

Abstract
Chronic pain is extremely difficult to manage, in part due to lack of progress in reversing the underlying pathophysiology. Since translation of mRNAs in the peripheral terminal of the nociceptor plays a role in the transition from acute to chronic pain, we tested the hypothesis that transient inhibition of translation in the peripheral terminal of the nociceptor could reverse hyperalgesic priming, a model of transition from acute to chronic pain. We report that injection of translation inhibitors, rapamycin and cordycepin, which inhibit translation by different mechanisms, at the peripheral terminal of the primed nociceptor, produce reversal of priming, in the rat, that outlasted the duration of action of these drugs to prevent the development of priming. These data support the suggestion that interruption of translation in the nociceptor can reverse a preclinical model of at least one form of chronic pain.
Keywords: peripheral protein translation, hyperalgesic priming, sensory neuron, mechanical hyperalgesia, rat
Introduction
Hyperalgesic priming, a model of the transition from acute to chronic pain, has been defined as a long-lasting latent hyperresponsiveness of nociceptors to inflammatory mediators subsequent to an inflammatory or neuropathic insult 1,35,39. Priming, which can be detected as an enhanced and prolonged hyperalgesic response to the proinflammatory cytokine prostaglandin E2 (PGE2), can be explained by a switch in the intracellular signaling pathway mediating PGE2-induced hyperalgesia 1,23. Since hyperalgesic priming is still present, unattenuated, for a period of months 1,10,35, it is likely associated with the formation of a type of molecular memory in primary afferent nociceptors. Given the continuous turnover of most cellular proteins it is difficult to conceive that a simple switch in the coupling of a G-protein coupled receptor to a different second messenger signaling pathway is sufficient to explain the endurance of the plasticity associated with the “primed” nociceptor.
We have recently shown that cytoplasmic polyadenylation element binding protein (CPEB), an RNA-binding molecule that regulates the translation of otherwise dormant mRNAs 41,53, is not only almost exclusively expressed by isolectin B4-positive (IB4(+))-nociceptors, the subset of nociceptors in which hyperalgesic priming occurs 22 and can be co-immunoprecipitated with PKCε, but that the downregulation of its level of expression by the intrathecal administration of antisense oligodeoxynucleotides (ODNs) targeting its mRNA can prevent, but not reverse, hyperalgesic priming 5. Because our antisense only decreased CPEB by 28%, in peripheral nerves, we might not be inhibiting peripheral translation adequately to reverse hyperalgesic priming. That is, the degree of attenuation required to produce reversal of priming may be far greater than that needed to prevent the development of priming. In addition, local injection of anisomycin, a protein translation inhibitor, also prevented the induction of priming 5, suggesting a role of local protein synthesis in our model. Therefore, in the present study, we evaluated the ability of peripheral administration of a translation inhibitor to reverse hyperalgesic priming. Our results are compatible with the idea that ongoing peripheral translation is necessary for maintenance of the hyperalgesic priming model of chronic pain.
Materials and Methods
Animals
All experiments were performed on adult male Sprague Dawley rats (220–400 g; Charles River Laboratories). Animals were housed, three per cage, under a 12 h light/dark cycle in a temperature- and humidity-controlled room in the animal care facility of the University of California, San Francisco. Food and water were available ad libitum. All nociceptive testing was done between 10:00 am and 5:00 pm and the experimental protocols were approved by the Institutional Animal Care and Use Committee at University of California at San Francisco and adhered to the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. All effort was made to minimize the number of animals used and their suffering.
Mechanical nociceptive threshold testing
Mechanical nociceptive threshold was quantified using an Ugo Basile Analgesymeter® (Randall-Selitto paw-withdrawal test, Stoelting, Chicago, IL), which applies a linearly increasing mechanical force to the dorsum of the rat’s hind paw, as previously described 10,11. The nociceptive threshold was defined as the force, in grams, at which the rat withdrew its paw, and baseline paw-pressure threshold was defined as the mean of the three readings taken before the test agents were injected. Each paw was treated as an independent measure and each experiment performed on a separate group of rats. Data are presented as mean change from baseline mechanical nociceptive threshold.
Drugs and their administration
Cordycepin 5′-triphosphate sodium salt, λ-carrageenan and prostaglandin E2 (PGE2) were purchased from Sigma-Aldrich (St. Louis, MO), and rapamycin from EMD Chemicals (Gibbstown, NJ). The selection of the drug doses used in this study was based on dose–response curves determined during our previous studies 1,34,35,48. Stock solutions of PGE2 in absolute ethanol (1 μg/μl) were diluted in 0.9% NaCl (1:50, Cfinal=0.2 μg/μl) immediately before injection. The ethanol concentration of the final PGE2 solution was ~2% and the injection volume was 5 μl. Stock solutions of rapamycin (20 μg/μl, dissolved in absolute DMSO) and cordycepin (10 μg/μl, dissolved in a 1:1 mixture of 0.9% NaCl and absolute ethanol) were further diluted in distilled water or 0.9% NaCl, respectively, immediately before injection. The DMSO and ethanol concentrations in the final rapamycin and cordycepin solutions were ~10% and ~2%, respectively. Carrageenan was dissolved and diluted in 0.9% NaCl (Cfinal=1%) immediately before injection. The injection volume was 5 μl. Carrageenan was injected using a 27-gauge hypodermic needle. Intradermal injection of carrageenan induces a decrease in the mechanical threshold in 30–60 min, reaching a maximum between 2 and 6 h, returning to baseline values within 72 h 1. All other drugs were administered via a beveled 30-gauge hypodermic needle attached to a Hamilton® microsyringe (Reno, NV, USA) by polyethylene tubing. Rapamycin, which inhibits the mammalian target of rapamycin (mTOR), a serinethreonine protein kinase involved in the local translation of mRNA, has been used to study peripheral 12,32,37 and spinal 2,31,42,57 mechanisms of inflammatory and neuropathic pain. Cordycepin, also a protein translation inhibitor, has a different mechanism of action than rapamycin 13,24,30,40,46. These protein translation inhibitors were administered on the dorsum of the hind paw with a preceding hypotonic shock to facilitate cell permeability to these agents (2 μl of distilled water, separated by a bubble of air to avoid mixing in the same syringe), to get compounds into the nerve terminal.
Hyperalgesic priming
Hyperalgesic priming, a model of transition from acute to chronic pain, was produced using a procedure previously used in other studies 1,33,35: to prime the sensory neuron terminals in the skin, carrageenan (1%) was intradermally injected on the dorsum of the hind paw. This induces mechanical hyperalgesia that resolves 3–5 days later, following which intradermal injection of PGE2 at the same site produces enhanced and prolonged mechanical hyperalgesia that lasts more than 4 h and is still significant at 24 h, as opposed to the effect of intradermal injection of PGE2 in intact skin, that lasts around 2 hours 1.
Statistics
In all experiments, the dependent variable was paw-withdrawal threshold, expressed as the percentage change from baseline. Average mechanical threshold immediately before the experiments was 115.1 ± 1.2 g (N=96). Paired Student’s t-test was used to compare the mechanical thresholds before and after the carrageenan injections, and as appropriate (as noted in the figure legends). When two different groups were compared (for instance, when comparing the mechanical thresholds of control and protein translation inhibitor treated groups 4 days after carrageenan treatment), unpaired Student’s t-test was used. To compare the hyperalgesia induced by PGE2 injection in different groups, two-way repeated-measures ANOVA, followed by Bonferroni post-test, was performed. Data are presented as mean ± SEM.
Results
Rapamycin
To test the hypothesis that inhibiting peripheral translation can reverse hyperalgesic priming we injected rapamycin (1 μg), which has been shown to inhibit peripheral translation 20,28,31, at the site of nociceptive testing, both 30 minutes before and again with carrageenan. We then tested for hyperalgesic priming, by injection of PGE2, 4 days later, at which time hyperalgesic priming should be established 1,5,33,39. In rats treated with rapamycin, before and again with carrageenan injection, priming, as indicated by prolongation of PGE2 hyperalgesia measured at 4 hours, did not develop (Fig. 1). Next, to determine if rapamycin can also reverse priming, it was injected 7 days after carrageenan, and PGE2 injected 30 minutes later. Again, PGE2 failed to produce prolonged hyperalgesia (Fig. 2). Thus, local administration of a translation inhibitor in the vicinity of the peripheral terminal of nociceptors whose mechanical threshold was being monitored can also reverse hyperalgesic priming.
Figure 1. Prevention of carrageenan-induced hyperalgesic priming by local injection of the mTOR inhibitor rapamycin.
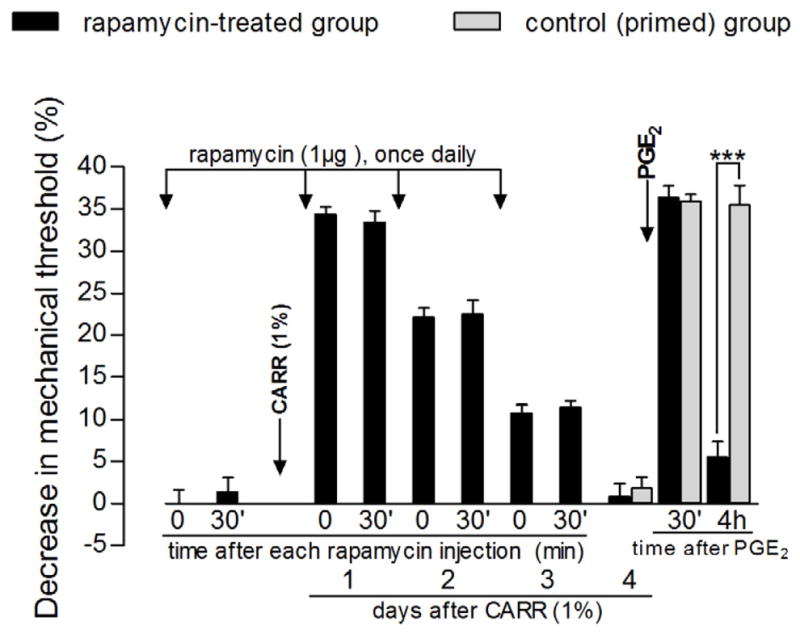
Rats received intradermal injection of rapamycin (1 μg) on the dorsum of the hind paw. Evaluation of the mechanical nociceptive threshold, by the Randall-Selitto paw-withdrawal test, before and 30 min after injection, showed no effect of rapamycin by itself [average paw withdrawal thresholds before and 30 min after rapamycin injection were 113.3 ± 1.7 g and 111.6 ± 1.5 g, respectively (paired Student’s t-test showed no significant difference between these values (t5=0.8811, p=0.4186, N=6)]. Carrageenan (CARR, 1%) was then injected at the same site and the mechanical threshold was evaluated daily, until the return to the baseline values (~4th day) [no significant difference on the mechanical nociceptive thresholds was observed immediately before and 4 days after injection of CARR (t5=0.2911, p=0.7827, paired Student’s t-test)]. Daily injections of rapamycin (1 μg) were performed and readings taken before and 30 min after each injection showed no effect of rapamycin on the hyperalgesia induced by CARR (day 1 after CARR: t5=0.5649, p=0.5965; day 2 after CARR: t5=1.976, p=0.1051; day 3 after CARR: t5=1.074, p=0.3318, paired Student’s t-test for each day). On the 4th day after CARR, test for priming was performed: PGE2 (100 ng) was injected at the same site as CARR and rapamycin and the mechanical thresholds were evaluated 30 min and 4 h later. Two-way repeated measures ANOVA followed by Bonferroni post-test showed significant mechanical hyperalgesia 30 min after PGE2 injection (p<0.001), with decrease on the 4th h (p>0.05, when compared to thresholds before PGE2), indicating an inhibitory effect of rapamycin on the prolongation of PGE2 hyperalgesia (black bars, F2,10=196.98, p<0.0001). Of note, in a control group of rats that received CARR 4 days before (gray bars), the PGE2-induced hyperalgesia was still present at that time point [two-way repeated measures ANOVA followed by Bonferroni post-test showed significant difference between both groups (F2,30=55.58, p<0.0001), with decrease in the mechanical hyperalgesia at the 4 h after PGE2 in the rapamycin-treated group, when compared to the control group (***p<0.001); N=6 paws].
Figure 2. Reversal of carrageenan-induced hyperalgesic priming by local injection of the mTOR inhibitor rapamycin.
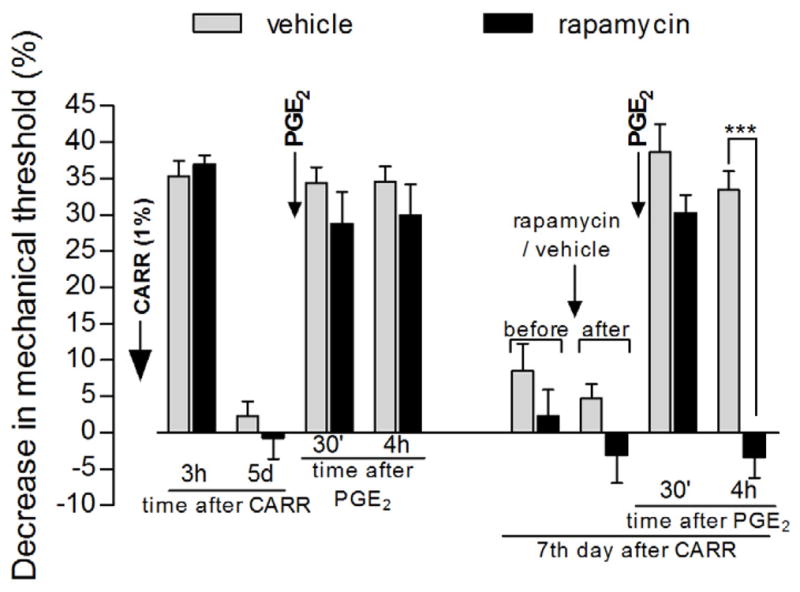
Different groups of rats received intradermal injection of carrageenan (CARR, 1%) on the dorsum of the hind paw. Mechanical nociceptive threshold evaluation, by the Randall-Selitto paw-withdrawal test, showed significant hyperalgesia 3 h after injection (p<0.0001, for both groups when compared to baseline thresholds, paired Student’s t-test, N=6 paws per group) and return to baseline values on the 5th day post CARR injection [average paw withdrawal thresholds before and on the 5th day after CARR injection: 115.0 ± 2.2 g and 116.6 ± 2.1 g, respectively, for the vehicle-treated group (t5=0.4152, p=0.6952, gray bars), and 108.3 ± 3.0 g and 109.3 ± 2.8 g, respectively, for the rapamycin-treated group (t5=0.3437, p=0.7451, black bars) (paired Student’s t-test showed no significant difference between these values). Also, there was no difference between both groups 5 days after CARR injection (t10=2.086, p=0.0635, unpaired Student’s t-test)]. Injection of PGE2 at the same site as CARR induced hyperalgesia that was still present after 4 h, indicating the presence of hyperalgesic priming. Two days later (7 days after CARR injection), vehicle (gray bars) or the mTOR inhibitor rapamycin (black bars) was injected at the same site. No significant change in the mechanical nociceptive threshold was observed 30 min after rapamycin injection (t5=1.126, p=0.3111, paired Student’s t-test). However, when tested for priming with PGE2, no hyperalgesia was observed 4 h later in the rats treated with rapamycin when compared to the rats treated with vehicle (***p<0.001, two-way repeated measures ANOVA followed by Bonferroni post-test), showing inhibition of CARR-induced priming by local injection of rapamycin (N=6 paws per group).
Cordycepin
To further test the hypothesis that inhibitors of peripheral translation can reverse hyperalgesic priming, we injected a second translation inhibitor, cordycepin, which inhibits translation by a different mechanism than rapamycin. Cordycepin was also injected at the site of nociceptive testing, both 30 minutes before and again with carrageenan. We then tested for hyperalgesic priming, by injection of PGE2, 5 days after carrageenan administration (Fig. 3). In contrast with the vehicle-treated group, in the rats treated with cordycepin, before and again with carrageenan, hyperalgesic priming, as indicated by prolongation of PGE2 hyperalgesia, at 4 hours, did not develop (Fig. 3). Next, to determine if cordycepin can also reverse priming, it was injected 4 days after carrageenan and PGE2 injected 30 minutes later. Again, PGE2 failed to produce prolonged hyperalgesia (Fig. 4). Thus, we have shown that a second translation inhibitor can reverse as well as prevent hyperalgesic priming.
Figure 3. Prevention of carrageenan-induced hyperalgesic priming by local injection of the protein translator inhibitor cordycepin.
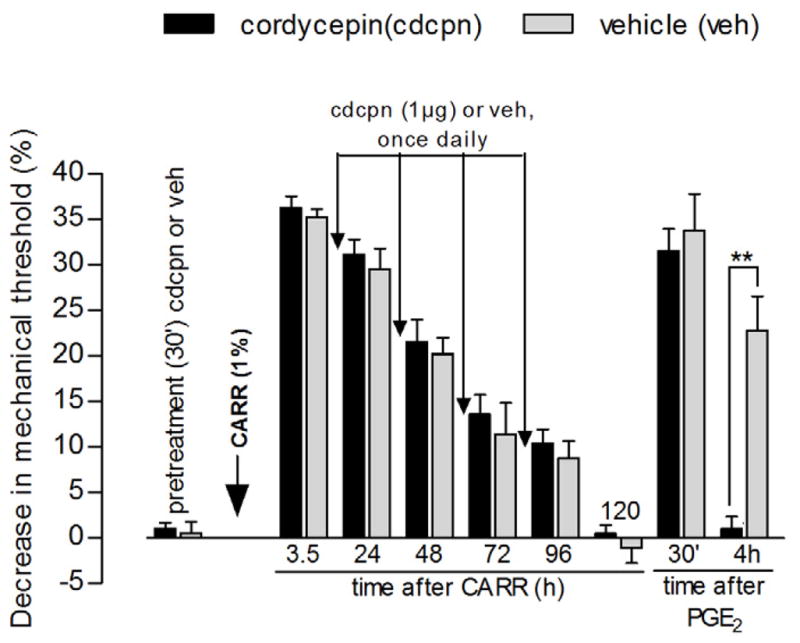
Different groups of rats received intradermal injection of vehicle (gray bars) or cordycepin (1 μg, black bars) on the dorsum of the hind paw. Evaluation of the mechanical nociceptive thresholds, by the Randall-Selitto paw-withdrawal test, before and 30 min after injection, showed no effect of cordycepin by itself [average paw withdrawal thresholds before and 30 min after injection: 106.6 ± 1.2 g and 105.6 ± 1.6 g, respectively, for the cordycepin group, and 106.6 ± 1.2 g and 106.0 ± 1.8 g, respectively, for the vehicle group (paired Student’s t-test for each group showed no significant difference between these values); also, unpaired Student’s t-test showed no difference between the groups after injections (t10=0.1334, p=0.8965)]. Carrageenan (CARR, 1%) was then injected at the same site and the mechanical thresholds were evaluated daily, until the return to the baseline values (~5th day) [no significant difference on the mechanical nociceptive threshold, for each group, was observed immediately before and 5 days after injection of carrageenan (cordycepin group: t5=0.3071, p=0.7711; vehicle group: t5=0.9325, p=0.3939, paired Student’s t-test)]. Daily injections of cordycepin (1 μg) or vehicle were performed; readings taken 30 min after each injection showed no difference in the hyperalgesia between both groups (F1,60=0.67, p=0.4321, two-way repeated measures ANOVA followed by Bonferroni post-test), i.e., no effect of cordycepin on the hyperalgesia induced by carrageenan. On the 5th day after carrageenan, test for priming was performed: PGE2 (100 ng) was injected at the same site as carrageenan and vehicle/cordycepin and the mechanical thresholds were evaluated 30 min and 4 h later. Two-way repeated measures ANOVA showed no hyperalgesia in the cordycepin-treated group, when compared to the vehicle-treated group, 4 h after PGE2 injection (F2,20=23.51, **p<0.0001, N=6 paws per group).
Figure 4. Reversal of carrageenan-induced hyperalgesic priming by local injection of the protein translator inhibitor cordycepin.
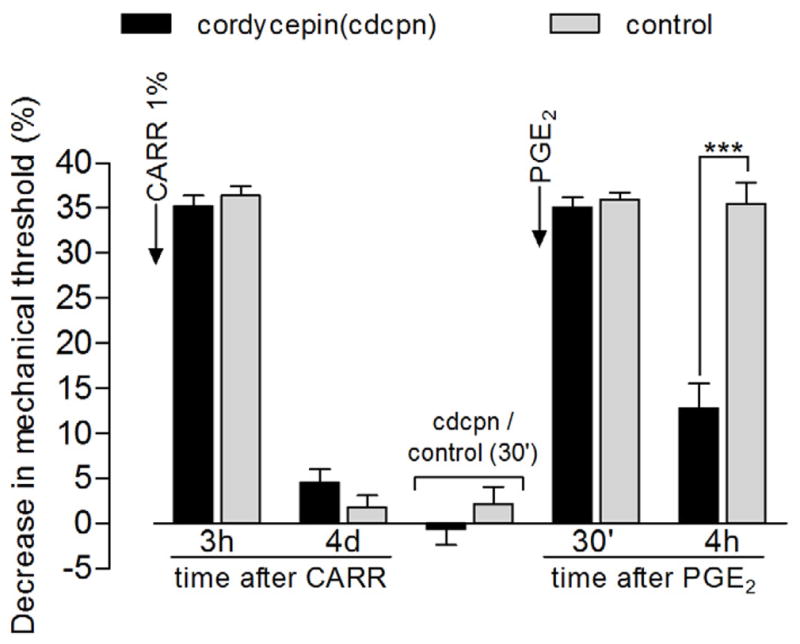
Different groups of rats received intradermal injection of carrageenan (CARR, 1%) on the dorsum of the hind paw. Mechanical nociceptive thresholds evaluation, by the Randall-Selitto paw-withdrawal test, showed significant hyperalgesia 3 h after injection and return to baseline values on the 4th day post CARR injection [average paw withdrawal thresholds before and 4 d after injection - cordycepin group: 116.8 ± 1.5 g and 113.3 ± 1.1 g, respectively; control group: 120.0 ± 2.0 g and 117.6 ± 1.7 g, respectively (paired Student’s t-test showed no significant difference between these values for each group: cordycepin: t11=2.116, p=0.0579, N=12 paws; control: t5=1.400, p=0.2204, N=6 paws); unpaired Student’s t-test showed no difference between the groups on the 4th day after carrageenan injection (t16=2.079, p=0.0540)]. No significant difference in the mechanical threshold was observed 30 min after injection of cordycepin, when compared to the control group either (unpaired Student’s t-test, t16=1.038, p=0.3148). Injection of PGE2 at the same site as carrageenan and cordycepin/vehicle induced significant hyperalgesia, evaluated 30 min and 4 h after injection (two-way repeated measures ANOVA followed by Bonferroni post-test, F2,32=205.62, p<0.0001). However, at the 4th h post-PGE2, the magnitude of the hyperalgesia was significantly decreased in the cordycepin-treated group when compared to the control group (***p<0.001), indicating an effect of cordycepin on hyperalgesic priming.
To confirm that the translation inhibitor had actually reversed priming, we wanted to demonstrate that the effect of the translation inhibitor was no longer present at the time of testing for hyperalgesic priming. First, we determined if the reversal of priming was still present 10 days after injection of cordycepin. A group of primed rats that received cordycepin 4 days after carrageenan (shown in Fig. 4) was tested again for priming 10 and 20 days later (Fig. 5). In either case, PGE2-induced hyperalgesia, although present 30 min after injection, was significantly decreased at the 4 h time point (Fig. 5). Next, in order to determine if the action of cordycepin on translation was still present 20 days after cordycepin treatment (when we still observed inhibition of the prolonged PGE2-induced hyperalgesia, Fig. 5), we injected cordycepin (1 μg) 20 days before carrageenan, and, then, tested with PGE2 5 days after carrageenan (Fig. 6). In this control experiment, hyperalgesic priming did develop, as we observed prolongation of the PGE2-induced hyperalgesia (Fig. 6). Thus, at a time after cordycepin administration that it no longer prevented hyperalgesic priming, its ability to reverse priming was detectable.
Figure 5. Duration of the effect of the protein translator inhibitor cordycepin on the carrageenan-induced hyperalgesic priming.

Panel A: Rats that received intradermal injection of carrageenan (CARR, 1%) on the dorsum of the hind paw were treated with cordycepin (1 μg) on the 4th day (after recovery from CARR-induced hyperalgesia). PGE2 was injected 30 min after cordycepin and the mechanical nociceptive thresholds were evaluated, by the Randall-Selitto paw-withdrawal test, 30 min and 4 h later. Paired Student’s t-test showed significant decrease in the PGE2-induced hyperalgesia on the 4th h after injection, indicating inhibition of hyperalgesic priming by condycepin (t11=8.748, **p<0.0001 when the magnitude of the hyperalgesia on 30 min and 4 h are compared, N=12 paws). When PGE2 was injected, again, 10 and 20 days later, at the same site where carrageenan/cordycepin were previously administered, it failed to induce prolonged hyperalgesia, evaluated at 4 h post injection [Paired Student’s t-test comparing the magnitude of the hyperalgesia on 30 min and 4 h: t11=8.799, **p<0.0001 (10 days after cordycepin injection), t11=10.05, **p<0.0001 (20 days after cordycepin injection)] indicating the long-term reversal of the hyperalgesic priming by cordycepin (N=12 paws); Panel B: Hyperalgesic priming is still present 3 weeks after carrageenan injection: rats that received carrageenan (CARR, 1%) on the dorsum of the hind paws were tested with PGE2 (100 ng) 2 and 3 weeks afterwards. Readings were taken 30 min and 4 h after PGE2 and, as expected, the magnitude of the hyperalgesia was still high at the 4th h, indicating the presence of hyperalgesic priming, induced by previous injection of CARR. This observation is important in order to evaluate the effect of cordycepin seen on Panel A [Paired Student’s t-test comparing the magnitude of the hyperalgesia on 30 min and 4 h: t5=1.101, p=0.3211 (2 weeks after CARR injection), t5=1.849, p=0.1237 (3 weeks after CARR injection)] (N=6 paws).
Figure 6. Duration of the effect of the protein translator inhibitor cordycepin.
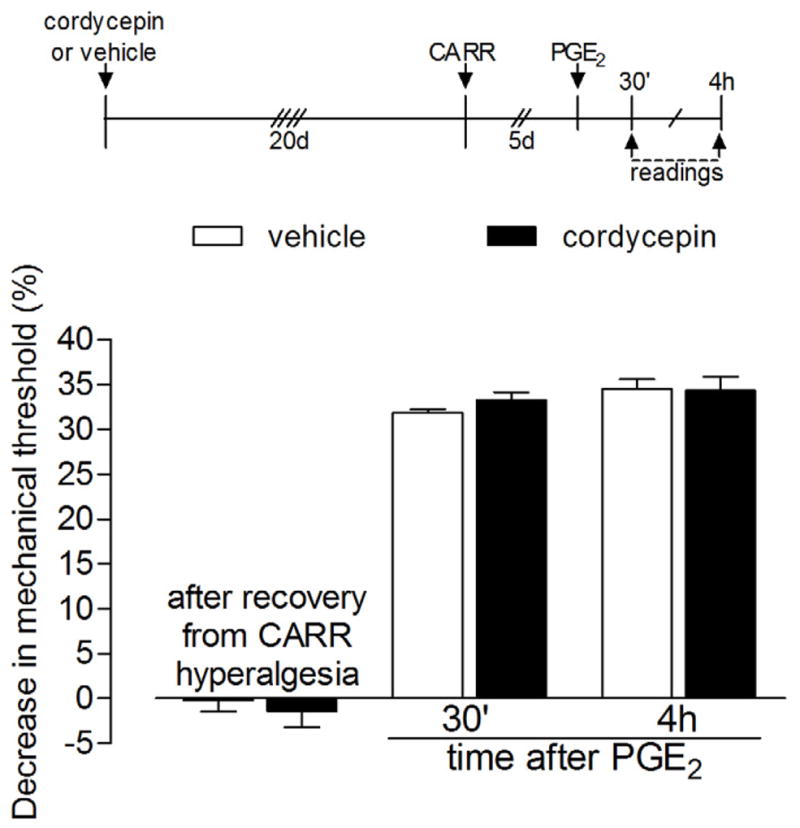
Rats received intradermal injection of vehicle (white bars) or cordycepin (1 μg, black bars) on the dorsum of the hind paws 20 days before injection of carrageenan (CARR, 1%) at the same site. There was no difference in the carrageenan-induced hyperalgesia, evaluated by the Randall-Selitto paw-withdrawal test, between both groups (data not shown). 5 days later, when the mechanical thresholds were back to pre-carrageenan values (vehicle group: t5=1.464, p=0.2031; cordycepin group: t5=0.4152, p=0.6952, paired Student’s t-test), PGE2 was injected at the same site and the mechanical thresholds were evaluated 30 min and 4 h later. Two-way repeated measures ANOVA followed by Bonferroni post-test showed no significant difference in the PGE2-induced hyperalgesia, both 30 min and 4 h after injection, between the vehicle- and the cordycepin-treated groups (F1,20=0.01, p=0.9067), indicating that, at that time point (20 days after injection), the effect of cordycepin was no longer present (N=6 paws per group).
Local effect of the protein translation inhibitors
In order to evaluate if the effect of the protein translation inhibitors was restricted to the site of injection, we performed a control experiment, in rats that had their right paw primed by carrageenan injection 4 days before, by administering rapamycin or cordycepin in the left paw. When PGE2 was injected in the right paw, it still induced prolonged hyperalgesia, i.e., there was no effect of the inhibitors injected in the contralateral paw on priming, confirming their lack of systemic effect (Fig. 7).
Figure 7. Local effect of the protein translation inhibitors rapamycin and cordycepin.
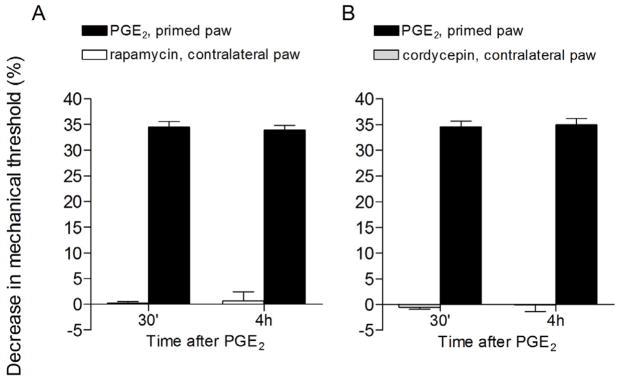
Rats that had their right hind paw primed with carrageenan (1%) 4 days before received intradermal injection of the protein translation inhibitors rapamycin (1 μg, panel A, white bars) or cordycepin (1 μg, panel B, gray bars) on the dorsum of the contralateral paw. 30 min after injecting the inhibitors, PGE2 was injected in the right hind paw and the mechanical nociceptive threshold was evaluated, by the Randall-Selitto paw-withdrawal test, 30 min and 4 h later (black bars, both panels). Paired Student’s t-test showed no change in the magnitude of PGE2-induced hyperalgesia at 30 min and 4 h (panel A: t5=0.5423, p=0.6109; panel B: t5=0.7006, p=0.5148), indicating lack of effect of rapamycin or cordycepin injected in the contralateral paw on the hyperalgesic priming in the right paws (N=6 paws per group).
Discussion
In this study we have examined the hypothesis that the persistence of chronic pain is dependent on ongoing translation in the peripheral terminal of the primary afferent nociceptor. We have previously shown, in a model of the transition from acute to chronic pain, hyperalgesic priming, that while the transition can be driven by activation of PKCε, intrathecal treatment with antisense ODN for PKCε, to decrease its level in the nociceptor, after the establishment of the chronic phase of hyperalgesic priming, could not permanently reverse the primed state (unpublished observation). In a subsequent study, to examine the signaling pathway downstream of PKCε in the induction of hyperalgesic priming, we demonstrated a role of CPEB, which is involved in protein translation in axons and dendrites 41,53 and has been implicated in neuroplasticity in Aplysia sensory neurons 4,44,45. However, while transient attenuation of CPEB, in the nociceptor, could prevent hyperalgesic priming, it was not able to reverse it 5.
Given that the downstream consequences of translation may be amplified 3,9,17,20,37,38,49,54, we considered the possibility that the intrathecal treatment with antisense ODN against CPEB mRNA would not adequately decrease CPEB to a level necessary to reverse the primed state. However, the fact that peripheral injection of anisomycin, a protein translation inhibitor, also prevented the development of priming 5, suggested that local protein synthesis plays an important role in this model. Therefore, in the present study we used translation inhibitors injected directly into the peripheral receptive field of the primed nociceptor. Using two translation inhibitors, rapamycin, which acts on the initiation of protein biosynthesis by blocking mTOR, a serine/threonine kinase that triggers the process of mRNA translation 15,43,55, and cordycepin, a derivative of the nucleoside adenosine shown to inhibit mRNA polyadenylation by preventing its incorporation into the poly-A tail, interrupting its elongation and, consequently, the initiation of protein translation 25,30,55, we were able, for the first time, to reverse hyperalgesic priming. To confirm that the translation inhibitor was no longer blocking translation as the explanation for the loss of prolongation of PGE2 hyperalgesia (i.e., the primed state) we determined how long after administration of a translation inhibitor, administration of a PKCε activator, carrageenan, would again be able to produce priming. When PGE2-induced hyperalgesia was examined in rats primed the same amount of time, after cordycepin administration, we still detected reversal of hyperalgesic priming. The results of these experiments support the hypothesis that a stronger inhibition of translation in the peripheral terminal of the primary afferent nociceptor is able to reverse priming.
While our results do support the suggestion that at least some forms of chronic pain may be interrupted by the transient attenuation of translation in the peripheral terminal of the nociceptor, it also raises the question of what protein(s) is downstream of the peripheral translation, which led to the reversal of hyperalgesic priming. Richter and colleagues have enumerated mRNA species in axonal processes/neural tissue that have polyadenylation tails and are thus downstream targets of CPEB 26,27,41,44. Amongst these, one, calcium/calmodulin-dependent protein kinase II (CaMKII), has also been implicated in neuroplasticity 6,7,8,14,19,58. Of note, CaMKII can be both upstream, regulating the activation of CPEB 3 as well as downstream of CPEB 18,56. Thus, CaMKII is a reasonable candidate as a potential positive feedback element that would mandate a stronger attenuation of mRNA translation in the peripheral terminal of the nociceptor, to reverse hyperalgesic priming. Cell biological experiments are currently in progress to test this hypothesis. To better understand the transition of acute to chronic pain it is useful to try to understand what is the normal function of peripheral translation of proteins in the peripheral terminal of a sensory neuron. Another approach is to consider the functions of the sensory neuron, over the lifespan of the organism. It has been suggested that mRNA translation in the peripheral terminal of sensory neurons is important in the developmental program and regeneration of these neurons 16,29,36,47,50,59. This is of more than passing interest with respect to chronic pain, as it has been noted that young children and animals are much less likely to develop chronic pain, especially after nerve injury 51,52. As neuropathic as well as inflammatory conditions produce hyperalgesic priming 1,21,34,39, we propose that alterations in peripheral translation between the perinatal and adult animal might help explain this differential, age dependent, predisposition to undergo the transition from acute to chronic pain. Elucidation of age dependent differences in peripheral translation will be needed to address this important clinical observation. In summary, in the present study we provide evidence that ongoing translation of mRNA in the peripheral terminal of the primary afferent nociceptor may be involved in the neuroplasticity underlying persistence in a model of chronic pain, that is, the transition from acute to chronic pain 1,33,39. Additional experiments will be required to elucidate the details of mechanism, which may involve mediators both upstream as well as downstream from the translation process in the peripheral terminal of the primary afferent nociceptor.
Perspectives.
This study provides evidence that ongoing protein translation in the sensory neuron terminals is involved in pain chronification and local treatment that transiently interrupts this translation may be a useful therapy to chronic pain.
Abbreviations
- CaMKII
calcium/calmodulin-dependent protein kinase II
- CPEB
cytoplasmic polyadenylation element binding protein
- IB4(+)
isolectin B4-positive
- mTOR
mammalian target of rapamycin
- ODN
oligodeoxynucleotide
- PGE2
prostaglandin E2
- PKCε
protein kinase C epsilon
- SEM
standard error of the mean
Footnotes
Disclosures
This study was funded by the National Institutes of Health (NIH). The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aley KO, Messing RO, Mochly-Rosen D, Levine JD. Chronic hypersensitivity for inflammatory nociceptor sensitization mediated by the epsilon isozyme of protein kinase C. J Neurosci. 2000;20:4680–5. doi: 10.1523/JNEUROSCI.20-12-04680.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asante CO, Wallace VC, Dickenson AH. Mammalian target of rapamycin signaling in the spinal cord is required for neuronal plasticity and behavioral hypersensitivity associated with neuropathy in the rat. J Pain. 2010;11:1356–67. doi: 10.1016/j.jpain.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Atkins CM, Nozaki N, Shigeri Y, Soderling TR. Cytoplasmic polyadenylation element binding protein-dependent protein synthesis is regulated by calcium/calmodulin-dependent protein kinase II. J Neurosci. 2004;24:5193–201. doi: 10.1523/JNEUROSCI.0854-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bailey CH, Kandel ER, Si K. The persistence of long-term memory: a molecular approach to self-sustaining changes in learning-induced synaptic growth. Neuron. 2004;44:49–57. doi: 10.1016/j.neuron.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 5.Bogen O, Alessandri-Haber N, Chu C, Gear RW, Levine JD. Generation of a pain memory in the primary afferent nociceptor triggered by PKCepsilon activation of CPEB. J Neurosci. 2012;32:2018–26. doi: 10.1523/JNEUROSCI.5138-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buard I, Coultrap SJ, Freund RK, Lee YS, Dell’Acqua ML, Silva AJ, Bayer KU. CaMKII “autonomy” is required for initiating but not for maintaining neuronal long-term information storage. J Neurosci. 2010;30:8214–20. doi: 10.1523/JNEUROSCI.1469-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cammarota M, Bevilaqua LR, Viola H, Kerr DS, Reichmann B, Teixeira V, Bulla M, Izquierdo I, Medina JH. Participation of CaMKII in neuronal plasticity and memory formation. Cell Mol Neurobiol. 2002;22:259–67. doi: 10.1023/A:1020763716886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coultrap SJ, Buard I, Kulbe JR, Dell’Acqua ML, Bayer KU. CaMKII autonomy is substrate-dependent and further stimulated by Ca2+/calmodulin. J Biol Chem. 2010;285:17930–7. doi: 10.1074/jbc.M109.069351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Du L, Richter JD. Activity-dependent polyadenylation in neurons. RNA. 2005;11:1340–7. doi: 10.1261/rna.2870505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferrari LF, Bogen O, Levine JD. Nociceptor subpopulations involved in hyperalgesic priming. Neuroscience. 2010;165:896–901. doi: 10.1016/j.neuroscience.2009.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferrari LF, Gear RW, Levine JD. Attenuation of activity in an endogenous analgesia circuit by ongoing pain in the rat. J Neurosci. 2010;30:13699–706. doi: 10.1523/JNEUROSCI.2867-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geranton SM, Jimenez-Diaz L, Torsney C, Tochiki KK, Stuart SA, Leith JL, Lumb BM, Hunt SP. A rapamycin-sensitive signaling pathway is essential for the full expression of persistent pain states. J Neurosci. 2009;29:15017–27. doi: 10.1523/JNEUROSCI.3451-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghoshal K, Jacob ST. Ara-ATP impairs 3′-end processing of pre-mRNAs by inhibiting both cleavage and polyadenylation. Nucleic Acids Res. 1991;19:5871–5. doi: 10.1093/nar/19.21.5871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gleason MR, Higashijima S, Dallman J, Liu K, Mandel G, Fetcho JR. Translocation of CaM kinase II to synaptic sites in vivo. Nat Neurosci. 2003;6:217–8. doi: 10.1038/nn1011. [DOI] [PubMed] [Google Scholar]
- 15.Guertin DA, Sabatini DM. The pharmacology of mTOR inhibition. Sci Signal. 2009;2:pe24. doi: 10.1126/scisignal.267pe24. [DOI] [PubMed] [Google Scholar]
- 16.Gumy LF, Tan CL, Fawcett JW. The role of local protein synthesis and degradation in axon regeneration. Exp Neurol. 2010;223:28–37. doi: 10.1016/j.expneurol.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hou L, Antion MD, Hu D, Spencer CM, Paylor R, Klann E. Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron. 2006;51:441–54. doi: 10.1016/j.neuron.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 18.Huang YS, Jung MY, Sarkissian M, Richter JD. N-methyl-D-aspartate receptor signaling results in Aurora kinase-catalyzed CPEB phosphorylation and alpha CaMKII mRNA polyadenylation at synapses. EMBO J. 2002;21:2139–48. doi: 10.1093/emboj/21.9.2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jama AM, Gabriel J, Al-Nagar AJ, Martin S, Baig SZ, Soleymani H, Chowdhury Z, Beesley P, Torok K. Lobe-specific functions of Ca2+.calmodulin in alphaCa2+.calmodulin-dependent protein kinase II activation. J Biol Chem. 2011;286:12308–16. doi: 10.1074/jbc.M110.157057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jimenez-Diaz L, Geranton SM, Passmore GM, Leith JL, Fisher AS, Berliocchi L, Sivasubramaniam AK, Sheasby A, Lumb BM, Hunt SP. Local translation in primary afferent fibers regulates nociception. PLoS One. 2008;3:e1961. doi: 10.1371/journal.pone.0001961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Joseph EK, Levine JD. Multiple PKCepsilon-dependent mechanisms mediating mechanical hyperalgesia. Pain. 2010;150:17–21. doi: 10.1016/j.pain.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Joseph EK, Levine JD. Hyperalgesic priming is restricted to isolectin B4-positive nociceptors. Neuroscience. 2010;169:431–5. doi: 10.1016/j.neuroscience.2010.04.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khasar SG, Burkham J, Dina OA, Brown AS, Bogen O, Alessandri-Haber N, Green PG, Reichling DB, Levine JD. Stress induces a switch of intracellular signaling in sensory neurons in a model of generalized pain. J Neurosci. 2008;28:5721–30. doi: 10.1523/JNEUROSCI.0256-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klumpen HJ, Beijnen JH, Gurney H, Schellens JH. Inhibitors of mTOR. Oncologist. 2010;15:1262–9. doi: 10.1634/theoncologist.2010-0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kondrashov A, Meijer HA, Barthet-Barateig A, Parker HN, Khurshid A, Tessier S, Sicard M, Knox AJ, Pang L, De Moor CH. Inhibition of polyadenylation reduces inflammatory gene induction. RNA. 2012;18:2236–50. doi: 10.1261/rna.032391.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu J, Schwartz JH. The cytoplasmic polyadenylation element binding protein and polyadenylation of messenger RNA in Aplysia neurons. Brain Res. 2003;959:68–76. doi: 10.1016/s0006-8993(02)03729-0. [DOI] [PubMed] [Google Scholar]
- 27.Liu LO, Li G, McCall MA, Cooper NG. Photoreceptor regulated expression of Ca(2+)/calmodulin-dependent protein kinase II in the mouse retina. Brain Res Mol Brain Res. 2000;82:150–66. doi: 10.1016/s0169-328x(00)00203-5. [DOI] [PubMed] [Google Scholar]
- 28.Mendelsohn AR, Larrick JW. Dissecting mammalian target of rapamycin to promote longevity. Rejuvenation Res. 2012;15:334–7. doi: 10.1089/rej.2012.1347. [DOI] [PubMed] [Google Scholar]
- 29.Mohr E, Richter D. Messenger RNA on the move: implications for cell polarity. Int J Biochem Cell Biol. 2001;33:669–79. doi: 10.1016/s1357-2725(01)00047-4. [DOI] [PubMed] [Google Scholar]
- 30.Muller WE, Seibert G, Beyer R, Breter HJ, Maidhof A, Zahn RK. Effect of cordycepin on nucleic acid metabolism in L5178Y cells and on nucleic acid-synthesizing enzyme systems. Cancer Res. 1977;37:3824–33. [PubMed] [Google Scholar]
- 31.Norsted Gregory E, Codeluppi S, Gregory JA, Steinauer J, Svensson CI. Mammalian target of rapamycin in spinal cord neurons mediates hypersensitivity induced by peripheral inflammation. Neuroscience. 2010;169:1392–402. doi: 10.1016/j.neuroscience.2010.05.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Obara I, Geranton SM, Hunt SP. Axonal protein synthesis: a potential target for pain relief? Curr Opin Pharmacol. 2012;12:42–8. doi: 10.1016/j.coph.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 33.Parada CA, Reichling DB, Levine JD. Chronic hyperalgesic priming in the rat involves a novel interaction between cAMP and PKCepsilon second messenger pathways. Pain. 2005;113:185–90. doi: 10.1016/j.pain.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 34.Parada CA, Yeh JJ, Joseph EK, Levine JD. Tumor necrosis factor receptor type-1 in sensory neurons contributes to induction of chronic enhancement of inflammatory hyperalgesia in rat. Eur J Neurosci. 2003;17:1847–52. doi: 10.1046/j.1460-9568.2003.02626.x. [DOI] [PubMed] [Google Scholar]
- 35.Parada CA, Yeh JJ, Reichling DB, Levine JD. Transient attenuation of protein kinase Cepsilon can terminate a chronic hyperalgesic state in the rat. Neuroscience. 2003;120:219–26. doi: 10.1016/s0306-4522(03)00267-7. [DOI] [PubMed] [Google Scholar]
- 36.Park KK, Liu K, Hu Y, Kanter JL, He Z. PTEN/mTOR and axon regeneration. Exp Neurol. 2010;223:45–50. doi: 10.1016/j.expneurol.2009.12.032. [DOI] [PubMed] [Google Scholar]
- 37.Price TJ, Geranton SM. Translating nociceptor sensitivity: the role of axonal protein synthesis in nociceptor physiology. Eur J Neurosci. 2009;29:2253–63. doi: 10.1111/j.1460-9568.2009.06786.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Price TJ, Rashid MH, Millecamps M, Sanoja R, Entrena JM, Cervero F. Decreased nociceptive sensitization in mice lacking the fragile X mental retardation protein: role of mGluR1/5 and mTOR. J Neurosci. 2007;27:13958–67. doi: 10.1523/JNEUROSCI.4383-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reichling DB, Levine JD. Critical role of nociceptor plasticity in chronic pain. Trends Neurosci. 2009;32:611–8. doi: 10.1016/j.tins.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ren Z, Cui J, Huo Z, Xue J, Cui H, Luo B, Jiang L, Yang R. Cordycepin suppresses TNF-alpha-induced NF-kappaB activation by reducing p65 transcriptional activity, inhibiting IkappaBalpha phosphorylation, and blocking IKKgamma ubiquitination. Int Immunopharmacol. 2012 doi: 10.1016/j.intimp.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 41.Richter JD. CPEB: a life in translation. Trends Biochem Sci. 2007;32:279–85. doi: 10.1016/j.tibs.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 42.Shih MH, Kao SC, Wang W, Yaster M, Tao YX. Spinal cord NMDA receptor-mediated activation of mammalian target of rapamycin is required for the development and maintenance of bone cancer-induced pain hypersensitivities in rats. J Pain. 2012;13:338–49. doi: 10.1016/j.jpain.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shor B, Gibbons JJ, Abraham RT, Yu K. Targeting mTOR globally in cancer: thinking beyond rapamycin. Cell Cycle. 2009;8:3831–7. doi: 10.4161/cc.8.23.10070. [DOI] [PubMed] [Google Scholar]
- 44.Si K, Giustetto M, Etkin A, Hsu R, Janisiewicz AM, Miniaci MC, Kim JH, Zhu H, Kandel ER. A neuronal isoform of CPEB regulates local protein synthesis and stabilizes synapse-specific long-term facilitation in aplysia. Cell. 2003;115:893–904. doi: 10.1016/s0092-8674(03)01021-3. [DOI] [PubMed] [Google Scholar]
- 45.Si K, Lindquist S, Kandel ER. A neuronal isoform of the aplysia CPEB has prionlike properties. Cell. 2003;115:879–91. doi: 10.1016/s0092-8674(03)01020-1. [DOI] [PubMed] [Google Scholar]
- 46.Sofroniadou S, Goldsmith D. Mammalian target of rapamycin (mTOR) inhibitors: potential uses and a review of haematological adverse effects. Drug Saf. 2011;34:97–115. doi: 10.2165/11585040-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 47.Sotelo-Silveira JR, Calliari A, Kun A, Koenig E, Sotelo JR. RNA trafficking in axons. Traffic. 2006;7:508–15. doi: 10.1111/j.1600-0854.2006.00405.x. [DOI] [PubMed] [Google Scholar]
- 48.Taiwo YO, Levine JD. Further confirmation of the role of adenyl cyclase and of cAMP-dependent protein kinase in primary afferent hyperalgesia. Neuroscience. 1991;44:131–5. doi: 10.1016/0306-4522(91)90255-m. [DOI] [PubMed] [Google Scholar]
- 49.Thakor DK, Lin A, Matsuka Y, Meyer EM, Ruangsri S, Nishimura I, Spigelman I. Increased peripheral nerve excitability and local NaV1.8 mRNA up-regulation in painful neuropathy. Mol Pain. 2009;5:14. doi: 10.1186/1744-8069-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Twiss JL, Fainzilber M. Ribosomes in axons--scrounging from the neighbors? Trends Cell Biol. 2009;19:236–43. doi: 10.1016/j.tcb.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 51.Vega-Avelaira D, Geranton SM, Fitzgerald M. Differential regulation of immune responses and macrophage/neuron interactions in the dorsal root ganglion in young and adult rats following nerve injury. Mol Pain. 2009;5:70. doi: 10.1186/1744-8069-5-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vega-Avelaira D, McKelvey R, Hathway G, Fitzgerald M. The emergence of adolescent onset pain hypersensitivity following neonatal nerve injury. Mol Pain. 2012;8:30. doi: 10.1186/1744-8069-8-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Villalba A, Coll O, Gebauer F. Cytoplasmic polyadenylation and translational control. Curr Opin Genet Dev. 2011;21:452–7. doi: 10.1016/j.gde.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 54.Willis D, Li KW, Zheng JQ, Chang JH, Smit AB, Kelly T, Merianda TT, Sylvester J, van Minnen J, Twiss JL. Differential transport and local translation of cytoskeletal, injury-response, and neurodegeneration protein mRNAs in axons. J Neurosci. 2005;25:778–91. doi: 10.1523/JNEUROSCI.4235-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wong YY, Moon A, Duffin R, Barthet-Barateig A, Meijer HA, Clemens MJ, de Moor CH. Cordycepin inhibits protein synthesis and cell adhesion through effects on signal transduction. J Biol Chem. 2010;285:2610–21. doi: 10.1074/jbc.M109.071159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu L, Wells D, Tay J, Mendis D, Abbott MA, Barnitt A, Quinlan E, Heynen A, Fallon JR, Richter JD. CPEB-mediated cytoplasmic polyadenylation and the regulation of experience-dependent translation of alpha-CaMKII mRNA at synapses. Neuron. 1998;21:1129–39. doi: 10.1016/s0896-6273(00)80630-3. [DOI] [PubMed] [Google Scholar]
- 57.Xu Q, Fitzsimmons B, Steinauer J, O’Neill A, Newton AC, Hua XY, Yaksh TL. Spinal phosphinositide 3-kinase-Akt-mammalian target of rapamycin signaling cascades in inflammation-induced hyperalgesia. J Neurosci. 2011;31:2113–24. doi: 10.1523/JNEUROSCI.2139-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yamauchi T. Neuronal Ca2+/calmodulin-dependent protein kinase II--discovery, progress in a quarter of a century, and perspective: implication for learning and memory. Biol Pharm Bull. 2005;28:1342–54. doi: 10.1248/bpb.28.1342. [DOI] [PubMed] [Google Scholar]
- 59.Yoo S, van Niekerk EA, Merianda TT, Twiss JL. Dynamics of axonal mRNA transport and implications for peripheral nerve regeneration. Exp Neurol. 2010;223:19–27. doi: 10.1016/j.expneurol.2009.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]