

Summary
Purpose
The management of epilepsy in children is particularly challenging when seizures are resistant to anti-epileptic medications, or undergo many changes in seizure type over time, or have comorbid cognitive, behavioral, or motor deficits. Despite efforts to classify such epilepsies based on clinical and electroencephalographic criteria, many children never receive a definitive etiological diagnosis. Whole exome sequencing (WES) is proving to be a highly effective method for identifying de novo variants that cause neurological disorders, especially those associated with abnormal brain development. Here we explore the utility of WES for identifying candidate causal de novo variants in a cohort of children with heterogeneous sporadic epilepsies without etiological diagnoses.
Methods
We performed WES (mean coverage ~40X) on 10 trios comprised of unaffected parents and a child with sporadic epilepsy characterized by difficult-to-control seizures and some combination of developmental delay, epileptic encephalopathy, autistic features, cognitive impairment, or motor deficits. Sequence processing and variant calling were performed using standard bioinformatics tools. A custom filtering system was used to prioritize de novo variants of possible functional significance for validation by Sanger sequencing.
Key Findings
In nine of ten probands, we identified one or more de novo variants predicted to alter protein function, for a total of 15. Four probands had de novo mutations in genes previously shown to harbor heterozygous mutations in patients with severe, early-onset epilepsies (two in SCN1A, and one each in CDKL5 and EEF1A2). In three children, the de novo variants were in genes with functional roles that are plausibly relevant to epilepsy (KCNH5, CLCN4 and ARHGEF15). The variant in KCNH5 alters one of the highly conserved arginine residues of the voltage sensor of the encoded voltage-gated potassium channel. In vitro analyses using cell-based assays revealed that the CLCN4 mutation greatly impaired ion transport by the ClC-4 2Cl−/H+-exchanger and that the mutation in ARHGEF15 reduced GEF exchange activity of the gene product, Ephexin5, by about 50%. Interestingly, these seven probands all presented with seizures within the first six months of life, and six of these have intractable seizures.
Significance
The finding that seven of ten children carried de novo mutations in genes of known or plausible clinical significance to neuronal excitability suggests that WES will be of use for the molecular genetic diagnosis of sporadic epilepsies in children, especially when seizures are of early onset and difficult to control.
Keywords: Epileptic encephalopathy, SCN1A, CDKL5, ARHGEF15, CLCN4
Introduction
For pediatric neurologists, some of the most challenging patients with epilepsy are those with early-onset (≤ 6 months of age) seizures that are difficult to control with medications, or mixed seizure types that change over time, and with cognitive impairment, motor deficits, or autism as comorbid features. Intractable seizures, especially when they begin early in life, disrupt brain development, causing regression of cognitive, language, and motor skills (Wirrell et al., 2005). However, good seizure control does not guarantee good cognitive and behavioral outcome, in part because some epilepsy disorders have co-occurring intellectual disability and/or autism (Tuchman and Cuccaro, 2011). Taken together, these patients are at considerable risk for long-term disability, reduced quality of life, and even early death (Berg et al., 2004). While many of these cases likely have an underlying genetic basis, the genetic heterogeneity of epilepsy makes Sanger-sequencing approaches for genetic testing impractical, with 265 epilepsy genes already identified (Lemke et al., 2012) and many thousands of genes potentially involved in brain development and function (Kang et al., 2011).
Fortunately, the rapid development of next-generation sequencing (NGS)—notably whole exome sequencing (WES)—now enables researchers to interrogate all known protein-coding genes in a single experiment. There is now great optimism that this technology will lead to the identification of new genes and new mutations that cause neurological disease (Bamshad et al., 2011; Bras and Singleton, 2011; The Epi4K Consortium, 2012). In particular, the application of NGS in parent-offspring trios and quartets has been successful in identifying candidate pathogenic de novo mutations in patients with neurodevelopmental disorders (Vissers et al., 2010; O’Roak et al., 2012; Neale et al., 2012; Sanders et al., 2012; Rauch et al., 2012; de Ligt et al., 2012; Bin Xu et al., 2012), including epilepsy (Veeramah et al., 2012).
In this study we explore the applicability of WES within a trio framework to identify de novo variants for molecular genetic diagnosis of children with sporadic cases of epileptic encephalopathies with no known etiology. These ten patients, from a single community-based pediatric neurology practice, also had a mix of other clinical manifestations, including developmental delay, autistic features, cognitive impairment, and motor deficits.
Subjects and Methods
Subjects
We identified 10 children being treated at the Tucson Center for Neurosciences who had epilepsy that was difficult to control as well as one or more complicating factors in their clinical history, either before or after seizure onset (Table 1). They presented with seizures between the ages of 3 weeks and 8.5 years (median age of onset = 6 months). At the time of the study, the probands had been followed for between 10 months and 17.5 years (median follow-up 7 years, 3 months). Many of the children have had mixed seizure types, often changing over time. Three of the probands (A, B, H) had clinical features of West syndrome with primary presentation of infantile spasms, but without a specific etiology, and two of the probands (D, E) were suspected of having Dravet syndrome on clinical grounds. As a group, their seizures were intractable to medical therapy, or inadequately controlled on anti-epileptic medication or required several medications or trials of medications to achieve adequate control. Six children have persistently refractory seizures (Probands A, B, C, D, E, G) and in 3 others seizures are infrequent but not completely controlled (Probands F, I, J). Seizures are currently controlled in only one patient (Proband H), who had late-onset infantile spasms, was treated with ACTH and subsequently stabilized on two anti-epileptic medications. Proband H was also the only child with congenital anomalies (hypospadias and ventriculoseptal defect) and significant prematurity (born at 34 weeks’ gestation), requiring a lengthy NICU stay.
Table 1.
Clinical summary of patients examined by WES
| Proband (Sex) | Age | Age at Sz onset | Gestational, Birth & Neonatal Hx | Initial Seizure type(s) | Subsequent Seizure History | EEG Findings and Evolution | Neurodevelomental History and Exam | Other |
|---|---|---|---|---|---|---|---|---|
| A (F) | 6 yrs | 3 wks | Full term; nuchal cord, brief O2 | Multifocal clonic, generalized clonic, myoclonic, & generalized tonic | Infantile spasms from 5 mos to 5 yrs; complex partial seizures @ 3 yrs; subsequently myoclonic, tonic, GTC. Now: refractory | Normal @ 3 wks; hypsarrhythmia @ 5 mos; then diffuse slowing, frequent multifocal spikes, bursts of generalized spike/polyspike-and-wave, runs of slow spike-wave & episodic diffuse suppression | Epileptic encephalopathy; nonverbal, limited comprehension; autism; hypotonia, but increased ankle tone; poor coordination; unsteady wide-based gait; L Babinski | Presentation consistent with West s. |
| B (M) | 14 yrs | 10 wks | Gestational diabetes (insulin) & HTN; full term, C-section | Multifocal myoclonic and generalized myoclonic | Infantile spasms @ 8 mos, responsive to pyridoxine; myoclonic @ 3 yrs; @ 4 yrs: myoclonic/tonic; later atonic & GTC. Now: refractory | Multifocal sharp waves & generalized spike-wave to hypsarrhythmia to pattern of Lennox-Gastaut s. (multifocal spikes, runs of slow spike-wave, generalized spike-wave bursts and periods of diffuse supression) | Epileptic encephalopathy; acquired microcephaly (<2nd %ile); severe delay with episodic regression; nonverbal, limited comprehension; hypotonia, with increased ankle tone; incoordination, gait instability | Presentation consistent with West s. PET scan - relative hypometabolism L temporal and parietal lobes. |
| C (M) | 14 mos | 4 mos | Detached placenta, mild fetal distress, C-section. | Complex partial seizures | Complex partial seizures and secondarily GTC, some prolonged, not fully responsive to medications. Now: refractory | 8 mos: bilateral independent high-amplitude spikes, L > R; bursts of generalized spike & polyspike-wave | Microcephaly (2nd %ile), delayed milestones, diffuse hypotonia, dystonic posturing of arms; smiles, follows, reaches; unable to sit, roll over. No regression. | MRI: corpus callosum hypoplasia, increased FLAIR signal in white matter |
| D (F) | 14 yrs | 5 mos | Full term after preterm labor, LGA, C-section (breech) | Prolonged febrile GTC | Generalized tonic-clonic; myoclonic; tonic. Dravet s. suspected. Now: refractory | Normal @ 5 mos; generalized spike-and-wave @ 2.5 yrs | Epileptic encephalopathy; normal @ 1 yr, then progressive delay; poor coordination, unsteady gait, speaks 2–3 word phrases, no reading, math | |
| E (M) | 4 yrs | 6 mos | Preeclampsia & gestational diabetes (insulin). Full term, nuchal cord, brief O2 | Vaccine-related febrile GTC and hemiclonic | GTC and hemiclonic febrile and nonfebrile, some prolonged. Dravet s. suspected. Now: refractory | Normal @ 6 mos; bifrontal spikes @ 4 yrs | Epileptic encephalopathy; normal motor development; language delay with limited progression, some regression (uses 2–3-word phrases); decreased coordination | (+) FH febrile Sz (maternal uncle) |
| F (M) | 13 yrs | 6 mos | Full term | Nonfebrile GTC | GTC or hemiclonic, prolonged & often in clusters with illness; relatively good control with valproate; occasional, brief clonic (facial). Now: partly controlled | Frequent multifocal spikes, almost continuous during sleep, even with good Sz control | Epileptic encephalopathy; normal until 6 mos; mild motor & severe language delay; regression @ 3 yrs - reduced social interaction & attention; nonverbal with limited comprehension; autism; hypotonia | |
| G (M) | 18 yrs | 6 mos | Full term; Apgar 4 | Infantile spasms, nonresponsive to ACTH, controlled with vigabatrin | Complex partial seizures @ 3.5 yrs, with recurrence of clusters of flexor spasms @ 14 yrs. Now: refractory | 6 mos: hypsarrhythmia (resolved on vigabatrin); subsequently bihemispheric slowing, multifocal sharp waves | Epileptic encephalopathy; mild motor delay, moderate to severe speech-language delay; 1st-grade level; movements slow with poor coordination; emotional lability & compulsive behaviors | PET: R > L frontal lobe hypometabolism |
| H (M) | 7 yrs | 23 mos | Born @ 34 wks, birthweight 3lb 11oz, NICU x 19d; VSD, hypospadius | Epileptic spasms with focal features - head down and to L, eyes to L. | Epileptic spasms; complex partial Szs, followed by spasms, controlled after ACTH and maintenance on valproate and ethosuximide. Now: controlled | Bitemporal independent spikes, L > R; bursts of generalized spike & polyspike-wave, sometimes followed by suppression | Epileptic encephalopathy; mild motor delay - walked @ 17 mos; severe language delay, mild autism, poor coordination, stereotypies; ADD, behavior problems (tantrums) | MRI: L temporal atrophy, ?neuron migration defect; PET: L temporal hypo-metabolism |
| I (F) | 7.5 yrs | 3.5 yrs | Full term | Complex partial seizures, with R clonic | Simple & complex partial: R face, R hemiclonic, hemisensory, some in clusters, postictal confusion; excellent response to prednisone, maintenance on valproate & zonisamide. Now: partly controlled | Frequent, L centrotemporal spikes, almost continuous during sleep; intermittent R centrotemporal synchronous spikes | Normal milestones & exam; ADHD and significant behavior problems after Sz onset. | PET: bitemporal hypometabolism R > L; Sz worse on carbamazepine; (+) FH intractable epilepsy (maternal 1st cousin) |
| J (M) | 17 yrs | 8.5 yrs | Gestational diabetes (diet-treated); born @ 37 wks; severe FTT | Complex partial seizures, giggling with decreased awareness, eye deviation | Complex partial seizures. Now: partly controlled (brief & infrequent, every few weeks, on valproate & zonisamide) | Diffuse slowing, multifocal spikes bifrontal & L occipital; clinical Sz originating from L frontal region | Acquired microcephaly (<2nd %ile); motor regression, progressing to spastic quadriparesis; choreoathetosis, dystonic posturing; severe ID, nonverbal; short stature | MRI progressed from normal to delayed myelination to perventricular leukomalacia |
AD(H)D, attention deficit (hyperactivity) disorder; FTT, failure to thrive; GTC, generalized tonic-clonic; HTN, hypertension; LGA, large for gestational age; L, left; R, right; VSD, ventriculoseptal defect
Seven of the ten patients have an epileptic encephalopathy causing regression in developmental abilities (Probands A, B, D, E, F, G, H). Two showed severe developmental delay with cognitive impairment without a clear regression in skills or cognition (Probands C, J). One child was showing regression in academic skills with increasing seizure frequency, but this stabilized and reversed with better seizure control (Proband I). In all probands, electroencephalograms revealed diffuse or multifocal bi-hemispheric abnormalities. Brain MRI studies were normal or showed mild non-specific abnormalities. None of the probands had an etiological diagnosis and all were considered to be sporadic (i.e. had no first-degree relatives with similar neurological problems). Blood was collected from each proband and their parents. At the time of sample collection, the probands ranged in age from 14 months to 18 years. Informed consent for participation of each trio was obtained from both parents, and the study was approved by the University of Arizona Institutional Review Board.
Whole exome sequencing, processing and analysis
WES was performed on all 10 families by array capture of 64MB of exome target sequence using the Illumina TruSeq Enrichment methodology followed by paired-end sequencing on an Illumina HiSeq 2000. Sequences were aligned to the human genome (hg19) using BWA (Li and Durbin, 2010). Base quality recalibration, indel realignment, and variant calling were performed using GATK as previously described (Depristo et al., 2011). Variants were annotated using ANNOVAR (Wang et al., 2010). Gene annotations were made against the UCSC KnownGenes database. Previously known variants were annotated with their allele frequencies (using all ethnic affiliations) from the 1000 Genomes Project Feb. 2012 release and the NHLBI GO Exome Sequencing Project (ESP) 5400 samples release. Non-synonymous variants were annotated with Polyphen2 (Adzhubei et al., 2010), MutationTaster (Schwarz et al., 2010) and EvoD (Kumar et al., 2012) scores.
Candidate de novo variants that were predicted to alter protein function (i.e., non-synonymous, stop-gain, stop-loss and splice-junction mutations) were identified in cases where only the proband, and neither parent, carried the non-reference allele. In order to winnow our list of candidates to a set of true de novo variants, we implemented a prioritizing system that hierarchically placed each candidate into one of 7 ‘levels’ according to sequence or genome architecture features that ranged from lowest (level 1) to highest (level 7) probability of being a false positive (Figure 1). All candidate de novo variants within level 1 (~3 candidates per trio, range 0–5 per trio, total 29) and level 2 (~1 candidate per trio, range 0–3 per trio, total 10) that were found at a frequency of <0.001% in the ESP and 0.01% in the 1000 Genomes project, were submitted for validation testing by Sanger sequencing and/or Restriction Fragment Length Polymorphism (RFLP) assays. We also submitted for validation a subset of candidate variants in lower levels (11 candidates total) that were chosen on the basis of variant properties and whether they were in genes previously implicated in severe epilepsies.
Figure 1. Automated hierarchical filtering system to identify real de novo mutations.

All variants that fit a de novo inheritance pattern (variant allele present in the proband but neither parent) are first examined for evidence of reads with the variant allele in either parent despite the actual genotype called (aa/aa/ab or aa/aa/bb). When <30% reads containing the alternate allele were present in the parent, variants are placed into Level 7 (Kong et al. (Kong et al., 2012)) because of a likely false heterozygote call in the proband. When >10% of reads with the variant allele were in one or both of the parents, variants are placed into Level 6 (because of a likely false homozygote reference call in the parent). Remaining variants within 5bp of an indel are assigned to Level 5 due to possible misalignment. Remaining variants in proximity to other (at least one) Mendelian Inheritance Errors (MIEs) (i.e. in the same gene) are assigned to Level 4 because of either possible misalignment or poor region capture/sequencing. Remaining variants within a coverage in the proband > 2 standard deviations from the mean are placed into Level 3 due to likely lying in repetitive sequence. Remaining variants found in large population genetic variation screens (i.e. 1000 Genomes and the 5400 exomes of ESP) are assigned to Level 2 because of likely non-pathogenicity or systematic sequencing artifacts. The remaining variants are assigned to Level 1 and represent those putative de novo variants that are most likely to be real and are Sanger validated. Variants with a frequency < 0.001 in Level 2 are also Sanger validated.
We also screened for inherited variants with a frequency <1% in the ESP, both in cases of homozygous recessive or compound heterozygous state (i.e., when the same variant was present as two copies in the proband but only in one copy in both parents, and when two different variants were present in the same gene in the proband and were inherited from different parents, respectively). Candidate X-linked mutations in male probands present in the mother but not the father and never previously observed in the 1000 Genomes or ESP datasets were also examined.
RhoA activity in cells with mutant ARHGEF15 (Ephexin5) constructs
All plasmids were sequenced for correct cDNA. pCMV-SPORT6-ARHGEF15 was purchased (OpenBiosystems). Ephexin5 mouse cDNA was previously described (Margolis et al., 2010). Human Ephexin5 R604C and mouse R612C were cloned using QuikChange Site Directed Mutagenesis (Stratagene). N-terminal Ephexin5 antibody was described previously (Margolis et al., 2010). LiCor Biosciences Odyssey Infrared detection software was used to quantify Ephexin5 protein expression.
Human Embrionic Kidney 293 (HEK 293T) cells were transfected in a 6-well dish for 36 hours with 1 ug of indicated plasmids using the calcium phosphate method. 0.5 mg/mL of protein lysates were subjected to the luminescence-format G-LISA RhoA Activation Assay Kit (Cytoskeleton, Inc; Cat# BK121) as per the manufacturer’s instructions. All lysate samples were run in duplicate and the GloMax Multi-plate Detection System was used for luminometer readings. Final values for RhoA activation were calculated as a fold-change over the plasmid control transfection.
ClC-4 constructs and immunocytochemistry of transfected cells
Constructs for human ClC-4 in pCIneo and pTLN were described previously (Friedrich et al., 1999). The Gly544Arg mutation was introduced by PCR and confirmed by sequencing the complete ORF. HeLa cells were transfected with pCIneo constructs using FuGENE6 (Roche) and analyzed after 2 days by confocal immunofluorescence as described (Stauber and Jentsch, 2010). We used an affinity-purified polyclonal rabbit antibody raised against a C-terminal ClC-4 peptide (EEPPELPANSPHPLK, (Poët et al., 2006)).
Voltage-clamp analysis of Xenopus laevis oocytes
Xenopus laevis oocytes were injected with 25 ng cRNA transcribed from the pTLN constructs. After 2–3 days at 17°C they were examined by two-electrode voltage clamp (TEVC) using a protocol that started from a holding potential of −30 mV and clamped the oocytes to voltages between −80 and +80 mV in steps of 20 mV essentially as described (Friedrich et al., 1999).
Results
De novo variant discovery
The average exome sequence coverage per nucleotide per individual was 40X with a mean of 64% of the target exome sequence captured at a minimum of 20X. The number of candidate de novo variants per proband ranged from 53–149. After applying our hierarchical prioritizing system (Figure 1), we validated 14 de novo variants (9 missense and 5 frameshift indels) across the 10 trios. Additional de novo candidate sequencing identified a nonsense variant in the gene CDKL5 with a 1:3 allele imbalance indicative of an early somatic mutation (we note that nine somatic mutations were in observed in a previous whole exome sequencing study of 200 trios with autism (O’Roak et al., 2012)). De novo variants were identified in 9 of the 10 trios, with an overall rate of 1.5 de novo variants per trio. Two trios possessed 2 de novo variants and two others possessed 3 de novo variants (Table 2). No de novo variants were identified in mtDNA.
Table 2.
De novo mutations observed in 10 trios
| Trio | De novo mutations genes | Mutation Type | cDNA change | Protein change | In silico prediction |
|---|---|---|---|---|---|
| A | CDKL5 | nonsense | C2494T | Q832X | np/D/np |
| B | EEF1A2 | non-synon | G208A | G70S | D/D/D |
| C | CLCN4 | non-synon | G1630A | G544R | D/D/D |
| D |
SCN1A KIAA1456 |
non-synon non-synon |
G3824C C277T |
G1275A L93F |
D/D/D D/D/D |
| E |
SCN1A OR10H2 MTMR11 |
frameshift non-synon frameshift |
3547delA G392A 1528insT |
I1183fs R131H L511fs |
np/D/np B/B/D np/D/np |
| F | KCNH5 | non-synon | G980A | R327H | D/D/D |
| G |
ARHGEF15 HADHB |
non-synon frameshift |
C1810T 1142insCTTT |
R604C A381fs |
D/D/D np/D/np |
| H | - | - | - | - | - |
| I |
LPHN2 ZNF182 ZMYND8 |
non-synon non-synon frameshift |
G1400C A128G 1203delC |
R467T N43S P401fs |
B/D/D B/B/B np/D/np |
| J | THAP1 | frameshift | 6insA | V2fs | np/D/np |
Bold type represents genes with known or likely clinical significance. fs = frameshift, damaging based on Polyphen2/Mutation Taster/EvoD. D = probably damaging (PP2), disease causing (MT), deleterious (EvoD); B = benign (PP2), polymorphism (MT), neutral (EvoD); np = no prediction
Genes previously associated with severe epilepsy
Two probands possess mutations in the voltage-gated sodium channel gene SCN1A, defects in which are known to cause Dravet Syndrome (DS) (Depienne et al., 2008), a rare and catastrophic form of intractable epilepsy that begins in infancy. Proband D carries a missense mutation in exon 21 that leads to a substitution of glycine with alanine at amino acid position 1275 (p.Gly1275Ala). This amino acid position has previously been shown to be altered in a patient with DS, though with a G>T change rather than a G>C mutation, resulting in p.Gly1275Val substitution (Depienne et al., 2008). Proband E possesses a 1bp deletion in exon 20 that is predicted to result in a substantially shorter amino acid sequence (i.e., 1207 amino acids rather than 2010) and thus nonsense-mediated decay. De novo nonsense or frameshift mutations are found in ~50% of DS patients with SCN1A mutations (Mulley et al., 2005).
A p.Gln832Stop premature stop-codon in exon 17 of Cyclin-dependent kinase-like 5 (CDKL5) gene, which is known to cause an atypical form of Rett Syndrome that results in early onset seizures as well as infantile spasms (Castrén et al., 2011), was found in proband A. Of particular interest is a previous report of a female with an atypical form of Rett Syndrome with seizures beginning 10 days after birth who possessed a p.Gln834Stop premature stop-codon, just two amino acids downstream of Proband A (Nectoux et al., 2006). Proband B possesses a non-synonymous mutation, p.Gly70Ser, in EEF1A2, the gene encoding eukaryotic translation elongation factor 1, alpha-2. A recent WES survey of 100 individuals with intellectual disability identified the same de novo missense mutation in EEF1A2 in a child with onset of epilepsy at 4 months of age followed by severe psychomotor development delay and autistic features (trio 91 of de Ligt et al. (de Ligt et al., 2012)).
New epilepsy candidate genes with relevant neurobiological functions
KCNH5, a gene not previously associated with any human disease, was found in Proband F to contain a non-synonymous mutation resulting in a p.Arg327His substitution in Kv10.2, the eag-related voltage-gated potassium channel. The amino acid change occurs in the S4 transmembrane segment, disrupting the first of four conserved Arg residues that constitute essential features of the channel voltage sensor. Voltage-gated potassium channels are essential regulators of neuronal excitability and previous studies have shown that mutations in some members of this gene superfamily, such as KCNA1, KCNQ2 and KCNQ3 (Graves, 2006), can cause epilepsy. However KCNH5 is the first member of the eag-related subfamily H to be associated with this phenotype. Eag K+ channels are named for their loss-of-function convulsion-like phenotype in Drosophila (ether-a-gogo) (Brüggemann et al., 1993). Their expression is selectively localized to layer IV of the cerebral cortex, in particular to the numerous excitatory interneurons (Saganich et al., 1999). Moreover, expression studies indicate a role for these channels in synaptic plasticity (Griffith et al., 1994).
Another ion transporter mutation was found in Proband C, this time in the X-chromosomal gene CLCN4 that encodes the voltage-dependent 2Cl−/H+-exchanger ClC-4 (Scheel et al., 2005). The mutation is a non-synonymous change causing a p.Gly544Arg substitution, and given that the proband is a male, any potential phenotypic effect is expected to manifest. CLCN4 expression is high in the nervous system where the exchanger may transport ions across intracellular membranes. The sequence change is within an intramembrane four-amino-acid-residue loop that connects intramembrane helices P and Q. While this gene has not previously been implicated in any human disorder, mutations in several CLCN genes underlie human pathologies (Jentsch, 2008). These include Dent’s disease that is caused by mutations in the endosomal 2Cl−/H+-exchanger ClC-5 (Lloyd et al., 1996), a close homolog of ClC-4.
When transfected into HeLa cells, both WT and mutant ClC-4 localized to structures resembling the endoplasmic reticulum (ER) (Figure 2A). ER localization of ClC-4 has been observed previously upon heterologous overexpression (Okkenhaug et al., 2006), but native ClC-4 might rather be found on endosomes (Jentsch, 2008). When expressed in Xenopus oocytes, the Gly544Arg mutation almost abolished the outwardly rectifying ClC-4 currents that are mediated by electrogenic 2Cl−/H+-exchange (Friedrich et al., 1999; Scheel et al., 2005) (Figure 2B), identifying it as a loss-of-function mutation.
Figure 2. Characterization of putative epilepsy-causing mutation in human CLC-4.
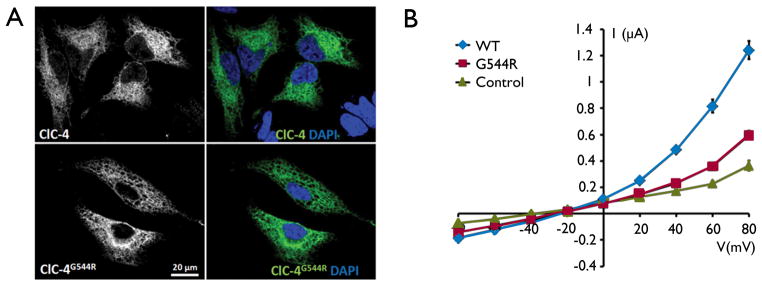
(A) Overexpression of human ClC-4 wildtype and mutant protein (green) in HeLa cells show that the G544R mutation does not alter the subcellular localization of the protein. Nuclei are stained with DAPI in blue. (B) Current voltage-relationships of ClC-4 WT and ClC-4G544R expressed in Xenopus oocytes. The results were obtained with at least five oocytes from four batches of oocytes. Control measurements were done on uninjected oocytes. Error bars indicate standard error of the mean (some error bars are smaller than symbol size).
Proband G possesses a de novo non-synonymous mutation in the gene Rho Guanine Nucleotide Exchange Factor 15 (ARHGEF15) resulting in a p.Arg604Cys substitution in the protein product. ARHGEF15 is also known as Ephexin5. While this gene has not previously been associated with a human disease, Ephexin5 is highly expressed in the brain (Sahin et al., 2005) and was recently shown to negatively regulate excitatory synapse formation during development (Margolis et al., 2010). In particular, Ephexin5 suppresses the function of EPH receptor B2 (EphB2), a mechanism believed to limit uncontrolled synapse formation. When EphB2 encounters its ligand Ephrin-B1/2, Ephexin5 is phosphorylated, ubiquinated and finally degraded, allowing synapse formation to occur. The process of degradation is mediated by ubiquitin protein ligase E3A (UBE3A). Interestingly, 90% of patients diagnosed with Angelman Syndrome, which is highly associated with seizures, lack expression of UBE3A (Dan, 2009). It has been suggested from Angelman mouse models that one mechanism of the disorder is due to a lack of degradation of Ephexin5 by UBE3A, which leads to a decrease in synaptic formation (Margolis et al., 2010).
Ephexin5’s GEF exchange activity is required to restrict synapse formation. Since Ephexin5 was previously shown to promote the activation of the small GTPase RhoA, we hypothesized that a de novo mutation in Ephexin5 may disrupt its GEF exchange activity towards RhoA. To test this we cloned an Ephexin5 human cDNA containing the p.Arg604Cys substitution (R604C). We transfected HEK 293T cells with a control plasmid, a plasmid driving the expression of wild-type (WT) human Ephexin5, or a plasmid driving the expression of mutant R604C. We prepared extracts from transfected cells and subjected them to a luminescence-based G-LISA RhoA activation assay. We find that RhoA activation by R604C is reduced by ~46% compared to WT (Figure 3A). We also find that a homologous amino acid in mouse Ephexin5 at Arg612Cys (R612C) shows a ~47% reduction as compared with mouse WT protein (Figure 3B). Importantly, we did not detect differences in mutant protein expression compared to the WT protein (Figure 3C and 3D), suggesting that the de novo mutation causes a deficit in GEF exchange activity, and not protein stability. We conclude that the R604C mutation may lead to decreased Ephexin5 GEF activity resulting in an increase in excitatory synapse number in the brain, which could be an underlying cause of the seizures observed in Proband G.
Figure 3. De novo mutation at Arg604Cys disrupts Ephexin5’s guanine nucleotide exchange activity.

A) Lysates from HEK 293T cells transfected with control plasmid, human WT-Ephexin5, or R604C were subjected to the G-LISA activation assay. Error bars indicate SEM (n=4) *p < .01, Student’s T-test. B) Mouse WT-Ephexin5, or R612C were assayed for RhoA activation similar to A). Error bars indicate SEM (n=3) *p < .05, Student’s T-test. C) Representative quantitative Western Blot using LiCor Odyssey IR software from lysates used in A) and B). Protein samples were run on an SDS-PAGE gel and Ephexin5 protein was detected using a rabbit polyclonal antibody raised against the N-terminus. D) Western Blot quantification for all samples. Error bars indicate SEM (n=4 per human conditions; n=3 per mouse conditions), p > .05, Student’s T-test.
Other de novo variants of unclear functional significance
Proband G also possesses a frameshift de novo mutation in the gene HADHB, which is known to cause mitochondrial trifunctional protein (MTP) deficiency when found in simple homozygous or compound heterozygous form (Park et al., 2009). However, there were no other coding variants found in this trio in this gene.
Proband I possesses 3 non-synonymous de novo mutations, the most interesting of which resulted in a p.Arg467Thr substitution in the protein Latrophilin-2 (LPHN2). Latrophilins were originally identified as G-protein-coupled receptors that bind α-Latrotoxin (black widow spider venom). Toxin binding at the synapse results in the mass release of neurotransmitters from synaptic vesicles (Silva and Ushkaryov, 2010). Latrophilin-2 interacts with two SH3-and-multiple-ankyrin-repeat-domains proteins (SHANK1 and SHANK2) (Kreienkamp, 2000) while Latrophilin-1 has recently been shown to bind neurexins (Boucard et al., 2012). Both SHANK and NRXN genes show strong associations with ASD and ID (Bourgeron, 2009). This proband also possesses a frameshift deletion at position 356 in the MYND-type containing 8 Zinc Finger gene (ZMYND8). ZMYND8 is expressed at high levels in the brain and is involved in early development of the nervous system in Xenopus (Zeng et al., 2010). While potentially interesting given their links to brain development, the functional mechanisms that might relate LPHN2 and ZMYND8 to epilepsy are not clear. It is noteworthy that this proband had later-onset seizures (3.5 years of age) that have been relatively well controlled by medication (Table 1).
Proband J was found to have a de novo frameshift mutation in the second codon of THAP domain-containing protein 1 (THAP1). THAP1 mutations, including frameshifts in the early part of the protein such as observed here, act in a dominant manner, causing torsion dystonia, DYT6 (Xiromerisiou et al., 2012). However, while Proband J did demonstrate dystonic posturing, the THAP1 mutation has not been shown to be associated with epilepsy or any of the other clinical features exhibited by this proband (Table 1).
Four other de novo mutations in the genes ZNF182 (Proband I), KIAA1456 (Proband D), OR10H2 (Proband E), MTMR11 (Proband E) were of unclear neurobiological significance.
Simple recessive, compound heterozygous and X-linked variants
We also scanned each trio’s WES data for variants fitting a simple recessive or compound heterozygous pattern (with an allele frequency cut-off of 1% in ESP) as well as X-linked variants in male probands that were not previously observed in any public database. After removing likely alignment errors, we manually curated these lists using Genecards and OMIM, looking for genes with known roles in neurological disorders, or known or hypothesized functions related to brain development or function and, where applicable, significant deleterious Polyphen2, MutationTaster, and EvoD (at least two of three) scores. While none of the inherited variants met this strict criterion, Proband H has two variants in the USP34 gene (p.R508S, p.H1671L) that are each predicted to be deleterious by two of the three in silico methods, although not in a concordant manner. This is of potential interest as this patient was the only proband in which we did not find a de novo variant. USP34 regulates Wnt signaling through Axins (Lui et al., 2011), and mouse mutations in its family members (UCHL1 (Cartier et al., 2012) and USP14 (Crimmins et al., 2006)) cause nervous system phenotypes.
Discussion
Our results suggest that there is substantial value of applying WES in a trio framework to patients with epileptic encephalopathies of unknown etiology. Of the 15 true de novo variants, 4 represent mutations in genes (SCN1A, CDKL5, EEF1A2) previously associated with severe epilepsies. Three of the true de novo variants represent mutations in genes (KCNH5, CLCN4, ARHGEF15) not previously associated with epilepsies in humans but with a highly relevant biological function. All seven highly functionally relevant de novo mutations caused substitutions at evolutionarily conserved amino acid positions and were predicted to be damaging by in silico methods (Table 2). Mutations in three genes may play a role in the proband’s phenotype (Table 1) based on what is known about their biological functions (LPHN2, ZMYND8, THAP1).
Four candidate mutations are found in genes not currently screened in commercial Sanger-sequencing assays or in a proposed panel of 265 epilepsy-associated genes interrogated using NGS at high coverage (Lemke et al., 2012). Clearly, more confirmatory information is required for three of these genes (KCNH5, ARHGEF15, CLCN4) before they can be accepted as truly pathogenic. Even the observation that the Gly544Arg mutation strongly impaired ClC-4 ion transport does not prove a role of CLCN4 in human epilepsy, in particular since Clcn4−/− mice do not display epilepsy or any other obvious phenotype (Rickheit et al., 2010). Interestingly, recent work investigating variants of seven CLCN genes in individuals with complex idiopathic epilepsy syndromes found enrichment of missense mutations of CLCN1 and CLCN2 (Chen et al, 2013). In addition to downstream functional studies, which are ongoing, large-scale sequencing efforts such as that being conducted by the Epi4K project (The Epi4K Consortium, 2012) should be useful for confirming the importance of these and related genes in epilepsy.
It is interesting to note that all seven of the probands in whose exomes we identified clear or promising candidate de novo mutations presented with seizures beginning within the first 6 months of life. On the other hand, we failed to identify plausible causal mutations in the three patients with seizures that presented later (i.e., at 23 month, 3.5 years, and 8.5 years). This suggests that patients with early-onset epileptic encephalopathies may be more likely to carry de novo mutations that cause abnormal excitability and disrupt synaptic plasticity in the developing brain (Brooks-Kayal, 2010). Future studies of larger numbers of patients with a range seizure onset ages will help to determine whether the trend hinted at here is statistically significant, which could have important implications for diagnosis, prognosis, and treatment.
We have presented evidence suggesting that WES is likely to be of use for diagnosing sporadic epilepsies within a research setting. However, the application of this technology in the clinical setting still requires thorough investigation. Lyon et al (Lyon and Wang, 2012) detail many of the issues generic to most disorders. Two issues that are particularly relevant to the applicability of this approach are (a) the utility of NGS for diagnosis in the absence of one or both parents given the large number of genes involved in brain development and the growing evidence for the importance of de novo mutations, and (b) the challenge of communicating results to clinicians, patients, and parents when mutations in novel and potentially causal candidate genes are identified. These two issues will be aided in some respects by large gene discovery screens such as that being conducted by the Epi4K project (The Epi4K Consortium, 2012). Despite these challenges, the results presented here give cause for optimism that NGS screening will soon aid pediatric neurologists to diagnose children with epilepsies of unknown etiology.
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines. You will find the policy on ethical publication in Epilepsia instructions for authors at http://onlinelibrary.wiley.com/journal/10.1111/(ISSN)1528-1167/homepage/ForAuthors.html
Acknowledgments
L.L.R. is funded by Autism Speaks and the Arizona Center for Biology of Complex Diseases. This work was partially supported by National Institute of Neurological Disorders and Stroke grant RO1 5R01NS045500 (M.E.G.) and by a Stuart H.Q. & Victoria Quan Predoctoral Fellowship (J.S.). This work is dedicated to the memory of Shay Emma Hammer.
Footnotes
Disclosure of Conflicts of Interest
None of the authors has any conflict of interest to disclose.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, Shendure J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12:745–755. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- Berg AT, Shinnar S, Testa FM, Levy SR, Smith SN, Beckerman B. Mortality in childhood-onset epilepsy. Arch Pediatr Adolesc Med. 2004;158:1147–1152. doi: 10.1001/archpedi.158.12.1147. [DOI] [PubMed] [Google Scholar]
- Bin Xu, Ionita-Laza I, Roos JL, Boone BE, Woodrick S, al E. De novo gene mutations highlight patterns of genetic and neural complexity in schizophrenia. Nat Genet. 2012;44:1365–1369. doi: 10.1038/ng.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucard AA, Ko J, Südhof TC. High Affinity Neurexin Binding to Cell Adhesion G-protein-coupled Receptor CIRL1/Latrophilin-1 Produces an Intercellular Adhesion Complex. J Biol Chem. 2012;287:9399–9413. doi: 10.1074/jbc.M111.318659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeron T. A synaptic trek to autism. Curr Opin Neurobiol. 2009;19:231–234. doi: 10.1016/j.conb.2009.06.003. [DOI] [PubMed] [Google Scholar]
- Bras JM, Singleton AB. Exome sequencing in Parkinson’s disease. Clin Genet. 2011;80:104–109. doi: 10.1111/j.1399-0004.2011.01722.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks-Kayal A. Epilepsy and autism spectrum disorders: Are there common developmental mechanisms? Brain Dev. 2010;32:731–738. doi: 10.1016/j.braindev.2010.04.010. [DOI] [PubMed] [Google Scholar]
- Brüggemann A, Pardo LA, Stühmer W, Pongs O. Ether-à-go-go encodes a voltage-gated channel permeable to K+ and Ca2+ and modulated by cAMP. Nature. 1993;365:445–448. doi: 10.1038/365445a0. [DOI] [PubMed] [Google Scholar]
- Cartier AE, Ubhi K, Spencer B, Vazquez-Roque RA, Kosberg KA, Fourgeaud L, Kanayson P, Patrick C, Rockenstein E, Patrick GN, Masliah E. Differential effects of UCHL1 modulation on alpha-synuclein in PD-like models of alpha-synucleinopathy. PLoS ONE. 2012;7:e34713. doi: 10.1371/journal.pone.0034713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castrén M, Gaily E, Tengström C, Lähdetie J, Archer H, Ala-Mello S. Epilepsy caused by CDKL5 mutations. Eur J Paed Neurol. 2011;15:65–69. doi: 10.1016/j.ejpn.2010.04.005. [DOI] [PubMed] [Google Scholar]
- Chen TT, Klassen TL, Goldman AM, Marini C, Guerrini R, Noebels JL. Novel brain expression of ClC-1 chloride channels and enrichment of CLCN1 variants in epilepsy. Neurology electr prepub. 2013 doi: 10.1212/WNL.0b013e31828868e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crimmins S, Jin Y, Wheeler C, Huffman AK, Chapman C, Dobrunz LE, Levey A, Roth KA, Wilson JA, Wilson SM. Transgenic rescue of ataxia mice with neuronal-specific expression of ubiquitin-specific protease 14. J Neurosci. 2006;26:11423–11431. doi: 10.1523/JNEUROSCI.3600-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan B. Angelman syndrome: Current understanding and research prospects. Epilepsia. 2009;50:2331–2339. doi: 10.1111/j.1528-1167.2009.02311.x. [DOI] [PubMed] [Google Scholar]
- de Ligt J, Willemsen MH, van Bon B, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012:367. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- Depienne C, Trouillard O, Saint-Martin C, Gourfinkel-An I, Bouteiller D, Carpentier W, Keren B, Abert B, Gautier A, Baulac S, Arzimanoglou A, Cazeneuve C, Nabbout R, LeGuern E. Spectrum of SCN1A gene mutations associated with Dravet syndrome: analysis of 333 patients. J Med Genet. 2008;46:183–191. doi: 10.1136/jmg.2008.062323. [DOI] [PubMed] [Google Scholar]
- Depristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, Mckenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich T, Breiderhoff T, Jentsch TJ. Mutational analysis demonstrates that ClC-4 and ClC-5 directly mediate plasma membrane currents. J Biol Chem. 1999;274:896–902. doi: 10.1074/jbc.274.2.896. [DOI] [PubMed] [Google Scholar]
- Graves TD. Ion channels and epilepsy. QJM. 2006;99:201–217. doi: 10.1093/qjmed/hcl021. [DOI] [PubMed] [Google Scholar]
- Griffith LC, Wang J, Zhong Y, Wu CF, Greenspan RJ. Calcium/calmodulin-dependent protein kinase II and potassium channel subunit eag similarly affect plasticity in Drosophila. Proc Natl Acad Sci USA. 1994;91:10044–10048. doi: 10.1073/pnas.91.21.10044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch TJ. CLC chloride channels and transporters: from genes to protein structure, pathology and physiology. Crit Rev Biochem Mol Biol. 2008;43:3–36. doi: 10.1080/10409230701829110. [DOI] [PubMed] [Google Scholar]
- Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu X, Li M, Sousa AMM, Pletikos M, Meyer KA, Sedmak G, Guennel T, Shin Y, Johnson MB, Krsnik Z, Mayer S, Fertuzinhos S, Umlauf S, Lisgo SN, Vortmeyer A, Weinberger DR, Mane S, Hyde TM, Huttner A, Reimers M, Kleinman JE, Šestan N. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Jonasdottir A, Wong WSW, Sigurdsson G, Walters GB, Steinberg S, Helgason H, Thorleifsson G, Gudbjartsson DF, Helgason A, Magnusson OT, Thorsteinsdottir U, Stefansson K. Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 2012;488:471–475. doi: 10.1038/nature11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreienkamp HJ. The Calcium-independent Receptor for alpha-Latrotoxin from Human and Rodent Brains Interacts with Members of the ProSAP/SSTRIP/Shank Family of Multidomain Proteins. J Biol Chem. 2000;275:32387–32390. doi: 10.1074/jbc.C000490200. [DOI] [PubMed] [Google Scholar]
- Kumar S, Sanderford M, Gray VE, Ye J, Liu L. Evolutionary diagnosis method for variants in personal exomes. Nat Methods. 2012;9:855–856. doi: 10.1038/nmeth.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemke JR, Riesch E, Scheurenbrand T, Schubach M, Wilhelm C, Steiner I, Hansen J, Courage C, Gallati S, Bürki S, Strozzi S, Simonetti BG, Grunt S, Steinlin M, Alber M, Wolff M, Klopstock T, Prott EC, Lorenz R, Spaich C, Rona S, Lakshminarasimhan M, Kröll J, Dorn T, Krämer G, Synofzik M, Becker F, Weber YG, Lerche H, Böhm D, Biskup S. Targeted next generation sequencing as a diagnostic tool in epileptic disorders. Epilepsia. 2012;53:1387–1398. doi: 10.1111/j.1528-1167.2012.03516.x. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd SE, Pearce SH, Fisher SE, Steinmeyer K, Schwappach B, Scheinman SJ, Harding B, Bolino A, Devoto M, Goodyer P, Rigden SP, Wrong O, Jentsch TJ, Craig IW, Thakker RV. A common molecular basis for three inherited kidney stone diseases. Nature. 1996;379:445–449. doi: 10.1038/379445a0. [DOI] [PubMed] [Google Scholar]
- Lui TTH, Lacroix C, Ahmed SM, Goldenberg SJ, Leach CA, Daulat AM, Angers S. The ubiquitin-specific protease USP34 regulates axin stability and Wnt/β-catenin signaling. Mol Cell Biol. 2011;31:2053–2065. doi: 10.1128/MCB.01094-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon GJ, Wang K. Identifying disease mutations in genomic medicine settings: current challenges and how to accelerate progress. Genome Med. 2012;4:58. doi: 10.1186/gm359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis SS, Salogiannis J, Lipton DM, Mandel-Brehm C, Wills ZP, Mardinly AR, Hu L, Greer PL, Bikoff JB, Ho H-YH, Soskis MJ, Sahin M, Greenberg ME. EphB-Mediated Degradation of the RhoA GEF Ephexin5 Relieves a Developmental Brake on Excitatory Synapse Formation. Cell. 2010;143:442–455. doi: 10.1016/j.cell.2010.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulley JC, Scheffer IE, Petrou S, Dibbens LM, Berkovic SF, Harkin LA. SCN1Amutations and epilepsy. Hum Mutat. 2005;25:535–542. doi: 10.1002/humu.20178. [DOI] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, Lin C-F, Stevens C, Wang L-S, Makarov V, Polak P, Yoon S, Maguire J, Crawford EL, Campbell NG, Geller ET, Valladares O, Schafer C, Liu H, Zhao T, Cai G, Lihm J, Dannenfelser R, Jabado O, Peralta Z, Nagaswamy U, Muzny D, Reid JG, Newsham I, Wu Y, Lewis L, Han Y, Voight BF, Lim E, Rossin E, Kirby A, Flannick J, Fromer M, Shakir K, Fennell T, Garimella K, Banks E, Poplin R, Gabriel S, DePristo M, Wimbish JR, Boone BE, Levy SE, Betancur C, Sunyaev S, Boerwinkle E, Buxbaum JD, Cook EH, Jr, Devlin B, Gibbs RA, Roeder K, Schellenberg GD, Sutcliffe JS, Daly MJ. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nectoux J, Heron D, Tallot M, Chelly J, Bienvenu T. Maternal origin of a novel C-terminal truncation mutation in CDKL5 causing a severe atypical form of Rett syndrome. Clin Genet. 2006;70:29–33. doi: 10.1111/j.1399-0004.2006.00629.x. [DOI] [PubMed] [Google Scholar]
- O’roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, Turner EH, Stanaway IB, Vernot B, Malig M, Baker C, Reilly B, Akey JM, Borenstein E, Rieder MJ, Nickerson DA, Bernier R, Shendure J, Eichler EE. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okkenhaug H, Weylandt KH, Carmena D, Wells DJ, Higgins CF, Sardini A. The human ClC-4 protein, a member of the CLC chloride channel/transporter family, is localized to the endoplasmic reticulum by its N-terminus. FASEB J. 2006;20:2390–2392. doi: 10.1096/fj.05-5588fje. [DOI] [PubMed] [Google Scholar]
- Park H-D, Kim SR, Ki C-S, Lee S-Y, Chang YS, Jin D-K, Park W-S. Two novel HADHB gene mutations in a Korean patient with mitochondrial trifunctional protein deficiency. Ann Clin Lab Sci. 2009;39:399–404. [PubMed] [Google Scholar]
- Poët M, Kornak U, Schweizer M, Zdebik AA, Scheel O, Hoelter S, Wurst W, Schmitt A, Fuhrmann JC, Planells-Cases R, Mole SE, Hübner CA, Jentsch TJ. Lysosomal storage disease upon disruption of the neuronal chloride transport protein ClC-6. Proc Natl Acad Sci USA. 2006;103:13854–13859. doi: 10.1073/pnas.0606137103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, Albrecht B, Bartholdi D, Beygo J, Di Donato N, Dufke A, Cremer K, Hempel M, Horn D, Hoyer J, Joset P, Röpke A, Moog U, Riess A, Thiel CT, Tzschach A, Wiesener A, Wohlleber E, Zweier C, Ekici AB, Zink AM, Rump A, Meisinger C, Grallert H, Sticht H, Schenck A, Engels H, Rappold GA, Schröck E, Wieacker P, Riess O, Meitinger T, Reis A, Strom TM. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. The Lancet. 2012:380. doi: 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
- Rickheit G, Wartosch L, Schaffer S, Stobrawa SM, Novarino G, Weinert S, Jentsch TJ. Role of ClC-5 in Renal Endocytosis Is Unique among ClC Exchangers and Does Not Require PY-motif-dependent Ubiquitylation. J Biol Chem. 2010;285:17595–17603. doi: 10.1074/jbc.M110.115600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saganich MJ, de Miera EV-S, Nadal MS, Baker H, Coetzee WA, Rudy B. Cloning of components of a novel subthreshold-activating K(+) channel with a unique pattern of expression in the cerebral cortex. J Neurosci. 1999;19:10789–10802. doi: 10.1523/JNEUROSCI.19-24-10789.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin M, Greer PL, Lin MZ, Poucher H, Eberhart J, Schmidt S, Wright TM, Shamah SM, O’connell S, Cowan CW, Hu L, Goldberg JL, Debant A, Corfas G, Krull CE, Greenberg ME. Eph-Dependent Tyrosine Phosphorylation of Ephexin1 Modulates Growth Cone Collapse. Neuron. 2005;46:191–204. doi: 10.1016/j.neuron.2005.01.030. [DOI] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, Walker MF, Ober GT, Teran NA, Song Y, El-Fishawy P, Murtha RC, Choi M, Overton JD, Bjornson RD, Carriero NJ, Meyer KA, Bilguvar K, Mane SM, Šestan N, Lifton RP, Günel M, Roeder K, Geschwind DH, Devlin B, State MW. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheel O, Zdebik AA, Lourdel S, Jentsch TJ. Voltage-dependent electrogenic chloride/proton exchange by endosomal CLC proteins. Nature. 2005;436:424–427. doi: 10.1038/nature03860. [DOI] [PubMed] [Google Scholar]
- Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- Silva J-P, Ushkaryov YA. The latrophilins, “split-personality” receptors. Adv Exp Med Biol. 2010;706:59–75. doi: 10.1007/978-1-4419-7913-1_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauber T, Jentsch TJ. Sorting motifs of the endosomal/lysosomal CLC chloride transporters. J Biol Chem. 2010;285:34537–34548. doi: 10.1074/jbc.M110.162545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Epi4K Consortium. Epi4K: Gene discovery in 4,000 genomes. Epilepsia. 2012;53:1457–1467. doi: 10.1111/j.1528-1167.2012.03511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuchman R, Cuccaro M. Epilepsy and Autism: Neurodevelopmental Perspective. Curr Neurol Neurosci Rep. 2011;11:428–434. doi: 10.1007/s11910-011-0195-x. [DOI] [PubMed] [Google Scholar]
- Veeramah KR, O’brien JE, Meisler MH, Cheng X, Dib-Hajj SD, Waxman SG, Talwar D, Girirajan S, Eichler EE, Restifo LL, Erickson RP, Hammer MF. De Novo Pathogenic SCN8A Mutation Identified by Whole-Genome Sequencing of a Family Quartet Affected by Infantile Epileptic Encephalopathy and SUDEP. Am J Hum Genet. 2012;90:502–510. doi: 10.1016/j.ajhg.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissers LELM, de Ligt J, Gilissen C, Janssen I, Steehouwer M, De Vries P, van Lier B, Arts P, Wieskamp N, del Rosario M, Van Bon BWM, Hoischen A, De Vries BBA, Brunner HG, Veltman JA. A de novo paradigm for mental retardation. Nat Genet. 2010;42:1109–1112. doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164–e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirrell E, Farrell K, Whiting S. The epileptic encephalopathies of infancy and childhood. Can J Neurol Sci. 2005;32:409–418. doi: 10.1017/s0317167100004388. [DOI] [PubMed] [Google Scholar]
- Xiromerisiou G, Houlden H, Scarmeas N, Stamelou M, Kara E, Hardy JA, Lees AJ, Korlipara P, Limousin P, Paudel R, Hadjigeorgiou GM, Bhatia KP. THAP1 mutations and dystonia phenotypes: Genotype phenotype correlations. Mov Disord. 2012:27. doi: 10.1002/mds.25146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng W, Kong Q, Li C, Mao B. Xenopus RCOR2 (REST corepressor 2) interacts with ZMYND8, which is involved in neural differentiation. Biochem Biophys Res Commun. 2010;394:1024–1029. doi: 10.1016/j.bbrc.2010.03.115. [DOI] [PubMed] [Google Scholar]