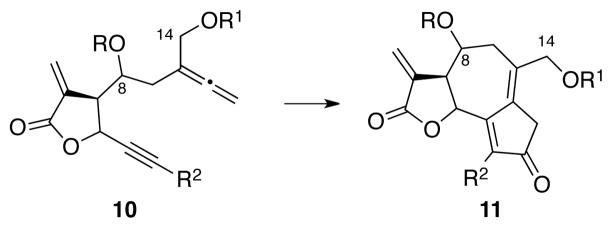
An APKR approach to highly oxidized guaianolides
Synthesis of alleneyne 10 was envisioned using the allylboration/lactonization chemistry developed by Hall and previously used by us to access less functionalized alleneyne precursors. Because there is only one report with functionality at a propargylic position, a model system was first examined.14 Compounds 12a-d were prepared and converted to the corresponding carbomethoxy allylboronates 13a-d by addition of DIBAL and subsequent trapping of the intermediate aluminum species with ClCH2BPin (Scheme 2). CuI was not required for the 1,4-addition reaction of hydride to the ynoate, possibly because the ether adjacent to the alkyne directs the addition. Moreover, Z:E ratios of allylboronates 13a-d were dependent upon the protecting group. For example, the reaction of 12a-b, with silyl protecting groups afforded 13a-b in Z:E ratios of 2–3:1. Whereas, reaction of methyl- and MOM-protected ethers, 12c and 12d, afforded the allylboronates 13c and 13d with Z:E ratios of 9:1 and 4:1, respectively. The stereochemical determining step is the addition of the electrophile to one face over the other of the intermediate allenoate 14. We propose that the Z:E ratios correlate with the degree of chelation of the respective ether groups with the aluminum species of the allenoate, where more chelation directs electrophilic addition to the α-face.15
Scheme 2.

Generation of the allylboronates, Z:E ratios
Next, the lactonization step was examined on these model systems (Scheme 3). Unfortunately, the E:Z isomers of allylboronate 13 were not readily separated by column chromatography so they were taken on to the lactonization step as a mixture. Reaction of allylboronates 13a or 13b, with either a TBS or TBDPS protecting group with boron trifluoride etherate, triflic acid or scandium triflate gave only decomposition. However, reaction of allylboronate 13c with either triflic acid or scandium triflate gave ~75% yield of 15c in a 3–4:1 trans:cis lactone ratio. For the MOM-protected ether 13d, purely thermal conditions gave the best results, whereby heating 13d and phenylpropiolaldehyde to 90 °C for 48 h gave an 82% yield of 15d as a trans:cis ratio of 2.7:1; acidic conditions led to decomposition of 13d. Next, the feasibility of allylation/lactonization chemistry was tested on a more functionalized substrate.
Scheme 3.
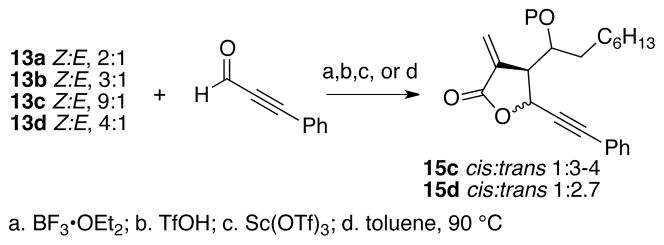
A model system for the lactonization protocol
To this end, allenyl ester 16 is obtained in 82% yield from the mono-protected butynediol using a Johnson-Claisen rearrangement (Scheme 4). Ester 16 is reacted with methoxymethylamine hydrochloride and isopropyl magnesium chloride to afford the corresponding Weinreb amide in 84% yield, which is taken on to alkynone 17 by reaction with ethynyl magnesium bromide. Reduction of the carbonyl of ynone 17 is accomplished with lithium aluminum hydride in 98% yield. The propargylic alcohol is not purified but taken directly on to the corresponding methyl ether in 90% yield. Deprotonation of the terminus of the alkyne with n-butyllithium followed by addition of chloromethylester gives the alkynoate 18 in 84% yield. Reaction of alkynoate 18 with Dibal-H, CuI, MeLi, and ClCH2BPin afforded allylboronate 19 in 78% yield with a Z:E ratio of 1.2:1. Performing this reaction in the absence of MeLi and CuI gave allylboronate 19 in 80% yield with a Z:E ratio of 2.2:1 with more byproduct contamination. The Z:E isomers were not separated, but taken on as a mixture to the allylboration/lactonization step. Reaction of 19 with phenylpropiolaldehyde using the acidic conditions described above all gave decomposition of the allylboronate. Purely thermal conditions in toluene afforded starting material at 50 °C and decomposition at 90 °C. Interestingly, heating allylboronate 19 with 3-phenylpropiolaldehyde in chloroform for 7 days afforded some of the desired lactones 20a-b, but the bulk of the material consisted of intermediate hydroxy esters.16 This complex mixture was reacted with PTSA to afford lactone trans-20a as 2:1 mixture of diastereomers in 40% yield. Uncyclized material was recovered after chromatography and reacted with NaH to afford lactone cis-20b as a single diastereomer in 14% yield. The cis- and trans-lactones were taken on independently to the Rh(I)-catalyzed cyclocarbonylation reaction. Reaction of 20a with rhodium biscarbonyl chloride dimer in toluene at 90 °C afforded the cyclocarbonylation product 21a as a mixture of diastereomers. The tert-butyldiphenylsilyl (TBDPS) group of 21a was removed using triethylamine hydrogen fluoride to give 22a in 64% yield for the two steps. Reaction of the cis-lactone 20b to the same sequence afforded 22b in 37% yield (two steps).
Scheme 4.

Synthesis of 6,12-guaianolide analogs 22a-b
Natural products bearing α-methylene butyrolactones are well-established bioactive molecules. Inhibition of the NF-κB signaling pathway, a hingepoint for the activation of the cellular inflammatory response, has been demonstrated by molecules of this class.17 In addition, it has been shown that a natural product analog, dimethylamino-parthenolide (DMAPT; LC-1), has the ability to simultaneously knockdown NF-κB levels and activate the p53 pathway, thus promoting the apoptosis of cancer cells.18 Inspired by these previous studies, we evaluated 22a-b for inhibition of induced NF-κB activity in cell cultures. A549 cells bearing a stably transfected NF-κB reporter construct were treated with each compound. Activation of NF-κB signaling yields an increase in reporter luminescence that is diminished in the presence of NF-κB inhibitors.19 Results from our study were benchmarked against parthenolide (PTL), a known NF-κB inhibitor bearing an α-methylene butyrolactone. Trans-22a and cis-22b were equipotent inhibitors in this assay, diminishing induced NF-κB activity to non-induced levels at 20 μM. Both analogs resulted in substantial decreases in NF-κB activity, with 57% (22a) and 59% (22b) residual activity measured at 10 μM. PTL was found to be slightly more potent, reducing NF-κB levels to 53% residual activity at 10 μM.
Inhibition of NF-κB signaling is an emerging strategy for developing novel anticancer agents. 20 Additionally, many α-methylene butyrolactone-containing natural products have documented antiproliferative activities.13 We evaluated 22a-b for growth inhibitory activity against a panel of cancerous and non-cancerous cell lines. Both compounds were benchmarked against PTL and clinically used drugs gemcitabine and doxorubicin (Figures S1 and S2). Antiproliferative data for PTL has been previously reported for HL-60, HeLa, U-87 MG, and Vero and our data is in close agreement to previous reports. 21 In general, 22a and 22b were similarly active when compared to each other, and slightly less active than PTL. Notable exceptions to this trend include HeLa breast carcinoma and HL-60 leukemia cells, in which 22a was approximately 2-fold more active than PTL (Table 1). Conversely, cis-22b was significantly more active than trans-22a in U-87 MG brain tumor cells (IC50 = 9.8 μM versus 27.1 μM) and has similar activity to PTL (IC50 = 8.8 μM). Interestingly, 22b (IC50 = 25.4 μM) was significantly more active than both 22a (IC50 = 80.9 μM) and PTL (IC50 = 57.6 μM) against the well-known NCI/ADR-RES cell line, which is a model of drug-resistant ovarian cancer due to overexpression of p-glycoprotein (P-gp) efflux pump. 22 NCI/ADR-RES is resistant to doxorubixin (adriamycin) and gemcitabine (IC50’s > 500 μM, Figure S2). These results suggest that molecules with covalent mechanisms of activity, such as the guaianolide analogs 22a-b, may be valuable scaffolds for targeting drug-resistant cells. Both compounds were screened against the non-cancerous cell line Vero and moderate toxicity was observed for all α-methylene butyrolactone analogs.
Table 1.
Antiproliferative activities of 22a-b and parthenolide (PTL)a
| compound | DU-145 | HeLa | HL-60 | U-87 MG | NCI/ADR-RES | Vero |
|---|---|---|---|---|---|---|
| 22a | 29.1 ± 4.7 | 20.3 ± 6.0 | 5.5 ± 0.4 | 27.1 ± 4.8 | 80.9 ± 24.0 | 32.2 ± 7.0 |
| 22b | 21.6 ± 1.9 | 39.7 ± 16.4 | 7.8 ± 2.3 | 9.8 ± 1.4 | 25.4 ± 1.0 | 30.1 ± 5.5 |
| PTL | 8.9 ± 4.6 | 45.1 ± 3.7 | 9.3 ± 3.8 | 8.8 ± 2.1 | 57.6 ± 8.9 | 22.4 ± 1.5 |
Compounds were dosed to cells and incubated for 48 h. Viability was measured by Alamar Blue staining. Mean IC50 values ± S.D. (μM) are shown.
In conclusion, the scope of the APKR has been extended to the preparation of highly oxygenated guaianolide analogs, 22a-b. Bioactivity data supports the potential of this class of compounds as regulators of NF-κB and cell proliferation, and further validates the medicinal relevancy of this region of chemical space. Our ability to modify the structure of these compounds de novo enables the optimization of analog solubility and pharmacokinetic properties for advanced biological applications. Furthermore, our strategy provides ready access to uniquely functionalized 6,12-guaianolide analogs with activities on par with the most studied member of the SLs, parthenolide. Studies are underway to establish structure activity relationships and the mechanism by which compounds 22a-b inhibit NF-κB, in addition to benchmarking their thiol reactivity.
Supplementary Material
Figure 2.

NF-κB luciferase reporter assay in A549 cells. Compounds 22a-b were dosed at 20, 10, and 1 μM and PTL was dosed at 10 and 1 μM. Cells were induced with TNF-α (15 ng/mL) 30 min after molecule treatment, except NI control. Shown is the mean of triplicate data and error bars represent propagated standard deviation. NI = non-induced, I = induced.
Acknowledgments
We thank The NIH for financial support (NIGMS P50GM067082 and GM54161). DAH acknowledges Hyundai Hope on Wheels (Hope Grant award) and The V Foundation for Cancer Research (V Scholar award to DAH) for financial support.
Footnotes
Supporting Information Available Detailed procedures and data for all compounds in Schemes 2–4, biochemical assays and Figures S1–S3 are available. This material is available free of charge via the Internet at http://pubs.acs.org.
Contributor Information
Daniel A. Harki, Email: daharki@umn.edu.
Kay M. Brummond, Email: kbrummon@pitt.edu.
References
- 1.Drew DP, Krichau N, Reichwald K, Simonsen HT. Phytochem Rev. 2009;8:581–599. [Google Scholar]
- 2.(a) Newman DJ, Cragg GM. J Nat Prod. 2012;75:311–335. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Welsch ME, Newman DJ, Cragg GM. J Nat Prod. 2007;70:461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]
- 3.Science and Technology in Kazakhstan: Current Status and Future Prospects, 2006. National Research Council, Office for Central Europe and Eurasia; 2006. [Google Scholar]
- 4.Ghantous A, Gali-Muhtasib H, Vuorela H, Saliba NA, Darwiche N. Drug Discov Today. 2010;15:668–678. doi: 10.1016/j.drudis.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 5.Rishton GM. Drug Discov Today. 1997;2:382–384. doi: 10.1016/s1359644602025722. [DOI] [PubMed] [Google Scholar]; Hogenauer K, Simic O, Antonello A, Hunger U, Smith MD, Ley SV. Chem Eur J. 2007;13:5688–5712. doi: 10.1002/chem.200700302. [DOI] [PubMed] [Google Scholar]; (11) Burns NZ, Baran PS, Hoffmann RW. Angew Chem Int Ed. 2009;48:2854–2867. doi: 10.1002/anie.200806086. [DOI] [PubMed] [Google Scholar]; (12) Grillet F, Huang C, Brummond KM. Org Lett. 2011;13:6304–6307. doi: 10.1021/ol2028515. [DOI] [PMC free article] [PubMed] [Google Scholar]; (13) Merfort I. Curr Drug Targets. 2011;12:1560–1573. doi: 10.2174/138945011798109437. [DOI] [PubMed] [Google Scholar]
- 6.(a) Potashman MH, Duggan ME. J Med Chem. 2009;52:1231–1246. doi: 10.1021/jm8008597. [DOI] [PubMed] [Google Scholar]; (b) Singh J, Petter RC, Kluge AF. Curr Opin Chem Biol. 2010;14:475–480. doi: 10.1016/j.cbpa.2010.06.168. [DOI] [PubMed] [Google Scholar]; (c) Singh J, Petter RC, Baillie TA, Whitty SA. Nat Rev Drug Discov. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]; (d) Liu Q, Sabnis Y, Zhao Z, Zang T, Buhrlage SJ, Jones LH, Gray NS. Chem Biol. 2013;20:146–159. doi: 10.1016/j.chembiol.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amslinger S. ChemMedChem. 2010;5:351–356. doi: 10.1002/cmdc.200900499. [DOI] [PubMed] [Google Scholar]
- 8.Rosen J, Gottfries J, Muresan S, Backlund A, Oprea TI. J Med Chem. 2009;52:1953–1962. doi: 10.1021/jm801514w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schall A, Reiser O. Eur J Org Chem. 2008:2353–2364. [Google Scholar]
- 10.(a) Ley SV, Antonello A, Balskus EP, Booth DT, Christensen SB, Cleator E, Gold H, Hogenauer K, Hunger U, Myeres RM, Oliver SF, Simic O, Smith MD, Sohoel H, Woolford AJA. Proc Natl Acad Sci USA. 2004;101:12073–12078. doi: 10.1073/pnas.0403300101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Andrews SA, Ball M, Wierschem F, Cleator E, Oliver S, Hogenauer K, Simic O, Antonello A, Hunger U, Smith MD, Ley SV. Chem Eur J. 2007;13:5688–5712. doi: 10.1002/chem.200700302. [DOI] [PubMed] [Google Scholar]
- 11.Burns NZ, Baran PS, Hoffmann RW. Angew Chem Int Ed. 2009;48:2854–2867. doi: 10.1002/anie.200806086. [DOI] [PubMed] [Google Scholar]
- 12.Grillet F, Huang C, Brummond KM. Org Lett. 2011;13:6304–6307. doi: 10.1021/ol2028515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Merfort I. Curr Drug Targets. 2011;12:1560–1573. doi: 10.2174/138945011798109437. [DOI] [PubMed] [Google Scholar]
- 14.Kennedy JWJ, Hall DG. J Org Chem. 2004;69:4412–4428. doi: 10.1021/jo049773m. [DOI] [PubMed] [Google Scholar]
- 15.Evans DA, Allison BD, Yang MG, Masse CE. J Am Chem Soc. 2001;123:10840–10852. doi: 10.1021/ja011337j. [DOI] [PubMed] [Google Scholar]
- 16.Huang Y, Rawal VH. J Am Chem Soc. 2002;124:9662–63. doi: 10.1021/ja0267627. [DOI] [PubMed] [Google Scholar]
- 17.Rungeler P, Castro V, Mora G, Goren N, Vichnewski W, Pahl HL, Merfort I, Schmidt TJ. Bioorg Med Chem. 1999;7:2343–52. doi: 10.1016/s0968-0896(99)00195-9. [DOI] [PubMed] [Google Scholar]
- 18.(a) Guzman M, Rossi R, Neelakantan S, Li X, Corbett C, Hassane D, Becker M, Bennett J, Sullivan E, Lachowicz J, Vaughan A, Sweeney C, Matthews W, Carrol M, Liesveld J, Crooks P, Jordan C. Blood. 2007;110:4427–4435. doi: 10.1182/blood-2007-05-090621. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dey A, Tergaonkar V, Lane DP. Nat Rev Drug Discov. 2008;7:1031–40. doi: 10.1038/nrd2759. [DOI] [PubMed] [Google Scholar]
- 19.Hexum JK, Tello-Aburto R, Struntz NB, Harned AM, Harki DA. ACS Med Chem Lett. 2012;3:459–464. doi: 10.1021/ml300034a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Karin M. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]; (b) Karin M, Greten FR. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]; (c) Karin M, Yamamoto Y, Wang M. Nat Rev Drug Discov. 2004;3:17–26. doi: 10.1038/nrd1279. [DOI] [PubMed] [Google Scholar]; (d) Naugler WE, Karin M. Curr Opin Gen Dev. 2008;18:19–26. doi: 10.1016/j.gde.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.(a) Han C, Barrios FJ, Riofski MV, Colby DA. J Org Chem. 2009;74:7176–23. doi: 10.1021/jo901533e. [DOI] [PubMed] [Google Scholar]; (b) Collu F, Bonsignore L, Casu M, Floris C, Gertsch J, Cottiglia F. Bioorg Med Chem Lett. 2008;18:1559–62. doi: 10.1016/j.bmcl.2008.01.078. [DOI] [PubMed] [Google Scholar]; (c) Zanotto-Filho A, Braganhol E, Schroder R, de Souza LH, Dalmolin RJ, Pasquali MA, Gelain DP, Battastini AM, Moreira JC. Biochem Pharmacol. 2011;81:412–24. doi: 10.1016/j.bcp.2010.10.014. [DOI] [PubMed] [Google Scholar]; (d) Onozato T, Nakamura CV, Cortez DA, Dias Filho BP, Ueda-Nakamura T. Phytother Res. 2009;23:791–6. doi: 10.1002/ptr.2638. [DOI] [PubMed] [Google Scholar]
- 22.(a) Batist G, Tulpule A, Sinha BK, Katki AG, Myers CE, Cowan KH. J Bio Chem. 1986;261:15544–15549. [PubMed] [Google Scholar]; (b) Ke W, Yu P, Wang J, Wang R, Guo C, Zhou L, Li C, Li K. Med Oncol. 2011;28:S135–S141. doi: 10.1007/s12032-010-9747-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.