

Abstract
The AP1 family transcription factor JUN is an important molecule in the neuronal response to injury. In retinal ganglion cells (RGCs), JUN is upregulated soon after axonal injury and disrupting JUN activity delays RGC death. JUN is known to participate in the control of many different injury response pathways in neurons, including pathways controlling cell death and axonal regeneration. The role of JUN in regulating genes involved in cell death, ER stress, and regeneration was tested to determine the overall importance of JUN in regulating RGC response to axonal injury. Genes from each of these pathways were transcriptionally controlled following axonal injury and Jun deficiency altered the expression of many of these genes. The differentially expressed genes included, Atf3, Ddit3, Ecel1, Gadd45α, Gal, Hrk, Pten, Socs3, and Sprr1a. Two of these genes, Hrk and Atf3, were tested for importance in RGC death using null alleles of each gene. Disruption of the prodeath Bcl2 family member Hrk did not affect the rate or amount of RGC death after axonal trauma. Deficiency in the ATF/CREB family transcription factor Atf3 did lessen the amount of RGC death after injury, though it did not provide long term protection to RGCs. Since JUN’s dimerization partner determines its transcriptional targets, the expression of several candidate AP1 family members were examined. Multiple AP1 family members were induced by axonal injury and had a different expression profile in Jun deficient retinas compared to wildtype retinas (Fosl1, Fosl2 and Jund). Overall, JUN appears to play a multifaceted role in regulating RGC response to axonal injury.
Keywords: glaucoma, cell death, regeneration, ER stress, trauma, axonal injury, AP1
Introduction
Numerous studies have shown that axonal injury is a critical insult to retinal ganglion cells (RGCs) in glaucoma (e.g. Anderson and Hendrickson, 1974; Buckingham et al., 2008; Howell et al., 2007; Howell et al., 2012; Li et al., 1999; Quigley et al., 1983; Schlamp et al., 2006). A major part of a neuron’s response to axonal injury is the activation of transcription factors, which result in large changes to the cell’s transcriptome (Hanz and Fainzilber, 2006; Michaelevski et al., 2010; Smith and Skene, 1997). These changes are critical for initiating regeneration and degeneration pathways in the injured cell (Leppa and Bohmann, 1999; Raivich, 2008). The ensuing gene expression changes ultimately decide whether a neuron will live or die following the axonal insult (Yang et al., 2007). The AP1 family member JUN (previously known as cJUN) is a transcription factor that is induced soon after neuronal injury and regulates diverse neuronal injury responses (Herdegen et al., 1997; Raivich and Behrens, 2006). Interestingly, JUN can promote both regenerative and degenerative states after axonal injury (Hull and Bahr, 1994; Isenmann and Bahr, 1997; Koistinaho et al., 1993; Levkovitch-Verbin et al., 2005) and has been proposed to be a central hub controlling the expression of axonal regeneration and cell death genes (Herdegen et al., 1997; Raivich and Behrens, 2006). JUN is upregulated in RGCs after several glaucoma-relevant insults such as excitotoxicity, mechanical optic nerve injury, and elevated intraocular pressure (Fernandes et al., 2012; Isenmann and Bahr, 1997; Levkovitch-Verbin et al., 2005; Munemasa et al., 2006). Importantly, inhibiting JUN expression or altering JUN activity significantly delays RGC death after axonal injury (Fernandes et al., 2012; Lingor et al., 2005; Yoshida et al., 2002). Given Jun’s role in mediating RGC viability after axonal injury, JUN-regulated transcriptional programs are likely to be major factors controlling RGC fate. Surprisingly given the importance of JUN, the number of known direct transcriptional targets of JUN are limited (Hartl et al., 2003), especially in the central nervous system (Freeman et al., 2004).
To determine if JUN played an extensive role in regulating RGC response to axonal injury at the transcriptional level, the expression of multiple genes implicated in this response were analyzed in wildtype and Jun deficient retinas after controlled optic nerve crush (CONC). Specifically, the transcriptional profile of genes implicated in determining neuronal survival (Bcl2 family members, ER stress pathway members) and ones implicated in regeneration were examined. Furthermore, the expression of known targets of JUN and other members of the JUN and FOS transcription family (AP1 family) were examined. Gene expression was examined at two time points: prior to the onset of cell death (2 days after CONC) to identify genes that may have a role in initiating cell death and/or promote viability/regeneration pathways; and at the peak of cell death (5 days after CONC) to determine if a gene’s expression is consistent with an involvement in RGC death. Furthermore, the importance of two of the genes found to be upregulated after CONC, Hrk and Atf3, were tested to determine if they were critical for RGC death after axonal injury. Overall, our results demonstrate that Jun regulates diverse signaling pathways that may ultimately influence RGC regeneration and survival following axonal injury.
2 Materials and Methods
2.1 Mice
Mice with conditional deletion of Jun in the retina were generated by crossing mice carrying a floxed allele of Jun (Junfl Behrens et al., 2002) with mice expressing cre recombinase under the control of an early retinal promoter, Six3 (Furuta et al., 2000). These mice were on a mixed genetic background of C57BL/6J and 129 origin. Note, there are ~10% of RGCs cells that do not have Six3-cre mediated recombination of the Junfl allele (see results and Fernandes et al., 2012). Traditional germ line null alleles of Atf3 (Hartman et al., 2004) and Hrk (Imaizumi et al., 2004) were also used. There were no differences observed between wildtype and heterozygous mice for either Atf3 or Hrk and the two genotypes were used as controls when studying these genes (referred to in the text as +/+). Mice were housed in a 12-hour light dark cycle and were fed chow and water ad libitum. All experiments were conducted in accordance with the Association for Research in Vision and Ophthalmology’s statement on the use of animals in ophthalmic research and were approved by the University of Rochester’s University Committee on Animal Resources.
2.2 Optic nerve injury
Controlled optic nerve crush (CONC) was performed as previously described (Libby et al., 2005). Briefly, mice were anaesthetized and the optic nerve was exposed. The optic nerve was clamped for 4 seconds approximately 0.5 mm from the globe using self-closing forceps (Roboz RS-5027).
2.3 Realtime PCR
All dissections were performed in RNAse free conditions. Eyes were dissected and placed in RNAse-free ice cold PBS. The retina was dissected free from the eye and submerged in RNA-later (Qiagen 76106). Retinas were stored in RNA-later at 4°C until RNA extraction. RNA was extracted as per manufacturer’s instructions using an RNAeasy Microkit (Qiagen 74004). The amount of RNA in each sample was estimated using Nanodrop. 500ng of RNA from each sample was reverse transcribed to cDNA using iScript (Biorad 170-8891). 20ul SYBR green (Biorad 170-8882) amplification reactions were prepared using 2ul of cDNA. Realtime PCR reactions were performed on a CFX Connect system (Biorad). Primer sequences for all genes tested are summarized in Table 1. Product sizes were validated on a gel. The geometric mean of CT values of the reference genes (Gapdh and Gad1) was subtracted from the CT value of the gene of interest to obtain the ΔCT value. The expression level for each gene was calculated using the ΔΔCT method as described previously (Livak and Schmittgen, 2001). The following genotypes: Jun+/+ Six3-cre−, Jun+/+ Six3-cre+, Jun+/fl Six3-cre−, or Junfl/fl Six3-cre− were used as controls and are collectively referred to as Jun+/+. Mice with retinal deletion of Jun (Junfl/fl Six3-cre+) are referred to as Jun−/−. No mice heterozygous for Jun deletion (Jun+/fl Six3-cre+) were used. At least 6 retinas of each genotype were assessed for gene expression at all time points examined (summarized in Table 2).
Table 1.
Primers used for qPCR
| Gene Name | Common Name | Forward Primer | Reverse Primer |
|---|---|---|---|
| Atf3 | Atf3 | CTGGAGTCAGTTACCGTCAACA | CAGGCACTCTGTCTTCTCCTTT |
| Atf6 | Atf6 | TGGGAGTGAGCTGCAAGTGT | ATAAGGGGGAACCGAGGAG |
| Bax | Bax | GGAGATGAACTGGACAGCAATATG | GATCAGCTCGGGCACTTTAG |
| Bcl2l1 | Bcl-x | GGAGAGCGTTCAGTGATCTAACAT | ACTTGCAATCCGACTCACCAATA |
| Ddit3 | Chop | CTGCCTTTCACCTTGGAGAC | CGTTTCCTGGGGATGAGATA |
| Ecel1 | Dine | ATGCCTACTATCTGCCCAACAA | GTCATAGCCATGGGTCAGTTC |
| Fos | cFos | CCTGTGAGCAGTCAGAGAAGG | TGGAAGAGGTGAGGACTGG |
| Fosl1 | Fra1 | GGAGACCGACAAGTTGGAGGAT | TGCAGTGCTTCCGGTTCAA |
| Fosl2 | Fra2 | TGCAGTCCTTGCGCGGTACGGG | GACAAGGTTTGAAGTGCCGGGAGTG |
| Gadd45a | Gadd45α | GAAGAAGGAAGCTGCGAGAAAA | CCTGGCCATCCTAAATTAGCAGT |
| Gad1 | Gad67 | TCTTCCACTCCTTCGCCTGC | GGAGAAGTCGGTCTCTGTGC |
| Gal | Gal | CAGTTTCTTGCACCTTAAAGAGG | GGTCTCAGGACTTCTCTAGGTCTTC |
| Gapdh | Gapdh | CAGGTTGTCTCCTGCGACTT | ATGTAGGCCATGAGGTCCAC |
| Hrk | Dp5 | AATTGTAAAGAGCTGATGGTGGA | AGTCTCAGAGTTCACATCGCAAG |
| Jun | cJun | CTGATCATCCAGTCCAGCAA | GACACTGGGAAGCGTGTTCT |
| Jund | JunD | GTCAAGACCCTCAAAAGCCAGA | TGTTGACGTGGCTGAGGACTT |
| Klf4 | Klf4 | GATTGCAAGTTCCGCCACTGAACA | AATTTCCACCCACAGCCGT |
| Pten | Pten | AATTCCCAGTCAGAGGCGCTATGT | GATTGCAAGTTCCGCCACTGAACA |
| Socs3 | Socs3 | GTTGAGCGTCAAGACCCAGT | ACAGTCGAAGCGGGGAACT |
| Sprr1a | Sprr1a | CCTGCTCTTCTCTGAGTATTAGGAC | GCTGCTTCACCTGCTGCT |
Table 2.
Delta Ct values and comparisons.
| Gene | Jun+/+ | Jun−/− | Jun+/+ vs Jun−/− | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ΔCT value ± SD (n) | Intra-geno P | ΔCT value ± SD (n) | Intra-geno P | Inter-Geno P | |||||||||
| Naïve | 2 day | 5 day | N vs 2 | N vs 5 | Naïve | 2 day | 5 day | N vs 2 | N vs 5 | N vs N | 2 vs 2 | 5 vs 5 | |
| Atf3 | 8.4 ± 1.1 (11) | 4.3 ± 1.0 (7) | 4.6 ± 0.5 (8) | 0.001 | 0.001 | 6.9 ± 1.1 (9) | 6.0 ± 1.3 (11) | 6.7 ± 1.3 (7) | 0.213 | 0.999 | 0.011 | 0.005 | 0.002 |
| Atf6 | 7.2 ± 1.9 (9) | 5.6 ± 1.1 (7) | 5.7 ± 0.9 (8) | 0.129 | 0.153 | 5.3 ± 2.3 (9) | 6.0 ± 1.5 (11) | 6.5 ± 1.9 (7) | 0.723 | 0.301 | 0.069 | 0.999 | 0.991 |
| Bax | 7.6 ± 1.1 (10) | 6.2 ± 1.0 (7) | 5.8 ± 1.1 (8) | 0.065 | 0.008 | 6.0 ± 0.9 (9) | 6.9 ± 1.4 (10) | 7.2 ± 1.7 (6) | 0.208 | 0.122 | 0.024 | 0.757 | 0.109 |
| Bbc3 | 10.1 ± 1.6 (11) | 8.7 ± 1.6 (7) | 8.6 ± 1.4 (8) | 0.428 | 0.334 | 8.5 ± 2.1 (9) | 8.9 ± 2.2 (10) | 12.5 ± 4.1 (6) | 0.999 | 0.003 | 0.385 | 0.999 | 0.007 |
| Bcl2l1 | 7.8 ± 1.3 (11) | 5.9 ± 0.4 (7) | 6.0 ± 0.3 (8) | 0.013 | 0.014 | 6.1 ± 1.3 (8) | 7.0 ± 2.2 (10) | 7.2 ± 1.2 (7) | 0.323 | 0.258 | 0.032 | 0.321 | 0.299 |
| Ddit3 | 3.7 ± 0.3 (11) | 2.1 ± 0.4 (7) | 2.8 ± 0.1 (8) | 0.001 | 0.001 | 3.1 ± 0.6 (9) | 2.5 ± 0.6 (10) | 3.1 ± 0.6 (7) | 0.022 | 0.999 | 0.081 | 0.224 | 0.610 |
| Ecel1 | 12.1 ± 1.3 (11) | 5.7 ± 1.0 (7) | 3.5 ± 0.6 (8) | 0.001 | 0.001 | 10.7 ± 1.2 (9) | 8.3 ± 1.4 (11) | 6.8 ± 1.0 (7) | 0.001 | 0.001 | 0.016 | 0.001 | 0.001 |
| Fos | 5.7 ± 0.9 (11) | 5.1 ± 1.2 (6) | 5.3 ± 0.4 (8) | 0.550 | 0.962 | 5.9 ±1.0 (8) | 5.8 ± 1.2 (10) | 6.7 ± 1.1 (7) | 0.999 | 0.254 | 0.999 | 0.580 | 0.033 |
| Fosl1 | 14.6 ± 2.2 (7) | 11.6 ± 1.1 (6) | 12.1 ± 1.2 (8) | 0.001 | 0.002 | 14.3 ± 1.1 (6) | 13.1 ± 1.3 (8) | 13.6 ± 0.6 (7) | 0.190 | 0.650 | 0.999 | 0.130 | 0.122 |
| Fosl2 | 9.1 ± 1.4 (11) | 8.5 ± 1.4 (6) | 8.5 ± 0.9 (8) | 0.850 | 0.784 | 9.4 ± 1.0 (8) | 8.0 ± 1.5 (10) | 9.7 ± 1.1 (7) | 0.035 | 0.999 | 0.999 | 0.999 | 0.221 |
| Gadd45a | 3.1 ± 0.6 (11) | 2.2 ± 0.2 (7) | 2.5 ± 0.6 (8) | 0.003 | 0.091 | 2.8 ± 0.5 (9) | 2.7 ± 0.7 (10) | 3.0 ± 0.4 (7) | 0.999 | 0.999 | 0.999 | 0.192 | 0.324 |
| Gal | 9.8 ± 0.6 (11) | 8.8 ± 0.4 (6) | 6.2 ± 0.6 (7) | 0.004 | 0.001 | 9.7 ± 0.8 (8) | 8.7 ± 0.8 (10) | 8.7 ± 0.6 (7) | 0.005 | 0.008 | 0.999 | 0.999 | 0.001 |
| Hrk | 11.2 ± 2.2 (11) | 9.4 ± 0.6 (7) | 9.4 ± 0.3 (8) | 0.035 | 0.028 | 11.4 ± 0.8 (9) | 9.4 ± 2.3 (10) | 11.5 ± 0.7 (7) | 0.011 | 0.998 | 0.999 | 0.999 | 0.036 |
| Jund | 4.7 ± 0.4 (11) | 3.8 ± 0.9 (6) | 3.8 ± 0.3 (8) | 0.009 | 0.004 | 4.5 ± 0.6 (8) | 4.2 ± 0.6 (10) | 4.7 ± 0.75 (7) | 0.471 | 0.999 | 0.999 | 0.711 | 0.016 |
| Klf4 | 6.2 ± 2.5 (11) | 7.7 ± 1.5 (7) | 7.8 ± 0.5 (8) | 0.168 | 0.110 | 7.2 ± 1.1 (9) | 5.7 ± 2.4 (11) | 8.2 ± 0.5 (6) | 0.134 | 0.623 | 0.615 | 0.075 | 0.999 |
| Pten | 2.4 ± 0.6 (11) | 1.8 ± 0.3 (7) | 1.1 ± 0.2 (8) | 0.096 | 0.001 | 1.6 ± 0.7 (9) | 2.2 ± 0.9 (10) | 2.2 ± 0.9 (7) | 0.140 | 0.127 | 0.008 | 0.237 | 0.003 |
| Socs3 | 7.6 ± 1.9 (11) | 7.6 ± 1.4 (7) | 7.8 ± 0.5 (8) | 0.999 | 0.999 | 7.8 ± 0.6 (9) | 6.2 ± 1.8 (11) | 9.4 ± 1.0 (7) | 0.029 | 0.065 | 0.999 | 0.143 | 0.105 |
| Sprr1a | 16.4 ± 1.1 (8) | 6.0 ± 1.1 (6) | 6.7 ± 0.4 (7) | 0.001 | 0.001 | 15.4 ± 1.1 (7) | 8.9 ± 1.4 (10) | 10.5 ± 0.8 (7) | 0.001 | 0.001 | 0.238 | 0.001 | 0.001 |
N, Naïve; n, number of samples; Bold, P<0.05; P values listed as 0.001 are actually ≤0.001 and those listed as 0.999 are actually ≥0.999.
2.4 Immunohistochemistry and Cell Counts
Eyes were processed as previously described (Harder and Libby, 2011; Libby et al., 2005). Briefly, following fixation in 4% paraformaldehyde (PFA), the anterior segment of each eye was removed and the posterior eye cup was processed for cryosectioning, whole mount immunostaining or whole mount Nissl staining. For immunohistochemistry rabbit anti-DDIT3 (CHOP; ABR, 1:250), rabbit anti-cCASP3 (RD, 1:1000), rabbit anti-JUN (Abcam, 1:250) and mouse anti-βIII tubulin (TUJ1; Covance, 1:1000) were used as primary antibodies. For cell counts, images were taken from eight 20x fields (for cCASP3+) or eight 40x fields (for TUJ1+ or Nissl+ cell counts) around the peripheral edge of whole mounted retinas. Each field was approximately 220 μm from the peripheral edge of the retina. Since Six3 mediated recombination varies with retinal eccentricity, for the JUN+ cell counts, images were obtained from four central 20x fields in addition to the eight 20x peripheral fields in order to obtain a fuller representation of the number of unrecombined cells throughout the retina. For Nissl counts, all ganglion cell layer cells within a field were counted with the exception of endothelial cells (which have an obvious elongated, non-neuronal morphology). The numbers of neurons immunolabeled with cCASP3, JUN or TUJ1 and the number of Nissl stained cells in each image were quantified using the cell-counter tool in ImageJ. Eyes from animals that underwent sham surgery were used as controls for all cell counts.
2.5 Statistical Analysis
P values < 0.05 were deemed to be significant for all experiments. Graphpad Prism was used for statistical analyses involving ANOVA. For experiments involving cell quantification the experimenter was masked to genotype and/or experimental group and a two way ANOVA was used to test for significance except for quantification of JUN positive cells which used a student’s t-test. For analysis of realtime PCR experiments involving multiple genotypes (Jun+/+ and Jun−/−) and time points (naïve, 2, and 5 days after CONC), two way ANOVA’s were performed followed by Bonferroni post hoc tests to determine whether gene expression changed within (intra-genotype comparison) and between (inter-genotype comparison) genotypes. A one way ANOVA followed by Bonferroni post hoc testing was used to determine if the expression of Jun changed in wildtype animals after CONC. ΔCT values of a gene were used in the statistical analyses.
Results
3.1 Jun is transcriptionally regulated following axonal injury
Consistent with previous reports (e.g. Johnson et al., 1993; Koistinaho et al., 1993), at 2 and 5 days following axonal injury (controlled optic nerve crush; CONC), there was a significant increase in Jun transcript expression in wildtype retinas (Fig 1A; P < 0.05). To study the effect of Jun deficiency on the RGC injury response, a floxed allele of Jun (Junfl) was deleted using an early retinal deleter cre, Six3-cre, since germline deletion of Jun results in embryonic lethality (Hilberg et al., 1993; Johnson et al., 1993). No differences were observed between Jun+/+ Six3cre−, Jun+/+ Six3cre+, Jun+/fl Six3cre−, or Junfl/fl Six3cre− retinas. All of these genotypes were used as controls and collectively referred to as Jun+/+ or wildtype (no mice heterozygous for Jun deletion were used). There was incomplete Six3-cre mediated recombination of the Junfl allele as a small number of cells still expressed JUN after CONC (10.3 ± 2.1% compared to wildtype; Fig. 1B,C). Consistent with high recombination efficiency, the level of Jun expression in Junfl/fl Six3cre+ retinas remained significantly below the level of Jun in Jun+/+ retinas at all time points examined (fold reduction in Jun−/− compared to Jun+/+: 0 days, 6.1; 2 days, 8.3; 5 days, 21.6). Therefore, JUN-dependent changes after CONC should be significantly attenuated in Junfl/fl Six3cre+ retinas (referred to as Jun−/− or Jun deficient retinas).
Figure 1. Axonal injury induces Jun upregulation.
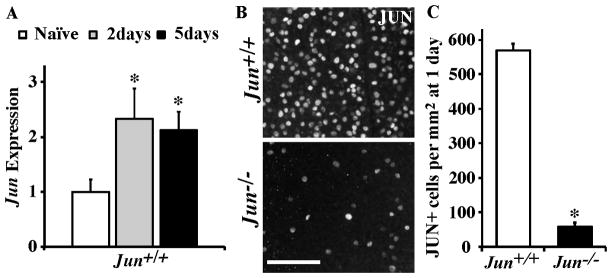
(A) The expression of Jun is significantly increased at both 2 and 5 days following CONC in Jun+/+ mice (represented as normalized fold expression). (B,C) To determine the recombination efficiency of the Jun floxed (Junfl/fl ) allele using Six3-cre, the number of JUN positive cells were counted in retinal flat mounts (RGC layer up) after CONC at a time when there is robust expression of JUN and before RGC cell death, 1 day after CONC. In Junfl/fl Six3cre+ (Jun−/−) retinas, the number of JUN+ cells was significantly reduced. *, P<0.05. Scale bar, 20 μm.
3.2 Jun deficiency alters the expression of an axonal injury response genes after CONC
To determine if Jun deficiency affected the pattern or level of a gene’s expression after CONC two biologically important comparisons were made. 1) To evaluate gene expression changes after CONC, expression levels were compared to the naïve condition of the same genotype (intra-genotype comparison). 2) To determine whether gene expression was Jun-dependent, expression levels were compared between genotypes (inter-genotype comparison) at each time point assessed. Importantly this comparison includes assessing whether there is a difference between Jun+/+ and Jun−/− retinas prior to injury (naïve comparison). The ΔCT values for all the genes assessed along with the P values for each of the comparisons listed above are summarized in Table 2.
To test if axonal injury responsive genes were regulated by Jun, the expression of Endothelin-Converting Enzyme-Like 1 (Ecel1), a gene known to be regulated by Jun after axonal injury in other neurons (Kiryu-Seo et al., 2008; Kiryu-Seo et al., 2000) was characterized following CONC. Interestingly, Jun deficiency appeared to have a relatively minor, but significant effect on basal Ecel1 expression. In unmanipulated eyes Ecel1 was increased 2.80 fold in Jun−/− compared to Jun+/+ retinas (Fig 2; Naïve comparison; P=0.016). This observation is consistent with basal JUN expression repressing the expression of some genes (Aguilera et al., 2011). Following CONC, the expression of Ecel1 progressively increased at 2 and 5 days in Jun+/+ retinas (Fig 2). While Ecel1 was upregulated in Jun−/− retinas, Ecel1 expression was significantly attenuated at both 2 and 5 days following CONC in Jun−/− retinas (Fig. 2; P < 0.001 for both comparisons). These results indicate that Jun deficiency alters the expression of transcriptionally regulated genes associated with axonal injury.
Figure 2. Jun deficiency alters the expression of an axon injury response gene, Ecel1, after CONC.
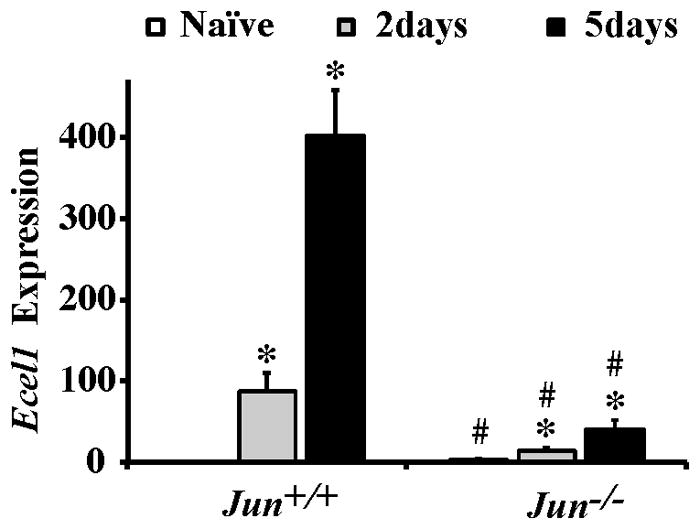
Realtime PCR analysis for Ecel1 from retinas of Jun+/+ and Jun−/− mice at the indicated time-points following CONC shown as normalized fold change. Expression of the neuronal injury responsive gene Ecel1 increased progressively in both Jun+/+ and Jun−/− animals following CONC (*, intra-genotype comparison, P < 0.001). However, comparing the change in gene expression between Jun+/+ and Jun−/− mice showed that in Jun deficient mice the CONC-induced expression of Ecel1 was attenuated at both time points (#, inter-genotype comparison, P < 0.05). Also, Ecel1 expression was significantly increased in Jun−/− retinas compared to Jun+/+ retinas prior to injury.
3.3 JUN and FOS family member regulation after axonal-injury
Immediate early genes belonging to the JUN and FOS families have been shown to be induced following axonal injury (e.g. Guo et al., 2010; Howell et al., 2011a; Howell et al., 2011b; Hull and Bahr, 1994; Koistinaho et al., 1993; Takeda et al., 2000; Yang et al., 2007). JUN dimerizes with itself and various other members of the Fos and Jun family to form the AP1 complex. Importantly, the transcriptional targets of AP1 dimers containing JUN change depending on JUN’s binding partner (Bakiri et al., 2002; Chinenov and Kerppola, 2001). The expression of several AP1 family members previously shown to be regulated in neurons following axonal injury was characterized following CONC. Jund was significantly upregulated in Jun+/+ retinas (P < 0.01 for each time point) but not in Jun−/− retinas at both 2 and 5 days after CONC. At 5 days post CONC Jund expression was significantly attenuated in Jun−/− retinas compared to wildtype (Fig. 3A; P = 0.016). Fos expression did not appear to be regulated following CONC in either genotype (Fig. 3B). Fosl1 expression increased significantly at both 2 and 5 days following CONC in wildtype retinas (Fig. 3C; P< 0.002 for each time point). In Jun deficient retinas, CONC did not lead to significant upregulation of Fosl1 expression, though Fosl1 expression levels were not significantly attenuated in Jun−/− mice compared to Jun+/+ mice. In contrast to the Jund and Fosl1 results where expression only increased in wildtype retinas, Fosl2 expression did not change in Jun+/+ retinas but did significantly increase in Jun−/− retinas at 2 days (Fig 3D; P = 0.035). Thus, it appears that JUN-dependent events are upstream of changes in expression of several AP1 family members after axonal injury. Since altered AP1 family member expression can alter the composition of AP1 dimers and thereby change transcriptional targets (Kaminska et al., 2000) it will be important to determine if different AP1 dimer combinations change with time and whether distinct AP1 dimers control cell death and cell regeneration programs.
Figure 3. JUN and FOS transcription factor expression after axonal-injury.

Realtime PCR analysis of JUN and FOS family members in Jun+/+ and Jun−/− mice expressed as normalized fold change. (A) Jund expression significantly increased at both 2 and 5 days after CONC in Jun+/+ mice (*, intra-genotype comparison, P < 0.05) and did not change in Jun−/− mice. Comparing expression level changes between genotypes showed that Jund expression was significantly attenuated in Jun−/− retinas compared to wildtype retinas at 5 days after CONC (#, inter-genotype comparison, P < 0.05) (B) Fos expression remained unchanged in Jun+/+ and Jun−/− mice at both time points examined, however, Fos expression was attenuated in Jun−/− retinas compared to Jun+/+ retinas 5 days after CONC. (C) A significant increase in Fosl1 expression was observed in Jun+/+ mice at 2 and 5 days after CONC. Fosl1 expression was not significantly changed at either time point examined in Jun−/− retinas. (D) Fosl2 expression did not change after CONC in wildtype mice at 2 and 5 days compared to naïve retinas. However, in Jun deficient retinas Fosl2 expression was significantly increased 2 days after CONC. *, P < 0.05 comparing 2 or 5 day time points to Naïve retinas of same genotype (Intra-genotype); #, P < 0.05, comparing same time points across genotypes (Inter-genotype).
3.4 JUN target Atf3 is involved in axonal-injury induced RGC death
Atf3 is a stress induced member of the ATF/CREB family of transcription factors (Chen et al., 1996; Hai and Hartman, 2001) and can dimerize with JUN in neurons (Nakagomi et al., 2003; Pearson et al., 2003). In neurons Atf3 is known to contribute to many aspects of response to injury and can be directly regulated by JUN (Mei et al., 2008; Nakagomi et al., 2003; Pearson et al., 2003). Consistent with previous studies Atf3 expression increases in RGCs after axonal injury (e.g. Guo et al., 2010; Takeda et al., 2000). At both 2 and 5 days after CONC, Atf3 was significantly increased in Jun+/+ retinas (Fig 4A; P<0.001 for both time points). Atf3 expression did not significantly change in Jun−/− retinas after CONC and the CONC-induced increase of Atf3 was significantly attenuated in Jun−/− retinas (Fig 4A, P < 0.005 for both time points). Thus, the induction of Atf3 expression after CONC appears to require JUN.
Figure 4. The Jun target, Atf3, suppresses RGC death following CONC.

(A) The expression of Atf3 significantly increased after CONC in Jun+/+ retinas at 2 and 5 days (shown as normalized fold change; *, intra-genotype comparison, P < 0.05). Atf3 expression was not significantly increased after CONC in Jun−/− retinas at 2 and 5 days. In fact, comparing the change in gene expression of Atf3 between Jun+/+ and Jun−/− mice showed that Atf3 expression was significantly attenuated in Jun−/− at both time points after CONC (#, inter-genotype comparison, P < 0.05). Also, Atf3 expression was significantly increased in Jun−/− retinas compared to Jun+/+ retinas prior to injury. (B) Representative images of cleaved caspase-3 (cCASP3) labeled, dying RGCs in Atf3+/+ and Atf3−/− retinal whole mounts. (C) The number of cCASP3 labeled cells was significantly reduced in Atf3−/− retinas at both 3 days and 5 days after CONC (*, P < 0.05 for both time points; N≥5 for both genotypes and time points). (D) Representative images of anti-βIII tubulin (TUJ1) positive RGCs in retinal whole mounts from sham injured and CONC injured eyes 14 days following the insult. (E) Despite the small decrease in cell death in Atf3 deficient mice, Atf3 deficiency did not increase the number of surviving RGCs 14 days after CONC (P = 0.82, N= 6 for genotypes). Scalebar, 25 μm.
To determine if ATF3 played a similar prodeath role in axonally injured RGCs as JUN, Atf3 null mice were subjected to CONC. Atf3 deficiency significantly reduced the number of dying cells (cleaved caspase 3+; cCASP3+) at 3 and 5 days after CONC by 45% and 27% respectively (Fig. 4B,C; P < 0.001 for both time points). However, this reduction of dying cells at the beginning of the CONC cell death window did not result in long term increase in the number of surviving RGCs, as judged by TUJ1+ cell counts 14 days post CONC (Fig 4D,E). Collectively, these results demonstrate that the JUN target, Atf3, has a minor proapoptotic role in RGCs following axonal injury.
3.5 Jun deficiency alters the expression of genes involved in RGC regeneration response
Although RGC axons do not naturally regenerate after injury in the mammalian retina, RGCs are responsive to numerous types of manipulations promoting regenerative outgrowth (de Lima et al., 2012; Fischer et al., 2004; Moore et al., 2009; Park et al., 2008; Sengottuvel et al., 2011; Smith et al., 2009; Yin et al., 2006). In combination these studies indicate that robust axon regeneration requires adding factors that promote regeneration and suppressing endogenous barriers to regeneration. JUN has been shown to promote axonal regeneration after injury (Raivich et al., 2004; Ruff et al., 2012; Smith and Skene, 1997). To test whether JUN could participate in controlling pro-regenerative genetic programs, the expression of two genes positively correlated with regenerative potential, Gal and Sprr1a (Holmes et al., 2000; Starkey et al., 2009), and known to be upregulated in an animal model of glaucoma (Howell et al., 2011a; Howell et al., 2011b) were assessed after CONC in wildtype and Jun deficient mice. At both 2 and 5 days after CONC, Gal and Sprr1a were significantly upregulated in both Jun+/+ and Jun−/− retinas (Fig. 5A,B P < 0.01 for all comparison). However, the upregulation of both of these proregenerative genes was significantly attenuated in Jun−/− retinas after CONC. Axonal injury also results in concurrent activation of cell-intrinsic suppressors of regeneration in RGCs. Knockout of two such suppressors, Klf4 and Socs3, has been shown to dramatically promote axon regeneration following optic nerve injury (Moore et al., 2009; Park et al., 2008; Smith et al., 2009). The expression of Klf4 and Socs3 were not significantly altered in wildtype retinas (Fig 5C,D) and Klf4 was not significantly changed in Jun−/− retinas after CONC. However, the expression of Socs3 significantly increased in Jun deficient retinas at 2 days following CONC (Fig 5D; P = 0.029), although the difference in expression between genotypes at this time point was not significant (P = 0.143). Collectively these data suggests that pro-regenerative pathways are suppressed in Jun deficient mice both through attenuation of pro-regenerative gene expression and through suppression of genes that inhibit regeneration.
Figure 5. JUN contributes to transcriptional control of RGC regeneration potential.

Realtime PCR analysis of genes involved in axon regeneration in Jun+/+ and Jun−/− mice shown as normalized fold change. (A,B) The expression of both Gal and Sprr1a positively correlate with regeneration and both significantly increased in Jun+/+ and Jun−/− mice after CONC (*, intra-genotype comparisons, P < 0.05). However, the increase in expression of both of these genes was significantly attenuated at least one time point in the Jun deficient mice (#, inter-genotype comparison, P < 0.05). (C–E) Genes that are known to suppress regeneration in injured RGCs were also examined. (C) The expression of Klf4 was not altered following CONC in either genotype. (D) Socs3 expression was not altered transcriptionally at 2 days or 5 days following CONC in Jun+/+ retinas but was significantly increased in Jun−/− retinas 2 days post CONC. (E) Pten was significantly upregulated at 5 days following CONC in Jun+/+ retinas. Pten expression did not change in Jun+/+ retinas, however, Pten expression was significantly attenuated at 5 days in Jun−/− deficient retinas compared to wildtype. Also, Pten expression was significantly increased in Jun−/− retinas compared to Jun+/+ retinas prior to injury. *, P < 0.05 comparing 2 or 5 day time points to Naïve retinas of same genotype (Intra-genotype); #, P < 0.05, comparing same time points across genotypes (Inter-genotype).
Pten deficiency has been shown to promote RGC regeneration by rescuing the deficit in protein synthesis that is observed following axonal injury (Park et al., 2008). Pten was upregulated in Jun+/+ retinas mice at 5 days after CONC (Fig. 5E, P = 0.001). This upregulation in expression was not observed in Jun−/− retinas and Pten expression was also significantly attenuated at 5 days following CONC in Jun−/− compared to Jun+/+ retinas (P < 0.003). Thus, in contrast to the other regeneration-associated genes examined for which pro-regenerative changes were attenuated in Jun−/− retinas, the pattern of Pten expression in Jun−/− retinas may favor regeneration compared to Jun+/+ retinas. However, the expression of Pten was significantly increased in Jun−/− compared to Jun+/+ retinas before injury (P = 0.008) and therefore basal Pten expression in Jun−/− retinas may negatively impact an RGC’s regenerative potential. Overall, these data suggest that JUN appears to prime RGCs for regeneration and/or may be important for making RGCs receptive to proregenerative manipulations.
3.6 Jun deficiency alters the expression of ER stress response genes after axonal injury
Axonal injury and ocular hypertension induce ER stress and the unfolded protein response (UPR) pathway in RGCs (Doh et al., 2010; Hu et al., 2012; Pernet et al., 2012). UPR activation has been shown promote apoptosis in RGCs (Hu et al., 2012). Specifically, the transcription of a key proapoptotic UPR target gene, Ddit3 (also know as Chop) was shown to increase in RGCs following optic nerve crush and Ddit3 deficiency reduced RGC death following axonal injury (Hu et al., 2012). Given the major role JUN-dependent pathways play in regulating RGC death following axonal injury, the possibility that JUN regulates key components of the ER stress and UPR activation pathways was tested (Lee et al., 2003; Schroder and Kaufman, 2005). Atf6 was not significantly regulated at 2 days or 5 days in either wildtype or Jun deficient eyes following CONC (Fig 6A). The expression of the ER stress marker, Gadd45a significantly increased in Jun+/+ retinas at 2 days post CONC (Fig 6B, P < 0.003), but did not change in Jun−/− retinas. In Jun+/+ retinas, the expression of Ddit3 increased at both 2 and 5 days following CONC (Fig 6C, P ≤ 0.001 for both comparisons). Ddit3 expression significantly increased in Jun−/− retinas but only at 2 days following CONC (P = 0.022). Immunohistochemical staining for DDIT3 confirmed the induction of DDIT3 in Jun+/+ and Jun−/− mice 3 days after CONC (Fig 6D). Collectively, these data demonstrate that upregulation of ER stress markers and the UPR pathway occurs retinas deficient in Jun, though JUN does appear to have a small role in transcriptionally regulating this pathway. Given that both of these pathways are ultimately prodeath, it will be interesting to determine if they interact to co-regulate downstream prodeath targets. In fact, DDIT3 has been shown to interact with JUN and other AP1 family members (Ubeda et al., 1999) and it is tempting to speculate that this dimer might control important prodeath pathways in RGCs. ER stress is also known to activate JNK signaling. Although upregulation of JUN in RGCs following axonal injury precedes the DDIT3 accumulation (data not shown), it is possible that ER stress signaling is required to sustain JNK activation in injured RGCs. Therefore, it will be important to determine whether JUN activation is sustained in mice where ER stress and/or UPR activation is altered.
Figure 6. After axonal injury ER stress occurs in the absence of Jun.

(A–C) Realtime PCR analysis of genes involved in ER stress response in Jun+/+ and Jun−/− mice shown as normalized fold change. (A) The expression of ER stress sensor Atf6 was not altered following CONC in either Jun+/+ or Jun−/− retinas. (B) The expression of the ER stress marker Gadd45a significantly increased at 2 days following CONC in Jun+/+ retinas (*, intra-genotype comparisons, P < 0.05). No other changes in Gadd45a expression were detected. (C) The expression of Ddit3, an ER stress target gene, significantly increased in Jun+/+ at both 2 and 5 days after CONC. In Jun−/− retinas Ddit3 expression was significantly increased only at 2 days following CONC. (D) Representative immunofluorescence staining for DDIT3 3 days following CONC confirms induction of DDIT3 in both Jun+/+ and Jun−/− retinas (DDIT3, green; DAPI, blue, was used to stain nuclei; the experiment was performed on 3 different mice for each genotype and condition). *, P < 0.05 comparing 2 or 5 day time points to naïve retinas of same genotype (Intra-genotype).
3.7 Jun deficiency alters the expression of Bcl2 family members after axonal injury
The Bcl2 family is a major regulator of RGC apoptosis (Bahr, 2000; Nickells et al., 2008) and multiple family members, including BAX, BCL2L1 (BCL-X), BBC3, and BIM, significantly contribute to RGC death after axonal injury (Harder et al., 2012a; Harder and Libby, 2011, 2013; Li et al., 2000; Libby et al., 2005; Semaan et al., 2010). We previously showed that accumulation of the Bcl2 family member BIM in axonally injured RGCs requires JUN (Harder et al., 2012b); however, Bim deficiency does not provide robust long term protection as is observed in Jun deficient retinas (Fernandes et al., 2012), suggesting JUN controls other genes important for RGC death. JUN is known to regulate the expression of numerous other Bcl2 family members, thus we assessed the requirement of JUN for the expression several Bcl2 family genes that have been implicated in RGC death after CONC. BBC3 is a Bcl2 family prosurvival gene that has been shown to play a role in RGC death after axonal injury (Harder and Libby, 2011, 2013). Bbc3 expression was not altered in Jun+/+ retinas. However, Bbc3 expression was significantly downregulated in Jun−/− retinas retinas 5 days after crush (Fig 7A; P = 0.003). Furthermore, at 5 days after CONC, Bbc3 expression was attenuated in Jun−/− retinas compared to Jun+/+ retinas (P = 0.007). This change in Bbc3 expression may have a role in RGC survival in Jun−/− retinas, however, it should be noted that BBC3 appears to play only a minor role in RGC death after axonal injury (Harder and Libby, 2011, 2013).
Figure 7. Bcl2 family expression after axonal-injury.
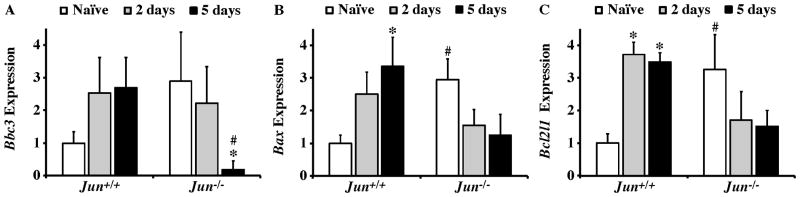
Results of realtime PCR analysis showing the normalized fold expression of a subset of Bcl2 family members that have been implicated in axonal injury induced RGC death in Jun+/+ and Jun−/− mice. (A) Bbc3 expression was not significantly altered after CONC in either genotype, though the expression was significantly attenuated in Jun−/− retinas compared to Jun+/+ retinas 5 days after CONC (#, inter-genotype comparison, P < 0.05). (B) Bax was significantly increased 5 days after CONC in Jun+/+ (*, intra-genotype comparisons, P < 0.05). There were no differences in Bax expression in Jun−/− animals after CONC. Also, Bax levels did not differ between Jun+/+ and Jun−/− retinas after CONC, but Bax levels were significantly higher in naïve Jun−/− retinas compared to Jun+/+ naïve retinas. (C) Interestingly, Bcl2l1 had a similar expression pattern to Bax after CONC even though they have opposite effects on the probability of a cell undergoing apoptosis. The expression of Bcl2l1 was significantly increased after CONC in Jun+/+ but not Jun−/− retinas. Though as with Bax, Bcl2l1 expression is significantly higher to begin with in Jun−/− retinas compared to controls. *, P < 0.05 comparing 2 or 5 day time points to Naïve retinas of same genotype (intra-genotype); #, P < 0.05, comparing same time points across genotypes (inter-genotype).
BAX is required for RGC death after axonal injury (Li et al., 2000; Libby et al., 2005; Semaan et al., 2010). Bax expression was significantly increased 5 days after CONC in Jun+/+ retinas (Fig 7B; P = 0.008). In Jun−/− retinas, Bax expression did not significantly change after axonal injury. The expression of the Bcl2 prosurvival family member Bcl2l1, which plays a major role in antagonizing RGC death after axonal injury (Harder et al., 2012a), was significantly increased in Jun+/+ but not in Jun−/− retinas at both 2 and 5 days after CONC (Fig. 7C). Thus, in wildtype retinas major prodeath and prosurvival members of the Bcl2 family were both upregulated after CONC. Interestingly, both Bax and Bcl2l1 were significantly upregulated in Jun−/− naïve retinas compared to Jun+/+ retinas (2.95 and 3.26 fold respectively; P < 0.05 for both comparison). Thus, for two of the major Bcl2 family members regulating RGC death after axonal injury, Bax and Bcl2l1, it appears that in Jun deficient mice the basal level of expression of these genes is similar to expression levels wildtype mice after injury. Collectively, our data indicate that transcriptional changes of Bcl2 family members are unlikely to contribute to the near complete prevention of cell death observed in Jun deficient retinas during the normal window of CONC induced RGC death (Fernandes et al., 2012). However, it is possible that Jun indirectly regulates the Bcl2 family by altering upstream events that initiate the cell death cascade and eventually funnel down to the Bcl2 family. Alternatively, Jun could also regulate the Bcl2 family translationally by altering expression of microRNAs that regulate expression of multiple BH3 only proteins (Kole et al., 2011).
3.8 HRK is not critical for RGC death after axonal injury
Despite numerous molecules being implicated in BAX activation in RGCs following axonal injury, the prodeath Bcl2 family members that activate BAX after axonal injury are not completely defined (Harder and Libby, 2013). The prodeath Bcl2 family member Hrk is known to kill neurons in a BAX-dependent manner (Harris and Johnson, 2001) and may have either a redundant or synergistic role with BIM in other neurons (Ghosh et al., 2011; Young et al., 2009). Therefore, Hrk may induce RGC death by complementing BIM (a JUN-dependent pathway). Hrk is known to be regulated by JUN and is important for injury induced cell death of some neurons (Besirli et al., 2005; Harder and Libby, 2011; Imaizumi et al., 2004; Ma et al., 2007). Hrk was significantly upregulated after CONC at both 2 and 5 days in Jun+/+ retinas (Fig. 8A; P < 0.05 for both comparisons). In Jun−/− retinas Hrk was only upregulated 2 days after CONC (P = 0.011). At 5 days after CONC Hrk expression appeared to return to baseline expression levels in Jun−/− retinas and was significantly attenuated compared to Jun+/+ retinas (Fig 8A; P = 0.036). Thus, Hrk expression was increased after axonal injury and Jun may participate in sustaining Hrk expression after axonal injury.
Figure 8. The proapoptotic gene Hrk is not required for RGC death after axonal injury.

(A) Expression of Hrk (a proapoptotic gene previously identified as a target of Jun) was significantly increased at both 2 days and 5 days following CONC (*, intra-genotype comparisons, P < 0.05). However, in Jun−/− retinas, Hrk expression was only significantly increased at 2 days following CONC and appeared to return to baseline levels of expression by 5 days after CONC. In fact, Hrk expression was significantly attenuated in Jun−/− retinas 5 days after CONC compared to Jun−/− retinas (#, inter-genotype comparison, P < 0.05). (B) Representative images of cleaved caspase 3 positive cells in retinal whole mounts from Hrk+/+ and Hrk−/− animals. At both 3 days and 5 days following CONC, the number of cleaved caspase-3 positive cells was unchanged by Hrk deficiency (N=4 for each genotype and time point; P ≥ 0.3). (C) Counts of anti-βIII tubulin (TUJ1) labeled RGCs in retinal whole mounts from sham injured and CONC injured eyes. Hrk deficiency does not alter the number of RGCs surviving 14 days following CONC (N=4 for each genotype; P = 0.37). (D) Counts of Nissl stained neurons in retinal whole mounts confirm the previous reported short term protection observed in Bim deficient mice compared to wildtype mice after CONC (*, P<0.01; Harder et al., 2012b). Combined deficiency of Bim and Hrk did not enhance RGC survival following CONC in comparison to single deficiency of Bim alone (P > 0.28 for both time points). Note, approximately 50% of RGC layer neurons are amacrine cells in mice (Jeon et al., 1998; Li et al., 1999; Li et al., 2007; Quigley et al., 2011) and do not die after CONC injury (Kielczewski et al., 2005), therefore a loss of 50% of RGC layer neurons reflects complete RGC loss. At least 4 retinas were examined at each time point for each genotype for the Nissl counts. Scale bar, 25 μm.
The importance HRK in axonal injury-induced RGC death was tested using Hrk deficient mice. RGC death was assessed by immunostaining for activated caspase 3 (cleaved CASP3, cCASP3) at the onset of RGC loss and at the peak of RGC death (3 and 5 days post CONC respectively; Harder et al., 2012b) and RGC survival was assessed by anti-βIII tubulin (TUJ1) immunostaining. Following axonal injury, the amount of RGC death did not significantly differ between wildtype and Hrk deficient mice. Similar to wildtype mice, in Hrk deficient mice substantial numbers of cCASP3+ cells were observed in the RGC layer at 3 and 5 days after injury (Fig 8B). In addition there was no increase in RGC survival at 14 days after injury (a time point when the majority of RGCs have died; Fernandes et al., 2012; Harder et al., 2012b) in Hrk deficient mice indicating that Hrk is not required for RGC death (Fig 8C). However, this result does not rule out the possibility that Hrk contributes to RGC death in this model, particularly given Hrk’s known involvement in neuronal cell death pathways involving Bim in neurons (Ghosh et al., 2011; Young et al., 2009). Therefore to further test whether Hrk plays a role in RGC death following axonal injury, RGC survival was assayed in Bim Hrk double knockout mice. Deleting Hrk and Bim together did not increase survival of RGCs beyond what was observed in Bim deficient mice (Fig. 8D). Thus, despite Hrk being a prodeath gene that is significantly upregulated early after axonal injury, it does not appear to play an important role role in RGC death.
3.9 Conclusion
While it is clear that JUN expression regulates prodeath pathways in RGCs (Fernandes et al., 2012; Lingor et al., 2005; Yoshida et al., 2002), JUN is also known to control various other aspects of injury response pathways in neurons (Herdegen et al., 1997). To determine the extent of JUN’s role in regulating the transcriptional response of RGCs to axonal injury, the expression of genes in multiple injury response pathways were analyzed in wildtype and Jun deficient retinas after injury. A representative subset of genes in pathways known to be important in RGC injury response, ER stress, regeneration, and cell death, were analyzed. Furthermore, the expression of JUN dimerization partners, which are known to alter JUN’s transcriptional targets were characterized. While this study only examined a small percent of the genome, it did reveal that JUN directly or indirectly regulated the expression of important genes in all of these pathways. Thus, these data support the hypothesis that JUN is a central hub in controlling RGC viability to axonal injury. It is important to note, that this study does not test whether JUN directly regulates the expression of a gene, merely whether changes in expression of the genes tested are downstream of JUN. Similarly, it is unclear, particularly at the 5 day time point where inter-genotype differences in gene expression are observed, if altered gene expression is because of a direct role of JUN or because Jun deficiency prevents RGC death (RGC cell death begins approximately 3 days after CONC). Therefore, follow up studies are required to determine if JUN binds to the promoters of the differentially regulated genes and directly affects their expression.
Sustained JUN transactivation activity has been linked with driving cells towards death. Analysis of the prodeath and prosurvival pathways did not show a transition of JUN targets from prosurvival/proregenerative pathways (2 day time point) to prodeath pathways when death was occurring (5 day time point). Ultimately, it will be important to understand the molecular pathways that JUN controls (either directly or indirectly) after axonal injury and whether these pathways change with time. This may not be an easy task since JUN can control transcription in multiple ways. In addition to direct transcriptional regulation, the expression of the microRNAs mir221/222 have been shown to be regulated by JUN expression (Galardi et al., 2011; Zhou et al., 2012) and these microRNAs are known to regulate genes involved in neuronal death and regeneration (Terasawa et al., 2009; Zhou et al., 2012). However, since JUN appears to control both prosurvival and prodeath pathways, understanding how it controls the transcriptome of a cell may provide important information about how RGCs remain viable when chronically insulted by ocular hypertension as occurs in glaucoma patients.
Highlights.
JUN appears to regulate axon regeneration pathways in RGCs after axonal injury.
JUN only has a minor role in regulating Bcl2 family genes in injured RGCs.
JUN appears to regulate other AP1 family members in axonally injured RGCs.
ATF3 has a minor prodeath role in RGCs.
Despite upregulation, HRK is not critical for RGC death after axonal injury.
Acknowledgments
The authors would like to thank Dr. Tsonwin Hai (Atf3) and Drs. Gabriel Nunez and Kevin Roth (Hrk) for generously providing mouse strains and Donna Shannon for technical help. This work was supported by EY018606 (RTL), T32 EY007125 (JMH), The Glaucoma Foundation, Research to Prevent Blindness Career Development Award (RTL) and a Research to Prevent Blindness unrestricted grant to the Department of Ophthalmology.
The Funding Bodies had no role in this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguilera C, Nakagawa K, Sancho R, Chakraborty A, Hendrich B, Behrens A. c-Jun N-terminal phosphorylation antagonises recruitment of the Mbd3/NuRD repressor complex. Nature. 2011;469:231–235. doi: 10.1038/nature09607. [DOI] [PubMed] [Google Scholar]
- Anderson DR, Hendrickson A. Effect of intraocular pressure on rapid axoplasmic transport in monkey optic nerve. Invest Ophthalmol. 1974;13:771–783. [PubMed] [Google Scholar]
- Bahr M. Live or let die - retinal ganglion cell death and survival during development and in the lesioned adult CNS. Trends Neurosci. 2000;23:483–490. doi: 10.1016/s0166-2236(00)01637-4. [DOI] [PubMed] [Google Scholar]
- Bakiri L, Matsuo K, Wisniewska M, Wagner EF, Yaniv M. Promoter specificity and biological activity of tethered AP-1 dimers. Mol Cell Biol. 2002;22:4952–4964. doi: 10.1128/MCB.22.13.4952-4964.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens A, Sibilia M, David JP, Mohle-Steinlein U, Tronche F, Schutz G, Wagner EF. Impaired postnatal hepatocyte proliferation and liver regeneration in mice lacking c-jun in the liver. The EMBO journal. 2002;21:1782–1790. doi: 10.1093/emboj/21.7.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besirli CG, Wagner EF, Johnson EM., Jr The limited role of NH2-terminal c-Jun phosphorylation in neuronal apoptosis: identification of the nuclear pore complex as a potential target of the JNK pathway. J Cell Biol. 2005;170:401–411. doi: 10.1083/jcb.200501138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckingham BP, Inman DM, Lambert W, Oglesby E, Calkins DJ, Steele MR, Vetter ML, Marsh-Armstrong N, Horner PJ. Progressive ganglion cell degeneration precedes neuronal loss in a mouse model of glaucoma. J Neurosci. 2008;28:2735–2744. doi: 10.1523/JNEUROSCI.4443-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BP, Wolfgang CD, Hai T. Analysis of ATF3, a transcription factor induced by physiological stresses and modulated by gadd153/Chop10. Mol Cell Biol. 1996;16:1157–1168. doi: 10.1128/mcb.16.3.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinenov Y, Kerppola TK. Close encounters of many kinds: Fos-Jun interactions that mediate transcription regulatory specificity. Oncogene. 2001;20:2438–2452. doi: 10.1038/sj.onc.1204385. [DOI] [PubMed] [Google Scholar]
- de Lima S, Koriyama Y, Kurimoto T, Oliveira JT, Yin Y, Li Y, Gilbert HY, Fagiolini M, Martinez AM, Benowitz L. Full-length axon regeneration in the adult mouse optic nerve and partial recovery of simple visual behaviors. Proc Natl Acad Sci U S A. 2012;109:9149–9154. doi: 10.1073/pnas.1119449109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doh SH, Kim JH, Lee KM, Park HY, Park CK. Retinal ganglion cell death induced by endoplasmic reticulum stress in a chronic glaucoma model. Brain Res. 2010;1308:158–166. doi: 10.1016/j.brainres.2009.10.025. [DOI] [PubMed] [Google Scholar]
- Fernandes KA, Harder JM, Fornarola LB, Freeman RS, Clark AF, Pang IH, John SW, Libby RT. JNK2 and JNK3 are major regulators of axonal injury-induced retinal ganglion cell death. Neurobiol Dis. 2012;46:393–401. doi: 10.1016/j.nbd.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer D, Petkova V, Thanos S, Benowitz LI. Switching mature retinal ganglion cells to a robust growth state in vivo: gene expression and synergy with RhoA inactivation. J Neurosci. 2004;24:8726–8740. doi: 10.1523/JNEUROSCI.2774-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman RS, Burch RL, Crowder RJ, Lomb DJ, Schoell MC, Straub JA, Xie L. NGF deprivation-induced gene expression: after ten years, where do we stand? Prog Brain Res. 2004;146:111–126. doi: 10.1016/S0079-6123(03)46008-1. [DOI] [PubMed] [Google Scholar]
- Furuta Y, Lagutin O, Hogan BL, Oliver GC. Retina- and ventral forebrain-specific Cre recombinase activity in transgenic mice. Genesis. 2000;26:130–132. [PubMed] [Google Scholar]
- Galardi S, Mercatelli N, Farace MG, Ciafre SA. NF-kB and c-Jun induce the expression of the oncogenic miR-221 and miR-222 in prostate carcinoma and glioblastoma cells. Nucleic Acids Res. 2011;39:3892–3902. doi: 10.1093/nar/gkr006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AP, Cape JD, Klocke BJ, Roth KA. Deficiency of pro-apoptotic Hrk attenuates programmed cell death in the developing murine nervous system but does not affect Bcl-x deficiency-induced neuron apoptosis. J Histochem Cytochem. 2011;59:976–983. doi: 10.1369/0022155411424311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Cepurna WO, Dyck JA, Doser TA, Johnson EC, Morrison JC. Retinal cell responses to elevated intraocular pressure: a gene array comparison between the whole retina and retinal ganglion cell layer. Invest Ophthalmol Vis Sci. 2010;51:3003–3018. doi: 10.1167/iovs.09-4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hai T, Hartman MG. The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: activating transcription factor proteins and homeostasis. Gene. 2001;273:1–11. doi: 10.1016/s0378-1119(01)00551-0. [DOI] [PubMed] [Google Scholar]
- Hanz S, Fainzilber M. Retrograde signaling in injured nerve--the axon reaction revisited. J Neurochem. 2006;99:13–19. doi: 10.1111/j.1471-4159.2006.04089.x. [DOI] [PubMed] [Google Scholar]
- Harder JM, Ding Q, Fernandes KA, Cherry JD, Gan L, Libby RT. BCL2L1 (BCL-x) promotes survival of adult and developing retinal ganglion cells. Mol Cell Neurosci. 2012a doi: 10.1016/j.mcn.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder JM, Fernandes KA, Libby RT. The Bcl-2 family member BIM has multiple glaucoma-relevant functions in DBA/2J mice. Sci Rep. 2012b;2:530. doi: 10.1038/srep00530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder JM, Libby RT. BBC3 (PUMA) regulates developmental apoptosis but not axonal injury induced death in the retina. Mol Neurodegener. 2011;6:50. doi: 10.1186/1750-1326-6-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder JM, Libby RT. Deficiency in Bim, Bid and Bbc3 (Puma) do not prevent axonal injury induced death. Cell Death Differ. 2013;20:182. doi: 10.1038/cdd.2012.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris CA, Johnson EM., Jr BH3-only Bcl-2 family members are coordinately regulated by the JNK pathway and require Bax to induce apoptosis in neurons. J Biol Chem. 2001;276:37754–37760. doi: 10.1074/jbc.M104073200. [DOI] [PubMed] [Google Scholar]
- Hartl M, Bader AG, Bister K. Molecular targets of the oncogenic transcription factor jun. Curr Cancer Drug Targets. 2003;3:41–55. doi: 10.2174/1568009033333781. [DOI] [PubMed] [Google Scholar]
- Hartman MG, Lu D, Kim ML, Kociba GJ, Shukri T, Buteau J, Wang X, Frankel WL, Guttridge D, Prentki M, Grey ST, Ron D, Hai T. Role for activating transcription factor 3 in stress-induced beta-cell apoptosis. Mol Cell Biol. 2004;24:5721–5732. doi: 10.1128/MCB.24.13.5721-5732.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herdegen T, Skene P, Bahr M. The c-Jun transcription factor--bipotential mediator of neuronal death, survival and regeneration. Trends Neurosci. 1997;20:227–231. doi: 10.1016/s0166-2236(96)01000-4. [DOI] [PubMed] [Google Scholar]
- Hilberg F, Aguzzi A, Howells N, Wagner EF. c-jun is essential for normal mouse development and hepatogenesis. Nature. 1993;365:179–181. doi: 10.1038/365179a0. [DOI] [PubMed] [Google Scholar]
- Holmes FE, Mahoney S, King VR, Bacon A, Kerr NC, Pachnis V, Curtis R, Priestley JV, Wynick D. Targeted disruption of the galanin gene reduces the number of sensory neurons and their regenerative capacity. Proc Natl Acad Sci U S A. 2000;97:11563–11568. doi: 10.1073/pnas.210221897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell GR, Libby RT, Marchant JK, Wilson LA, Cosma IM, Smith RS, Anderson MG, John SW. Absence of glaucoma in DBA/2J mice homozygous for wild-type versions of Gpnmb and Tyrp1. BMC Genet. 2007;8:45. doi: 10.1186/1471-2156-8-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell GR, Macalinao DG, Sousa GL, Walden M, Soto I, Kneeland SC, Barbay JM, King BL, Marchant JK, Hibbs M, Stevens B, Barres BA, Clark AF, Libby RT, John SW. Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. J Clin Invest. 2011a;121:1429–1444. doi: 10.1172/JCI44646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell GR, Soto I, Libby RT, John SW. Intrinsic axonal degeneration pathways are critical for glaucomatous damage. Exp Neurol. 2012 doi: 10.1016/j.expneurol.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell GR, Walton DO, King BL, Libby RT, John SW. Datgan, a reusable software system for facile interrogation and visualization of complex transcription profiling data. BMC Genomics. 2011b;12:429. doi: 10.1186/1471-2164-12-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Park KK, Yang L, Wei X, Yang Q, Cho KS, Thielen P, Lee AH, Cartoni R, Glimcher LH, Chen DF, He Z. Differential effects of unfolded protein response pathways on axon injury-induced death of retinal ganglion cells. Neuron. 2012;73:445–452. doi: 10.1016/j.neuron.2011.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull M, Bahr M. Regulation of immediate-early gene expression in rat retinal ganglion cells after axotomy and during regeneration through a peripheral nerve graft. J Neurobiol. 1994;25:92–105. doi: 10.1002/neu.480250109. [DOI] [PubMed] [Google Scholar]
- Imaizumi K, Benito A, Kiryu-Seo S, Gonzalez V, Inohara N, Lieberman AP, Kiyama H, Nunez G. Critical role for DP5/Harakiri, a Bcl-2 homology domain 3-only Bcl-2 family member, in axotomy-induced neuronal cell death. J Neurosci. 2004;24:3721–3725. doi: 10.1523/JNEUROSCI.5101-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isenmann S, Bahr M. Expression of c-Jun protein in degenerating retinal ganglion cells after optic nerve lesion in the rat. Exp Neurol. 1997;147:28–36. doi: 10.1006/exnr.1997.6585. [DOI] [PubMed] [Google Scholar]
- Jeon CJ, Strettoi E, Masland RH. The major cell populations of the mouse retina. The Journal of neuroscience. 1998;18:8936–8946. doi: 10.1523/JNEUROSCI.18-21-08936.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RS, van Lingen B, Papaioannou VE, Spiegelman BM. A null mutation at the c-jun locus causes embryonic lethality and retarded cell growth in culture. Genes Dev. 1993;7:1309–1317. doi: 10.1101/gad.7.7b.1309. [DOI] [PubMed] [Google Scholar]
- Kaminska B, Pyrzynska B, Ciechomska I, Wisniewska M. Modulation of the composition of AP-1 complex and its impact on transcriptional activity. Acta Neurobiol Exp (Wars) 2000;60:395–402. doi: 10.55782/ane-2000-1358. [DOI] [PubMed] [Google Scholar]
- Kielczewski JL, Pease ME, Quigley HA. The effect of experimental glaucoma and optic nerve transection on amacrine cells in the rat retina. Investigative ophthalmology & visual science. 2005;46:3188–3196. doi: 10.1167/iovs.05-0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiryu-Seo S, Kato R, Ogawa T, Nakagomi S, Nagata K, Kiyama H. Neuronal injury-inducible gene is synergistically regulated by ATF3, c-Jun, and STAT3 through the interaction with Sp1 in damaged neurons. J Biol Chem. 2008;283:6988–6996. doi: 10.1074/jbc.M707514200. [DOI] [PubMed] [Google Scholar]
- Kiryu-Seo S, Sasaki M, Yokohama H, Nakagomi S, Hirayama T, Aoki S, Wada K, Kiyama H. Damage-induced neuronal endopeptidase (DINE) is a unique metallopeptidase expressed in response to neuronal damage and activates superoxide scavengers. Proc Natl Acad Sci U S A. 2000;97:4345–4350. doi: 10.1073/pnas.070509897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koistinaho J, Hicks KJ, Sagar SM. Long-term induction of c-jun mRNA and Jun protein in rabbit retinal ganglion cells following axotomy or colchicine treatment. J Neurosci Res. 1993;34:250–255. doi: 10.1002/jnr.490340213. [DOI] [PubMed] [Google Scholar]
- Kole AJ, Swahari V, Hammond SM, Deshmukh M. miR-29b is activated during neuronal maturation and targets BH3-only genes to restrict apoptosis. Genes Dev. 2011;25:125–130. doi: 10.1101/gad.1975411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23:7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppa S, Bohmann D. Diverse functions of JNK signaling and c-Jun in stress response and apoptosis. Oncogene. 1999;18:6158–6162. doi: 10.1038/sj.onc.1203173. [DOI] [PubMed] [Google Scholar]
- Levkovitch-Verbin H, Quigley HA, Martin KR, Harizman N, Valenta DF, Pease ME, Melamed S. The transcription factor c-jun is activated in retinal ganglion cells in experimental rat glaucoma. Exp Eye Res. 2005;80:663–670. doi: 10.1016/j.exer.2004.11.016. [DOI] [PubMed] [Google Scholar]
- Li Y, Schlamp CL, Nickells RW. Experimental induction of retinal ganglion cell death in adult mice. Invest Ophthalmol Vis Sci. 1999;40:1004–1008. [PubMed] [Google Scholar]
- Li Y, Schlamp CL, Poulsen KP, Nickells RW. Bax-dependent and independent pathways of retinal ganglion cell death induced by different damaging stimuli. Exp Eye Res. 2000;71:209–213. doi: 10.1006/exer.2000.0873. [DOI] [PubMed] [Google Scholar]
- Li Y, Semaan SJ, Schlamp CL, Nickells RW. Dominant inheritance of retinal ganglion cell resistance to optic nerve crush in mice. BMC neuroscience. 2007;8:19. doi: 10.1186/1471-2202-8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby RT, Li Y, Savinova OV, Barter J, Smith RS, Nickells RW, John SW. Susceptibility to neurodegeneration in a glaucoma is modified by Bax gene dosage. PLoS Genet. 2005;1:17–26. doi: 10.1371/journal.pgen.0010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingor P, Koeberle P, Kugler S, Bahr M. Down-regulation of apoptosis mediators by RNAi inhibits axotomy-induced retinal ganglion cell death in vivo. Brain. 2005;128:550–558. doi: 10.1093/brain/awh382. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Ma C, Ying CY, Yuan ZM, Song B, Li D, Liu YL, Lai BQ, Li WM, Chen RZ, Ching YP, Li MT. dp5/HRK is a c-jun target gene and required for apoptosis induced by potassium deprivation in cerebellar granule neurons. J Biol Chem. 2007;282:30901–30909. doi: 10.1074/jbc.M608694200. [DOI] [PubMed] [Google Scholar]
- Mei Y, Yuan Z, Song B, Li D, Ma C, Hu C, Ching YP, Li M. Activating transcription factor 3 up-regulated by c-Jun NH(2)-terminal kinase/c-Jun contributes to apoptosis induced by potassium deprivation in cerebellar granule neurons. Neuroscience. 2008;151:771–779. doi: 10.1016/j.neuroscience.2007.10.057. [DOI] [PubMed] [Google Scholar]
- Michaelevski I, Segal-Ruder Y, Rozenbaum M, Medzihradszky KF, Shalem O, Coppola G, Horn-Saban S, Ben-Yaakov K, Dagan SY, Rishal I, Geschwind DH, Pilpel Y, Burlingame AL, Fainzilber M. Signaling to transcription networks in the neuronal retrograde injury response. Sci Signal. 2010;3:ra53. doi: 10.1126/scisignal.2000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore DL, Blackmore MG, Hu Y, Kaestner KH, Bixby JL, Lemmon VP, Goldberg JL. KLF family members regulate intrinsic axon regeneration ability. Science. 2009;326:298–301. doi: 10.1126/science.1175737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munemasa Y, Ohtani-Kaneko R, Kitaoka Y, Kumai T, Hayashi Y, Watanabe M, Takeda H, Hirata K, Ueno S. Pro-apoptotic role of c-Jun in NMDA-induced neurotoxicity in the rat retina. J Neurosci Res. 2006;83:907–918. doi: 10.1002/jnr.20786. [DOI] [PubMed] [Google Scholar]
- Nakagomi S, Suzuki Y, Namikawa K, Kiryu-Seo S, Kiyama H. Expression of the activating transcription factor 3 prevents c-Jun N-terminal kinase-induced neuronal death by promoting heat shock protein 27 expression and Akt activation. J Neurosci. 2003;23:5187–5196. doi: 10.1523/JNEUROSCI.23-12-05187.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickells RW, Semaan SJ, Schlamp CL. Involvement of the Bcl2 gene family in the signaling and control of retinal ganglion cell death. Prog Brain Res. 2008;173:423–435. doi: 10.1016/S0079-6123(08)01129-1. [DOI] [PubMed] [Google Scholar]
- Park KK, Liu K, Hu Y, Smith PD, Wang C, Cai B, Xu B, Connolly L, Kramvis I, Sahin M, He Z. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science. 2008;322:963–966. doi: 10.1126/science.1161566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson AG, Gray CW, Pearson JF, Greenwood JM, During MJ, Dragunow M. ATF3 enhances c-Jun-mediated neurite sprouting. Brain Res Mol Brain Res. 2003;120:38–45. doi: 10.1016/j.molbrainres.2003.09.014. [DOI] [PubMed] [Google Scholar]
- Pernet V, Joly S, Dalkara D, Schwarz O, Christ F, Schaffer D, Flannery JG, Schwab ME. Neuronal Nogo-A upregulation does not contribute to ER stress-associated apoptosis but participates in the regenerative response in the axotomized adult retina. Cell Death Differ. 2012;19:1096–1108. doi: 10.1038/cdd.2011.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley HA, Cone FE, Gelman SE, Yang Z, Son JL, Oglesby EN, Pease ME, Zack DJ. Lack of neuroprotection against experimental glaucoma in c-Jun N-terminal kinase 3 knockout mice. Experimental eye research. 2011;92:299–305. doi: 10.1016/j.exer.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley HA, Hohman RM, Addicks EM, Massof RW, Green WR. Morphologic changes in the lamina cribrosa correlated with neural loss in open-angle glaucoma. Am J Ophthalmol. 1983;95:673–691. doi: 10.1016/0002-9394(83)90389-6. [DOI] [PubMed] [Google Scholar]
- Raivich G. c-Jun expression, activation and function in neural cell death, inflammation and repair. J Neurochem. 2008;107:898–906. doi: 10.1111/j.1471-4159.2008.05684.x. [DOI] [PubMed] [Google Scholar]
- Raivich G, Behrens A. Role of the AP-1 transcription factor c-Jun in developing, adult and injured brain. Prog Neurobiol. 2006;78:347–363. doi: 10.1016/j.pneurobio.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Raivich G, Bohatschek M, Da Costa C, Iwata O, Galiano M, Hristova M, Nateri AS, Makwana M, Riera-Sans L, Wolfer DP, Lipp HP, Aguzzi A, Wagner EF, Behrens A. The AP-1 transcription factor c-Jun is required for efficient axonal regeneration. Neuron. 2004;43:57–67. doi: 10.1016/j.neuron.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Ruff CA, Staak N, Patodia S, Kaswich M, Rocha-Ferreira E, Da Costa C, Brecht S, Makwana M, Fontana X, Hristova M, Rumajogee P, Galiano M, Bohatschek M, Herdegen T, Behrens A, Raivich G. Neuronal c-Jun is required for successful axonal regeneration, but the effects of phosphorylation of its N-terminus are moderate. J Neurochem. 2012;121:607–618. doi: 10.1111/j.1471-4159.2012.07706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlamp CL, Li Y, Dietz JA, Janssen KT, Nickells RW. Progressive ganglion cell loss and optic nerve degeneration in DBA/2J mice is variable and asymmetric. BMC Neurosci. 2006;7:66. doi: 10.1186/1471-2202-7-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- Semaan SJ, Li Y, Nickells RW. A single nucleotide polymorphism in the Bax gene promoter affects transcription and influences retinal ganglion cell death. ASN Neuro. 2010;2:e00032. doi: 10.1042/AN20100003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengottuvel V, Leibinger M, Pfreimer M, Andreadaki A, Fischer D. Taxol facilitates axon regeneration in the mature CNS. J Neurosci. 2011;31:2688–2699. doi: 10.1523/JNEUROSCI.4885-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DS, Skene JH. A transcription-dependent switch controls competence of adult neurons for distinct modes of axon growth. J Neurosci. 1997;17:646–658. doi: 10.1523/JNEUROSCI.17-02-00646.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PD, Sun F, Park KK, Cai B, Wang C, Kuwako K, Martinez-Carrasco I, Connolly L, He Z. SOCS3 deletion promotes optic nerve regeneration in vivo. Neuron. 2009;64:617–623. doi: 10.1016/j.neuron.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkey ML, Davies M, Yip PK, Carter LM, Wong DJ, McMahon SB, Bradbury EJ. Expression of the regeneration-associated protein SPRR1A in primary sensory neurons and spinal cord of the adult mouse following peripheral and central injury. J Comp Neurol. 2009;513:51–68. doi: 10.1002/cne.21944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda M, Kato H, Takamiya A, Yoshida A, Kiyama H. Injury-specific expression of activating transcription factor-3 in retinal ganglion cells and its colocalized expression with phosphorylated c-Jun. Invest Ophthalmol Vis Sci. 2000;41:2412–2421. [PubMed] [Google Scholar]
- Terasawa K, Ichimura A, Sato F, Shimizu K, Tsujimoto G. Sustained activation of ERK1/2 by NGF induces microRNA-221 and 222 in PC12 cells. FEBS J. 2009;276:3269–3276. doi: 10.1111/j.1742-4658.2009.07041.x. [DOI] [PubMed] [Google Scholar]
- Ubeda M, Vallejo M, Habener JF. CHOP enhancement of gene transcription by interactions with Jun/Fos AP-1 complex proteins. Mol Cell Biol. 1999;19:7589–7599. doi: 10.1128/mcb.19.11.7589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Quigley HA, Pease ME, Yang Y, Qian J, Valenta D, Zack DJ. Changes in gene expression in experimental glaucoma and optic nerve transection: the equilibrium between protective and detrimental mechanisms. Invest Ophthalmol Vis Sci. 2007;48:5539–5548. doi: 10.1167/iovs.07-0542. [DOI] [PubMed] [Google Scholar]
- Yin Y, Henzl MT, Lorber B, Nakazawa T, Thomas TT, Jiang F, Langer R, Benowitz LI. Oncomodulin is a macrophage-derived signal for axon regeneration in retinal ganglion cells. Nat Neurosci. 2006;9:843–852. doi: 10.1038/nn1701. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Behrens A, Le-Niculescu H, Wagner EF, Harada T, Imaki J, Ohno S, Karin M. Amino-terminal phosphorylation of c-Jun regulates apoptosis in the retinal ganglion cells by optic nerve transection. Invest Ophthalmol Vis Sci. 2002;43:1631–1635. [PubMed] [Google Scholar]
- Young JE, Garden GA, Martinez RA, Tanaka F, Sandoval CM, Smith AC, Sopher BL, Lin A, Fischbeck KH, Ellerby LM, Morrison RS, Taylor JP, La Spada AR. Polyglutamine-expanded androgen receptor truncation fragments activate a Bax-dependent apoptotic cascade mediated by DP5/Hrk. J Neurosci. 2009;29:1987–1997. doi: 10.1523/JNEUROSCI.4072-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Shen D, Wang Y, Gong L, Tang X, Yu B, Gu X, Ding F. microRNA-222 targeting PTEN promotes neurite outgrowth from adult dorsal root ganglion neurons following sciatic nerve transection. PLoS One. 2012;7:e44768. doi: 10.1371/journal.pone.0044768. [DOI] [PMC free article] [PubMed] [Google Scholar]