

Key Points
CD30 expression defines a novel and unique subgroup of DLBCL with favorable clinical outcome and distinct gene expression signature.
Abstract
CD30, originally identified as a cell-surface marker of Reed-Sternberg and Hodgkin cells of classical Hodgkin lymphoma, is also expressed by several types of non-Hodgkin lymphoma, including a subset of diffuse large B-cell lymphoma (DLBCL). However, the prognostic and biological importance of CD30 expression in DLBCL is unknown. Here we report that CD30 expression is a favorable prognostic factor in a cohort of 903 de novo DLBCL patients. CD30 was expressed in ∼14% of DLBCL patients. Patients with CD30+ DLBCL had superior 5-year overall survival (CD30+, 79% vs CD30–, 59%; P = .001) and progression-free survival (P = .003). The favorable outcome of CD30 expression was maintained in both the germinal center B-cell and activated B-cell subtypes. Gene expression profiling revealed the upregulation of genes encoding negative regulators of nuclear factor κB activation and lymphocyte survival, and downregulation of genes encoding B-cell receptor signaling and proliferation, as well as prominent cytokine and stromal signatures in CD30+ DLBCL patients, suggesting a distinct molecular basis for its favorable outcome. Given the superior prognostic value, unique gene expression signature, and significant value of CD30 as a therapeutic target for brentuximab vedotin in ongoing successful clinical trials, it seems appropriate to consider CD30+ DLBCL as a distinct subgroup of DLBCL.
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most common and one of most heterogeneous types of non-Hodgkin lymphoma. It is categorized into different morphologic variants, immunohistochemical and molecular subgroups, and distinct subtypes/entities in the current World Health Organization classification.1 According to gene expression signatures or immunohistochemistry (IHC) using several algorithms as surrogates, most DLBCL cases can be placed into prognostically favorable germinal center B-cell–like (GCB) or prognostically unfavorable activated B-cell–like (ABC) subtypes.2-9 However, these immunophenotypic algorithms may not always correlate well with the gene expression profiling (GEP) results and may show poor prognostic power.10-13 Although the cell of origin (COO) stratification by gene expression signatures is able to provide a general perspective of clinical outcome, currently it is not practical to perform GEP routinely in the clinical setting. Furthermore, both subtypes of DLBCL are heterogeneous and contain additional subgroups, which have different prognoses and may require different therapeutic approaches. For example, the GCB subtype generally carries a favorable prognosis, but it also includes MYC/BCL2 double-hit B-cell lymphoma, which has an extremely aggressive clinical course.14,15 Therefore, it is critical to further stratify cases of DLBCL into biologically similar and clinically meaningful subgroups, which will not only guide prognostic assessment and facilitate therapeutic decisions, but also stimulate further research to understand pathogenesis and develop potential novel treatments.
CD30, a member of the tumor necrosis factor receptor (TNFR) superfamily, was originally identified as a cell-surface marker of Reed-Sternberg and Hodgkin cells in classical Hodgkin lymphoma.16 CD30 is also expressed by several types of T- and B-cell non-Hodgkin lymphoma, such as anaplastic large cell lymphoma (ALCL), primary mediastinal large B-cell lymphoma (PMBCL), and Epstein-Barr virus (EBV)–driven clonal lymphoproliferative disorders, as well as in reactive conditions, such as infectious mononucleosis.17 CD30 is an activation marker inducible in vitro by mitogenic signals and is normally expressed by T and B immunoblasts in the parafollicular region and the peripheral rim of germinal centers in healthy individuals. This highly restricted distribution of CD30 expression makes it an ideal target for monoclonal antibody therapy in patients with CD30+ lymphomas. Brentuximab vedotin, an anti-CD30 monoclonal antibody-drug conjugate recently approved by the US Food and Drug Administration, has produced marked responses in relapsed and refractory Hodgkin lymphoma and systemic ALCL.18-22
Compared with CD30– T-cell or B-cell lymphoma, the favorable prognoses of ALCL and PMBCL are well established.23-27 However, the significance of CD30 expression in DLBCL, not otherwise specified (DLBCL-NOS), remains unknown. Early studies in small cohorts of patients treated with cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) chemotherapy reported inconsistent results.28,29 The addition of rituximab to the standard CHOP regimen (R-CHOP) has resulted in improved survival of patients with DLBCL.30-32 In this study, we assessed CD30 expression in a large cohort of 903 de novo DLBCL patients who were treated with R-CHOP immunochemotherapy and correlated CD30 expression with patients’ clinicopathological features, molecular and genetic changes, gene expression profiles, and clinical outcome. Our results indicate that CD30 expression identifies a novel subgroup of DLBCL patients with superior clinical outcome.
Materials and methods
Patient selection
We studied 903 cases of de novo DLBCL patients who were treated with R-CHOP chemotherapy (including 461 cases in the training set and 442 in the validation set). These cases were organized as a part of the International DLBCL Rituximab-CHOP Consortium Program Study.9,33 All cases were diagnosed according to the World Health Organization classification criteria and were centrally reviewed by a group of hematopathologists (primary pathologists, A.T., S.M.M., M.B.M., M.A.P., and K.H.Y.). Cases were excluded if the patients had a history of low-grade B-cell lymphoma, acquired immunodeficiency, primary cutaneous DLBCL, primary central nervous system DLBCL, and PMBCL. Cases of EBV+ DLBCL were analyzed separately from those of EBV– DLBCL given their different clinical behavior. The study, conducted in accordance with the Declaration of Helsinki, was reviewed and approved by the Institutional Review Boards of each participating center, and the comprehensive collaborative study was approved by the Institutional Review Board at The University of Texas MD Anderson Cancer Center.9,33
Tissue microarray (TMA) IHC and in situ hybridization for EBV-encoded RNA
Hematoxylin-eosin stained slides from each of the 903 cases of DLBCL were reviewed, and TMAs were constructed from tumor cell–rich areas. IHC studies for a variety of markers using a streptavidin-biotin complex technique and in situ hybridization for EBV-encoded RNA (EBER) were performed on 4-μm TMA sections in all cases. The markers assessed included CD30, CD10, B-cell lymphoma 6 protein (Bcl-6), Germinal Center B cell-expressed Transcript-1 (GCET1), Multiple Myeloma Oncogene 1 (MUM1), Forkhead box protein P1 (FOXP1), B-cell lymphoma 2 (Bcl-2), Myc, p53, p21, and Ki-67. Staining was not always achieved for each marker owing to tissue exhaustion. A cutoff value for each marker was established from analysis of receiver-operating characteristic curves to achieve maximum specificity and sensitivity as described previously.9,33 The cutoff scores for these markers were as follows: 20% for CD30, 30% for CD10, 30% for Bcl-6, 60% for GCET1, 60% for MUM1, 60% for FOXP1, 40% for Myc, 70% for Bcl-2, 20% for p53, and 20% for p21. The cutoff value for Ki-67 was 70%, the median value of Ki-67 for all cases in the training set.
GEP
Total RNA was extracted from each of the 445 cases in the training set using the HighPure RNA Extraction Kit (Roche Applied Science, Indianapolis, IN) and subjected to GEP as previously described.9,33 For data analysis and classification, the microarray DQN signals were generated and normalized to the quantiles of β distribution. DQN is an ideal expression algorithm used for expression microarray analysis and represents the non-central trimmed mean of differences between perfect match and mismatch intensities with quantile normalization. A Bayesian model was also used to determine the classification probability.34 The methodology developed in this study has been validated with the Lymphoma Leukemia Molecular Profiling Program dataset in the Gene Expression Omnibus Genomic Spatial Event database #10846, which has 181 CHOP-treated and 233 R-CHOP–treated DLBCL patients. We obtained an 80% concordance rate of classification for all 3 classes (GCB, ABC, and unclassified) and a 97% rate for 2 classes (GCB and ABC), if excluding the unclassified. We required a percentage call rate ≥12% for the project with a failure rate of 10.64%. The 445 cases with GEP in this study were part of a larger data set on which profiling was successfully performed and validated.
COO classification
COO classification was achieved by combining GEP (considered the “gold standard”) and IHC data. Briefly, GEP was performed in 445 cases in the training set, and 442 cases in the validation set. IHC was performed for all 461 cases in the training set and all 442 cases in the validation set. In the training set, 38 cases not classifiable by GEP and 16 additional cases for which GEP was not performed, as well as all the cases in the validation set were classified by IHC methods according to the Visco-Young algorithm.9 The GEP-IHC correlation of COO classification was 87.3% in GCB, 86.1% in ABC, and 86.7% overall.34
Fluorescence in situ hybridization (FISH) for MYC, BCL2, and BCL6
FISH analysis was performed on formalin-fixed paraffin-embedded tissue sections in each of the 903 cases of DLBCL using BCL2 and BCL6 dual-color break-apart probes (Vysis, Downers Grove, IL) as described previously.9 FISH for MYC was performed with a locus-specific IGH/MYC/CEP8 tricolor dual-fusion probes and a locus-specific MYC dual-color break-apart probe (Vysis). A copy number of threefold to sixfold increase was considered as low-level amplification, and greater than sixfold increase or tight clusters of at least 5 gene signals as high-level amplification.
Sequencing of TP53
Genomic DNA and total RNA were extracted from formalin-fixed paraffin-embedded tissue samples from each of the 461 cases in the training set. DNA samples were used for TP53 exon sequencing using p53 AmpliChip (Roche Molecular Systems, Pleasanton, CA).33
Statistical analysis
Clinical and laboratory features at time of presentation between different DLBCL subgroups were compared using the χ2 test and the Spearman rank correlation. Overall survival (OS) duration was calculated from the date of diagnosis to the date of last follow-up or death. Progression-free survival (PFS) duration was calculated from the date of diagnosis to the time of progression or death. Kaplan-Meier survival curves were used to estimate OS and PFS rates, and the log-rank (Mantel-Cox) test was used to assess differences in survival between groups. Multivariate analysis for survival was performed on IBM statistics SPSS 19 using the Cox proportional hazards regression model. All differences with P ≤ .05 were considered to be statistically significant.
Results
A total of 461 cases of de novo DLBCL patients treated with R-CHOP were included in the training set for further analysis. These cases were selected from a data set of 503 cases of de novo DLBCL in the consortium program after excluding cases of EBV+ DLBCL (19 cases), cases with unknown CD30 expression status owing to tissue exhaustion in the TMA (11 cases), patients with a mediastinal mass regardless of the pathological diagnosis (10 cases), and those with a cervical mass but for whom a diagnosis of PMBCL was entertained (2 cases).
Clinical and pathological features
The clinical and pathological features of 461 cases of EBER– DLBCL in the training set are listed in Table 1. Briefly, there were 270 men and 191 women (male/female, 1.4:1) with a median age of 64 years (range, 16-92). Approximately one-third of the patients presented with B symptoms, and slightly over half (53%) had advanced Ann Arbor stages. Most of the patients had good performance status (Eastern Cooperative Oncology Group score 0-1, 87%), elevated serum lactate dehydrogenase level (61%), and low or low-intermediate International Prognostic Index (IPI) risk (IPI score 0-2, 63%). Involvement of multiple extranodal sites (>2) was seen in 22% of cases, and bulky tumor size >6 cm was seen in 32% of cases. The vast majority (89%) of the patients achieved complete or partial remission after R-CHOP therapy. The median follow-up time was 57 months.
Table 1.
Clinical characteristics and outcome of 461 cases of de novo DLBCL treated with R-CHOP immunochemotherapy
| Overall | CD30+ | CD30– | ||||
|---|---|---|---|---|---|---|
| N (%) | OS (P value) | PFS (P value) | N (%) | N (%) | P value | |
| Patients | 461 (100) | 65 (100) | 396 (100) | |||
| Gender | ||||||
| Male | 270 (59) | .9688 | .6409 | 38 (58) | 232 (59) | .9850 |
| Female | 191 (41) | 27 (42) | 164 (41) | |||
| Age | ||||||
| ≤60 | 188 (41) | .0011 | .0047 | 24 (37) | 164 (41) | .4947 |
| >60 | 273 (59) | 41 (63) | 232 (59) | |||
| B symptoms* | ||||||
| Absence | 276 (69) | .0003 | .0003 | 41 (68) | 235 (70) | .8534 |
| Presence | 122 (31) | 19 (32) | 103 (30) | |||
| ECOG performance status* | ||||||
| <2 | 344 (87) | <.0001 | <.0001 | 50 (93) | 294 (86) | .1942 |
| ≥2 | 51 (13) | 4 (7) | 47 (14) | |||
| Stage* | ||||||
| I-II | 207 (47) | <.0001 | <.0001 | 34 (53) | 173 (46) | .2674 |
| III-IV | 236 (53) | 30 (47) | 206 (54) | |||
| Extranodal sites* | ||||||
| <2 | 341 (78) | <.0001 | <.0001 | 51 (85) | 290 (77) | .1605 |
| ≥2 | 96 (22) | 9 (15) | 87 (23) | |||
| LDH* | ||||||
| Normal | 160 (39) | .0017 | .0002 | 23 (40) | 137 (39) | .8775 |
| Elevated | 253 (61) | 35 (60) | 218 (61) | |||
| IPI risk group* | ||||||
| 0-2 | 256 (63) | <.0001 | <.0001 | 40 (73) | 216 (61) | .1047 |
| 3-5 | 151 (37) | 15 (27) | 136 (39) | |||
| Tumor size (cm)* | ||||||
| <6 | 222 (68) | .0296 | .0279 | 29 (71) | 193 (68) | .6988 |
| ≥6 | 104 (32) | 12 (29) | 92 (32) | |||
| Treatment response | ||||||
| CR/PR | 409 (89) | <.0001 | <.0001 | 62 (95) | 347 (88) | .0669 |
| No response | 52 (11) | 3 (5) | 49 (12) | |||
CR, complete remission; LDH, lactate dehydrogenase; PR, partial remission; ECOG, eastern cooperative oncology group.
Clinical information not available in some cases.
Of the 461 cases, 235 (51%) and 223 (49%) were stratified as GCB and ABC subtypes, respectively, by GEP supplemented by IHC (Table 2). The COO classification of 3 cases was not known because GEP was not done, and IHC studies were not successful owing to TMA tissue exhaustion.
Table 2.
Pathogenetic characteristics of 461 cases of de novo DLBCL treated with R-CHOP immunochemotherapy
| Overall | CD30+ | CD30– | P value | |
|---|---|---|---|---|
| COO classification | ||||
| GCB | 235 (51%) | 38 (59%) | 197 (50%) | .1640 |
| ABC | 223 (49%) | 26 (41%) | 197 (50%) | |
| BCL2 expression | 228/454 (50%) | 26/65 (40%) | 202/389 (52%) | .0750 |
| BCL6 expression | 369/449 (82%) | 51/62 (82%) | 318/387 (82%) | .9867 |
| MYC expression | 295/455 (65%) | 38/64 (59%) | 257/391 (66%) | .3237 |
| MYC/BCL2 coexpression | 155/452 (34%) | 15/64 (23%) | 140/388 (36%) | .0483 |
| MYC/BCL6 coexpression | 249/445 (56%) | 33/64 (52%) | 216/381 (57%) | .4443 |
| BCL2 aberration | 129/395 (33%) | 14/59 (24%) | 115/336 (34%) | .1128 |
| MYC aberration | 33/303 (11%) | 0/46 (0%) | 33/257 (13%) | .0100 |
| BCL6 aberration | 202/294 (69%) | 24/42 (57%) | 178/252 (71%) | .0808 |
| TP53 mutation | 107/461 (23%) | 11/65 (17%) | 96/396 (24%) | .1952 |
| P53 expression | 163/449 (36%) | 28/62 (45%) | 135/387 (35%) | .1182 |
| P21 expression | 57/449 (13%) | 15/62 (24%) | 42/387 (11%) | .0034 |
| Ki-67 | 303/454 (67%) | 36/64 (56%) | 267/390 (68%) | .0546 |
Cutoff for each of the biomarkers is detailed in the “Materials and methods” section.
CD30 expression was associated with favorable prognosis in DLBCL
Of the 461 cases in the training set, CD30 was positive in 65 cases (14%), of which 38 were stratified as GCB, 26 as ABC, and 1 was unclassified (Figure 1A-H; Table 2). The training set excluded those with concurrent CD30 expression and EBV infection, which were analyzed separately because of apparently different clinical behavior (Figure 1I-L).
Figure 1.
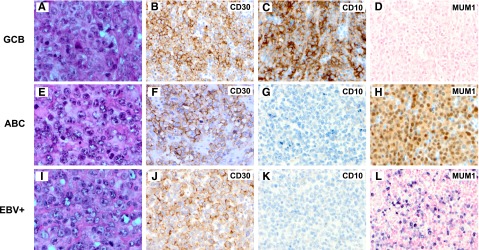
Morphology and immunophenotype of CD30+ de novo DLBCL. (A-D) A case of CD30+ DLBCL of the GCB subtype (panel A, H&E; panel B, CD30; panel C, CD10; and panel D, EBER in situ hybridization). (E-H) A case of CD30+ DLBCL of the ABC subtype (panel E, H&E; panel F, CD30; panel G, CD10; and panel H, MUM1). (I-L) A case of CD30+EBV+ DLBCL (panel I, H&E; panel J, CD30; panel K, CD10; and panel L, EBER in situ hybridization). Original magnification ×1000 (A,E,I) and ×500 (all others).
Morphologically, most (56/65, 85%) CD30+ DLBCL cases showed centroblastic features. Eight cases demonstrated pleomorphic centroblastic/anaplastic morphology. However, it was inherently difficult to reliably distinguish them. One case showed immunoblastic features. Most of these cases were diagnosed on the biopsy specimens of lymph nodes (49/65, 75%), obtained from the abdominal/retroperitoneal, inguinal, neck (nonsupraclavicular), or axillary regions. The extranodal biopsies of the remaining 16 cases included those from soft tissue (5); tonsil (2); gastrointestinal tract (2); and breast, kidney, liver, nasal sinus, orbit, salivary gland, and spleen (1 case each).
After a median follow-up time of 57 months, patients with CD30+ DLBCL showed significantly superior OS and PFS compared with those with CD30– DLBCL (Figure 2A-B). The 5-year OS was 79% in patients with CD30+ vs 59% in CD30– DLBCL (P = .0013). The 5-year PFS was 73% in patients with CD30+ vs 57% in CD30– DLBCL (P = .0030). The favorable prognostic impact of CD30 expression was validated in an independent cohort of de novo DLBCL patients treated with R-CHOP and was more significant when combined training and validation sets were analyzed (Figure 2C-D; supplemental Table 1, see the Blood Web site).
Figure 2.

Prognostic impact of CD30 expression in de novo DLBCL. (A-B) OS (A) and PFS (B) of patients with CD30+ vs CD30– DLBCL in the training set. (C) OS of patients with CD30+ vs CD30– DLBCL in the validation set. These patients were part of an independent cohort of 442 patients with available survival information (supplementary Table 1). (D) OS of patients with CD30+ vs CD30– DLBCL in combined training and validation sets.
When the GCB and ABC subtypes were analyzed separately, in the GCB subtype, patients with CD30+ DLBCL had significantly better OS (P = .0177) and PFS (P = .0188) than those with CD30– DLBCL (Figure 3A-B). There was a trend toward better OS (P = .0623) and PFS (P = .1498) in patients with CD30+ DLBCL of the ABC subtype (Figure 3D-E). The lack of statistical significance in the ABC subtype was apparently due to the lower number of CD30+ cases in this subtype. When combining the training and validation sets, CD30 expression was a significant predictor of superior survival in both COO subtypes (Figure 3C,F).
Figure 3.

Prognostic impact of CD30 expression in COO subtypes of DLBCL. (A-B) OS (A) and PFS (B) of patients with CD30+ DLBCL of the GCB subtype in the training set. (C) OS of patients with CD30+ DLBCL of the GCB subtype in the combined training and validation sets. (D-E) OS (D) and PFS (E) of patients with CD30+ DLBCL of the ABC subtype in the training set. (F) OS of patients with CD30+ DLBCL of the ABC subtype in the combined training and validation sets.
Concurrent Myc and Bcl-2 protein expression was previously shown to be a strong predictor of patient survival in DLBCL.35,36 When all DLBCL cases were stratified into 2 subgroups with or without Myc/Bcl-2 coexpression, patients with CD30+ DLBCL had a significant better survival than patients with CD30– DLBCL in both subgroups (supplemental Figure 1). However, CD30+ DLBCL with Myc/Bcl-2 coexpression shows similar survival to CD30– DLBCL without Myc/Bcl-2 coexpression, consistent with the antagonistic prognostic impact of CD30 expression and Myc/Bcl-2 double positivity in DLBCL.
In multivariate analysis, controlling other variables, including B symptoms, tumor size, IPI risk, COO classification, and TP53 mutational status, the lack of CD30 expression remained a significant independent predictor of inferior OS (hazard ratio [HR] of 3.03; 95% confidence interval [CI], 1.33-6.89; P = .0082) and PFS (HR of 2.89; 95% CI, 1.35-6.20; P = .0064) (Table 3).
Table 3.
Multivariate analysis of clinicopathological parameters in de novo DLBCL treated with R-CHOP immunochemotherapy
| OS | PFS | |||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P value | HR | 95% CI | P value | |
| B symptoms | 1.36 | 0.95-1.94 | .0975 | 1.38 | 0.97-1.95 | .0712 |
| Tumor size ≥6 cm | 1.16 | 0.82-1.64 | .4154 | 1.13 | 0.81-1.58 | .4757 |
| IPI score >2 | 2.83 | 1.97-4.05 | <.0001 | 2.59 | 1.84-3.66 | <.0001 |
| ABC subtype | 1.56 | 1.08-2.24 | .0165 | 1.54 | 1.08-2.18 | .0157 |
| Negative CD30 IHC | 3.03 | 1.33-6.89 | .0082 | 2.89 | 1.35-6.20 | .0064 |
| TP53 mutation | 1.96 | 1.33-2.90 | .0007 | 1.84 | 1.26-2.69 | .0016 |
Clinical, pathological, and genetic features of CD30+ vs CD30– DLBCL
Various clinical characteristics were compared between patients with CD30+ and CD30– DLBCL (Table 1). These 2 groups did not significantly differ in any of the clinical parameters examined. CD30+ DLBCL was associated with a trend toward a lower rate of treatment failure (no response: 5% vs 12%, P = .0669; no response and partial response: 15% vs 26%, P = .0769).
We then assessed a variety of molecular and genetic factors, including expression of Bcl-2, Bcl6, Myc, p53, and p21 by IHC; gene alterations of BCL2, BCL6, and MYC by FISH; and TP53 mutations by sequencing in the 2 groups (Table 2). Compared with CD30– DLBCL, CD30+ DLBCL showed a lower frequency of Myc/Bcl-2 coexpression (23% vs 36%, P = .0483), a higher frequency of p21 expression (24% vs 11%, P = .0034), and no MYC alterations detected by FISH (rearrangement or amplification) (0% vs 13%, P = .0100). However, when all cases with Myc/Bcl-2 coexpression, p21 expression, or MYC gene alterations were excluded from both groups, patients with CD30+ DLBCL still maintained a favorable OS and PFS (supplemental Figure 2). These results suggest that these 3 factors may not be major factors contributing to the favorable prognosis of patients with CD30+ DLBCL. There was no significant difference in the frequency of p53 protein expression or TP53 gene mutation between the 2 groups (Table 2). A trend toward a lower frequency of Bcl-2 expression and BCL6 rearrangement or amplification, and a lower Ki-67 index were seen in CD30+ DLBCL. However, Bcl-2 expression alone, Ki-67 index, or BCL6 alterations did not affect patient survival (data not shown).
Gene expression signature of CD30+ DLBCL
To identify additional molecular mechanisms underlying the favorable prognosis of patients with CD30+ DLBCL, we compared the GEP signatures of CD30+ and CD30– DLBCL (Figure 4). A total of 164 independent genes corresponding to 231 array probe sets were differentially expressed between the 2 groups (P < .001), including 99 genes upregulated and 65 downregulated in CD30+ DLBCL compared with CD30– DLBCL (supplemental Table 2).
Figure 4.

Gene expression profiles and GSEA analysis of CD30+ vs CD30– DLBCL. (A) Comparison of gene expression profiles between CD30+ and CD30– DLBCL. (B) GSEA for differentially expressed genes of cytokine and TNFR pathways was performed on Kyoto Encyclopedia of Genes and Genomes pathway gene sets using all the probe sets that are associated with a known gene. The gene sets with a false discovery rate q value <0.05 after performing 1000 permutations were considered to be significantly enriched.
Among the genes with high-level expression in CD30+ DLBCL, 9 were cytokines/cytokine receptors and were involved in cytokine pathways, including IL-1R1, IL-13-Rα1, IL-21R, IL-12B, CCR7, SOCS1, IRF5, NF1B, and IER2 (Table 4; supplemental Table 2A). Seven genes encoded TNFR family members, signaling proteins, or induced proteins. CD30 and FAS induce apoptosis. TRAF1, TNFAIP3, TNFAIP6, TNIP2, and IKBα encode inhibitors of nuclear factor κB (NF-κB) activation. Fifteen genes were involved in cell-growth regulation; SLP76/LCP2, LMO2, and CDK6 positively regulated cell growth, whereas DUSP4, NXN, and SAMSN1 negatively regulated mitogen-activated protein kinase, Wnt, and B-cell receptor signaling pathways, respectively. Two HLA genes (HLA-A/F/J and HLA-DQB1) were also upregulated. Twenty genes encoded various extracellular matrix proteins, cell adhesion molecules, and molecules involved in cytoskeletal reorganization.
Table 4.
Differentially expressed genes in CD30+ DLBCL vs CD30– DLBCL
| Gene functional categories | No. of genes | Representative genes |
|---|---|---|
| Genes upregulated in CD30+ DLBCL | ||
| Cytokines and cytokine pathways | 9 | IL-1R1, IL-12B, IL-13RA1, IL-21R, CCR7, IRF5, SOCS1, IER2, NFIB |
| TNFR family and pathways | 7 | CD30, FAS, TRAF1, NFKBIA, TNFAIP3, TNFAIP6, TNIP2 |
| Cell growth regulation | 15 | LMO2, SLP76/LCP2, PBX4, DUSP ID2, TUSC1, MIR21, CDK6 |
| MHC class II components | 2 | HLA-A /F/J, HLA-DQB1 |
| Extracelluar matrix, adhesion, and cytoskeletal organization | 20 | CDH11, THY1, CD58, CD99, TIAM2 |
| Others, including unknown | 46 | CHIC2, SOX4, SMARCA2, SUMO3 |
| Genes downregulated in CD30+ DLBCL | ||
| B-cell receptor signaling and proliferation | 14 | IGHM, IGL@, PKCB, NF-ATC1, BACH2, TCL1A, FOXP1, MIR17HG, SH2D3C |
| Cell growth regulation | 9 | CDCA7L, FZD5, PTCHD1, BTG2 |
| Extracellular matrix, adhesion, and cytoskeletal organization | 7 | ST14, SSX2IP, GPR110 |
| Others, including unknown | 35 | ARF5, GABRB2, XK, ARF5 |
Among these downregulated genes in CD30+ DLBCL, the most prominent signature was 14 genes involved in B-cell receptor signaling and proliferation/differentiation, including IGHM, IGL@, CD72, PKCβ, SH2D3C, NFATc1, BACH2, FOXP1, and MIR17HG, as well as 9 genes involved in cell growth (less lymphocyte specific), such as CDCA7L and FZD5 (Wnt receptor) (Table 4; supplemental Table 2B).
Gene set enrichment analysis (GSEA) reinforced the molecular association of CD30+ DLBCL with the enrichment of genes related to cytokine and TNFR pathways, cell adhesion molecules, and B-cell receptor signaling, although the association with cytokine/TNFR pathways and cell adhesion was more striking (Figure 4B and data not shown).
We further compared the GEPs of CD30+ vs CD30– DLBCL according to COO subtypes (supplemental Tables 3 and 4). A total of 52 genes (corresponding to 80 probe sets) were differentially expressed in GCB CD30+DLBCL, and 87 genes (corresponding to 139 probe sets) in ABC CD30+ DLBCL. The majority of these genes were also present in the GEP of CD30+ DLBCL when all GCB and ABC cases were combined with a few exceptions. CD40LG and CD80 were upregulated, and IGJ downregulated in CD30+ DLBCL of the GCB subtype (supplemental Table 3). CD30+ DLBCL of the ABC subtype showed increased expression of additional genes in the cytokine pathway (CXCL13, CCL17, IL7R, and IRAK2) and decreased expression of BCL2 and CD24 (supplemental Table 4).
CD30+EBV+ DLBCL is a unique subset of lymphoma with aggressive clinical course
Of the total 503 cases in the original data set from which the training set was derived, 484 were successfully evaluated for EBV infection by in situ hybridization, and 19 cases (4%) were positive for EBER. Eight of these 19 cases (42%) were also positive for CD30. Thus, EBV+ DLBCL more frequently showed CD30 expression than EBV– DLBCL (42% vs 14%, P = .0009). Realizing the prognostic impact of EBV infection in CD30+ DLBCL in this data set, 30 additional CD30+EBV+ DLBCL cases were included from the consortium program. Patients with CD30+EBV+ DLBCL had markedly worse OS (P < .0001; Figure 5A) and PFS (P < .0001) than those with CD30+EBER– DLBCL.
Figure 5.
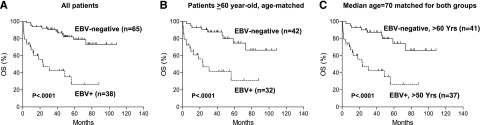
Prognostic impact of concurrent EBV infection in CD30+ DLBCL. (A) OS of all CD30+EBER+ vs all CD30+EBER– DLBCL patients. Age range, 52 to 91 for EBV+ group; 16 to 84 for EBV– group. (B) OS of CD30+EBER+ vs CD30+EBER– DLBCL patients in >60-year-old age group. Median age, 73 for EBV+ group; 70 for EBV– group. (C) OS of CD30+EBV+ DLBCL patients in >50-year-old age group vs CD30+EBV– DLBCL in >60-year-old age group. Median age, 70 for both groups.
Because CD30+EBV+ DLBCL cases were associated with advanced age, we then stratified patients with CD30+EBV+ or CD30+EBV– DLBCL into subgroups according to age (>50 years old, >60 years old, and >70 years old). Compared with CD30+EBV– DLBCL, CD30+EBV+ DLBCL had significantly poorer survival in all age subgroups (Figure 5B and data not shown). Given that the median age of CD30+EBV+ DLBCL was 1 to 4 years older than that of CD30+EBV– DLBCL in each age subgroup, we further compared >50-year-old patients with CD30+EBV+ DLBCL vs >60-year-old patients with CD30+EBV– DLBCL because both groups had the same median ages. Clearly, CD30+EBV+ DLBCL showed inferior OS (P < .0001; Figure 5C) and PFS (P = .0001). Furthermore, >60-year-old patients CD30+EBV+ DLBCL had a significantly worse survival than >70-year-old patients with CD30+EBV– DLBCL despite a lower median age in the CD30+EBV+ subgroup. These studies suggest that advanced age is not the major contributing factor to the dismal survival of patients with CD30+EBV+ DLBCL.
Furthermore, patients with CD30+EBV+ DLBCL had inferior clinical outcome compared with those with CD30–EBER+ DLBCL despite the favorable prognosis conferred by CD30 expression, suggesting a synergistic adverse impact between CD30 expression and EBV infection in this group of patients (data not shown).
Discussion
In this study, we demonstrated that patients with CD30+ DLBCL had superior OS and PFS regardless of COO stratification in a cohort of 903 patients with de novo DLBCL treated with R-CHOP. CD30 was expressed in ∼14% of DLBCL cases. CD30+ and CD30– groups of DLBCL had similar age distributions as well as many other similar clinicopathological characteristics. It is unlikely that the observed prognostic difference between these 2 groups is due to a difference in patient selection.
Unlike Fas or TNFR1, 2 other members of TNFR superfamily, CD30 does not contain a cytoplasmic death domain. Interestingly, studies performed in various cell lines showed that activation of CD30 signals cell death or cell-cycle arrest in addition to cell activation.37-39 Multiple in vivo studies in mouse models found that CD30 is required for lymphocyte apoptosis.40,41 The findings that expression of CD30 and CD30 ligand correlates with the regression of lymphomatoid papulosis and cutaneous ALCL further support the antiproliferative role of CD30.42 However, many of these studies were performed in T cells and Hodgkin lymphoma cells in addition to B cells.
The molecular mechanism of the antiproliferative function of CD30 is not known. In our GEP studies of CD30+ DLBCL, we observed the upregulation of death receptor FAS and downregulation of some oncogenes, such as CDCA7L, FZD5, and DTX1. Most importantly, multiple genes encoding negative regulators of the NF-κB pathway and lymphocyte survival were upregulated, including I-κBα, TNFAIP3, TNFRAIP6, TNIP2, TRAF1, DUSP4, NXN, SAMSN1, and SOCS1. Furthermore, multiple genes encoding components of B-cell receptor signaling and proliferation were downregulated, including IgH μ, IgL@, CD72, PKCβ, SH2D3C, NF-ATc1, TCL1A, FOXP1, and BACH2. The inhibition of these pathways may render lymphoma cells susceptible to chemotherapy-induced apoptosis.
Interestingly, the GEP of CD30+ DLBCL partially overlaps with that of PMBCL reported by Savage et al43 and Rosenwald et al. 44 IL-13R, FAS, TRAF1, TNFAIP3, and TNFRAIP6 are among those upregulated, and IgM, IgL@, PKCβ, NF-ATc1, and FOXP1 among those downregulated in both types of lymphoma. However, the gene expression signatures of these 2 types of lymphoma were apparently different. First, they demonstrated different cytokine profiles. Of the total 21 differentially expressed genes in this functional category (12 genes upregulated in PMBCL and 9 upregulated in CD30+ DLBCL), IL-13Rα1 was the only one present in both types of lymphoma. Second, several major histocompatibility complex (MHC) components were downregulated in PMBCL but upregulated in CD30+ DLBCL. Loss of MHC components is associated with poor outcome. Third, although both showed an incomplete B-cell program, PMBCL appeared to lose more B-cell markers. These markers downregulated in PMBCL but not in CD30+ DLBCL included CD22; cytoplasmic signaling molecules BLK, SAB, BLNK, and AKT-1; as well as the transcription factors SPIB and IKAROS. Fourth, multiple costimulatory molecules upregulated in PMBCL, such as CD86 and SLAM, were not upregulated in CD30+ DLBCL. Rosenwald et al44 used a 46-gene signature to distinguish PMBCL from de novo DLBCL, including 35 genes highly expressed in PMBCL and 11 highly expressed in DLBCL. Of these 46 differentially expressed genes, only 6 were identified in our CD30+ DLBCL GEP studies, including 5 (CD30, FAS, TRAF1, SAMSN1, and FNBP1) upregulated and 1 (TCL1A) downregulated in CD30+ DLBCL. These results show that CD30+ DLBCL is more related to CD30– DLBCL than to PMBCL in gene expression signature. Recurrent genetic alterations in PMBCL, such as CIITA break and STAT6 mutation, are rare in DLBCL.45,46 Despite the distant relatedness between CD30+ DLBCL and PMBCL, it will be interesting to investigate whether CD30+ DLBCL also harbors these genetic changes.
The finding of an incomplete B-cell program in CD30+ DLBCL is quite intriguing. In addition to Hodgkin lymphoma and PMBCL, an incomplete B-cell program is also seen in several other types of B-cell lymphoma, such as primary effusion lymphoma, plasmablastic lymphoma, some cases of EBV+ DLBCL, and here CD30+ DLBCL. All of them are associated with CD30 expression. Similarly, several types of CD30+ peripheral T-cell lymphoma also show variable loss of the T-cell program, including a subset of CD30+ peripheral T-cell lymphoma NOS, primary cutaneous CD30+ lymphoproliferative disorders, as well as systemic ALCL (ALK+ and ALK–).47-49 However, it is not clear whether there is causal relationship between CD30 expression and the loss of B- or T-cell program.
Regardless, CD30 expression seems to be associated with lymphomas with generally favorable outcomes, such as ALCL (vs peripheral T-cell lymphoma NOS) and PMBCL (vs DLBCL-NOS), and here, we have demonstrated this trend for CD30+ DLBCL (vs CD30– DLBCL). However, CD30+EBV+ DLBCL had an extremely aggressive clinical course despite the favorable prognostic impact conferred by CD30 expression. EBV+ DLBCL has been shown to have prominent NF-κB activation.50 How CD30 synergistically functions with EBV infection is not known. Further investigation in a larger cohort of patients is warranted.
Great strides have been made recently in the management of refractory/relapsed classical Hodgkin lymphoma and ALCL with the development of the antibody-drug conjugate brentuximab vedotin. The agent’s dramatic effectiveness and manageable toxicity raise the issue of its potential application to other CD30+ lymphomas, including CD30+ DLBCL. In light of the unique gene expression signature, the prognostic value associated with CD30 expression, and the ongoing successful clinical trials of brentuximab vedotin in DLBCL, it seems advisable to consider CD30+ DLBCL as a distinct subgroup of DLBCL.
Supplementary Material
Acknowledgments
The authors thank the consortium program team of pathologists, hematologists, and clinicians, and each of the contributing center principal physicians for their support. The authors thank the patients and former and current hematopathology and hematology/oncology fellows for their support. The DLBCL Rituximab-CHOP Consortium Program’s principal investigation center is at The University of Texas MD Anderson Cancer Center, Houston, TX, and includes 29 collaboration centers for the study. A material transfer agreement was established and approved by each of the participating Consortium Collaborative Centers.
This work was supported by a Pathology Research Fellowship Award (S.H.); a Shannon Timmins Fellowship Award (Z.Y.X.-M.); the Zurich Stiftung zur Krebsbekaempfung (A.T.); The University of Texas MD Anderson Cancer Center Institutional R & D Fund, an Institutional Research Grant Award, an MD Anderson Lymphoma SPORE Research Development Program Award, an MD Anderson Myeloma SPORE Research Development Program Award, and MD Anderson Collaborative Research Funds with Daiichi Sankyo Pharmaceuticals, High-Throughput Molecular Diagnostics and Roche Molecular System (K.H.Y.). This work was also partially supported by National Cancer Institute and National Institute of Health (R01CA138688, 1RC1CA146299, P50CA136411, and P50CA142509).
Footnotes
The online version of this article contains a data supplement.
There is an Inside Blood commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: S.H. and K.H.Y. were responsible for study conception and design, performing research, and data analysis and interpretation; S.H., Z.Y.X.-M., A.B., G.C.M., C.V., A.T., W.L., L.Z., R.N.M., S.M.-M., K.D., A.C., A.O., Y.Z., G.B., K.L.R., E.D.H., W.W.L.C., J.H.v.K., Q.H., J.H., W.A., M.P., A.J.M.F., X.Z., J.N.W., M.Z., L.L., M.B.M., M.A.P., Y.L., R.S.G., L.W., L.J.M., and K.H.Y. provided study materials, key reagents, or patients, and collected and assembled data under approved institutional review boards and material transfer agreement; S.H., L.J.M., and K.H.Y. wrote the manuscript; and all authors gave final approval of manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ken H. Young, Department of Hematopathology, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd, Houston, TX 77030-4009; e-mail: khyoung@mdanderson.org.
References
- 1.Stein H, Warnke RA, Chan WC, et al. Diffuse large B-cell lymphoma, not otherwise specified. In: Swerdlow SH, Campo E, Harris NL, et al., editors. WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: IARC Press; 2008. pp. 233–237. [Google Scholar]
- 2.Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403(6769):503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 3.Hans CP, Weisenburger DD, Greiner TC, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood. 2004;103(1):275–282. doi: 10.1182/blood-2003-05-1545. [DOI] [PubMed] [Google Scholar]
- 4.Muris JJ, Meijer CJ, Vos W, et al. Immunohistochemical profiling based on Bcl-2, CD10 and MUM1 expression improves risk stratification in patients with primary nodal diffuse large B cell lymphoma. J Pathol. 2006;208(5):714–723. doi: 10.1002/path.1924. [DOI] [PubMed] [Google Scholar]
- 5.Nyman H, Adde M, Karjalainen-Lindsberg ML, et al. Prognostic impact of immunohistochemically defined germinal center phenotype in diffuse large B-cell lymphoma patients treated with immunochemotherapy. Blood. 2007;109(11):4930–4935. doi: 10.1182/blood-2006-09-047068. [DOI] [PubMed] [Google Scholar]
- 6.Natkunam Y, Farinha P, Hsi ED, et al. LMO2 protein expression predicts survival in patients with diffuse large B-cell lymphoma treated with anthracycline-based chemotherapy with and without rituximab. J Clin Oncol. 2008;26(3):447–454. doi: 10.1200/JCO.2007.13.0690. [DOI] [PubMed] [Google Scholar]
- 7.Choi WW, Weisenburger DD, Greiner TC, et al. A new immunostain algorithm classifies diffuse large B-cell lymphoma into molecular subtypes with high accuracy. Clin Cancer Res. 2009;15(17):5494–5502. doi: 10.1158/1078-0432.CCR-09-0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meyer PN, Fu K, Greiner TC, et al. Immunohistochemical methods for predicting cell of origin and survival in patients with diffuse large B-cell lymphoma treated with rituximab. J Clin Oncol. 2011;29(2):200–207. doi: 10.1200/JCO.2010.30.0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Visco C, Li Y, Xu-Monette ZY, et al. Comprehensive gene expression profiling and immunohistochemical studies support application of immunophenotypic algorithm for molecular subtype classification in diffuse large B-cell lymphoma: a report from the International DLBCL Rituximab-CHOP Consortium Program Study. Leukemia. 2012;26(9):2103–2113. doi: 10.1038/leu.2012.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ott G, Ziepert M, Klapper W, et al. Immunoblastic morphology but not the immunohistochemical GCB/nonGCB classifier predicts outcome in diffuse large B-cell lymphoma in the RICOVER-60 trial of the DSHNHL. Blood. 2010;116(23):4916–4925. doi: 10.1182/blood-2010-03-276766. [DOI] [PubMed] [Google Scholar]
- 11.Thieblemont C, Briere J, Mounier N, et al. The germinal center/activated B-cell subclassification has a prognostic impact for response to salvage therapy in relapsed/refractory diffuse large B-cell lymphoma: a bio-CORAL study. J Clin Oncol. 2011;29(31):4079–4087. doi: 10.1200/JCO.2011.35.4423. [DOI] [PubMed] [Google Scholar]
- 12.Gutiérrez-García G, Cardesa-Salzmann T, Climent F, et al. Grup per l’Estudi dels Limfomes de Catalunya I Balears (GELCAB) Gene-expression profiling and not immunophenotypic algorithms predicts prognosis in patients with diffuse large B-cell lymphoma treated with immunochemotherapy. Blood. 2011;117(18):4836–4843. doi: 10.1182/blood-2010-12-322362. [DOI] [PubMed] [Google Scholar]
- 13.Gu K, Weisenburger DD, Fu K, et al. Cell of origin fails to predict survival in patients with diffuse large B-cell lymphoma treated with autologous hematopoietic stem cell transplantation. Hematol Oncol. 2012;30(3):143–149. doi: 10.1002/hon.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin P, Medeiros LJ. High-grade B-cell lymphoma/leukemia associated with t(14;18) and 8q24/MYC rearrangement: a neoplasm of germinal center immunophenotype with poor prognosis. Haematologica. 2007;92(10):1297–1301. doi: 10.3324/haematol.11263. [DOI] [PubMed] [Google Scholar]
- 15.Aukema SM, Siebert R, Schuuring E, et al. Double-hit B-cell lymphomas. Blood. 2011;117(8):2319–2331. doi: 10.1182/blood-2010-09-297879. [DOI] [PubMed] [Google Scholar]
- 16.Dürkop H, Latza U, Hummel M, et al. Molecular cloning and expression of a new member of the nerve growth factor receptor family that is characteristic for Hodgkin’s disease. Cell. 1992;68(3):421–427. doi: 10.1016/0092-8674(92)90180-k. [DOI] [PubMed] [Google Scholar]
- 17.de Leval L, Gaulard P. CD30+ lymphoproliferative disorders. Haematologica. 2010;95(10):1627–1630. doi: 10.3324/haematol.2010.029256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Younes A, Bartlett NL, Leonard JP, et al. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med. 2010;363(19):1812–1821. doi: 10.1056/NEJMoa1002965. [DOI] [PubMed] [Google Scholar]
- 19.Fanale MA, Forero-Torres A, Rosenblatt JD, et al. A phase I weekly dosing study of brentuximab vedotin in patients with relapsed/refractory CD30-positive hematologic malignancies. Clin Cancer Res. 2012;18(1):248–255. doi: 10.1158/1078-0432.CCR-11-1425. [DOI] [PubMed] [Google Scholar]
- 20.Pro B, Advani R, Brice P, et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: results of a phase II study. J Clin Oncol. 2012;30(18):2190–2196. doi: 10.1200/JCO.2011.38.0402. [DOI] [PubMed] [Google Scholar]
- 21.Gopal AK, Ramchandren R, O’Connor OA, et al. Safety and efficacy of brentuximab vedotin for Hodgkin lymphoma recurring after allogeneic stem cell transplantation. Blood. 2012;120(3):560–568. doi: 10.1182/blood-2011-12-397893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen R, Palmer JM, Thomas SH, et al. Brentuximab vedotin enables successful reduced-intensity allogeneic hematopoietic cell transplantation in patients with relapsed or refractory Hodgkin lymphoma. Blood. 2012;119(26):6379–6381. doi: 10.1182/blood-2012-03-418673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Falini B, Pileri S, Zinzani PL, et al. ALK+ lymphoma: clinico-pathological findings and outcome. Blood. 1999;93(8):2697–2706. [PubMed] [Google Scholar]
- 24.Medeiros LJ, Elenitoba-Johnson KS. Anaplastic large cell lymphoma. Am J Clin Pathol. 2007;127(5):707–722. doi: 10.1309/r2q9ccuvtlrycf3h. [DOI] [PubMed] [Google Scholar]
- 25.Savage KJ, Harris NL, Vose JM, et al. International Peripheral T-Cell Lymphoma Project. ALK- anaplastic large-cell lymphoma is clinically and immunophenotypically different from both ALK+ ALCL and peripheral T-cell lymphoma, not otherwise specified: report from the International Peripheral T-Cell Lymphoma Project. Blood. 2008;111(12):5496–5504. doi: 10.1182/blood-2008-01-134270. [DOI] [PubMed] [Google Scholar]
- 26.Inghirami G, Pileri SA European T-Cell Lymphoma Study Group. Anaplastic large-cell lymphoma. Semin Diagn Pathol. 2011;28(3):190–201. doi: 10.1053/j.semdp.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 27.Savage KJ, Al-Rajhi N, Voss N, et al. Favorable outcome of primary mediastinal large B-cell lymphoma in a single institution: the British Columbia experience. Ann Oncol. 2006;17(1):123–130. doi: 10.1093/annonc/mdj030. [DOI] [PubMed] [Google Scholar]
- 28.Noorduyn LA, de Bruin PC, van Heerde P, et al. Relation of CD30 expression to survival and morphology in large cell B cell lymphomas. J Clin Pathol. 1994;47(1):33–37. doi: 10.1136/jcp.47.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maes B, Anastasopoulou A, Kluin-Nelemans JC, et al. EORTC Lymphoma Group. Among diffuse large B-cell lymphomas, T-cell-rich/histiocyte-rich BCL and CD30+ anaplastic B-cell subtypes exhibit distinct clinical features. Ann Oncol. 2001;12(6):853–858. doi: 10.1023/a:1011195708834. [DOI] [PubMed] [Google Scholar]
- 30.Sehn LH, Donaldson J, Chhanabhai M, et al. Introduction of combined CHOP plus rituximab therapy dramatically improved outcome of diffuse large B-cell lymphoma in British Columbia. J Clin Oncol. 2005;23(22):5027–5033. doi: 10.1200/JCO.2005.09.137. [DOI] [PubMed] [Google Scholar]
- 31.Fu K, Weisenburger DD, Choi WW, et al. Addition of rituximab to standard chemotherapy improves the survival of both the germinal center B-cell-like and non-germinal center B-cell-like subtypes of diffuse large B-cell lymphoma. J Clin Oncol. 2008;26(28):4587–4594. doi: 10.1200/JCO.2007.15.9277. [DOI] [PubMed] [Google Scholar]
- 32.Coiffier B, Thieblemont C, Van Den Neste E, et al. Long-term outcome of patients in the LNH-98.5 trial, the first randomized study comparing rituximab-CHOP to standard CHOP chemotherapy in DLBCL patients: a study by the Groupe d’Etudes des Lymphomes de l’Adulte. Blood. 2010;116(12):2040–2045. doi: 10.1182/blood-2010-03-276246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu-Monette ZY, Wu L, Visco C, et al. Mutational profile and prognostic significance of TP53 in diffuse large B-cell lymphoma patients treated with R-CHOP: report from an International DLBCL Rituximab-CHOP Consortium Program Study. Blood. 2012;120(19):3986–3996. doi: 10.1182/blood-2012-05-433334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wright G, Tan B, Rosenwald A, et al. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc Natl Acad Sci USA. 2003;100(17):9991–9996. doi: 10.1073/pnas.1732008100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Green TM, Young KH, Visco C, et al. Immunohistochemical double-hit score is a strong predictor of outcome in patients with diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol. 2012;30(28):3460–3467. doi: 10.1200/JCO.2011.41.4342. [DOI] [PubMed] [Google Scholar]
- 36.Johnson NA, Slack GW, Savage KJ, et al. Concurrent expression of MYC and BCL2 in diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol. 2012;30(28):3452–3459. doi: 10.1200/JCO.2011.41.0985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gruss HJ, Boiani N, Williams DE, et al. Pleiotropic effects of the CD30 ligand on CD30-expressing cells and lymphoma cell lines. Blood. 1994;83(8):2045–2056. [PubMed] [Google Scholar]
- 38.Mir SS, Richter BW, Duckett CS. Differential effects of CD30 activation in anaplastic large cell lymphoma and Hodgkin disease cells. Blood. 2000;96(13):4307–4312. [PubMed] [Google Scholar]
- 39.Wright CW, Rumble JM, Duckett CS. CD30 activates both the canonical and alternative NF-kappaB pathways in anaplastic large cell lymphoma cells. J Biol Chem. 2007;282(14):10252–10262. doi: 10.1074/jbc.M608817200. [DOI] [PubMed] [Google Scholar]
- 40.Amakawa R, Hakem A, Kundig TM, et al. Impaired negative selection of T cells in Hodgkin’s disease antigen CD30-deficient mice. Cell. 1996;84(4):551–562. doi: 10.1016/s0092-8674(00)81031-4. [DOI] [PubMed] [Google Scholar]
- 41.Chiarle R, Podda A, Prolla G, et al. CD30 overexpression enhances negative selection in the thymus and mediates programmed cell death via a Bcl-2-sensitive pathway. J Immunol. 1999;163(1):194–205. [PubMed] [Google Scholar]
- 42.Mori M, Manuelli C, Pimpinelli N, et al. CD30-CD30 ligand interaction in primary cutaneous CD30(+) T-cell lymphomas: a clue to the pathophysiology of clinical regression. Blood. 1999;94(9):3077–3083. [PubMed] [Google Scholar]
- 43.Savage KJ, Monti S, Kutok JL, et al. The molecular signature of mediastinal large B-cell lymphoma differs from that of other diffuse large B-cell lymphomas and shares features with classical Hodgkin lymphoma. Blood. 2003;102(12):3871–3879. doi: 10.1182/blood-2003-06-1841. [DOI] [PubMed] [Google Scholar]
- 44.Rosenwald A, Wright G, Leroy K, et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med. 2003;198(6):851–862. doi: 10.1084/jem.20031074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Steidl C, Shah SP, Woolcock BW, et al. MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature. 2011;471(7338):377–381. doi: 10.1038/nature09754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ritz O, Guiter C, Castellano F, et al. Recurrent mutations of the STAT6 DNA binding domain in primary mediastinal B-cell lymphoma. Blood. 2009;114(6):1236–1242. doi: 10.1182/blood-2009-03-209759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bonzheim I, Geissinger E, Roth S, et al. Anaplastic large cell lymphomas lack the expression of T-cell receptor molecules or molecules of proximal T-cell receptor signaling. Blood. 2004;104(10):3358–3360. doi: 10.1182/blood-2004-03-1037. [DOI] [PubMed] [Google Scholar]
- 48.de Leval L, Rickman DS, Thielen C, et al. The gene expression profile of nodal peripheral T-cell lymphoma demonstrates a molecular link between angioimmunoblastic T-cell lymphoma (AITL) and follicular helper T (TFH) cells. Blood. 2007;109(11):4952–4963. doi: 10.1182/blood-2006-10-055145. [DOI] [PubMed] [Google Scholar]
- 49.Geissinger E, Sadler P, Roth S, et al. Disturbed expression of the T-cell receptor/CD3 complex and associated signaling molecules in CD30+ T-cell lymphoproliferations. Haematologica. 2010;95(10):1697–1704. doi: 10.3324/haematol.2009.021428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Montes-Moreno S, Odqvist L, Diaz-Perez JA, et al. EBV-positive diffuse large B-cell lymphoma of the elderly is an aggressive post-germinal center B-cell neoplasm characterized by prominent nuclear factor-kB activation. Mod Pathol. 2012;25(7):968–982. doi: 10.1038/modpathol.2012.52. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.