

Abstract
We evaluated the acute (up to 24 h) pathophysiological response to primary blast using a rat model and helium driven shock tube. The shock tube generates animal loadings with controlled pure primary blast parameters over a wide range and field-relevant conditions. We studied the biomechanical loading with a set of pressure gauges mounted on the surface of the nose, in the cranial space, and in the thoracic cavity of cadaver rats. Anesthetized rats were exposed to a single blast at precisely controlled five peak overpressures over a wide range (130, 190, 230, 250, and 290 kPa). We observed 0% mortality rates in 130 and 230 kPa groups, and 30%, 24%, and 100% mortality rates in 190, 250, and 290 kPa groups, respectively. The body weight loss was statistically significant in 190 and 250 kPa groups 24 h after exposure. The data analysis showed the magnitude of peak-to-peak amplitude of intracranial pressure (ICP) fluctuations correlates well with mortality rates. The ICP oscillations recorded for 190, 250, and 290 kPa are characterized by higher frequency (10–20 kHz) than in other two groups (7–8 kHz). We noted acute bradycardia and lung hemorrhage in all groups of rats subjected to the blast. We established the onset of both corresponds to 110 kPa peak overpressure. The immunostaining against immunoglobulin G (IgG) of brain sections of rats sacrificed 24-h post-exposure indicated the diffuse blood-brain barrier breakdown in the brain parenchyma. At high blast intensities (peak overpressure of 190 kPa or more), the IgG uptake by neurons was evident, but there was no evidence of neurodegeneration after 24 h post-exposure, as indicated by cupric silver staining. We observed that the acute response as well as mortality is a non-linear function over the peak overpressure and impulse ranges explored in this work.
Key words: cellular membrane permeability, lung injury, mortality, pathophysiological response, primary blast, traumatic brain injury
Introduction
Improvised explosive devices are widely used by insurgents, increasing the risk of sustaining blast-induced injury by military personnel and civilians. The blast injury is described by four causative categories.1–3 Primary blast injury is caused by the sharp rising pressure of the shockwave with duration of a few milliseconds, passing through the tissue. Secondary blast injury results from the impact of high-velocity fragments and debris. Tertiary blast injuries originate from the blast wind and the resulting body acceleration, displacement, and impact, and quaternary blast injuries are consequences of the heat and toxic smoke exposure associated with explosive detonations.4
Based on limited available field data, it can be estimated that the effect of pure primary air blast may occur in the range of 2 kg to 100 kg of explosive C4 when the object is in the range of 2 m to 10 m distance. In such a range, the peak overpressure is in the range of 60 kPa to 400 kPa, time duration of 2 to 8 msec, and impulse of 70 to 400 kPa/msec.5 In this article, we are concerned only with the primary blasts when the head and its contents (along with the entire body) is subjected to primary blast loadings. The loading modes and mechanisms of injury are well documented for blunt (tertiary) and ballistic (secondary) impacts. Whether and how the primary blast (pure shockwaves) causes mechanical insult to the brain and triggers initiation of biochemical sequelae resulting in dysfunction is presently not known and is the chief interest of this article.
The primary injury and associated physiological effects gained prominent interest during the aerial bombing of World War II when soldiers sustained shock or death with no external signs of injury.6 The primary blast injury is most significant in air-containing organs: ears,7 lungs,8–11 upper respiratory tract,9 and intestines,12 thanks to direct coupling of stress waves with these organs.13 The advent of advanced personal protective equipment diminished these types of injuries, but resulted in a vastly increased number of reports on blast-induced brain trauma.
The wide prevalence of blast traumatic brain injury (bTBI) in conflicts in Iraq and Afghanistan14–16 is a driving force behind a research effort to characterize biomedical effects of blast waves. When studying the mechanical and physiological effects of shockwaves, it is desirable to produce a shockwave with precisely controlled parameters for a given field explosive condition, specified a priori: the maximum peak overpressure, positive and negative phase duration, and impulse. This level of control is achieved with a compressed gas (helium, nitrogen, or air) driven air shock tube.17 The in vivo models are used frequently in conjunction with shock tubes to simulate field conditions, to establish injury thresholds, and discern pathological mechanisms.18–24
Traditionally, there are two approaches to develop blast exposure animal models using shock-wave generators: (1) the test specimen is secured on the outside, or inside near the exit,6,8,11,20,21,25–31 or (2) placed in the test section located deep inside of the shock tube.32 In a recently published article,33 we demonstrated that very different loading conditions are characteristic for these two configurations. Internal organs of the rat placed inside experienced Friedlander waveform type of loading characteristic of primary blast conditions. On the outside, the blast wave is degraded to sub-sonic jet winds (with relatively high velocity and long duration), the incident pressure is decreased, and these conditions result in acceleration-deceleration type of injury, classified as the tertiary blast injury.33 Moreover, the choice of the location of the test specimen inside of the shock tube is not arbitrary, but governed by the physics of the supersonic shock traveling inside the confined space.5 Only deep inside the shock tube are primary blast loading conditions generated and not elsewhere. Hence, our blast injury animal model accurately replicates the primary blast loading conditions, unaffected by three-dimensional jet winds and other artifacts. This is the key contribution of this work.
This article is focused on characterization of biomechanical response and major selected pathologies associated with primary blast exposure in vivo, in the acute phase (up to 24 h post-exposure). We focused our studies on the effects associated with the positive-pressure phase of the blast wave exclusively, with absent blast wind and no head acceleration. Our long-term goal is to develop a reliable blast injury scale—i.e., to quantify pathologies at systemic, organ, tissue, and cellular levels and relate them to appropriate blast exposure metrics.
The first and most important step to realize this goal in the current work is evaluation of mortality rates in the rodent model at five discrete blast intensities. Concurrently, we have quantified the primary blast biomechanical loading experienced by the body—i.e., we correlated external load (incident and reflected pressures) with biomechanical response (intracranial and intrathoracic pressures). Further, we evaluated the corresponding response at systemic and organ levels (lung injury, heart rate decrease, and body weight [BW] loss) and finally focused on selected brain pathologies (blood-brain barrier [BBB] damage, immunoglobulin G [IgG] extravasation, neurodegeneration).
Methods
Adult 10-week-old male Sprague-Dawley (Charles River Laboratories) rats weighing 320–360 g were used in all the studies. The animals were housed with free access to food and water in a 12-h dark-light cycle at 22°C. All procedures followed the guidelines established in the Guide for the Care and Use of Laboratory Animals and were approved by the University of Nebraska-Lincoln Institutional Animal Care and Use Committee (IACUC) before experiments.
Three studies with separate groups of rats were performed. In the first study, three cadaver rats were used to record the pressure on the surface of the nose (reflected pressure), in the lungs, and in the brain. In the second study, the rats were sacrificed immediately after blast exposure for gross lung pathology and histology evaluation (27 rats). In the third study, monitoring of physiological vital signs such as heart rate, blood oxygen saturation (spO2,) and perfusion index was performed for two periods of 30 min, before and after the blast injury (50 animals). The 14 rats in this group were transcardially perfused 24 h after the blast exposure, and the brain sections were evaluated by histological and immunohistological methods.
Exposure to primary blast
We developed modular, multi-size shock tubes that are capable of reproducing complex shock-wave signature seen in theaters.5,33,34 The rats were anesthetized with a mixture of ketamine and xylazine (10:1 (100 mg/10 mg/kg), 0.1 mL/100 g) administered via intraperitoneal injection. Rats were exposed to the blast wave in the test section located inside the shock tube—i.e., 2.80 m from the breech, or 3.05 m from the exit. The tests were performed at five discrete incident peak overpressures detailed in Table 1. The incident pressure was controlled by adjusting the number of Mylar membrane, and keeping the breech length constant at 17.625 inches (0.4477 m).
Table 1.
Peak Overpressure and Impulse Average Values Measured by Side-On Sensor (Incident Pressure)*
| Group alias | Peak overpressure, kPaa | Impulse, Pa·sa,b | Survival, % | Number of animalsc | Lung injury score | Number of animalsd | |
|---|---|---|---|---|---|---|---|
| 1 | 130e | 127±8 | 184±19 | 100 | 18 | 1.8±0.7 | 5 |
| 2 | 190 | 195±19 | 335±25 | 70 | 20 | 8.5±2.9 | 6 |
| 3 | 230 | 223±20 | 393±44 | 100 | 10 | 10.8±3.0 | 5 |
| 4 | 250 | 243±21 | 437±31 | 76 | 17 | 14.4±2.4 | 5 |
| 5 | 290f | 288±17 | 452±45 | 0 | 6 | 13.5±4.0 | 6 |
Corresponding survival and lung injury score, and the numbers of animals used for both test, are presented.
Standard deviations are reported for peak overpressure and impulse.
Impulse is the pressure-time integral of the positive phase of the shockwave.
Number of animals used in survival tests.
Number of animals used to evaluate blast lung injury.
Equivalent of a pressure-impulse profile of a 10 kg of C4 at a distance of 5.62 m from the explosion epicenter.
Equivalent of a pressure-impulse profile of a 50 kg of C4 at a distance of 6.72 m from the explosion epicenter.
An aluminum bed was designed and fabricated for holding the rat during the application of blast wave. The aerodynamic riser is attached to the bed to hold the sample in the center of the shock tube. Typically, rats are tested in a prone position and are strapped securely to the bed with a thin cotton cloth wrapped around the body (Fig. 1A). High-speed videos were recorded for all the tested animals as a quality control measure, and those animals with substantial head motion were excluded from further examination. Sham control rats received anesthesia and noise exposure but without blast exposure, and naïve control rats were given just anesthesia.
FIG. 1.

Schematic representation of the shock tube at UNL blast facility. (A) Left inset illustrates the side-on pressure sensor location (arrow) and the position and strapping of the animal in the holder; right inset: close up high-speed video still images demonstrate the typical head displacement during the blast exposure in our model. Representative overpressure profiles (B), side-on overpressure (C), and impulse values (D) of five blast waves with increasing intensities used in our experiments. These data show significant difference in both overpressure values and impulse, (p<0.05). Box range: 25–75%, whisker range: 1–99% (median: 50%; mean: ►).
Biomechanical loading evaluation in cadaver rats
Five 10-week-old male Sprague Dawley rats (320–360 g) were sacrificed using carbon dioxide (CO2) until all movements had ceased. The death of the animal was confirmed before the experiment—i.e. by ensuring no reaction to a noxious stimulus. Immediately after the sacrifice, a surface mount Kulite sensor (LE-080-250A) was mounted on the nose to measure the reflected pressure, and two Kulite probe sensors (XCL-072-500A, diameter: 1.9 mm, length: 9.5 mm) were implanted in the thoracic cavity and in the brain. Figure 2A shows the schematic of the approximate positions of these sensors. For the brain sensor implantation, the tip of the sensor was backfilled with water to ensure good contact with tissue, and the sensor was inserted through the foramen magnum 4–5 mm into the brain tissue (detailed description of methodology of sensor implantation and sensor locations are specified in reference34).
FIG. 2.

The diagram of sensor locations (A) used in the cadaver rat experiments. The rightmost inset shows the brain remains intact after the experiments. Representative pressure profiles of the 190 kPa group (B), and average peak overpressures recorded by side-on, nose (reflected pressure), brain, and lungs pressure sensors (C) are presented (no statistically significant differences are noted for 190 and 230 kPa groups (marked with ampersand)). There are no statistical differences between intracranial pressure (ICP) impulse of 230 kPa group (100% survival, Table 1) and ICP impulses of the two lethal groups (190 and 250 kPa) (D).
All pressure sensors were calibrated individually, using the 101 mm diameter shock tube specifically designated for that purpose. We accurately calibrated sensors via precise measurements of the shock-wave velocity (with the application of a series of gauges placed along the shock tube) and invoking Rankine-Hugoniot jump conditions to relate shock-wave velocity and overpressure. Cadaver rats were exposed to five shots per animal at 130 kPa (two rats) and 190, 230, 250, and 290 kPa (two rats). One rat with lungs filled with ballistic gel to ameliorate inconsistent pressure readings was exposed at five blast overpressures (130–290 kPa). A Fast Fourier Transformation (FFT) analysis was performed on the 50 msec of the intracranial pressure (ICP) signal acquired at the sampling rate of 1 MHz (50,000 points) using Origin 7.5 software with rectangle window and amplitude power normalization using the mean square amplitude algorithm.
Mortality evaluation
The anesthetized animals underwent various levels of injury, characterized by the peak overpressure ranging from 130 to 290 kPa (Table 1). No resuscitation was performed after trauma.
Physiological parameters
BW measurements were performed before and 24 h post-exposure. Multiple vital signs were measured non-invasively using clinically validated MouseSTAT, a rodent physiological monitoring system (Kent Scientific Corp., Torrington, CT). Briefly, the anesthetized rat was placed in a supine position on the warming pad attached to the control unit to maintain natural body temperature and prevent hypothermia. The temperature was measured rectally at a rate of 1 Hz over the 30-min period before and after the blast exposure. Simultaneously, heart rate, spO2, and perfusion index were recorded by a y-clip sensor clamped to the back paws of the rat.
Evaluation of pulmonary injury severity (IS)
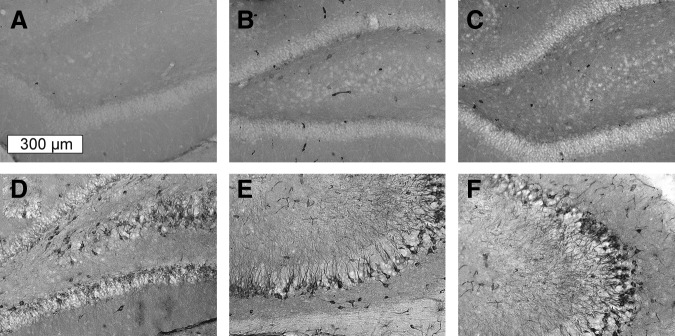
Immediately after the blast exposure, the lungs were extracted from the thoracic cavity, placed in 30–40 mL of a freshly prepared solution of 10% formalin and stored at 4°C for further evaluation. Lung IS associated with the primary blast exposure was performed using the Pathology Scoring System for Blast Injuries developed by Yelveton.35 The extent of injury is defined by the elements of the IS according to the equation:
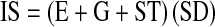 |
(1) |
where E is the extent of injury to the lungs (range 0–5); G is the injury grade including the surface area of the lesions (range 0–4); ST, severity type elements, which classify the type of the worst-case lesions (range 0–4); and SD is the severity depth element, indicating the depth or the degree of disruption of the worst-case lesion (range 1–4).
Transcardial perfusion
At 24 h post-exposure, rats were anesthetized with a ketamine/xylazine mixture and transcardially perfused with wash solution (0.8% NaCl, 0.4% dextrose, 0.8% sucrose, 0.023% CaCl2, and 0.034% sodium cacodylate), followed by fresh fix solution (4% paraformaldehyde solution supplemented with 4% of sucrose in 1.4% sodium cacodylate buffer, pH=7.3). Immediately after perfusion, heads were decapitated and stored for an additional 16–18 hours in the excess of fix solution. The brains were subsequently extracted the following day and stored refrigerated (4°C) in the sodium cacodylate storage buffer. Brains were shipped in phosphate-buffered saline (pH=7.2) to Neuroscience Associates (Knoxville, TN) for sectioning and staining.
Histological procedures
At Neuroscience Associates, the brains were embedded into one gelatin matrix block using MultiBrain Technology.™ The block was then allowed to cure and subsequently rapidly frozen in isopentane chilled with crushed dry ice (−70°C). The block was mounted on a freezing stage of a sliding microtome and coronal sections with a thickness of 40 μm were prepared. All sections were collected sequentially into an array of 4”×6” containers that were filled with antigen preserve (buffered 50% ethylene glycol). At the completion of sectioning, each container held a serial set of one of every twelfth sections—i.e., single section at every 480 μm intervals). These sections were stained with an antibody against rat IgG to assess the BBB damage and with amino cupric silver staining to detect neurodegeneration.
Immunostain
For immunohistochemistry, free-floating sections were stained. Sections were rinsed with Tris Buffered Saline (TBS) and incubated in 0.3% H2O2 to block endogenous peroxidase activity. Next, sections were blocked in 10% block serum with 0.3% Triton X-100 in TBS for 1 h at room temperature, followed by incubation in primary antibody overnight at 4°C. After rinses, a secondary biotinylated antibody was applied. IgG immunoreactivity was visualized by incubation of sections with diaminobenzidine tetrahydrochloride and hydrogen peroxide. The sections were mounted on gelatinized glass slide, air dried, dehydrated in alcohol, cleared in xylene, and a cover slip applied.
Amino cupric silver stain
The amino cupric silver staining was performed according to the modified protocol described by de Olmos and associates36 and described in detail elsewhere.32 Briefly, the 40-μm free-floating sections were taken through the following major steps: pre-impregnation, impregnation, reduction, bleaching, and fixing. The sections were removed from the cacodylate buffered formaldehyde and rinsed with deionized water. They were then placed in dishes containing the pre-impregnation solution (for detailed composition refer to the work of Garman and coworkers32) and heated in the microwave to 45–50°C. The sections were kept in this solution overnight. The sections were then incubated in impregnation solution (AgNO3, LiOH, NH4OH, ethanol, and acetone in deionized water) for 50 min. The sections were transferred from the impregnation solution into the reducer solution (ethanol, formalin, citric acid in deionized water), placed in a water bath (32–35°C) for 25 min, and then rinsed with deionized water.
The sections were rapidly transferred through bleaching solutions: (1) potassium ferricyanide in potassium chlorate with lactic acid, (2) potassium permanganate with sulfuric acid, and (3) sodium thiosulfate, and then fixed in rapid fixer solution for 1.5 min. Finally, the sections were rinsed in deionized water, mounted on gelatin-subbed glass slides, and counterstained with neutral red to reveal normal cell bodies.
Microscopic evaluation
The slides with anti-IgG stained coronal sections were digitized at 1200×1200 dpi resolution. A total number of 54 slides was used in this study, but some sections from the anterior and the posterior parts of the brain parenchyma were omitted because of smaller size and lower staining intensity and only slides from 13 to 52 were used in data analysis. Images were loaded into ImageJ software version 1.46h (http://rsbweb.nih.gov/ij/), and the intensity of each entire coronal section was measured with Gels plug-in to evaluate quantities of IgG extravasated into brain parenchyma (uncalibrated optical density [OD] option in the Image J). The OD was subsequently divided by the area of respective brain section and compared with averaged OD of three control sections from the same slide (relative OD). The average relative OD (AROD) per brain was calculated using relative OD values of 40 sections.
For the visualization of neurodegeneration and IgG uptake, the slides stained with amino cupric silver and anti-IgG were inspected using Nikon Plan Apo 10× and Nikon Plan Fluor 40× magnification objectives mounted on a Nikon Eclipse Ti-E inverted microscope. Representative images for observed pathologies were recorded using a QImaging EXi Aqua™ camera attached to the microscope with the resolution of 1392×1040 pixels.
Data analysis
Unless otherwise stated, the data are presented as mean±standard error of the mean. Unpaired two-tailed Student t tests were performed to assess statistical significance between samples, and p values below 0.05 were considered significant. One-way analysis of variance with a Bonferroni-Holm post-hoc test was performed to evaluate the differences between test groups. Power analysis was performed using freely available G*Power software, version 3.1.5.37
Results
Metrics of the primary blast
The experimental setup used in this study is presented in Figure 1A. Rats were exposed to primary blast in the 9” square cross-section shock generator using helium as a driver gas. Animals were tested in the prone position. Five different blast overpressure intensities were used with representative examples of overpressure profiles presented in Figure 1B. Statistical evaluation of 20 shots per group revealed that they were normally distributed and independent (Fig. 1C). Noticeably, groups with the average peak overpressure difference of merely 20 kPa (groups 230 and 250 kPa, Table 1) are also independent (p<0.0001, power>0.99). Similar analysis performed on overpressure-time integrals revealed all groups are also independent (p<0.001, power>0.95). Thus, it appears peak overpressure and impulse are equally sensitive metrics to describe blast-related biomechanical loading and associated pathologies. In the present case, we will refer to different groups studied within the framework of this research effort using peak overpressure values (see Group Alias column in Table 1).
Biomechanical loading evaluation
For evaluation of biomechanical loading in the rat, a single pressure sensor was mounted on the nose of the sacrificed rat and two more pressure probe sensors were implanted in the brain and lungs (Fig. 2A), to monitor the reflected, intracranial, and intrathoracic pressures. Typical pressure profiles for all four sensors used (side-on, nose, brain, and lung) are presented in Figure 2B. The intensity of the reflected pressure measured on the rat's nose is higher than the intensity of side-on pressure, and both are characterized by gradually increasing values (Fig. 2C). The reflected (nose sensor), and intracranial pressures are higher than the incident pressure (p<0.001) as observed in Figure 2C, but there are no clear differences between reflected pressure and ICP. Moreover, the ICP not only is higher than the incident pressure but also shows an oscillatory tendency.
While the reflected pressure shows a monotonic increase with side-on pressure for all the pressure groups (130, 190, 230, 250, and 290), the same cannot be said for the ICP: there are no statistically significant differences between groups190 and 230 kPa (p=0.70) (Fig. 2C). The impulse values for brain sensor indicate there are statistically significant correlations between non-lethal group 230 kPa and two lethal groups—i.e., 190 and 250 kPa. This indicates the outcome (mortality) is not a function of the total energy transferred to the brain but depends on the specific response of the cranium (vide infra).
Primary blast induced mortality
The mortality rates among animals exposed to different levels of blast overpressures are shown in Table 1. Exposure to 130 kPa (n=18) and 230 kPa (n=10) resulted in no animal death, whereas exposure to 190 kPa (n=20), 250 kPa (n=17), and 290 kPa (n=6) peak overpressure resulted in 30%, 24%, and 100% mortality, respectively. All animals died immediately after blast exposure, and there was no delayed death.
Intracranial pressure analysis
To elucidate various modes of blast loading, we performed a set of analyses on the data reported by the brain pressure sensor. Using peak-trough analysis, we correlated biomechanical loading at the early, the most violent stages of blast wave-cranium interactions (initial 0.4 msec, Fig. 3A,B), with the blast induced mortality (Table 1). The monotonically increasing peak overpressure and impulse values measured outside (side-on sensor) or inside of the cranium are not following the same trend as mortality data in the Table 1. There is strong correlation, however, between the sum of the first two peak-to-peak amplitudes and mortality (Fig. 3C). The intracranial pressure data subjected to FFT analysis indicated the oscillations have harmonic characteristics, and the high frequency component (10–20 kHz) is present exclusively in the case of the lethal groups (190, 250, and 290 kPa, Fig. 3D). Moreover, the maximum ICP frequency increases with increasing blast intensity: peaks at 12.6 kHz (190 kPa), 16.4 kHz (250 kPa), and 19.3 kHz (290 kPa) marked with arrows in Fig. 3D. In 290 kPa group, two characteristic frequencies are noted (16.4 and 19.3 kHz).
FIG. 3.

Representative pressure traces for five exposure groups (A). The signal was smoothed using Fast Fourier Transformation (FFT) band pass filter for the clarity of presentation. (B) Schematic representation of peak-trough analysis on overpressure recorded by brain sensor: Δpt is a sum of the first two peak-to-peak amplitudes. Results of peak-trough analysis: (C) Lethal groups (190, 250 and 290 kPa) have significantly higher Δpt (p<0.05) compared with non-lethal groups (130 and 230 kPa). There are no statistically significant differences between Δpt in respective non-lethal and lethal groups (p>0.05, marked with ampersand). (D) FFT analysis of the intracranial pressure profiles recorded by the sensor implanted in the brain. The high frequency component (peaks marked with arrows, >10 kHz) is present in the signal of lethal groups (190, 250, and 290 kPa).
Physiological parameters
We noted statistically significant weight loss in both 190 kPa and 250kPa injury groups 24 hours after blast exposure (Fig. 4).
FIG. 4.

Exposure to different intensity of primary blast results in the body weight decrease in groups 190 and 250 kPa. In these groups, statistically significant differences (marked with asterisk) were established immediately before and 24 h after the exposure (p<0.05). Numbers of animals used in respective tests are provided in the bars corresponding to each group.
The heart rate monitoring was performed over the period of 30 minutes before and after the blast exposure (Fig. 5A, B). Observed changes in the heart rate difference between control groups were non-significant (Fig. 5C, p>0.05). The blast groups, as expected, showed an onset of bradycardia occurring immediately after the blast exposure (Fig. 5C, p<0.05): the heart rates decreased by 40±9 (130 kPa), 62±7 (190 kPa), 62±15 (230 kPa), and 62±10 (250 kPa) beats per minute (bpm). It appears blast-induced bradycardia—i.e., expressed as the difference of average heart rate measured during 30-min intervals before and after exposure—follows a simple dose-response model, with upper value of 62±4 bpm and inflection point at 126 kPa (Fig. 5D). All the groups of rats exposed to blast are correlated, however, with 130 and 190 kPa groups being borderline correlated (p=0.065).
FIG. 5.

Representative experimental data demonstrating blast-related bradycardia in the rat model: (A) naïve control; (B) rat exposed to 190 kPa peak overpressure (sampling rate 1 Hz in both cases). (C) The heart rate monitoring in the acute phase post-exposure revealed the average heartbeat decrease was statistically significant (p<0.05) in animals exposed to blast at peak overpressure of 130 kPa (ΔHR=40±9 beats per minute [bpm]) and higher (average ΔHR=62±11 bpm for 190, 230, and 250 kPa groups). (D) The heart rate difference is not statistically significant between two control and four exposed groups (p>0.05, marked with ampersand) and follows dose response model in the acute phase post-exposure.
There are no differences in spO2 between control and blast-exposed rats (data not shown).
Shock-wave–induced acute lung injury
No animals showed external signs of trauma. At necropsy (performed immediately after the blast exposure), animals subjected to a blast wave showed typical evidence of moderate pulmonary hemorrhage (Fig. 6), associated with vascular damage, direct alveolar injury, and edema, and generally described as “blast lung.”10,38–40 As illustrated in Figure 6, animals subjected to a higher blast overpressure (190–290 kPa groups) were found to have statistically significant pulmonary hemorrhage compared with sham control. The onset of injury in our model takes place at 130 kPa peak overpressure. Modeling of the pulmonary injury as a function of peak overpressure with dose-response function revealed the inflection point is located at 184.2±15.7 kPa, with a plateau (the highest score) at 14.32±1.85 (Fig. 6G). This value is rather low: the maximum possible value in the Yelverton blast injury index for lungs is 64.35 Interestingly, pathological evaluation of pulmonary injuries in one rat from group 290 kPa (100% acute mortality) revealed virtually no signs of lung injury. In the areas most severely affected by hemorrhage, approximately 15–60% of the alveoli are filled with acute pools of hemorrhage (data not shown). Near these pools, there were respiratory bronchioles, which contained small amounts of edema fluid. The more normal sections of lung have moderate atelectasis and very sparse to absent emphysema.
FIG. 6.

Blast-induced lung injury immediately post-exposure: (A) control, (B) 130 kPa, (C) 190 kPa, (D) 230 kPa, (E) 250 kPa, (F) 290 kPa. The extent of injury was quantified using the Pathology Scoring System for Blast Injuries.35 The extent of injury is defined by the Elements of the Injury Severity according to the equation (1) and was modeled with dose-response function (G). Groups with no statistically significant differences (p>0.05) are marked with an ampersand. Color image is available online at www.liebertpub.com/neu
BBB damage and IgG uptake by neurons
Staining to reveal IgG in the brain parenchyma was performed for every twelfth section (each section was 40 μm thick) equally spaced across the entire rat brain (Fig. 7). The purpose of this staining was twofold: (1) to measure the extent of BBB damage and (2) to visualize the accumulation of IgG in the intracellular space of neurons. Figure 7A presents coronal sections where elevated levels of IgG with respect to control samples are obvious. The slides with mounted sections were first digitized, and then we integrated the staining intensity to quantify the amount of IgG. The olfactory bulb region (initial 12 sections, 5.76 mm from the brain anterior, see inset in Fig. 7B) was omitted in this analysis.
FIG. 7.

(A) Immunostaining for rat immunoglobulin G (IgG) as an indicator for compromised blood-brain barrier. (B) Relative optical density (OD) of full coronal sections from the brain of a single rat exposed at 190 kPa. The OD of each section was divided by average OD of three controls (two naïve and one sham). Inset illustrates brain region covered in this study. (C) Average relative OD of IgG across the brain parenchyma for rats exposed to primary blast and sacrificed 24 h post-exposure. Error bars are standard deviation.
The OD of each section was divided by average OD of three control samples resulting in relative OD (ROD). Figure 7B presents RODs of 40 sections as a function of section number for a single rat from 190 kPa group. At lower blast intensities (190 and 230 kPa), in some rats the ROD has higher value in the rostral than in the dorsal brain region (Fig. 7B). The ROD gradient is not a general pattern and typically the variations are not substantial. In one rat exposed at 250 kPa blast intensity, however, the highest RODs (2.3–2.7) are noted in the slides number 28–35 (not shown).
The RODs pertain to the same brain were averaged and expressed as AROD. The results for rats exposed at three different blast intensities are presented in Figure 7C. In the 190 kPa group, two rats have higher AROD than the control group (AROD>1), but two others are comparable. Similar results are noted for the 230 kPa group, but only one rat has higher average intensity of IgG staining than rats in the 190 kPa group. The level of BBB damage appears to be correlated with the blast intensity (Fig. 7C).
In animals with compromised BBB, numerous IgG-positive cells of neuronal morphology were noted across brain parenchyma. The IgG accumulated predominantly in the cells localized in various parts of the cerebral cortex (Fig. 8) and the hippocampus (Fig. 9). We have also evaluated brain sections of rats exposed to 190, 230, and 250 kPa blast intensity using amino cupric silver staining. These tests gave negative results, indicating 24 h is an insufficient period to induce neurodegeneration. This result is consistent with findings of others.32
FIG. 8.
The IgG uptake in cortical neurons 24 h after the blast. Coronal sections of the brain were collected from brains of sham control (A) and surviving rats exposed to blast with (B) 190 kPa, (C) 230 kPa, and (D) 250 kPa peak overpressure. The scale bar (300 μm) is the same for all samples. Arrows indicate faintly stained neurons.
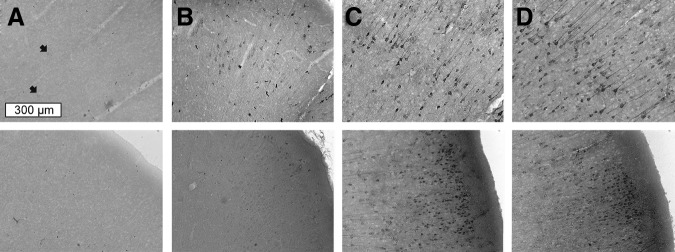
FIG. 9.
The IgG positive cells in the hippocampus of sham control (A) and in rats exposed to blast overpressure of 190 (B), 230 (C), and 250 kPa (D–F). The scale bar (300 μm) is the same for all samples.
Discussion
When the blast wave impacts the human body, part of the shock is reflected and diffracted, while part of it is transmitted. Reflected pressure is the pressure acting on the body and is always higher than the incident pressure (the details are discussed in reference 33). The mechanical load experienced by the animal is based on the reflected pressure (that is directly acting on the surface of the animal) and not incident pressure. An additional factor that needs to be considered is that the structure may be deformed because of the external load; this deflection will, in turn, induce additional pressure in the contained fluid. Thus, the ICP will depend on the directly transmitted energy and that induced from the pressure surges associated with the skull deflection. Further, the magnitude of deflection itself depends on the structural rigidity of the skull and the magnitude of external biomechanical loading. If the skull structure is thin (e.g., rat skull), the skull will deflect more, pressurizing the brain higher compared with a thicker skull (e.g., pig skull) for the level of external pressure. Hence, the ICP should be viewed in terms of transmission and deflection-induced pressure surges. It is possible that both these factors acting together or alone can generate high-frequency stress waves and lower-frequency shear waves, and both types are potentially injurious to the tissue.13,41
Experimental evidence collected in the past suggests that blast-related injury is governed by three key input parameters associated with the shockwave: (1) peak overpressure, (2) the overpressure (positive phase) duration, and (3) positive phase impulse (the integral of overpressure in the time domain). To evaluate the effect of blast exposure, various animal models were exposed to different forms of blast ranging from direct exposure to live explosives and controlled blast waves.24 Although testing animal models using live explosives provides an alternative loading condition, this does not provide a controlled setting for studying primary blast injuries. Because of complications in controlling blast parameters, it is difficult to investigate separately the effects of primary blast,15 and repeatability of the blast conditions is rather poor.42
In the present study, we demonstrate precise control of shock-wave characteristics (Fig. 1) combined with an appropriate test specimen location in the shock tube,33 and facilitate an inquiry into the mechanisms of pure primary blast injury.
Quantification of the biomechanical loading was achieved via monitoring of the incident, reflected, intracranial, and lung pressure histories (denoted as the side-on, nose, brain, and lungs, respectively, in Fig. 2). We noticed that the reflected pressure is higher than incident pressure (Fig. 2B,C), which is typical for a scenario when a high-velocity pressure wave is brought to a sudden stop.33,43 Further, the average magnitude of ICP is generally higher than incident pressure, and ICP profiles have characteristic harmonic pressure oscillations with amplitude of 100–250 kPa (Fig. 2B, 3A,B).
Our experimental data are consistent with the results reported by Bolander and coworkers18 and Leonardi and colleagues.20 The ICP pressure oscillations are caused by the specific material response of the rat skull, which was also demonstrated in a simplified numerical model of in vivo response to blast proposed by Moss and associates.44 The intrathoracic pressures (lungs) were substantially lower than the incident overpressure and ICPs. We experienced substantial difficulties, however, during pressure measurements in lungs caused by uncontrolled interactions between the pressure sensor and walls of internal organs in the abdomen. It is clearly seen in experimental data as high variability of peak overpressure values between data sets and a relatively high experimental errors (Fig. 2C). In an attempt to mitigate these issues, we filled up lungs with ballistic gel (20% gelatin), but the recorded pressure profiles in this configuration were of much higher magnitude—i.e., comparable with pressures recorded in the brain.
The mechanisms of the primary blast waves-tissue interactions are at present not completely understood.20 Frequently, the immediate or delayed mortality is used as an index to quantify the outcome of blast exposure.21,24,31 Thus, we decided to evaluate mortality of rats as a function of exposure to a primary blast at five discrete peak overpressure values (Table 1, Fig. 1). In our model, all rats survived exposure to 130 kPa peak overpressure, compared with 37.5% at 126 kPa in the work of Long and coworkers,21 which further decreased to 0% when rats were protected by a Kevlar vest. We should exercise caution, however, when comparing these two studies, because we measure the incident pressure at the animal location, whereas in the experiments by Long and colleagues,21 the pressure is measured inside the tube, and the animal is located outside (non-primary blast conditions). In the latter, the pressure experienced by the animal is different and, further, the jet wind effect may also cloud the comparison. Also, in our case, only the rat's head is directly exposed to the shockwave, and the internal organs are partially screened by the specimen holder (Fig. 1A, insets). In the tests by Long and associates,21 rats are placed perpendicular to the shock tube, and injuries sustained by internal organs are thus predominantly responsible for lethality.
At higher shock intensities, mortality reached 30% and 24% for 190 and 250 kPa groups, respectively. This is similar to mortality rates reported by Garman and colleagues32: ∼25% at 240 kPa peak overpressure. We found the mortality is not simply a linear function of the blast intensity, which, to the best of our knowledge, was not reported before. To elucidate the possible etiology and mechanisms of mortality, we focused on the brain and the lungs pathology. The detailed analysis of the ICP profiles indicates the peak-to-peak amplitude (Δpt, Fig. 3) of the two initial peaks is statistically significant between lethal (190, 250, and 290 kPa) and non-lethal (130 and 230 kPa) groups (p<0.05, Fig. 3C). Moreover, the FFT analysis of ICP profiles revealed the characteristic oscillation frequencies of more than 10 kHz (indicated with arrows in Fig. 3D) are exclusive for lethal groups. In non-lethal groups (100% survival at 130 and 230 kPa), only low frequency oscillations are noted.
ICP frequencies observed in our studies, however, are of much higher magnitude than those reported in the literature related to blast exposure, typically below 1.5 kHz.11,13,45 It appears that in our model, there exist strong correlation between ICP amplitude, ICP oscillation frequency, and mortality (and also a weight loss, as discussed below). The skull flexures are closely associated with the blast exposure; we observed ICP oscillations in other animal models tested in our laboratory—i.e., mice and mini-pig, and also in experiments with cadaver heads (data not published).
In a recent article, Vandevord and coworkers23 report a specific response to blast overpressure complementary to our findings. Cognitive and histological deficits were noted at relatively low peak overpressure values (117 kPa) and were more severe than in rats exposed to 153 kPa blast.23 The authors associate their unusual range-specific susceptibility to blast with biomechanical response of the skull. Our data suggest similar response exists also at higher overpressures. We observed increased mortality at 190 kPa (Table 1) and a statistically significant BW loss (weight loss proportional to the blast intensity was reported in mice,19,31 and this is typically a manifestation of impairment of the neuroendocrine system (hypothalamic-pituitary-target organ axes46) associated with the damage to the hypothalamic region of the brain.47
Despite the fact that there are pretty compelling correlations between mortality rates, peak-to-peak amplitudes, and ICP frequencies, more experimental evidence is needed to establish definitive relationships between these variables. These results thus need to be treated with extreme caution, because comparison between the shockwave with living and dead tissues could be affected by several factors: lack of blood circulation in the brain parenchyma, or effects associated with post-mortem rigor mortis. Moreover, the effect of variability of skull shape between different animals (within the same age group), which might ultimately be a decisive factor between survival and death, will necessitate more extensive evaluation than presented in this study.
Animals surviving blast exposure had a significantly decreased heart rate, compared with control (Fig. 5). At high peak overpressure values— i.e., 190 kPa or higher—the heart rate is independent of the blast intensity (Fig. 5C, D). The blast-induced bradycardia in our model is similar to that reported in other studies using rats.6,48,49 In the blast trauma mouse model, however, the opposite relationship was observed: exposure to the mild blast resulted in heart rate increase immediately after the blast.19 Bilateral cervical vagotomy and intraperitoneal injection of atropine methyl bromide completely prevented the bradycardia, thus confirming the vasovagal reflex is responsible for bradycardia in the acute phase after blast exposure.6
Gross pathological evaluation revealed that the lung injury immediately after blast exposure is mild to moderate (Fig. 6), and at higher blast intensities, the injury score is independent of peak overpressure (Fig. 6G). Cernak and colleagues19 reported that mice tested in the prone position sustained lower lung injury than when tested in the supine position, and these results were correlated with mortality data. Mortality rates were higher for animals tested in the supine (10% and 32%, for moderate and severe groups, respectively) versus the prone position (38% and 52%, for moderate and severe groups, respectively). It indicates respiratory tract injury is likely solely responsible or a very important contributing factor for mortality in this model. It appears in our case that because of the different orientation of the rat's body (Fig. 1), lungs are well shielded and thus interaction with the shock is markedly diminished.
Hence, it is unlikely that lung injury is the cause of death in our model. The mortality rates do not correlate with the lung injury scoring data (Fig. 6G). The intrapulmonary hemorrhage and edema are major factors contributing to the initial respiratory insufficiency, which is accompanied by cardiovascular irregularities.11 In agreement with others, we observed scattered petechiation and hemorrhage in rats exposed to 130 kPa.8,39 Interestingly, in the group of rats exposed to 290 kPa, there was still one animal without any noticeable gross damage in the lungs. These data suggest that in our model, lung injury can be a contributing factor for the outcome but is not solely responsible for the mortality rates observed. Only in the case of massive pulmonary hemorrhage and edema, lung injury is potentially a life threatening event; however, this is not supported by our data.3,39
Gross pathological observation of the brains after the blast exposure revealed an absence of focal lesions, contusion, or subarachnoid hemorrhage, similar to findings in the blast mice model.19 Cernak and colleagues19 reported that after improving the head restraint on the animal holder, no signs of subdural and subarachnoid bleeding caused by acceleration/deceleration were seen. This does not rule out the possibility that pure blast can inflict diffuse BBB damage at higher blast intensities.
To test this hypothesis, we stained coronal brain sections of rats exposed at 190, 230, and 250 kPa to detect the presence of IgG. The BBB breakdown observed in the acute phase is a hallmark of TBI leading to brain edema and secondary ischemic injuries.50 Endogenous proteins such as IgG are typically detected in the brain parenchyma on loss of integrity of the BBB.51–53 In our bTBI model, there were no obvious signs of injuries at gross pathological evaluation, but we detected widespread presence of IgG in the brain parenchyma 24 h after exposure (Fig. 7). Approximately half of the tested animals in each group, however, had negligible levels of IgG in the brain when compared with control (Fig. 7C), indicating BBB damage is not an omnipresent universal pathology after bTBI, even at relatively high shock intensities. The “random” character of the BBB damage indicates also the complexity of blast injuries and should be taken into account to devise successful diagnostic and remediating strategies.
In our bTBI model, the intracerebral bleeding in the 24-h period post-exposure has diffuse character; there are no obvious localized regions of high levels of IgG in the tested samples. Readnower and coworkers22 reported increased levels of IgG at 0.5, 3, and 24 h post-blast in brains of rats exposed to 120 kPa of blast overpressure. In their model, the outermost layer of the cortex was affected, and the maximum levels of IgG were detected after 3 h post-exposure. Rats exposed to blast of 240 kPa peak overpressure had widespread presence of IgG in the neuropil of the cortex, particularly the piriform and entorhinal cortices, and within the underlying striatum contralateral to the blast.32 These authors reported also relatively high variability for IgG extravasation, similar to our results (Fig. 7B). In none of these studies, however, was the IgG detected inside the neuronal cells. Microscopic analysis at high magnification revealed IgG penetrated into the intracellular space of numerous neurons located in the cortex and hippocampus (Fig. 8,9).
To the best of our knowledge, this is the first report of ubiquitous IgG accumulation in neurons in a blast exposure in vivo model. It appears neurons are the only cell type in which the uptake of this extravasated protein takes place. Similar findings were reported in the model of focal ischemia,54 and authors associated accumulation of IgG in the intracellular space with increased membrane permeability. Hoshino and coworkers55 observed time dependent extracellular IgG accumulation in cortices and hippocampi for up to 2 weeks after the injury in the model of severe fluid percussion injury. They concluded that IgG immunostaining was most prominent after 24 h. These authors also observed intracellular IgG uptake in the brain regions where extracellular IgG was present.
Selective accumulation of IgG in neurons was recently observed in the rat model of temporal lobe epilepsy.56 In the cold injury model,57 uptake of blood plasma proteins (albumin, fibrinogen, and fibronectin) into damaged neurons was demonstrated by immunostains, and these proteins were retained in the injured neurons for 2–4 days. The labeling of normal neurons, whose axons or their collaterals passed through or terminated in the lesion, was reported, through retrograde axonal transport. The authors noted, however, that this labeling was weaker in non-damaged neurons, and neuronal nuclei were not labeled in these cells in contrast to those of damaged cells. The intracerebral hemorrhage in rats exposed to blast has diffuse character, and thus it is more likely that dominant mechanism of IgG uptake into neurons is from IgG transport through openings in damaged or dysfunctional plasma membrane. This is corroborated by recent findings of increased levels of neuron-specific enolase in blood circulation in our model of blast exposure.58 Accumulation of serum proteins in neurons is linked to brain proteinopathies; their aggregation alters neuronal function and adversely affects survival.59
Conclusion
During the course of this study, we established a relationship between the blast overpressure intensity and acute injury outcomes. We have demonstrated with our shock tube that it is possible to precisely control the loading conditions, generate shockwaves resembling field conditions in a wide range of intensities and with peak overpressure difference of merely 20 kPa. We report pressure profiles at the test location of animals, which should be a standard practice for the research reports in blast injury models. On the contrary, it is infrequently exercised as evidenced by a review of the contemporary literature, which makes virtually impossible comparison of the results between different laboratories.
Using our primary blast injury model, we established that the mortality rates and acute responses are not a simple linear function of shock-wave intensity. We found the mortality rates are correlated to ICP peak-to-peak amplitude and frequency, indicating the specific response of the skull to the blast loading is affecting the outcome. These findings are corroborated by the BW loss, 24 h post-exposure, which was statistically significant in rats exposed to blast at 190 kPa and 250 kPa. The bradycardia and lung injury are independent of the blast intensity at peak overpressures higher than 190 kPa. In both cases the onset at 110 kPa was established, according to models used to characterize both pathologies.
The described primary blast injury model is an attempt to standardize the research in the field and provides the basis for further exploration of the etiology of complex primary blast-induced systemic injuries, testing of injury thresholds, and mitigation strategies.
Acknowledgments
Financial support under the U.S. Army Research Office project “Army-UNL Center of Trauma Mechanics” (Contract no. W911NF-08-10483, Project Manager: Larry Russell, P.I.: Namas Chandra) is gratefully acknowledged. We are thankful to Holly Reiling, Vicky Samek, and Katherine Ellenbolt-Pinkerton (Office of Research Responsibility, UNL) for invaluable help received during the realization of this research effort. Dr. Annette Fleckenstein's (University of Utah) useful comments on experimental procedures are gratefully acknowledged. We would like to acknowledge assistance received from Mr. Brandon Perry and Mr. Kurtis Palu during rat exposure experiments.
Author Disclosure Statement
Dr. Switzer has commercial interests in a company (NeuroScience Associates) that performs immuno- and amino cupric silver stains for academia, government, and industry. For the remaning authors, no competing financial interests exist.
References
- 1.Chen Y.C. Smith D.H. Meaney D.F. In-vitro approaches for studying blast-induced traumatic brain injury. J. Neurotrauma. 2009;26:861–876. doi: 10.1089/neu.2008.0645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Phillips Y.Y. Primary blast injuries. Ann. Emerg. Med. 1986;15:1446–1450. doi: 10.1016/s0196-0644(86)80940-4. [DOI] [PubMed] [Google Scholar]
- 3.DePalma R.G. Burris D.G. Champion H.R. Hodgson M.J. Blast injuries. N. Engl. J. Med. 2005;352:1335–1342. doi: 10.1056/NEJMra042083. [DOI] [PubMed] [Google Scholar]
- 4.Zhang J. Wang Z. Leng H. Yang Z. Studies on lung injuries caused by blast underpressure. J. Trauma. 1996;40(Suppl 3):S77–S80. [PubMed] [Google Scholar]
- 5.Chandra N. Ganpule S. Kleinschmit N.N. Feng R. Holmberg A.D. Sundaramurthy A. Selvan V. Alai A. Evolution of blast wave profiles in simulated air blasts: experiment and computational modeling. Shock Waves. 2012;22:403–415. [Google Scholar]
- 6.Irwin R.J. Lerner M.R. Bealer J.F. Mantor P.C. Brackett D.J. Tuggle D.W. Shock after blast wave injury is caused by a vagally mediated reflex. J. Trauma. 1999;47:105–110. doi: 10.1097/00005373-199907000-00023. [DOI] [PubMed] [Google Scholar]
- 7.Mellor S.G. Cooper G.J. Analysis of 828 servicemen killed or injured by explosion in Northern Ireland 1970-84: the Hostile Action Casualty System. Br. J. Surg. 1989;76:1006–1010. doi: 10.1002/bjs.1800761006. [DOI] [PubMed] [Google Scholar]
- 8.Chavko M. Prusaczyk W.K. McCarron R.M. Lung injury and recovery after exposure to blast overpressure. J. Trauma. 2006;61:933–942. doi: 10.1097/01.ta.0000233742.75450.47. [DOI] [PubMed] [Google Scholar]
- 9.Yang Z. Wang Z. Tang C. Ying Y. Biological effects of weak blast waves and safety limits for internal organ injury in the human body. J. Trauma. 1996;40(Suppl 3):S81–S84. doi: 10.1097/00005373-199603001-00019. [DOI] [PubMed] [Google Scholar]
- 10.Pizov R. Oppenheim-Eden A. Matot I. Weiss Y.G. Eidelman L.A. Rivkind A.I. Sprung C.L. Blast lung injury from an explosion on a civilian bus. Chest. 1999;115:165–172. doi: 10.1378/chest.115.1.165. [DOI] [PubMed] [Google Scholar]
- 11.Guy R.J. Glover M.A. Cripps N.P. The pathophysiology of primary blast injury and its implications for treatment. Part I: The thorax. J. R. Nav. Med. Serv. 1998;84:79–86. [PubMed] [Google Scholar]
- 12.Peters P. Primary blast injury: an intact tympanic membrane does not indicate the lack of a pulmonary blast injury. Mil. Med. 2011;176:110–114. doi: 10.7205/milmed-d-10-00300. [DOI] [PubMed] [Google Scholar]
- 13.Cooper G.J. Townend D.J. Cater S.R. Pearce B.P. The role of stress waves in thoracic visceral injury from blast loading: modification of stress transmission by foams and high-density materials. J. Biomech. 1991;24:273–285. doi: 10.1016/0021-9290(91)90346-o. [DOI] [PubMed] [Google Scholar]
- 14.Hoge C.W. McGurk D. Thomas J.L. Cox A.L. Engel C.C. Castro C.A. Mild traumatic brain injury in U.S. Soldiers returning from Iraq. N. Engl. J. Med. 2008;358:453–463. doi: 10.1056/NEJMoa072972. [DOI] [PubMed] [Google Scholar]
- 15.Elder G.A. Cristian A. Blast-related mild traumatic brain injury: mechanisms of injury and impact on clinical care. Mt. Sinai J. Med. 2009;76:111–118. doi: 10.1002/msj.20098. [DOI] [PubMed] [Google Scholar]
- 16.Moore D.F. Jaffee M.S. Military traumatic brain injury and blast. NeuroRehabilitation. 2010;26:179–181. doi: 10.3233/NRE-2010-0553. [DOI] [PubMed] [Google Scholar]
- 17.Celander H. Clemedson C.J. Ericsson U.A. Hultman H.I. The use of a compressed air operated shock tube for physiological blast research. Acta Physiol. Scand. 1955;33:6–13. doi: 10.1111/j.1748-1716.1955.tb01188.x. [DOI] [PubMed] [Google Scholar]
- 18.Bolander R. Mathie B. Bir C. Ritzel D. VandeVord P. Skull flexure as a contributing factor in the mechanism of injury in the rat when exposed to a shock wave. Ann. Biomed. Eng. 2011;39:2550–2559. doi: 10.1007/s10439-011-0343-0. [DOI] [PubMed] [Google Scholar]
- 19.Cernak I. Merkle A.C. Koliatsos V.E. Bilik J.M. Luong Q.T. Mahota T.M. Xu L. Slack N. Windle D. Ahmed F.A. The pathobiology of blast injuries and blast-induced neurotrauma as identified using a new experimental model of injury in mice. Neurobiol. Dis. 2011;41:538–551. doi: 10.1016/j.nbd.2010.10.025. [DOI] [PubMed] [Google Scholar]
- 20.Leonardi A.D. Bir C.A. Ritzel D.V. VandeVord P.J. Intracranial pressure increases during exposure to a shock wave. J. Neurotrauma. 2011;28:85–94. doi: 10.1089/neu.2010.1324. [DOI] [PubMed] [Google Scholar]
- 21.Long J.B. Bentley T.L. Wessner K.A. Cerone C. Sweeney S. Bauman R.A. Blast overpressure in rats: recreating a battlefield injury in the laboratory. J. Neurotrauma. 2009;26:827–840. doi: 10.1089/neu.2008.0748. [DOI] [PubMed] [Google Scholar]
- 22.Readnower R.D. Chavko M. Adeeb S. Conroy M.D. Pauly J.R. McCarron R.M. Sullivan P.G. Increase in blood-brain barrier permeability, oxidative stress, and activated microglia in a rat model of blast-induced traumatic brain injury. J. Neurosci. Res. 2010;88:3530–3539. doi: 10.1002/jnr.22510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vandevord P.J. Bolander R. Sajja V.S. Hay K. Bir C.A. Mild neurotrauma indicates a range-specific pressure response to low level shock wave exposure. Ann. Biomed. Eng. 2012;40:227–236. doi: 10.1007/s10439-011-0420-4. [DOI] [PubMed] [Google Scholar]
- 24.Richmond D.R. Goldizen V.C. Clare V.R. Pratt D.E. Sherping F. Sanchez R.T. Fischer C.C. White C.S. The biologic response to overpressure. III. Mortality in small animals exposed in a shock tube to sharprising overpressures of 3 to 4 msec duration. Aerosp. Med. 1962;33:1–27. [PubMed] [Google Scholar]
- 25.Alley M.D. Schimizze B.R. Son S.F. Experimental modeling of explosive blast-related traumatic brain injuries. Neuroimage. 2011;54(Suppl 1):S45–S54. doi: 10.1016/j.neuroimage.2010.05.030. [DOI] [PubMed] [Google Scholar]
- 26.Chavko M. Watanabe T. Adeeb S. Lankasky J. Ahlers S.T. McCarron R.M. Relationship between orientation to a blast and pressure wave propagation inside the rat brain. J. Neurosci. Methods. 2011;195:61–66. doi: 10.1016/j.jneumeth.2010.11.019. [DOI] [PubMed] [Google Scholar]
- 27.Koliatsos V.E. Cernak I. Xu L. Song Y. Savonenko A. Crain B.J. Eberhart C.G. Frangakis C.E. Melnikova T. Kim H. Lee D. A mouse model of blast injury to brain: initial pathological, neuropathological, and behavioral characterization. J. Neuropathol. Exp. Neurol. 2011;70:399–416. doi: 10.1097/NEN.0b013e3182189f06. [DOI] [PubMed] [Google Scholar]
- 28.Svetlov S.I. Prima V. Kirk D.R. Gutierrez H. Curley K.C. Hayes R.L. Wang K.K. Morphologic and biochemical characterization of brain injury in a model of controlled blast overpressure exposure. J. Trauma. 2010;69:795–804. doi: 10.1097/TA.0b013e3181bbd885. [DOI] [PubMed] [Google Scholar]
- 29.Svetlov S.I. Prima V. Glushakova O. Svetlov A. Kirk D.R. Gutierrez H. Serebruany V.L. Curley K.C. Wang K.K. Hayes R.L. Neuro-glial and systemic mechanisms of pathological responses in rat models of primary blast overpressure compared to “composite” blast. Front. Neurol. 2012;3:15. doi: 10.3389/fneur.2012.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reneer D.V. Hisel R.D. Hoffman J.M. Kryscio R.J. Lusk B.T. Geddes J.W. A multi-mode shock tube for investigation of blast-induced traumatic brain injury. J. Neurotrauma. 2011;28:95–104. doi: 10.1089/neu.2010.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y. Wei Y. Oguntayo S. Wilkins W. Arun P. Valiyaveettil M. Song J. Long J.B. Nambiar M.P. Tightly coupled repetitive blast-induced traumatic brain injury: development and characterization in mice. J. Neurotrauma. 2011;28:2171–2183. doi: 10.1089/neu.2011.1990. [DOI] [PubMed] [Google Scholar]
- 32.Garman R.H. Jenkins L.W. Switzer R.C., 3rd Bauman R.A. Tong L.C. Swauger P.V. Parks S.A. Ritzel D.V. Dixon C.E. Clark R.S. Bayir H. Kagan V. Jackson E.K. Kochanek P.M. Blast exposure in rats with body shielding is characterized primarily by diffuse axonal injury. J. Neurotrauma. 2011;28:947–959. doi: 10.1089/neu.2010.1540. [DOI] [PubMed] [Google Scholar]
- 33.Sundaramurthy A. Alai A. Ganpule S. Holmberg A. Plougonven E. Chandra N. Blast-induced biomechanical loading of the rat: experimental and anatomically accurate computational blast injury model. J. Neurotrauma. 2012;29:2352–2364. doi: 10.1089/neu.2012.2413. [DOI] [PubMed] [Google Scholar]
- 34.Chandra N. Holmberg A. Feng R. Controlling the shape of the shock wave profile in a blast facility. 2011. U.S. Provisional patent application no. 61542354. 61542354, U.S.P.p.a.n. (ed).
- 35.Yelveton J.T. Pathology scoring system for blast injuries. J. Trauma. 1996;40(Suppl 3):S111–115. doi: 10.1097/00005373-199603001-00025. [DOI] [PubMed] [Google Scholar]
- 36.de Olmos J.S. Beltramino C.A. de Olmos de Lorenzo S. Use of an amino-cupric-silver technique for the detection of early and semiacute neuronal degeneration caused by neurotoxicants, hypoxia, and physical trauma. Neurotoxicol. Teratol. 1994;16:545–561. doi: 10.1016/0892-0362(94)90033-7. [DOI] [PubMed] [Google Scholar]
- 37.Faul F. Erdfelder E. Lang A.G. Buchner A. G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav. Res. Methods. 2007;39:175–191. doi: 10.3758/bf03193146. [DOI] [PubMed] [Google Scholar]
- 38.Sasser S.M. Sattin R.W. Hunt R.C. Krohmer J. Blast lung injury. Prehosp. Emerg. Care. 2006;10:165–172. doi: 10.1080/10903120500540912. [DOI] [PubMed] [Google Scholar]
- 39.Tsokos M. Paulsen F. Petri S. Madea B. Puschel K. Turk E.E. Histologic, immunohistochemical, and ultrastructural findings in human blast lung injury. Am. J. Respir. Crit. Care Med. 2003;168:549–555. doi: 10.1164/rccm.200304-528OC. [DOI] [PubMed] [Google Scholar]
- 40.Brown R.F. Cooper G.J. Maynard R.L. The ultrastructure of rat lung following acute primary blast injury. Int. J. Exp. Pathol. 1993;74:151–162. [PMC free article] [PubMed] [Google Scholar]
- 41.Gorbunov N.V. McFaul S.J. Van Albert S. Morrissette C. Zaucha G.M. Nath J. Assessment of inflammatory response and sequestration of blood iron transferrin complexes in a rat model of lung injury resulting from exposure to low-frequency shock waves. Crit. Care Med. 2004;32:1028–1034. doi: 10.1097/01.ccm.0000120051.79520.b6. [DOI] [PubMed] [Google Scholar]
- 42.Risling M. Plantman S. Angeria M. Rostami E. Bellander B.M. Kirkegaard M. Arborelius U. Davidsson J. Mechanisms of blast induced brain injuies, experimental studies in rats. Neuroimage. 2011;54(Suppl 1):S89–S97. doi: 10.1016/j.neuroimage.2010.05.031. [DOI] [PubMed] [Google Scholar]
- 43.Bass C.R. Rafaels K.A. Salzar R.S. Pulmonary injury risk assessment for short-duration blasts. J. Trauma. 2008;65:604–615. doi: 10.1097/TA.0b013e3181454ab4. [DOI] [PubMed] [Google Scholar]
- 44.Moss W.C. King M.J. Blackman E.G. Skull flexure from blast waves: a mechanism for brain injury with implications for helmet design. Phys. Rev. Lett. 2009;103:108702. doi: 10.1103/PhysRevLett.103.108702. [DOI] [PubMed] [Google Scholar]
- 45.Cernak I. Noble-Haeusslein L.J. Traumatic brain injury: an overview of pathobiology with emphasis on military populations. J. Cereb. Blood Flow Metab. 2010;30:255–266. doi: 10.1038/jcbfm.2009.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raghupathi R. Cell death mechanisms following traumatic brain injury. Brain Pathol. 2004;14:215–222. doi: 10.1111/j.1750-3639.2004.tb00056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rothman M.S. Arciniegas D.B. Filley C.M. Wierman M.E. The neuroendocrine effects of traumatic brain injury. J. Neuropsychiatry Clin. Neurosci. 2007;19:363–372. doi: 10.1176/jnp.2007.19.4.363. [DOI] [PubMed] [Google Scholar]
- 48.Guy R.J. Kirkman E. Watkins P.E. Cooper G.J. Physiologic responses to primary blast. J. Trauma. 1998;45:983–987. doi: 10.1097/00005373-199812000-00001. [DOI] [PubMed] [Google Scholar]
- 49.Knoferl M.W. Liener U.C. Seitz D.H. Perl M. Bruckner U.B. Kinzl L. Gebhard F. Cardiopulmonary, histological, and inflammatory alterations after lung contusion in a novel mouse model of blunt chest trauma. Shock. 2003;19:519–525. doi: 10.1097/01.shk.0000070739.34700.f6. [DOI] [PubMed] [Google Scholar]
- 50.Unterberg A.W. Stover J. Kress B. Kiening K.L. Edema and brain trauma. Neuroscience. 2004;129:1021–1029. doi: 10.1016/j.neuroscience.2004.06.046. [DOI] [PubMed] [Google Scholar]
- 51.Sullivan P.G. Bruce-Keller A.J. Rabchevsky A.G. Christakos S. Clair D.K. Mattson M.P. Scheff S.W. Exacerbation of damage and altered NF-kappaB activation in mice lacking tumor necrosis factor receptors after traumatic brain injury. J. Neurosci. 1999;19:6248–6256. doi: 10.1523/JNEUROSCI.19-15-06248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dietrich W.D. Alonso O. Halley M. Early microvascular and neuronal consequences of traumatic brain injury: a light and electron microscopic study in rats. J. Neurotrauma. 1994;11:289–301. doi: 10.1089/neu.1994.11.289. [DOI] [PubMed] [Google Scholar]
- 53.Tanno H. Nockels R.P. Pitts L.H. Noble L.J. Breakdown of the blood-brain barrier after fluid percussive brain injury in the rat. Part 1: Distribution and time course of protein extravasation. J. Neurotrauma. 1992;9:21–32. doi: 10.1089/neu.1992.9.21. [DOI] [PubMed] [Google Scholar]
- 54.Lo E.H. Pan Y. Matsumoto K. Kowall N.W. Blood-brain barrier disruption in experimental focal ischemia: comparison between in vivo MRI and immunocytochemistry. Magn. Reson. Imaging. 1994;12:403–411. doi: 10.1016/0730-725x(94)92533-x. [DOI] [PubMed] [Google Scholar]
- 55.Hoshino S. Kobayashi S. Nakazawa S. Prolonged and extensive IgG immunoreactivity after severe fluid-percussion injury in rat brain. Brain Res. 1996;711:73–83. doi: 10.1016/0006-8993(95)01329-6. [DOI] [PubMed] [Google Scholar]
- 56.Michalak Z. Lebrun A. Di Miceli M. Rousset M.C. Crespel A. Coubes P. Henshall D.C. Lerner-Natoli M. Rigau V. IgG leakage may contribute to neuronal dysfunction in drug-refractory epilepsies with blood-brain barrier disruption. J. Neuropathol. Exp. Neurol. 2012;71:826–838. doi: 10.1097/NEN.0b013e31826809a6. [DOI] [PubMed] [Google Scholar]
- 57.Loberg E.M. Torvik A. Uptake of plasma proteins into damaged neurons. An experimental study on cryogenic lesions in rats. Acta Neuropathol. 1991;81:479–485. doi: 10.1007/BF00310126. [DOI] [PubMed] [Google Scholar]
- 58.Abdul-Muneer P.M. Schuetz H. Wang F. Skotak M. Jones J. Gorantia S. Zimmerman M.C. Chandra N. Haorah J. Induction of oxidative and nitrosative damage leads to cerebrovascular inflammation in animal model of mild traumatic brain injury induced by primary blast. Free Radical Biol. Med. 2013;60:282–291. doi: 10.1016/j.freeradbiomed.2013.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Uversky V.N. Oldfield C.J. Midic U. Xie H. Xue B. Vucetic S. Iakoucheva L.M. Obradovic Z. Dunker A.K. Unfoldomics of human diseases: linking protein intrinsic disorder with diseases. BMC Genomics. 2009;10(Suppl 1):S7. doi: 10.1186/1471-2164-10-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]