

Abstract
We proposed recently that induction of delayed activation of trigeminovascular neurons by cortical spreading depression (CSD) can explain the delayed onset of headache after the aura. This prompted us to search for ways to block the neuronal activation by the CSD; a preclinical correlate of an attempt to find a drug that can block the initiation of the headache when administered shortly after onset of aura (i.e., pre-emptively). Because migraine headache and epileptic seizures are comorbid chronic neurological disorders characterized by hyperexcitable brain, we began the search for such goal with an M-type potassium channel opener. We opted to use ezogabine, recently approved by the FDA as adjunctive treatment of partial onset seizures in adults, because it is a selective KCNQ2/3 channels opener. When CSD was induced before ezogabine injection (8.25 mg/kg, i.p.), 40% (6/15) of the units doubled their firing rate about 45 min later for about 95 min. Similarly, when the CSD was induced before vehicle was injected (4% DMSO, 0.5% methylcellulose), 50% (3/6) of the units doubled their firing rate about 30 min later for about 120 min. When CSD was triggered 1 h after ezogabine injection, it activated only 8% of the units. By itself, ezogabine injection resulted in a 30% attenuation of ongoing firing in all 10 control units. Thus, activation of KCNQ2/3 channels during the aura is unlikely to preempt the onset of headache, but may reduce the incidence of migraine if given during prodromes that precede the headache by hours. Given the mechanistic similarities between migraine aura and epileptic seizures, it may be worthwhile to determine whether preemptive administration of ezogabine can prevent incoming seizures in patients whose warning signs precede their attacks by more than an hour.
1. Introduction
Clinical data suggest that the cortex of patients experiencing migraine aura is hyperexcitable [1]. Aura is a manifestation of cortical spreading depression (CSD)—a slowly propagating wave of abnormal cortical activity associated with initial hyperemia and sustained oligemia [2-6]. Migraine headache, on the other hand, appears to be mediated by activation of a trigeminovascular pathway originating with nociceptors that innervate the cranial meninges [7]. In most cases, the aura begins 30–40 min before the onset of migraine headache, suggesting that molecular and physiological events that occur in the cortex during aura might initiate headache by activating meningeal nociceptors. Along this line, we have shown that induction of CSD in the rat can lead 15–25 min later to onset of long-lasting activation in peripheral and central trigeminovascular neurons [8, 9]. This time lag led us to search for an effective means of intervention during the early stage of aura that would preempt the initiation of migraine headache. Our candidate for such preemptive intervention was ezogabine, which was recently approved by the FDA as adjunctive treatment of partial onset seizures in adults. Ezogabine is a M-type potassium channel opener that reduces neuronal excitability through its action on KCNQ2/3 channels [10-13].
Epileptic seizures and migraine headache are comorbid chronic neurological disorders characterized by recurrent episodes [14, 15]. In fact, migraine is two times more common among individuals with epilepsy than in the general population [16] and vise versa, epilepsy is two times more common among individuals with migraine than in the general population. [17] Convincing evidence that like the epileptic brain [18, 19], the migraine brain is hyperexcitable between, as well as during attacks [20, 21] have led the attempts to explain the comorbidity between the two disorders [17].
Potassium channel openers such as ezogabine have been implicated in pain relief through their action to reduced excitability in central and peripheral neurons [22-24]. While ezogabine is not approved for treating any aspect of migraine or any other pain condition, we thought it would be suitable to test in this study whether delayed activation of meningeal nociceptors in response to CSD can be intercepted by injecting ezogabine at the end of the CSD wave (preemptive intervention) or 1 h before CSD (prophylactic intervention).
2. Materials and Methods
2.1. Surgical preparation
Experiments were approved by the standing committee on animals of Harvard Medical School and Beth Israel Deaconess Medical Center, in accordance with the U.S. National Institutes of Health Guide for the Care and Use of Laboratory Animals. Male Sprague-Dawley rats weighing 250–350 g were deeply anesthetized with urethane (1.8 g/kg i.p.) and mounted on a stereotaxic frame. Core temperature was kept at 370C using a heating blanket. End-tidal CO2 was continuously monitored and kept within a physiological range of 3.5-4.5%. The dura over the left hemisphere was exposed using an opening in the roof of the skull spanning from Bregma to 2 mm caudal to Lambda. The exposed dura was kept moist throughout the experiment, using modified synthetic interstitial fluid (SIF, pH 7.2) containing 135 mM NaCl, 5 mM KCl, 1 mM MgCl2, 5 mM CaCl2, 10 mM glucose, and 10 mM HEPES.
Recording and identification of meningeal nociceptors
Single-unit activity of meningeal nociceptors (1 unit/rat) was recorded in the trigeminal ganglion as described in detail elsewhere [25, 26]. A platinum-coated tungsten microelectrode (impedance 50 kΩ; FHC Inc., Bowdoinham, ME) was lowered toward the left trigeminal ganglion through an incision made in the dura, approximately 2 mm caudal to the Bregma suture and 2 mm lateral to the midline. Included in the study were meningeal nociceptors that exhibited discrete bursts of activity in response to mechanical stimulation of the dura overlying the visual cortex (indentation with von-Frey monofilaments), plus consistent response latencies to repeated electrical stimulation of the dura (0.5 ms pulse, 5 mA, 0.5 Hz). The tip of the stimulating electrode was moved to different sites within the dural receptive field to find a point at which electrical pulses yielded the shortest neuronal response latency. That response latency was divided by the average distance between the dural receptive field and the trigeminal ganglion (12.5 mm), yielding the conduction velocity (CV) of the neuron under study. Slow-conducting neurons (CV ≤1.5 m/sec) were classified as C-units; faster conducting neurons (CV >1.5 m/sec) were classified as Aδ-units. Real-time waveform discriminator was used to create and store a template for the action potential evoked in the neuron under study by electrical pulses on the dura; spikes of activity matching the template waveform were acquired and analyzed online and offline using Spike 2 software (CED, Cambridge, UK).
2.2. Induction and recording of CSD
Single waves of CSD were induced using mechanical (pin-prick) or chemical stimulation (a KCl granule) of the visual cortex, about 6 mm away from the dural receptive field of the neuron under study. Changes in steady potential were recorded on the surface of the cortex using a glass micropipette filled with 150 mM NaCl (impedance 70–120 kΩ), about halfway between the dural receptive field and the site of cortical stimulation. At a propagation rate of 3–5 mm/min [27], a wave of CSD was expected to be registered by the recording electrode and enter the neuronal receptive field within 1–2 min of cortical stimulation. Pin prick was delivered by inserting a glass micropipette (diameter 25 μm) about 1 mm into the visual cortex for 10 sec. Chemical stimulation was delivered by placing a granule of crystalline KCl on the surface of the cortex; the granule was washed away with SIF at the end of the CSD wave.
2.3. Drug treatment
Ezogabine (GlaxoSmithKline), a KCNQ2/3 potassium channel opener, was administered intraperitoneally (i.p.) at a dose of 8.25 mg/kg body weight (0.25–0.35 ml per rat) which was compatible with the dose allowed for human subjects. We confirmed that this dose of ezogabine did not increase ongoing activity of the meningeal nociceptors. Due to poor aqueous solubility at natural pH, ezogabine was first dissolved in 100% DMSO then diluted 20X in 0.5% methylcellulose (Sigma-Aldrich). Control rats received an equivalent volume of the vehicle alone (4% DMSO in 0.5% methylcellulose solution).
2.4. Experimental paradigm
Four group of rats were used. The potential for preemptive intervention was assessed by timing the injection of ezogabine (n = 15) or vehicle (n = 6) immediately after registering a CSD wave passing through dural receptive field of the neuron under study (usually within 2 min of cortical irritation). Ongoing neuronal activity was recorded continuously over 30 min before CSD/ezogabine or CSD/vehicle (baseline) and over 120 min thereafter. The potential for prophylactic intervention was assessed by injecting ezogabine 60 min before triggering CSD (n = 13). Ongoing neuronal activity was recorded continuously over 30 min before ezogabine (baseline), over the 60 min leading up to the induction of CSD, and over 120 min thereafter. Finally, the effects of ezogabine was tested in the absence of CSD (n = 12) by monitoring ongoing neuronal firing over 30 min before the injection (baseline) and over 120 min thereafter.
2.5. Data analysis
Data were analyzed using nonparametric statistics with a two-tailed level of significance set at 0.05. Wilcoxon matched-pairs signed-ranks test was used to compare neuronal activity (mean spikes/sec) over 30 min of baseline with the activity recorded over 120 min after CSD induction and/or ezogabine injection. A neuron was considered to be activated if its firing rate after cortical stimulation increased ≥25% compared to baseline and remained elevated for a period longer than 30 min. Response magnitude was calculated from the 30-min period at which activity increase was maximal. Response duration was calculated from the entire period during which neuronal activity was ≥25% higher than baseline activity. Response latency was defined as the time interval between the end of the CSD wave and the onset of a ≥25% increase over baseline activity.
3. Results
Using single-unit recording in the trigeminal ganglion (1 unit/rat), a total of 46 meningeal nociceptors were isolated after exhibiting brief bouts of activity in response to electrical and mechanical stimulation of the dura overlying the transverse sinus and/or the visual cortex. Based on their conduction velocity, 23 of the units were classified as C-fiber nociceptors and 21 as Aδ-fibers. Baseline spontaneous activity was 0.94 ± 0.18 and 0.45 ± 0.13 spikes/sec (mean ± SE) for the C-fibers and the Aδ-fibers, respectively.
3.1. Ezogabine after CSD
When ezogabine was injected immediately after a wave of CSD was registered, 40% (6/15) of the meningeal nociceptors exhibited delayed activation (Table 1, Fig. 1A, B). Beginning 47.5 ± 11.0 min after CSD, these neurons nearly doubled their firing rate from 0.60 ± 0.25 spikes/sec at baseline to 1.14 ± 0.42 spikes/sec (P = 0.028) over an interval of 95.0 ± 14.3 min. In the remaining neurons, mean firing rate was 0.38 ± 0.22 spikes/sec at baseline and 0.21 ± 0.11 spikes/sec after the CSD (P = 0.23).
Table 1.
Ezogabine effects on CSD-induced activation of meningeal nociceptors
| Increased activity
|
Decreased activity
|
|||||
|---|---|---|---|---|---|---|
| Experimental group | C-fibers | Aδ-fibers | % | C-fibers | Aδ-fibers | % |
| Preemptive design (ezogabine given 2 min after CSD) | 4 | 2 | 40% | 4 | 5 | 60% |
| Preemptive design – control I (vehicle given 2 min after CSD) | 1 | 2 | 50% | 3 | 0 | 50% |
| Prophylactic design (ezogabine given 60 min before CSD) | 1 | 0 | 8% | 6 | 6 | 92% |
| Drug effect – control II | 0 | 0 | 4 | 6 | ||
| Spontaneous activity before CSD | 0.94±0.34 | 0.24±0.12 | 0.83±0.22 | 0.49±0.16 | ||
| Spontaneous activity after CSD | 1.76±0.55 | 0.53±0.20 | 0.61±0.17 | 0.33±0.11 | ||
Fig. 1.
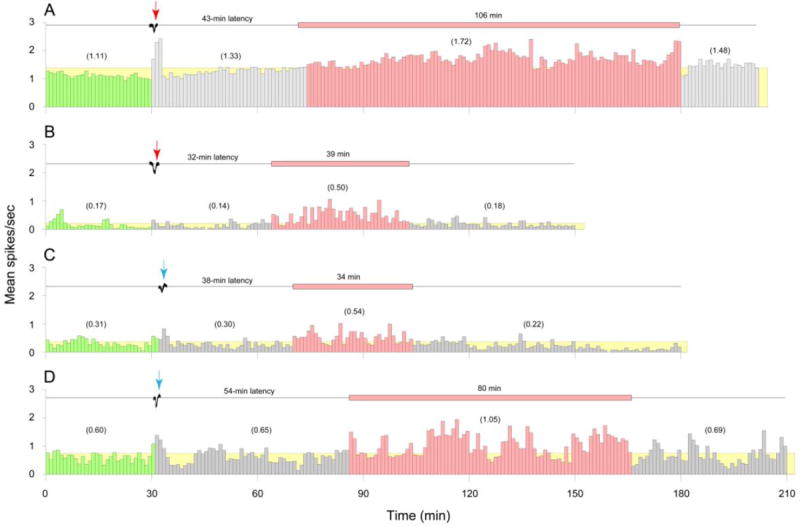
Delayed activation of meningeal nociceptors by CSD in the face of preemptive injection of ezogabine. (A–D) Individual examples of delayed neuronal activation following waves of CSD (black traces) and immediate intravenous administration of ezogabine (A, B, red arrows) or vehicle (C, D, blue arrows). Mean firing rate of the activated neurons (red histograms) and the duration of their activation phase (red horizontal bars) are shown for C-units (A, C) and Aδ-units (B, D). Green histograms show baseline neuronal activity before CSD. Grey histograms depict neuronal activity during the latency between the wave of CSD and the onset of neuronal activation. The threshold for neuronal activation (yellow) was defined as an increase in mean firing rate that was >25% above the pre-CSD baseline. Numbers in parentheses depict mean spikes/sec for the corresponding intervals.
3.2. Vehicle after CSD
Control vehicle injection immediately after a wave of CSD was registered resulted in delayed activation of 50% (3/6) of the meningeal nociceptors (Table 1, Fig. 1C, D). Beginning 30 min after CSD, these neurons increased their mean firing rate from 0.27 ± 0.16 spikes/sec at baseline to 0.68 ± 0.22 spikes/sec after the CSD, which did not reach level of significance due to sample size. This activation lasted over 2 h. In the remaining neurons, mean firing rate was 1.0 ± 0.34 spikes/sec at baseline and 1.05 ± 0.37 spikes/sec after the CSD.
3.3. Ezogabine before CSD
Ezogabine injection 1 h before cortical stimulation, although incapable of preventing the induction of CSD, resulted in delayed activation of only one out of thirteen meningeal nociceptor (Table 1). In the remaining 12 neurons (92%), the mean numbers of spikes/sec were 0.95 ± 0.26 at baseline, 0.69 ± 0.21 one hour after ezogabine, and 0.65 ± 0.20 after CSD. This 30% attenuation of neuronal firing was significant among the 6 Aδ-units (P = 0.028), but not in the 6 C-fiber units (P = 0.5)
3.4. Ezogabine without CSD
Ezogabine injection, in and of itself, resulted in a 30% decrease of ongoing firing rate in all 10 nociceptors (Table 1, Fig. 3A, B). This attenuation was significant in 6 Aδ-fibers (0.19 ± 0.1 vs. 0.15 ± 0.08 spikes/sec; P = 0.046) but fell short of the level of significance among 4 C-fibers (0.86 ± 0.53 vs. 0.58 ± 0.41; P = 0.068).
Fig. 3.
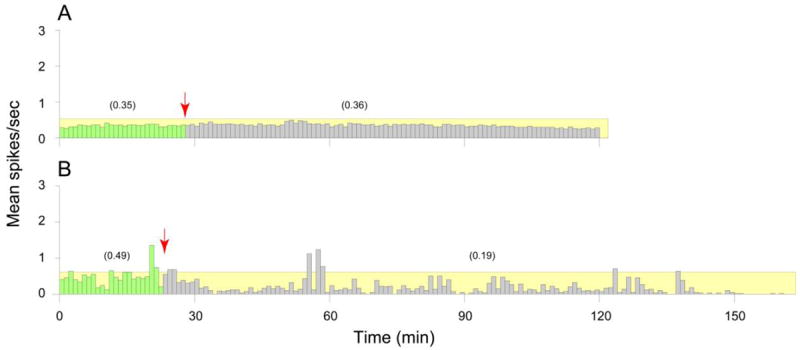
Effects of systemic administration of ezogabine on ongoing activity of meningeal nociceptors. (A) An example of a C-unit that maintained the same rate of activity after ezogabine injection (red arrow, grey bars) compared to baseline (green bars). (B) An example of an Aδ-unit that decreased its firing rate after ezogabine administration compared to baseline. Numbers in parentheses depict mean spikes/sec for the corresponding intervals.
4. Discussion
Patients diagnosed with episodic migraine are usually offered a variety of medications used to abort their attacks. Patients diagnosed with chronic migraine are usually offered a variety of prophylactic medication used to reduce the frequency of attacks. The clinical dilemma of using prophylactic medications is the need to maintain a certain level of the drug in circulation. Since all drugs have some unwanted side effects, the prophylactic approach dictates that patients must choose between unwanted side effects and reduced migraine frequency.
Attempting to capitalize on the therapeutic opportunity presented to us by the delayed onset of headache after occurrence of aura and delayed activation of meningeal nociceptors after occurrence of cortical spreading depression, we sought to determine whether it is feasible to develop yet another approach to migraine therapy – the preemptive approach. By definition, such an approach dictates that an agent should be introduced only when the patient knows with certainty that an attack is imminent. Tailoring our experimental paradigm to address this challenge, we first induced a wave of CSD in the visual cortex and immediately after, administered ezogabine, a KCNQ2/3 channel opener approved recently by the FDA for adjunctive treatment of partial onset seizures in adults, but not for any aspect of migraine (http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2011/ucm258834.htm). We chose this drug because both epilepsy and migraine are chronic neurological disorders with episodic manifestation (i.e., neuronal dysfunction return to baseline between attacks) that frequently occur together [28], and because both migraine and epilepsy are viewed as disorders of neuronal hyperexcitability [17, 18, 20], and as such, may benefit from attenuation of neuronal excitability [13, 29] by activation of a resting potassium current (KCNQ or M-current) [13, 29].
Unfortunately, our results with ezogabine suggest that a KCNQ channel opener does not prevent the delayed activation of the meningeal nociceptors by CSD when given preemptively: there was no significant difference in the proportion of meningeal nociceptors activated by CSD in drug treated animals (40%) compared to that observed in vehicle treated animals (50%). Importantly, the proportion of nociceptors activated by CSD in vehicle treated animals is comparable to that observed with CSD alone [8]. There are three likely explanations for the negative results obtained with the pre-emptive administration of ezogabine. First, because of the marked heterogeneity among sensory neurons that includes the distribution of ion channels critical for the regulation of neuronal excitability [30] it is possible that KCNQ channels are not present in meningeal nociceptors at a density necessary to enable the therapeutic efficacy of ezogagine, despite evidence that KCNQ channels are present in at least some nociceptive neurons [24]. Second, the dose of ezogabine used in the present study may have not been high enough for the drug-induced increase in KCNQ channel activity to block the CSD-induced increase in excitability. Third, because of pharmacokinetic profile of ezogabine, there was not enough time between the induction of CSD and ezogabine administration to achieve the concentration of the drug necessary to block the delayed CSD-induced activation of meningeal nociceptors. The results of our prophylactic treatment group argue against the first two possibilities. The observation that ezogabine administration one hour prior to induction of CSD was sufficient to attenuate the delayed activation of meningeal nociceptors (from 50% to 8%) and the spontaneous activity (at least in A-delta fibers) suggests that KCNQ channels are not only present in meningeal nociceptors, but at a density sufficient to underlie the “therapeutic” efficacy of the drug at the dose used. Conversely, the results of the prophylactic treatment experiment argue in favor of the third possibility, clearly indicating that if given enough time, ezogabine can prevent the delayed activation of the nociceptors in spite of the occurrence of CSD. Additional support of the third possibility is provided by data indicating that while rapidly absorbed, peak concentration of ezogabine is only achieved within 1-2 hours of dosing [31-33] likely due to enterohepatic recirculation that delays the occurrence of an earlier Tmax. Furthermore, the 2-4 hr half-life of the compound would not be relevant to the different results obtained with prophylactic and preemptive use of the drug in the present study.
In the context of epilepsy, the findings raise the possibility that ezogabine may be able to preempt incoming attacks in the estimated 7-12% of patients whose prodromes predict reliably an approaching episode [34, 35]. We propose to consider this novel approach for the treatment of partial onset seizure because unlike the migraine aura, where the prodrome precedes the attack by 20-30 min, seizure-related prodromes often precede an attack by 90 minutes (median estimated time, range 30 min - 24 hrs) [34] and thus should allow sufficient time for the drug to reach its Tmax. Suggesting that, we must acknowledge, though, that the viability of this proposal depends on the extent to which common warning signs and prodromes such as restlessness, malaise, nausea, impaired concentration, dizziness and tiredness are reliable in predicting onset of seizure attacks. Developing and validating a tool with adequate sensitivity and specificity to assess the reliability of prodromes in predicting onset of seizure attack may thus be timely.
While the relatively slow bioavailability of ezogabine can account for the failure of this drug to block the CSD-induced increase in meningeal nociceptor activity preemptively, it cannot account for the failure to influence the duration of the CSD-induced increase in activity. This suggests that there may be important differences in the processes that underlie the initial increase in afferent activity and those that underlie its maintenance. That is, if KCNQ channels are localized in the afferent in such a way as to either attenuate the generator potential responsible for the initiation of the action potential ultimately recorded within the trigeminal ganglia, or the electrophysiological impact of the generator potential (e.g., its electrotonic spread, action potential initiation and/or action potential propagation), then there should have been a delayed decrease in afferent activity following preemptive administration of ezogabine. The fact that no such decrease was detected, suggests that KCNQ channels are adequately localized in meningeal afferents to disrupt processes underling the initiation of the increase in afferent activity. Evidence from a study of isolated meningeal afferents in vitro suggests that a coincident increase in intracellular Ca2+ and inflammatory mediator-induced activation of a second messenger cascade is necessary for the activation of a Cl- current that contributes to an increase in dural afferent excitability [36]. As KCNQ channel activity should attenuate a depolarization-induced increase in intracellular Ca2+ secondary to the activation of voltage-gated Ca2+ channels, an increase in KCNQ channel activity prior to the release of inflammatory mediators associated with CSD could block the activation of the Cl- current. Once activated, however, the current is no longer dependent on an increase intracellular Ca2+, and consequently, KCNQ channel activity would no longer be able to attenuate the increase in afferent activity.
Given the wide distribution of KCNQ2/3 channels in the CNS, which account for their therapeutic efficacy in the treatment of epilepsy, it was surprising that administration of ezogabine one hour prior to cortical stimulation did not prevent induction and propagation of CSD. There are at least three explanations for this observation. The first is that the channels are differentially distributed within the CNS and are consequently not present in the superficial layers of the visual cortex at a density sufficient to block the CSD [37]. The second is that both the initiation and propagation of CSD involve changes in excitability that exceed the modulatory influence of KNCQ channels. And the third is that potassium current plays a role in the initiation but not the propagation of CSD [38-40].
In the context of isolated seizures, where therapy is directed toward the underlying cause, our results suggest that a preemptive reduction in neuronal network hyperexcitability, through enhancement of the outward potassium current (as one of several ways to reduce neuronal excitability), may ‘push’ the seizure threshold above the level required to initiate the classical high-frequency bursts of action potentials and the consequential hypersynchronization of neuronal firing.
While a KCNQ channel opener with a more rapid onset of action may still be a viable approach to preemptive migraine therapy, there are at least two additional implications of the positive results obtained with administration of ezogabine one hour prior to the induction of CSD. First, it provides yet more evidence in support of the already accepted notion that epilepsy and migraine share common mechanisms. Second, it provides a rational for testing the possibility that KCNQ channel openers could be used prophylactically for migraine therapy. Such an approach would complement the only antiepileptic drugs approved for migraine prophylaxis in the US [41-43], as neither valproic acid nor topiramate, appear to have activity at potassium channels. Valproic acid enhances GABAergic neurotransmission and possibly inhibits voltage-gated sodium channels [44-46], whereas topiramate blocks voltage-gated sodium channels, enhances GABAergic neurotransmission, and potentially reduces activation of AMPA/kainite glutamate receptors [47-51]. Because the enhancement of GABAergic neurotransmission is believed to be responsible for unwanted CNS side effects such as somnolence, dizziness, ataxia, confusion, speech disorder, vertigo, tremor, amnesia and abnormal thinking, associated with the valproic acid and topiramate, it may be possible to avoid these side effects with KCNQ channel openers. That said, it must be noted that ezogabine can enhance GABA-activated chloride current, though at a concentration higher than those affecting KCNQ channels [52, 53].
Fig. 2.
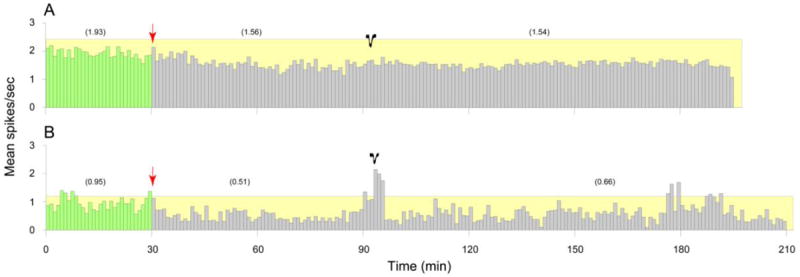
Blockade of meningeal nociceptors activation by ezogabine injection that preceded the induction of CSD. Mean firing rate of a C-unit (A) and an Aδ-unit (B) at baseline (green) and after systemic administration of ezogabine (red arrow) and the subsequent induction of CSD (black traces). Numbers in parentheses depict mean spikes/sec for the corresponding intervals. Note that preemptive ezogabine intervention did not interfere with the induction of CSD.
Highlights.
Activation of meningeal nociceptors by CSD is a preclinical model of migraine aura
Pretreatment with ezogabine blocks CSD-induced activation of meningeal nociceptors
Preemptive (post-CSD) treatment does not block activation of meningeal nociceptors
Possible prophylactic treatment of migraine with KCNQ2/Q3 openers is discussed
Possible preemptive treatment of epileptic seizure with KCNQ2/Q3 opener is discussed
Acknowledgments
This research was supported by a GlaxoSmithKline grant 01025007, and by NIH grants NS-079678 and NS-069847 (RB). Dr. Buettner was supported by an NIH grant K23AR055664.
Footnotes
Conflict of interest statement
Ezogabine and funding for this study were provided to RB by GlaxoSmithKline. RB has received honoraria from GlaxoSmithKline for consulting on headache. All other authors report no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aurora SK, Cao Y, Bowyer SM, Welch KM. The occipital cortex is hyperexcitable in migraine: experimental evidence. Headache. 1999;39:469–76. doi: 10.1046/j.1526-4610.1999.3907469.x. [DOI] [PubMed] [Google Scholar]
- 2.Leao A. Spraeding depression of activity in cerebral cortex. J Neurophysiol. 1944;7:359–390. doi: 10.1152/jn.1947.10.6.409. [DOI] [PubMed] [Google Scholar]
- 3.Leao AA. Spreading depression. Funct Neurol. 1986;1:363–6. [PubMed] [Google Scholar]
- 4.Lauritzen M, Olesen J. Regional cerebral blood flow during migraine attacks by Xenon-133 inhalation and emission tomography. Brain. 1984;107:447–461. doi: 10.1093/brain/107.2.447. [DOI] [PubMed] [Google Scholar]
- 5.Cao Y, Welch KM, Aurora S, Vikingstad EM. Functional MRI-BOLD of visually triggered headache in patients with migraine. Arch Neurol. 1999;56:548–54. doi: 10.1001/archneur.56.5.548. [DOI] [PubMed] [Google Scholar]
- 6.Hadjikhani N, Sanchez Del Rio M, Wu O, Schwartz D, Bakker D, Fischl B, Kwong KK, Cutrer FM, Rosen BR, Tootell RB, Sorensen AG, Moskowitz MA. Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci U S A. 2001;98:4687–92. doi: 10.1073/pnas.071582498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olesen J, Burstein R, Ashina M, Tfelt-Hansen P. Origin of pain in migraine: evidence for peripheral sensitisation. Lancet Neurol. 2009;8:679–90. doi: 10.1016/S1474-4422(09)70090-0. [DOI] [PubMed] [Google Scholar]
- 8.Zhang XC, Levy D, Noseda R, Kainz V, Jakubowski M, Burstein R. Activation of meningeal nociceptors by cortical spreading depression: implications to migraine with aura. J Neurosci. 2010;30:8807–8814. doi: 10.1523/JNEUROSCI.0511-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang X, Levy D, Kainz V, Noseda R, Jakubowski M, Burstein R. Activation of central trigeminovascular neurons by cortical spreading depression. Ann Neurol. 2011;69:855–65. doi: 10.1002/ana.22329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wickenden AD, Yu W, Zou A, Jegla T, Wagoner PK. Retigabine, a novel anticonvulsant, enhances activation of KCNQ2/Q3 potassium channels. Mol Pharmacol. 2000;58:591–600. doi: 10.1124/mol.58.3.591. [DOI] [PubMed] [Google Scholar]
- 11.Tatulian L, Delmas P, Abogadie FC, Brown DA. Activation of expressed KCNQ potassium currents and native neuronal M-type potassium currents by the anti-convulsant drug retigabine. J Neurosci. 2001;21:5535–45. doi: 10.1523/JNEUROSCI.21-15-05535.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Main MJ, Cryan JE, Dupere JR, Cox B, Clare JJ, Burbidge SA. Modulation of KCNQ2/3 potassium channels by the novel anticonvulsant retigabine. Mol Pharmacol. 2000;58:253–62. doi: 10.1124/mol.58.2.253. [DOI] [PubMed] [Google Scholar]
- 13.Camerino DC, Tricarico D, Desaphy JF. Ion channel pharmacology. Neurotherapeutics. 2007;4:184–98. doi: 10.1016/j.nurt.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 14.Welch KM, Lewis D. Migraine and epilepsy. Neurol Clin. 1997;15:107–14. doi: 10.1016/s0733-8619(05)70297-8. [DOI] [PubMed] [Google Scholar]
- 15.Belcastro V, Striano P, Kasteleijn-Nolst Trenite DG, Villa MP, Parisi P. Migralepsy, hemicrania epileptica, post-ictal headache and “ictal epileptic headache”: a proposal for terminology and classification revision. J Headache Pain. 2011;12:289–94. doi: 10.1007/s10194-011-0318-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tellez-Zenteno JF, Matijevic S, Wiebe S. Somatic comorbidity of epilepsy in the general population in Canada. Epilepsia. 2005;46:1955–62. doi: 10.1111/j.1528-1167.2005.00344.x. [DOI] [PubMed] [Google Scholar]
- 17.Haut SR, Bigal ME, Lipton RB. Chronic disorders with episodic manifestations: focus on epilepsy and migraine. Lancet Neurol. 2006;5:148–57. doi: 10.1016/S1474-4422(06)70348-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koch UR, Musshoff U, Pannek HW, Ebner A, Wolf P, Speckmann EJ, Kohling R. Intrinsic excitability, synaptic potentials, and short-term plasticity in human epileptic neocortex. J Neurosci Res. 2005;80:715–26. doi: 10.1002/jnr.20498. [DOI] [PubMed] [Google Scholar]
- 19.McNamara JO. Emerging insights into the genesis of epilepsy. Nature. 1999;399:A15–22. doi: 10.1038/399a015. [DOI] [PubMed] [Google Scholar]
- 20.Aurora SK, Wilkinson F. The brain is hyperexcitable in migraine. Cephalalgia. 2007;27:1442–53. doi: 10.1111/j.1468-2982.2007.01502.x. [DOI] [PubMed] [Google Scholar]
- 21.Coppola G, Pierelli F, Schoenen J. Is the cerebral cortex hyperexcitable or hyperresponsive in migraine? Cephalalgia. 2007;27:1427–39. doi: 10.1111/j.1468-2982.2007.01500.x. [DOI] [PubMed] [Google Scholar]
- 22.McCleskey EW, Gold MS. Ion channels of nociception. Annu Rev Physiol. 1999;61:835–56. doi: 10.1146/annurev.physiol.61.1.835. [DOI] [PubMed] [Google Scholar]
- 23.Marrion NV. Control of M-current. Annu Rev Physiol. 1997;59:483–504. doi: 10.1146/annurev.physiol.59.1.483. [DOI] [PubMed] [Google Scholar]
- 24.Passmore GM, Selyanko AA, Mistry M, Al-Qatari M, Marsh SJ, Matthews EA, Dickenson AH, Brown TA, Burbidge SA, Main M, Brown DA. KCNQ/M currents in sensory neurons: significance for pain therapy. J Neurosci. 2003;23:7227–36. doi: 10.1523/JNEUROSCI.23-18-07227.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strassman AM, Raymond SA, Burstein R. Sensitization of meningeal sensory neurons and the origin of headaches. Nature. 1996;384:560–564. doi: 10.1038/384560a0. [DOI] [PubMed] [Google Scholar]
- 26.Levy D, Burstein R, Strassman AM. Calcitonin gene-related peptide does not excite or sensitize meningeal nociceptors: Implications for the pathophysiology of migraine. Ann Neurol. 2005;58:698–705. doi: 10.1002/ana.20619. [DOI] [PubMed] [Google Scholar]
- 27.Lauritzen M. Pathophysiology of the migraine aura. The spreading depression theory. Brain. 1994;117:199–210. doi: 10.1093/brain/117.1.199. [DOI] [PubMed] [Google Scholar]
- 28.Ottman R, Lipton RB. Is the comorbidity of epilepsy and migraine due to a shared genetic susceptibility? Neurology. 1996;47:918–24. doi: 10.1212/wnl.47.4.918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 1998;282:1890–3. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- 30.Harriott AM, Gold MS. Electrophysiological properties of dural afferents in the absence and presence of inflammatory mediators. J Neurophysiol. 2009;101:3126–34. doi: 10.1152/jn.91339.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Porter RJ, Partiot A, Sachdeo R, Nohria V, Alves WM. Randomized, multicenter, dose-ranging trial of retigabine for partial-onset seizures. Neurology. 2007;68:1197–204. doi: 10.1212/01.wnl.0000259034.45049.00. [DOI] [PubMed] [Google Scholar]
- 32.Ferron GM, Paul J, Fruncillo R, Richards L, Knebel N, Getsy J, Troy S. Multiple-dose, linear, dose-proportional pharmacokinetics of retigabine in healthy volunteers. J Clin Pharmacol. 2002;42:175–82. doi: 10.1177/00912700222011210. [DOI] [PubMed] [Google Scholar]
- 33.Large CH, Sokal DM, Nehlig A, Gunthorpe MJ, Sankar R, Crean CS, Vanlandingham KE, White HS. The spectrum of anticonvulsant efficacy of retigabine (ezogabine) in animal models: implications for clinical use. Epilepsia. 2012;53:425–36. doi: 10.1111/j.1528-1167.2011.03364.x. [DOI] [PubMed] [Google Scholar]
- 34.Schulze-Bonhage A, Kurth C, Carius A, Steinhoff BJ, Mayer T. Seizure anticipation by patients with focal and generalized epilepsy: a multicentre assessment of premonitory symptoms. Epilepsy Res. 2006;70:83–8. doi: 10.1016/j.eplepsyres.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 35.Lee SA, No YJ. Perceived self-control of seizures in patients with uncontrolled partial epilepsy. Seizure. 2005;14:100–5. doi: 10.1016/j.seizure.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 36.Vaughn AH, Gold MS. Ionic mechanisms underlying inflammatory mediator-induced sensitization of dural afferents. J Neurosci. 30:7878–88. doi: 10.1523/JNEUROSCI.6053-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saganich MJ, Machado E, Rudy B. Differential expression of genes encoding subthreshold-operating voltage-gated K+ channels in brain. J Neurosci. 2001;21:4609–24. doi: 10.1523/JNEUROSCI.21-13-04609.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Herreras O, Largo C, Ibarz JM, Somjen GG, Martin del Rio R. Role of neuronal synchronizing mechanisms in the propagation of spreading depression in the in vivo hippocampus. J Neurosci. 1994;14:7087–98. doi: 10.1523/JNEUROSCI.14-11-07087.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Herreras O, Somjen GG. Propagation of spreading depression among dendrites and somata of the same cell population. Brain Res. 1993;610:276–82. doi: 10.1016/0006-8993(93)91411-k. [DOI] [PubMed] [Google Scholar]
- 40.Sugaya E, Takato M, Noda Y. Neuronal and glial activity during spreading depression in cerebral cortex of cat. J Neurophysiol. 1975;38:822–41. doi: 10.1152/jn.1975.38.4.822. [DOI] [PubMed] [Google Scholar]
- 41.Brandes JL, Saper JR, Diamond M, Couch JR, Lewis DW, Schmitt J, Neto W, Schwabe S, Jacobs D. Topiramate for migraine prevention: a randomized controlled trial. JAMA. 2004;291:965–73. doi: 10.1001/jama.291.8.965. [DOI] [PubMed] [Google Scholar]
- 42.Freitag FG, Collins SD, Carlson HA, Goldstein J, Saper J, Silberstein S, Mathew N, Winner PK, Deaton R, Sommerville K. A randomized trial of divalproex sodium extended-release tablets in migraine prophylaxis. Neurology. 2002;58:1652–9. doi: 10.1212/wnl.58.11.1652. [DOI] [PubMed] [Google Scholar]
- 43.Loder E, Burch R, Rizzoli P. The 2012 AHS/AAN guidelines for prevention of episodic migraine: a summary and comparison with other recent clinical practice guidelines. Headache. 2012;52:930–45. doi: 10.1111/j.1526-4610.2012.02185.x. [DOI] [PubMed] [Google Scholar]
- 44.Rosenberg G. The mechanisms of action of valproate in neuropsychiatric disorders: can we see the forest for the trees? Cell Mol Life Sci. 2007;64:2090–103. doi: 10.1007/s00018-007-7079-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rho JM, Sankar R. The pharmacologic basis of antiepileptic drug action. Epilepsia. 1999;40:1471–83. doi: 10.1111/j.1528-1157.1999.tb02029.x. [DOI] [PubMed] [Google Scholar]
- 46.Johannessen CU. Mechanisms of action of valproate: a commentatory. Neurochem Int. 2000;37:103–10. doi: 10.1016/s0197-0186(00)00013-9. [DOI] [PubMed] [Google Scholar]
- 47.McLean MJ, Bukhari AA, Wamil AW. Effects of topiramate on sodium-dependent action-potential firing by mouse spinal cord neurons in cell culture. Epilepsia. 2000;41(Suppl 1):S21–4. [PubMed] [Google Scholar]
- 48.Czapinski P, Blaszczyk B, Czuczwar SJ. Mechanisms of action of antiepileptic drugs. Curr Top Med Chem. 2005;5:3–14. doi: 10.2174/1568026053386962. [DOI] [PubMed] [Google Scholar]
- 49.Taverna S, Sancini G, Mantegazza M, Franceschetti S, Avanzini G. Inhibition of transient and persistent Na+ current fractions by the new anticonvulsant topiramate. J Pharmacol Exp Ther. 1999;288:960–8. [PubMed] [Google Scholar]
- 50.Simeone TA. Mechanisms of antiepileptic drug action. In: Rho JM, Sankar R, Stafstrom CE, editors. Epilepsy:Mechanisms, models, and translational perspectives. CRC Press; 2010. pp. 123–141. [Google Scholar]
- 51.Simeone TA, Wilcox KS, White HS. cAMP-dependent protein kinase A activity modulates topiramate potentiation of GABA(A) receptors. Epilepsy Res. 2011;96:176–9. doi: 10.1016/j.eplepsyres.2011.05.009. [DOI] [PubMed] [Google Scholar]
- 52.Rundfeldt C. The new anticonvulsant retigabine (D-23129) acts as an opener of K+ channels in neuronal cells. Eur J Pharmacol. 1997;336:243–9. doi: 10.1016/s0014-2999(97)01249-1. [DOI] [PubMed] [Google Scholar]
- 53.van Rijn CM, Willems-van Bree E. Synergy between retigabine and GABA in modulating the convulsant site of the GABAA receptor complex. Eur J Pharmacol. 2003;464:95–100. doi: 10.1016/s0014-2999(03)01426-2. [DOI] [PubMed] [Google Scholar]