

Abstract
Purpose
To create a poly(lactic-co-glycolic acid) (PLGA) nanoparticles (NPs), where a drug-encapsulating NP core is covered with polyethylene glycol (PEG) in a normal condition but exposes a cell-interactive TAT-modified surface in an environment rich in matrix metalloproteinases (MMPs).
Methods
PLGA NPs were modified with TAT peptide (PLGA-pDA-TAT NPs) or dual-modified with TAT peptide and a conjugate of PEG and MMP-substrate peptide (Peritumorally activatable NPs, PANPs) via dopamine polymerization. Cellular uptake of fluorescently-labeled NPs was observed with or without a pre-treatment of MMP-2 by confocal microscopy and flow cytometry. NPs loaded with paclitaxel (PTX) were tested against SKOV-3 ovarian cancer cells to evaluate the contribution of surface modification to cellular delivery of PTX.
Results
While the size and morphology did not significantly change due to the modification, NPs modified with dopamine polymerization were recognized by their dark color. Moreover, TAT-containing NPs (PLGA-pDA-TAT NPs and PANPs) showed changes in surface charge, indicative of effective conjugation of TAT peptide on the surface. PLGA-pDA-TAT NPs and MMP-2-pre-treated PANPs showed relatively good cellular uptake as compared to PLGA NPs, MMP-2-non-treated PANPs, and NPs with non-cleavable PEG. After 3 hour treatment with cells, PTX loaded in cell-interactive NPs showed greater toxicity than that in non-interactive ones as the former could enter cells during the incubation period. However, due to the initial burst drug release, the difference was not as clear as microscopic observation.
Conclusions
PEGylated polymeric NPs that exposed cell-interactive surface in response to MMP-2 were successfully created by dual modification of PLGA NPs using dopamine polymerization.
Keywords: dopamine polymerization, surface modification, polymeric nanoparticles, PEG cleavage, TAT peptide
1. Introduction
Polymeric nanoparticles (NPs) are widely explored as a carrier of chemotherapeutic drugs as they can reach solid tumors selectively via leaky vasculature surrounding the tumors (1). Once the NPs arrive at tumors, equally important is to retain and/or internalize the NPs, which may otherwise be removed from the tumors and returned to circulation. To improve tumoral retention and uptake of NPs, their surface is often decorated with cell-interactive ligands, such as antibodies, peptides, nucleic acids, and small molecules, which can enhance interactions between NPs and target cells (2–7). However, the ligand-modified NPs face a greater chance of removal by the reticuloendothelial system (RES) during circulation (8, 9), subjecting the RES organs to toxic effects of chemotherapeutic agents.
An approach to avoid RES recognition of the ligands is to “hide” them under protective polyethylene glycol (PEG) layer until the NPs arrive at target tissues and the ligands are supposed to play a role. Here, the PEG layer is attached to NP surface via a linker that may be cleaved by a condition unique to solid tumors such as weakly acidic environment (10, 11) or enzymes abundant in the tumor extracellular matrix such as matrix metalloproteinases (MMPs) (12–15). For example, liposomes with removable PEG were prepared using a PEG-peptide-phospholipid ternary conjugate, where the peptide was sensitive to MMPs. These liposomes were used for the delivery of an anti-cancer drug (16), plasmid DNA (17), or siRNA (18), showing greater cellular uptake and biological activities with the removal of PEG by MMPs. The same principle was used in imaging agents such as quantum dots (19) or gold nanorods (12). These NPs were covered with a conjugate of PEG and MMP-substrate peptide via the avidin/biotin interaction (19) or a conjugate of PEG and a peptide substrate of urokinase-type plasminogen activator via gold-sulfur bond (12). These imaging agents showed increased cellular uptake/binding or tumor accumulation as PEG was removed by enzymatic cleavage of the peptide linkers.
One of the challenges in extending this approach to polymeric NPs is the increasing complexity of conjugation chemistry. Unless the NP surface is inherently reactive, decorating the surface with more than one type of functional groups is quite cumbersome and inefficient. It also requires multi-step reactions and exhaustive purification process (20, 21), which are often detrimental to the integrity of NPs and the production yield. An alternative approach is to pre-functionalize a polymer and produce NPs with the modified polymer. However, the synthesis is usually lengthy and inefficient, and needs to be tailored to each functional group (22). Sometimes the modified polymer ends up with altered chemical properties, losing the ability to encapsulate a therapeutic agent.
To overcome this problem, we employed a simple surface modification method based on dopamine polymerization (23). In an alkaline condition (~pH 8), the catechol group of dopamine is oxidized to quinone and reacts with other catechols/quinones to form a water-insoluble polymer film similar in composition to mussel adhesive proteins on solid surfaces (24). The polymerized dopamine (pDA) possesses latent reactivity toward nucleophiles like amine and thiol groups, allowing a facile immobilization of functional ligands on different surfaces (23, 24). Since the discovery, this method has been widely exploited in modifying a variety of substrates, such as magnetic nanoparticles (25, 26), synthetic polymers including polystyrene, polyurethane, polyethylene (27–29), stainless steel (30), and metals and oxides (24, 31).
Capitalizing on the simplicity and versatility of this technique, we immobilized a cell-interactive peptide and a PEG-peptide conjugate on PLGA NPs, thereby obtaining cell-interactive NPs with a removable PEG layer (Fig. 1). We modified the surface of PLGA NPs with (i) TAT peptide, a membrane-translocating peptide sequence (RKKRRQRRR) of the HIV TAT protein, as a cell-interactive ligand to promote cellular uptake of the NPs (22), and (ii) PEG conjugated to a MMP-substrate peptide (GPLGVRGC), where the peptide served as a MMP-2 cleavable linker. As the cleavage of PEG in the tumoral environment means the exposure of TAT peptide, which enables the NPs to actively engage with tumor cells, we call the dual-modified NPs “peritumorally activtable NPs” or PANPs. We first tested the applicability of the dopamine polymerization technique in modifying PLGA NPs with TAT peptide and extended the modification to create the dual-modified PANPs. To test the effect of TAT peptide and MMP-2 sensitivity, cellular uptake and biological activity of the NPs were tested in vitro.
Fig. 1.

Schematic diagram of a peritumorally activatable nanoparticle (PANP).
2. Materials and Methods
2.1 Materials
Paclitaxel (PTX) was a gift from Samyang Genex Corp. (Seoul, Korea). PLGA (LA:GA = 50:50, carboxylic acid end group, viscosity range: 0.15–0.25 dL/g, molecular weight: 4.2 kDa according to the vendor) was purchased from Lactel Absorbable Polymers (Durect Corporation, AL, USA). TAT peptide (GRKKRRQRRRGYKC-NH2) was purchased from Biomatik (Cambridge, Ontario, Canada) or New England Peptide (MA, USA). An MMP-2 substrate peptide (GPLGVRGC) was purchased from Biomatik, and Fmoc-NH-PEG-O-C3H6-CONHS was from Rapp Polymere GmbH (Tuebingen, Germany). PLGA-PEG (LA:GA = 50:50; PLGA: 4000 Da; PEG: 5000 Da) was purchased from PolySciTech® (Akina Inc., West Lafayette, IN, USA). Purified human recombinant MMP-2 was purchased from Calbiochem® (EMD Chemicals, NJ, USA). Dopamine hydrochloride was purchased from VWR (PA, USA). All other materials were purchased from Sigma Aldrich (St. Louis, MO, USA), unless specified otherwise.
2.2 MMP-2 mediated cleavage of a MMP-2 substrate peptide (GPLGVRGC)
The sensitivity of GPLGVRGC to MMP-2 was determined via High Pressure Liquid Chromatography (HPLC). First, purified human recombinant MMP-2 was activated using p-aminophenylmercuric acetate (APMA) following the vendor’s protocol. Briefly, APMA solution was made in 0.1M sodium hydroxide (NaOH) solution in a concentration ranging from 10–50 mM. MMP-2 (1.4 μM) was activated by mixing the MMP solution with APMA solution in a 10:1 volume ratio and incubating at 37°C for 2–3 hours, vortexing every 30 minutes. After incubation, the activated MMP-2 was diluted to 20 μg/mL with storage buffer (150 mM NaCl, 20 mM Tris-HCl, 5 mM CaCl2, and 0.05% Brij®-35 at pH 7.4) and stored in aliquots at −80°C.
The cleavage of GPLGVRGC peptide was tested with the activated MMP-2. Two μmol of GPLGVRGC was dissolved in a reaction buffer (pH 7.4) consisting of 50 mM HEPES, 200 mM NaCl, 10 mM CaCl2, and 1 mM ZnCl2. Three hundred nanograms of MMP-2 (4.2 pmol) was added to the peptide solution to determine whether MMP-2 could cleave the peptide. As a negative control, a reaction buffer without MMP-2 was added to the peptide. The mixture was incubated for 24 hours at 37°C and analyzed with HPLC (HPLC 1100 series, Agilent Technologies, Palo Alto, CA) and an Ascentis C18 Column (25 cm × 4.6 mm, 5 μm). The mobile phase was a mixture of water and acetonitrile containing 0.1% trifluoroacetic acid (with an increasing ratio of acetonitrile from 5% to 50% over 15 min). The flow rate was 0.5 mL/min, and the detection wavelength was 222 nm.
2.3 Synthesis of PEG5000-GPLGVRGC
A MMP-2 substrate peptide conjugated to PEG (5000 Da) (PEG5000-GPLGVRGC) was custom-synthesized at Biomatik (Cambridge, Ontario, Canada). Briefly, the MMP substrate peptide (GPLGVR(pbf)GC-resin) was first synthesized via the standard Fmoc-amino acid coupling process. After confirming successful synthesis of the peptide by mass spectroscopy, Fmoc-NH-PEG-O-C3H6-CONHS was coupled to obtain a Fmoc-NH-PEG-O-CH3H6-CONH-GPLGVR(pbf)GC-resin. The PEG-peptide conjugate was then removed from the resin, and all protecting groups were cleaved to reveal the final product, NH2-PEG-O-CH3H6-CONH-GPLGVRGC (PEG5000-GPLGVRGC).
2.4 NP Preparation (Fig. 2)
Fig. 2.

Schematic diagram of preparation of NPs used in this study.
To make PLGA NPs or PLGA-PEG NPs, 20 mg of PLGA or a mixture of PLGA (15 mg) and PLGA-PEG (5 mg) was dissolved in 600 μL of dichloromethane (DCM). The polymer solution was added to 2 mL of a 5% polyvinyl alcohol solution and immediately homogenized using a Sonics Vibracell™ probe sonicator for 1 min, pulsing 4 sec on and 2 sec off at an amplitude of 80%. After sonication, the NP solution was added to 5 mL of deionized water and spun overnight to evaporate the remaining DCM. The NPs were then collected via centrifugation at 10,000 rpm for 1 hour at 4°C and washed twice in deionized water with centrifugation at 8000 rpm for 30 min at 4°C.
Polydopamine-coated NPs (PLGA-pDA) were created by incubating PLGA NPs in 0.5 mg/mL dopamine hydrochloride dissolved in a 10 mM Tris buffer (pH 8.5) for 3 hours at room temperature with rotation. Following this primary incubation, the NPs were collected by centrifugation at 8000 rpm for 30 min at 4°C, resuspended in a fresh dopamine solution, and incubated for additional 2 hours at room temperature with rotation. The PLGA-pDA NPs were collected by centrifugation at 8000 rpm for 30 min at 4°C, resuspended with 1 mM phosphate buffer (pH 7.4), and rotated for 30 min prior to centrifugation.
To make TAT-coated NPs (PLGA-pDA-TAT), the PLGA-pDA NPs were resuspended in 1 mM phosphate buffer with the TAT peptide (2 mg/mL). After 30 min incubation at room temperature with rotation, the NPs were collected by centrifugation.
PANPs (PLGA-pDA-TAT/MMP substrate-PEG), the NPs dual-modified with TAT peptide and a conjugate of PEG and a MMP substrate (PEG5000-GPLGVRGC), were prepared by incubating the PLGA-pDA NPs in 1 mM phosphate buffer containing the TAT peptide (2 mg/mL) and PEG5000-GPLGVRGC (1 mg/mL) for 30 min at room temperature with rotation, followed by centrifugation.
For fluorescently labeled NPs, 5 mg of fluoresceinamine-conjugated PLGA (32) was added to replace an equivalent amount of PLGA. NPs loaded with PTX (PTX/NPs) were prepared by replacing 2 mg of PLGA with 2 mg of PTX (theoretical drug loading: 10%). The fluorescently labeled NPs were designated with an asterisk (*), such as *PLGA NPs, *PLGA-pDA NPs, or *PANPs. All NPs were stored as a pellet at 4°C and used in less than 36 hours.
2.5 NP Characterization
NPs were characterized for their size and zeta potential as previously described (22). Briefly, NPs were prepared as 0.5 mg/mL suspension in 1 mM phosphate buffer (pH 7.4). The size and zeta potential were measured on a Malvern Zetasizer Nano ZS90 (Worcestershire, UK). The PTX loading efficiency (LE, weight percentage of PTX in NPs) was determined by analyzing a known mass of NPs for the drug concentration via HPLC according to the previously described method (22). Briefly, a known weight of NPs was dissolved in 50:50 mixture of water and acetonitrile and analyzed with HPLC equipped with an Ascentis C18 Column (25 cm × 4.6 mm, 5um). The mobile phase was a mixture of water and acetonitrile (50:50). The flow rate was 1 mL/min, and the detection wavelength was 227 nm.
NPs were sputter-coated with platinum for 60 sec and visualized with a FEI NOVA nanoSEM field emission scanning electron microscope (FEI Company, Hillsboro, Oregon) using a high resolution through-the-lens detector (TLD) operating at a 7 kV accelerating voltage, ~4 mm working distance, spot 2.5 or 3, and 30 μm aperture.
2.6 Release Kinetics
PTX-loaded NPs equivalent to 8.75 μg of PTX were suspended in1 mL Dulbecco’s phosphate-buffered saline (DPBS) (Invitrogen, CA, USA) supplemented with 100 mg/L calcium chloride, 100 mg/L magnesium chloride, and 0.2% Tween 80. The NPs were incubated in the release media at 37°C with rotation. At designated time points, the NP suspensions were centrifuged at 10,000 rpm for 10 minutes at 4°C. Eight hundred microliters of the supernatant was sampled and replaced with fresh release medium. The supernatants were analyzed via HPLC.
2.7 Cellular uptake of NPs
SKOV-3 ovarian cancer cells (ATCC) were maintained in complete RPMI-1640 medium supplemented with 10% FBS, 100 units/mL penicillin, and 100 μg/mL streptomycin. The cells were subcultured at a ratio of 1:20 when they were 70–80% confluent. For confocal imaging, SKOV-3 cells were plated in 35 mm dishes at a density of 80,000 cells/cm2, grown overnight, and treated with different *NPs at a concentration of 0.1 mg/mL. *PLGA, *PLGA-pDA, and *PLGA-PEG NPs were prepared as a suspension in serum-free or serum-containing medium. In the case of *PANPs and *PLGA-PEG NPs, the NPs (0.3 mg) were pre-treated in a reaction buffer with or without activated MMP-2 for 24 hours at 37°C, collected by centrifugation for 30 minutes at 8,000 rpm at 4°C, and resuspended in a small volume of sterile water (100–200 μL), then in serum-free media to make a total volume of 3 mL. After incubation with *NPs for 3 hours, the *NP-containing medium was replaced with 2 mL of fresh serum-free medium. For nuclear staining, 1 μL of Draq5 (Biostatus Limited, UK) was added prior to imaging. Cells were imaged as previously described (22) with an Olympus X81 confocal microscope equipped with argon FV5-LA-MAR lasers or Nikon A1R confocal microscope equipped with a Spectra Physics 163C argon ion laser and a Coherent CUBE diode laser. The NPs were excited with a 488-nm laser, and the emission was read from 500 to 600 nm (green). Cell nuclei were excited with a 633-nm laser, and the emission was read from 650 to 750 nm (blue).
Flow cytometry was performed to quantify cellular uptake of NPs. SKOV-3 cells were plated in 24-well plates at a density of 50,000 cells per well, grown overnight, and incubated with different *NPs suspended in serum-free medium at a concentration of 0.5 mg/mL. As a negative control, a group of cells were treated with fresh medium with no NPs. After 3 hours of incubation, cells were then collected by trypsinization and analyzed with a flow cytometer (Beckman Coulter, FC 500, Indianapolis, IN USA) with a FL-1 detector (Ex: 488 nm; Em: 525 nm). In all FACS analysis, cell debris and free NPs were excluded by setting a gate on the plot of side-scattered light vs. forward-scattered light. A total of 10,000 gated events were acquired for each analysis. The fluorescent amplifier of the FL-1 detector filter was adjusted to ensure that the negative cell population appeared in the first logarithmic decade. All experiments were performed in triplicate.
2.8 Cytotoxicity
Cytotoxicity of the PTX-loaded NPs was tested using the MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. SKOV-3 cells were plated in a 96-well plate at a density of 8,000 cells per well in 200 μL of complete medium. After overnight incubation, 2 μL of a concentrated NP suspension was added to each well to provide PTX in the final concentration ranging from 1 to 10,000 nM. When PTX/PANPs and PTX/PLGA-PEG NPs were tested, the NPs were pre-treated in a reaction buffer with or without MMP-2 for 4 hours prior to the addition.
Cells were incubated with NPs for 3 hours, washed twice with fresh medium, and incubated for additional 3 days in NP-free complete medium. At the end of incubation, the medium was replaced with 100 μL of fresh medium and 15 μL of 5 mg/mL MTT solution and incubated for 3.5 hours. One hundred microliters of solubilization/stop solution was then added, and the plate was left in dark overnight. The absorbance of solubilized formazan in each well was read with a microplate reader at a wavelength of 562 nm. The absorbance of medium was subtracted from the absorbance of all wells. The measured sample absorbance was then normalized to the absorbance of control cells, which did not receive PTX.
2.9 Statistical Analysis
All data were expressed as means ± standard deviations. Statistics were performed using ANOVA with a Tukey test for means comparison. A value of p<0.05 was considered statistically significant.
3. Results
3.1 MMP-2 mediated cleavage of a MMP substrate peptide (GPLGVRGC)
GPLGVRGC peptide was chosen as an MMP-2 substrate according to previous studies (33–35). To confirm the enzymatic cleavage of GPLGVRGC, the peptide was incubated with or without 20 nM of activated MMP-2 for 24 hours and analyzed with HPLC. Only 5.6±1.6% of GPLGVRGC remained after the incubation with MMP-2, whereas the reaction buffer alone had little effect on the peptide.
3.2 NP Characterization
TAT and/or PEG5000-GPLGVRGC peptides were added to the surface of PLGA NPs via the dopamine polymerization process. NPs that had been incubated with dopamine (PLGA-pDA, PLGA-pDA-TAT NPs, or PANPs) turned black in color, indicative of dopamine polymerization. On the other hand, PLGA-pDA or PLGA-pDA-TAT NPs showed no noticeable difference from PLGA NPs on a microscopic level (Fig. 3), which suggests that the pDA coating did not contribute a significant mass. The particle size estimated from scanning electron microscopy was ~100 nm irrespective of particle type, which was significantly smaller than the hydrodynamic diameters measured by dynamic light scattering (170–200 nm) (Table 1). This observation is attributable to NP aggregation in suspension and consistent with our previous study (36).
Fig. 3.
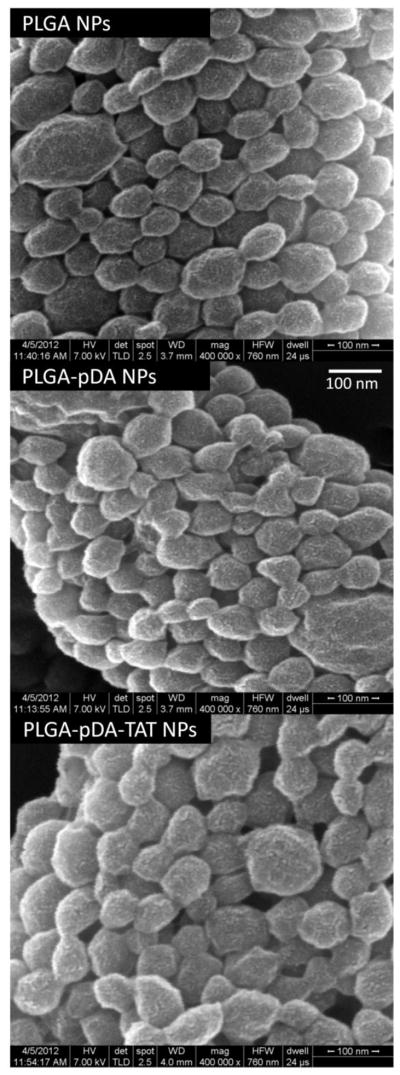
Scanning electron micrographs of PLGA, PLGA-pDA, and PLGA-pDA-TAT NPs.
Table 1.
Particle size, zeta potential, PTX loading content of different NPs
| NPs | Properties | Blank | Fluorescently labeled | PTX-loaded |
|---|---|---|---|---|
| PLGA | Size (d, nm) | 173.4 (5.6) | 177.9 (16.2) | 325.0 (64.4) |
| Zeta potential (mV) | −9.8 (1.9) | −8.3 (2.4) | −9.0 (4.5) | |
| PTX loading (wt%) | - | - | 21.8 (7.1) | |
| PLGA-pDA | Size (d, nm) | 171.2 (8.0) | 185.7 (23.1) | NA |
| Zeta potential (mV) | −11.7 (2.1) | −12.0 (2.3) | NA | |
| PTX loading (wt%) | - | - | NA | |
| PLGA-pDA-TAT | Size (d, nm) | 191.0 (11.0) | 192.2 (35.8) | 291.8 (72.8) |
| Zeta potential (mV) | −1.2 (3.4) | +1.6 (0.5) | +0.3 (0.9) | |
| PTX loading (wt%) | - | - | 18.8 (7.7) | |
| PANP | Size (d, nm) | NA | 210.8 (37.9) | 341.8 (86.8) |
| Zeta potential (mV) | NA | −0.2 (6.2) | −3.5 (5.9) | |
| PTX loading (wt%) | - | - | 20.0 (3.7) | |
| PLGA-PEG | Size (d, nm) | NA | 179.8 (24.4) | 248.8 (67.3) |
| Zeta potential (mV) | NA | −11.2 (4.6) | −7.5 (2.7) | |
| PTX loading (wt%) | - | - | 20.6 (11.4) |
PANP: Peritumorally activatable NPs, PLGA-pDA-TAT/MMP-substrate PEG NPs; NA: Not available. Data are expressed as averages (standard deviations) of 3–9 identically but independently prepared samples.
Table 1 summarizes particle sizes, surface charges, and loading efficiencies of the NPs. PTX-loaded NPs had an increased size of 250–350 nm, larger than blank or fluorescently labeled NPs. Surface charges of PLGA, PLGA-pDA, and PLGA-PEG NPs at pH 7.4 were approximately -10 mV, but NPs conjugated to the TAT peptide (PLGA-pDA-TAT NPs or PANPs) showed a reduced zeta potential, ranging from −3.5 to +1.6 mV.
PTX encapsulated NPs were tested for their drug release kinetics in PBS containing 0.2% Tween 80 (Fig. 4). PLGA NPs released ~70% of PTX in 4 hours, and all other NPs released drug in a similar pattern with no significant difference between the formulations at any time point.
Fig. 4.

Drug release kinetics from PTX/PLGA NPs, PTX/PLGA-pDA-TAT NPs, PTX/PLGA-PEG NPs, and PTX/PANPs. Each data point represents an average and standard deviation of at least 3 identically and independently prepared samples (ANOVA, Tukey test means comparison, p<0.05).
3.3 Cellular uptake of PLGA, PLGA-pDA, and PLGA-pDA-TAT NPs
To examine the effect of polydopamine coating and TAT conjugation on cellular uptake of NPs, fluorescently labeled *PLGA, *PLGA-pDA, and *PLGA-pDA-TAT NPs were incubated with SKOV-3 cells for 3 hours and observed with confocal microscopy. Since all three NPs were based on *PLGA NPs, the initial level of fluorescence was comparable across the NPs. As shown in Fig. 5, *PLGA NPs showed little cellular uptake, consistent with our previous reports (22, 32, 36). *PLGA-pDA NPs showed some cellular binding and/or uptake, which is likely due to the latent reactivity of pDA with proteins on the cell membrane. *PLGA-pDA-TAT NPs showed greater cellular uptake/association than *PLGA-pDA NPs, confirming additional contribution of TAT to the cellular interaction with the NPs. This trend remained the same irrespective of the presence of serum in the medium (Fig. 5 vs. Supporting Fig. 1), indicating that the NP-cell interaction reflects direct effects of the surface functional groups rather than serum proteins adsorbed to the NP surfaces. Quantitative analysis using flow cytometry corroborated the microscopic observation. As shown in Fig. 6, the cells incubated with *PLGA-pDA-TAT NPs showed the greatest fluorescence intensity, followed by those with *PLGA-pDA and *PLGA NPs.
Fig. 5.
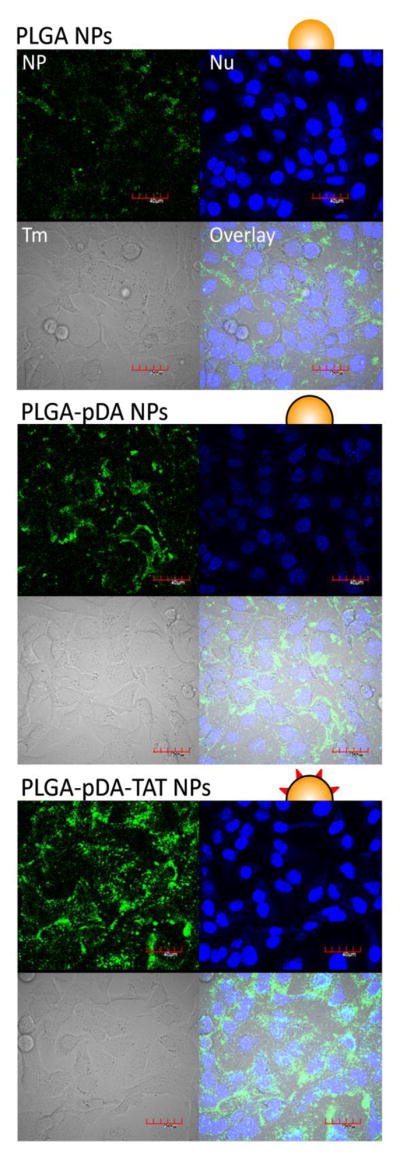
Cellular uptake of *PLGA, *PLGA-pDA, and *PLGA-pDA-TAT NPs. NPs were added to SKOV-3 cells in culture medium supplemented with 10% FBS. NP: green fluorescence signal from NPs; Nu: nuclei stained with Draq5; Tm: transmission image; Overlay: overlay of NP, Nu and Tm. Magnification: 600×. Scale bar = 40 μm.
Fig. 6.

(A) A representative flow cytometry overlay histogram showing fluorescence intensity of SKOV-3 cells treated with *PLGA, *PLGA-pDA, and *PLGA-pDA-TAT NPs. (B) Geometric mean fluorescence intensity (GMFI) of SKOV-3 cells treated with *PLGA, *PLGA-pDA, and *PLGA-pDA-TAT NPs. Data are expressed averages and standard deviations of 3 tests performed with identically and independently prepared samples. *: p<0.05 by Tukey test means comparison (difference from control was not indicated).
3.4 Cytotoxicity of PTX/PLGA NPs and PTX/PLGA-pDA-TAT NPs
To evaluate if differential NP uptake translated to drug delivery, PTX was loaded in PLGA and PLGA-pDA-TAT NPs and incubated with SKOV-3 cells. Since NP-cell interaction was established in 3 hours as shown in confocal microscopy, the incubation was limited to 3 hours, and then the cells were kept in NP-free medium so that they could grow under the influence of NPs that managed to remain with the cells. Fig. 7 shows that PTX/PLGA-pDA-TAT NPs had slightly greater activity than PTX/PLGA NPs, showing a decrease in the half maximal inhibitory concentration (IC50) of PTX from 258.7 nM (PTX/PLGA NPs) to 72.6 nM (PTX/PLGA-pDA-TAT NPs). The presence of polymerized dopamine caused no additional toxicity (data not shown), and the TAT peptide was shown to be non-toxic at the level used for surface modification (22, 37). Therefore, the relatively high cytotoxicity of PTX/PLGA-pDA-TAT NPs is attributable to their ability to establish interaction with cells in the 3 hour incubation period rather than the vehicle toxicity.
Fig. 7.

Viability of SKOV-3 cells treated with PTX/PLGA NPs or PLGA/PLGA-pDA-TAT NPs. Data are expressed as averages with standard deviations of 3 identically and independently prepared samples. (For IC50 calculation, see Supporting Table).
3.5 MMP-2 dependence of PANP uptake
To examine the responsiveness of PANPs to MMP-2, their cellular uptake was examined before and after MMP-2 treatment and compared with the uptake of PLGA-PEG NPs. When added without MMP-2 treatment, neither *PANPs nor *PLGA-PEG NPs showed interactions with SKOV-3 cells (Fig. 8), indicating that PEGylation was effective in both NPs. Although SKOV-3 cells naturally produce MMP-2 (38) (Supporting Fig. 2), the amount produced during the 3 hour incubation was apparently insufficient to cleave the peptide linker. On the other hand, *PANPs pre-treated with MMP-2 (20 nM) showed increased cellular uptake compared to non-treated *PANPs. *PLGA-PEG NPs did not show such MMP-2 sensitivity. Moreover, the majority of MMP-2-treated *PANPs were seen in the cytoplasm, whereas the few MMP-2-treated *PLGA-PEG NPs were located outside the cell boundaries. Quantitative analysis was performed with flow cytometry (Fig. 9). Since *PANPs and *PLGA-PEG NPs had different levels of fluorescence, comparison between the two groups was not made. Within each group, cells treated with *PLGA-PEG NPs showed no difference due to MMP-2 treatment, but those treated with *PANPs did, consistent with the confocal microscope observation. These results demonstrate that PANPs responded to MMP-2 and revealed the cell-interactive TAT peptide as the enzyme cleaved GPLGVRGC and removed PEG5000.
Fig. 8.
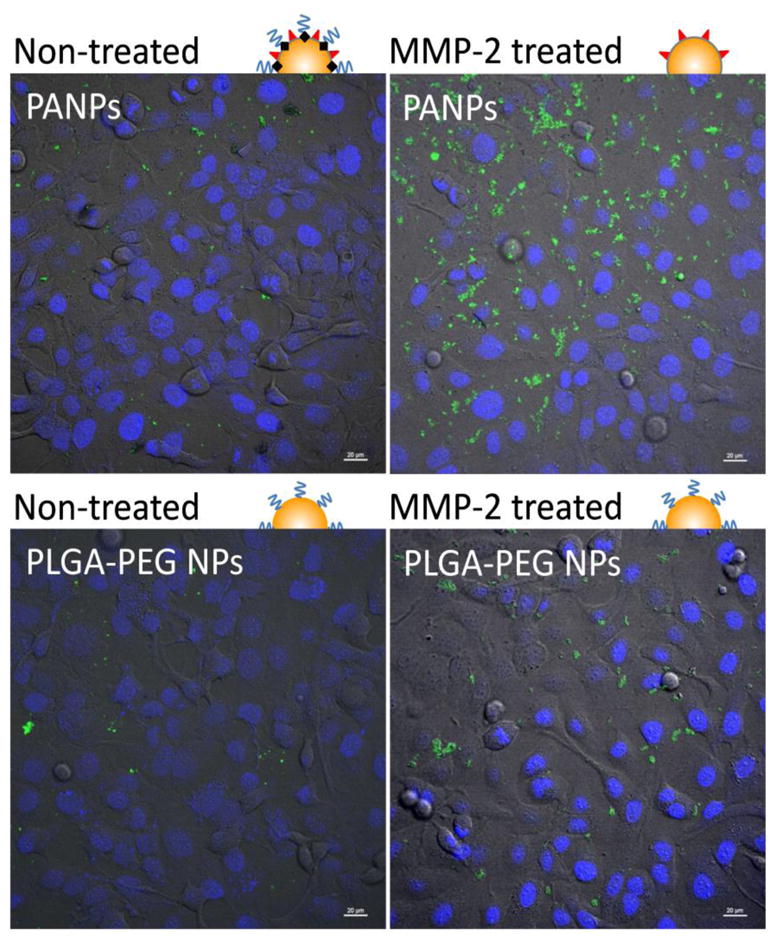
Cellular uptake of *PANPs and *PLGA-PEG NPs with or without MMP-2 pre-treatment. Scale bar = 20 μm.
Fig. 9.

A representative flow cytometry overlay histograms showing fluorescence intensity of SKOV-3 cells treated with *PLGA-PEG and *PANP NPs. (A) Without MMP-2 treatment and (B) with MMP-2 treatment. (C) Geometric mean fluorescence intensity (GMFI) of SKOV-3 cells treated with *PLGA-PEG and *PANP NPs. Data are expressed averages and standard deviations of 3 tests performed with identically and independently prepared samples. *: p<0.05 by one-tailed t-test.
3.6 MMP-dependence of drug delivery by PANPs
To test whether drug delivery by PANPs were enhanced by removal of PEG, as expected from NP uptake observation, PTX was loaded in PANPs and incubated with SKOV-3 cells with or without MMP-2 treatment. For comparison, PLGA-PEG NPs were used as a carrier of PTX and tested in the same manner. PTX/PLGA-PEG NPs showed similar cytotoxicity irrespective of MMP-2 treatment (Fig. 10). PTX/PANPs appeared to show relatively higher bioactivity after MMP-2 treatment, although statistical significance was not obtained. Notably, PTX/PANPs pre-treated with MMP-2 showed significantly greater cytotoxicity than PTX/PLGA-PEG NPs at 10,000 nM, corroborating the microscopic observation.
Fig. 10.

Viability of SKOV-3 cells treated with PTX/PANPs or PTX/PLGA-PEG NPs with or without MMP-2 pre-treatment. Data are expressed as averages with standard deviations of 3 identically and independently prepared samples. *: p<0.05 by Tukey test means comparison.
4. Discussion
TAT peptide has been widely used to enhance transport of NPs across cell membranes (37, 39–42). We previously reported a three-step synthesis of TAT-conjugated PLGA and production of TAT-modified PLGA NPs (22). While the NPs were able to enter cells efficiently, the production of TAT-conjugated PLGA was a lengthy and inefficient process with a total yield of ~10%. In contrast, functionalization of NPs using dopamine polymerization could be performed on the pre-formed NPs, irrespective of the chemical reactivity of the NP surface, in a mild condition with a minimal number of steps. Dopamine polymerization was visually recognized by the characteristic dark color. However, the size, zeta potential, and surface morphology of pDA-coated PLGA NPs were not different from those of PLGA NPs, suggesting that pDA content in the NPs was not substantial. On the other hand, when TAT peptide was added, the negative charge of NPs was neutralized or converted to slightly cationic values, reflecting the presence of the TAT peptide on the NP surface (Table 1). Previously, we did not observe the change of surface charge with the NPs prepared with TAT-conjugated PLGA, which was attributed to low efficiency of TAT conjugation to PLGA (22). The surface charge change observed in this study indicates that dopamine polymerization is a robust conjugation method that can deposit more TAT peptide on the NP surface.
PLGA-pDA-TAT NPs showed a greater uptake by SKOV-3 cells than PLGA NPs, consistent with our previous report (22). The contribution of enhanced cellular uptake of NPs to drug delivery was examined by comparing cytotoxicity of PTX-loaded in PLGA-pDA-TAT and PLGA NPs. The cell exposure to PTX-loaded NPs was limited to 3 hours for two reasons. First of all, a short-term exposure better represents dynamic in vivo situation, where NPs are continuously removed and returned to the circulation. Secondly, drug release increases with a prolonged incubation of NPs in culture medium. The released drug can kill cells irrespective of the location of NPs relative to the cells; thus, it becomes increasingly difficult to appreciate the contribution of the enhanced NP uptake. As anticipated from the cellular uptake of PLGA-pDA-TAT NPs, PTX delivered with the TAT-modified NPs showed slightly greater cytotoxicity than unmodified NPs (Fig. 7). Even though there were substantial drug release during the 3 hour incubation and large variation in the amount of released drug (Fig. 4), the 3.5-fold decrease in IC50 demonstrates the cytotoxic advantage of NPs with the TAT peptide. However, the premature drug release remains a significant issue in systemic application of the NPs, which may nullify their functionality. This problem may be overcome in the future by the use of covalent conjugation of a drug to a component of NPs via a linker that hydrolyzes over time or cleaves by enzymes at target tissues (43).
After we confirmed the effectiveness of dopamine polymerization process, we produced PANPs modified with two functional groups, TAT peptide and a conjugate of PEG and MMP-2 substrate peptide, where (i) PEG provides protection from non-specific cellular interaction of TAT peptide and latent reactivity of pDA coating and (ii) the peptide allows removal of the protection in the MMP-2 rich environment. Dual modification of NPs was simple and easy with the only difference from the previous NPs being the addition of a PEG-peptide conjugate. Since the PEG-peptide has nucleophiles in both termini (NH2 on PEG terminus, and NH2 and SH on cysteine terminus of the peptide), it can be incorporated into pDA layer in either direction, which means that a portion of PEG-peptide conjugated to the NP surface might have been attached in a reverse direction via the NH2 on PEG terminus. In this case, enzymatic cleavage of peptide would not result in removal of PEG. In other words, the MMP-2 dependent dePEGylation would have been more evident, had the NPs been modified with a PEG-peptide conjugate where the free terminus of PEG was non-reactive with pDA.
Despite the suboptimal choice of PEG, the dual-modified PANPs showed MMP-2 dependent cellular uptake (Fig. 8). In the absence of MMP-2 treatment, virtually no uptake of PANPs was observed due to the added PEG layer. On the other hand, MMP-2-treated PANPs were found in the cytoplasm of cells, indicating the exposure of cell-interactive TAT peptide on the NP surface after cleavage of PEG--albeit incomplete due to the PEG conjugated in a reverse direction. In contrast, PLGA-PEG NPs, which had non-cleavable PEG layer, did not show MMP-2 dependence in cellular uptake. To evaluate the MMP-2 dependence of drug delivery by the NPs, cytotoxicity of PTX loaded in PANPs and PLGA-PEG NPs was tested with or without a pre-treatment with MMP-2. As anticipated from the confocal microscopy, PTX/PLGA-PEG NPs show no difference due to MMP-2 pre-treatment. On the other hand, cytotoxicity of PTX/PANPs was apparently higher with the MMP-2 pre-treatment than without, but no statistical significance was observed. Multiple factors may account for the lack of significant difference, including the incomplete removal of PEG, premature drug release from the NPs, and large variation in the amount of released drug. The latter two must have been even more serious than PLGA-pDA-TAT NPs, given the 4 hours of pre-treatment with MMP-2 in the reaction buffer in addition to 3 hour incubation with cells, which additively contributed to the premature drug release.
5. Conclusion
This study demonstrates modification of PLGA NPs with dual functional groups, TAT peptide and a PEG-peptide conjugate, where the MMP-2 cleavable peptide linker allows for conditional removal of PEG. We expect that the dual-modified PLGA NPs has advantages of both long-circulating PEGylated NPs and cell-interactive ligand-bound NPs without a tradeoff between the two features. We observed that an increased cellular uptake of NPs did not always translate to a significant increase in cellular delivery of PTX, which is likely due to initial burst release of the drug. The early drug release has been a consistent problem in systemic application of PLGA NPs, which remains to be overcome in future studies. Finally, we note that dopamine polymerization was a simple and versatile method, which accomplished the dual surface modification with great ease and flexibility. This method may be used for functionalizing a variety of NP platforms irrespective of their surface reactivity as long as the ligands contain amine or thiol groups.
Supplementary Material
Acknowledgments
The authors thank Dr. Gaurav Bajaj for the help with quantitative RT-PCR. This work was supported by NIH R21 CA135130, NSF DMR-1056997, a Grant from the Lilly Endowment, Inc. to College of Pharmacy, Purdue University, Intramural Research Program (Global RNAi Carrier Initiative) of Korea Institute of Science and Technology, the P.E.O. Scholar Award (EG), and the Bilsland Dissertation Fellowship (EG).
Abbreviations
- MMPs
Matrix metalloproteinases
- NPs
Nanoparticles
- PANPs
Peritumorally activatable nanoparticles
- PLGA NPs
dual-modified with TAT peptide and a conjugate of PEG and MMP-substrate via dopamine polymerization (PLGA-pDA-TAT/MMP-substrate PEG NPs)
- pDA
Polymerized dopamine
- PEG
Polyethylene glycol
- PLGA
Poly(lactic-co-glycolic acid)
- PLGA-pDA NPs
PLGA NPs with pDA coating
- PLGA-pDA-TAT NPs
PLGA NPs modified with TAT peptide via dopamine polymerization
- PLGA-PEG NPs
NPs prepared with a PLGA-PEG conjugate
- PTX
Paclitaxel
References
- 1.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12):6387–92. [PubMed] [Google Scholar]
- 2.Gabizon AA, Shmeeda H, Zalipsky S. Pros and cons of the liposome platform in cancer drug targeting. J Liposome Res. 2006;16(3):175–83. doi: 10.1080/08982100600848769. [DOI] [PubMed] [Google Scholar]
- 3.Yokoyama M. Drug targeting with nano-sized carrier systems. J Artif Organs. 2005;8(2):77–84. doi: 10.1007/s10047-005-0285-0. [DOI] [PubMed] [Google Scholar]
- 4.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2(12):751–60. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 5.Wang M, Thanou M. Targeting nanoparticles to cancer. Pharmacol Res. 62(2):90–9. doi: 10.1016/j.phrs.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 6.Gullotti E, Yeo Y. Extracellularly activated nanocarriers: A new paradigm of tumor targeted drug delivery. Mol Pharm. 2009;6(4):1041–51. doi: 10.1021/mp900090z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu B, Tai HC, Xue W, Lee LJ, Lee RJ. Receptor-targeted nanocarriers for therapeutic delivery to cancer. Mol Membr Biol. 2010;27(7):286–98. doi: 10.3109/09687688.2010.521200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gu F, Zhang L, Teply BA, Mann N, Wang A, Radovic-Moreno AF, et al. Precise engineering of targeted nanoparticles by using self-assembled biointegrated block copolymers. Proc Natl Acad Sci U S A. 2008;105(7):2586–91. doi: 10.1073/pnas.0711714105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng J, Teply BA, Sherifi I, Sung J, Luther G, Gu FX, et al. Formulation of functionalized PLGA-PEG nanoparticles for in vivo targeted drug delivery. Biomaterials. 2007;28(5):869–76. doi: 10.1016/j.biomaterials.2006.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Montcourrier P, Silver I, Farnoud R, Bird I, Rochefort H. Breast cancer cells have a high capacity to acidify extracellular milieu by a dual mechanism. Clin Exp Metastasis. 1997;15(4):382–92. doi: 10.1023/a:1018446104071. [DOI] [PubMed] [Google Scholar]
- 11.Swallow CJ, Grinstein S, Rotstein OD. A vacuolar type h(+)-atpase regulates cytoplasmic ph in murine macrophages. J Biol Chem. 1990;265(13):7645–54. [PubMed] [Google Scholar]
- 12.Niidome T, Ohga A, Akiyama Y, Watanabe K, Niidome Y, Mori T, et al. Controlled release of peg chain from gold nanorods: Targeted delivery to tumor. Bioorg Med Chem. 2010;18(12):4453–8. doi: 10.1016/j.bmc.2010.04.070. [DOI] [PubMed] [Google Scholar]
- 13.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2(3):161–74. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 14.Roomi MW, Ivanov V, Kalinovsky T, Niedzwiecki A, Rath M. Inhibition of matrix metalloproteinase-2 secretion and invasion by human ovarian cancer cell line sk-ov-3 with lysine, proline, arginine, ascorbic acid and green tea extract. J Obstet Gynaecol Res. 2006;32(2):148–54. doi: 10.1111/j.1447-0756.2006.00389.x. [DOI] [PubMed] [Google Scholar]
- 15.Rabinovich A, Medina L, Piura B, Segal S, Huleihel M. Regulation of ovarian carcinoma SKOV-3 cell proliferation and secretion of mmps by autocrine IL-6. Anticancer Res. 2007;27(1A):267–72. [PubMed] [Google Scholar]
- 16.Terada T, Iwai M, Kawakami S, Yamashita F, Hashida M. Novel PEG-matrix metalloproteinase-2 cleavable peptide-lipid containing galactosylated liposomes for hepatocellular carcinoma-selective targeting. J Control Release. 2006;111(3):333–42. doi: 10.1016/j.jconrel.2005.12.023. [DOI] [PubMed] [Google Scholar]
- 17.Hatakeyama H, Akita H, Kogure K, Oishi M, Nagasaki Y, Kihira Y, et al. Development of a novel systemic gene delivery system for cancer therapy with a tumor-specific cleavable PEG-lipid. Gene Ther. 2007;14(1):68–77. doi: 10.1038/sj.gt.3302843. [DOI] [PubMed] [Google Scholar]
- 18.Hatakeyama H, Akita H, Ito E, Hayashi Y, Oishi M, Nagasaki Y, et al. Systemic delivery of sirna to tumors using a lipid nanoparticle containing a tumor-specific cleavable PEG-lipid. Biomaterials. 2011;32(18):4306–16. doi: 10.1016/j.biomaterials.2011.02.045. [DOI] [PubMed] [Google Scholar]
- 19.Mok H, Bae KH, Ahn C-H, Park TG. PEGylated and mmp-2 specifically depegylated quantum dots: Comparative evaluation of cellular uptake. Langmuir. 2009;25(3):1645–50. doi: 10.1021/la803542v. [DOI] [PubMed] [Google Scholar]
- 20.Narayanan S, Binulal NS, Mony U, Manzoor K, Nair S, Menon D. Folate targeted polymeric ‘green’ nanotherapy for cancer. Nanotechnology. 2010;21(28):285107. doi: 10.1088/0957-4484/21/28/285107. [DOI] [PubMed] [Google Scholar]
- 21.Rao KS, Reddy MK, Horning JL, Labhasetwar V. TAT-conjugated nanoparticles for the CNS delivery of anti-HIV drugs. Biomaterials. 2008;29(33):4429–38. doi: 10.1016/j.biomaterials.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gullotti E, Yeo Y. Beyond the imaging: Limitations of cellular uptake study in the evaluation of nanoparticles. J Control Release. 2012;164(2):170–6. doi: 10.1016/j.jconrel.2012.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee H, Rho J, Messersmith PB. Facile conjugation of biomolecules onto surfaces via mussel adhesive protein inspired coatings. Adv Mater. 2009;21(4):431–4. doi: 10.1002/adma.200801222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee H, Dellatore SM, Miller WM, Messersmith PB. Mussel-inspired surface chemistry for multifunctional coatings. Science. 2007;318(5849):426–30. doi: 10.1126/science.1147241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang M, Zhang X, He X, Chen L, Zhang Y. A self-assembled polydopamine film on the surface of magnetic nanoparticles for specific capture of protein. Nanoscale. 2012;4(10):3141–7. doi: 10.1039/c2nr30316g. [DOI] [PubMed] [Google Scholar]
- 26.Ni K, Lu H, Wang C, Black KCL, Wei D, Ren Y, et al. A novel technique for in situ aggregation of gluconobacter oxydans using bio-adhesive magnetic nanoparticles. Biotechnol Bioeng. 2012;109(12):2970–7. doi: 10.1002/bit.24582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee H, Dellatore SM, Miller WM, Messersmith PB. Mussel-inspired surface chemistry for multifunctional coatings. Science. 2007;318(5849):426–30. doi: 10.1126/science.1147241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsai WB, Chen WT, Chien HW, Kuo WH, Wang MJ. Poly(dopamine) coating of scaffolds for articular cartilage tissue engineering. Acta Biomater. 2011;7(12):4187–94. doi: 10.1016/j.actbio.2011.07.024. [DOI] [PubMed] [Google Scholar]
- 29.Ryou MH, Lee YM, Park JK, Choi JW. Mussel-inspired polydopamine-treated polyethylene separators for high-power li-ion batteries. Adv Mater. 2011;23(27):3066–70. doi: 10.1002/adma.201100303. [DOI] [PubMed] [Google Scholar]
- 30.Lu L, Li QL, Maitz MF, Chen JL, Huang N. Immobilization of the direct thrombin inhibitor-bivalirudin on 316l stainless steel via polydopamine and the resulting effects on hemocompatibility in vitro. J Biomed Mater Res A. 2012;100(9):2421–30. doi: 10.1002/jbm.a.34143. [DOI] [PubMed] [Google Scholar]
- 31.Kang K, Choi IS, Nam Y. A biofunctionalization scheme for neural interfaces using polydopamine polymer. Biomaterials. 2011;32(27):6374–80. doi: 10.1016/j.biomaterials.2011.05.028. [DOI] [PubMed] [Google Scholar]
- 32.Xu P, Gullotti E, Tong L, Highley CB, Errabelli DR, Hasan T, et al. Intracellular drug delivery by poly(lactic-co-glycolic acid) nanoparticles, revisited. Mol Pharmaceut. 2009;6(1):190–201. doi: 10.1021/mp800137z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y, So MK, Rao J. Protease-modulated cellular uptake of quantum dots. Nano Lett. 2006;6(9):1988–92. doi: 10.1021/nl0611586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bremer C, Tung CH, Weissleder R. In vivo molecular target assessment of matrix metalloproteinase inhibition. Nat Med. 2001;7(6):743–8. doi: 10.1038/89126. [DOI] [PubMed] [Google Scholar]
- 35.Lee S, Cha EJ, Park K, Lee SY, Hong JK, Sun IC, et al. A near-infrared-fluorescence-quenched gold-nanoparticle imaging probe for in vivo drug screening and protease activity determination. Angew Chem Int Ed Engl. 2008;47(15):2804–7. doi: 10.1002/anie.200705240. [DOI] [PubMed] [Google Scholar]
- 36.Amoozgar Z, Park J, Lin Q, Yeo Y. Low molecular-weight chitosan as a pH-sensitive stealth coating for tumor-specific drug delivery. Molecular Pharmaceut. 2012;9(5):1262–70. doi: 10.1021/mp2005615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berry CC. Intracellular delivery of nanopartides via the HIV-1 tat pepticle. Nanomedicine. 2008;3(3):357–65. doi: 10.2217/17435889.3.3.357. [DOI] [PubMed] [Google Scholar]
- 38.Sood AK, Fletcher MS, Coffin JE, Yang M, Seftor EA, Gruman LM, et al. Functional role of matrix metalloproteinases in ovarian tumor cell plasticity. Am J Obstet Gynecol. 2004;190(4):899–909. doi: 10.1016/j.ajog.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 39.Torchilin VP. Tat peptide-mediated intracellular delivery of pharmaceutical nanocarriers. Adv Drug Deliv Rev. 2008;60(4–5):548–58. doi: 10.1016/j.addr.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 40.Nam YS, Park JY, Han SH, Chang IS. Intracellular drug delivery using poly(d,l-lactide-co-glycolide) nano- particles derivatized with a peptide from a transcriptional activator protein of HIV-1. Biotechnol Lett. 2002;24(24):2093–8. [Google Scholar]
- 41.Torchilin VP, Rammohan R, Weissig V, Levchenko TS. TAT peptide on the surface of liposomes affords their efficient intracellular delivery even at low temperature and in the presence of metabolic inhibitors. Proc Natl Acad Sci U S A. 2001;98(15):8786–91. doi: 10.1073/pnas.151247498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koch AM, Reynolds F, Merkle HP, Weissleder R, Josephson L. Transport of surface-modified nanoparticles through cell monolayers. Chem Bio Chem. 2005;6(2):337–45. doi: 10.1002/cbic.200400174. [DOI] [PubMed] [Google Scholar]
- 43.Tong R, Cheng J. Paclitaxel-initiated, controlled polymerization of lactide for the formulation of polymeric nanoparticulate delivery vehicles. Angew Chem Int Ed Engl. 2008;47(26):4830–4. doi: 10.1002/anie.200800491. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.