

Summary
Factor X (FX) is a vitamin K-dependent coagulation zymogen, which upon activation to factor Xa assembles into the prothrombinase complex to activate prothrombin to thrombin. FX can be activated by either factor VIIa-tissue factor or factor IXa-factor VIIIa in extrinsic and intrinsic pathways, respectively. In this study, we identified a bleeding patient with moderate FX deficiency who exhibits a clotting defect only in the intrinsic pathway. Exome sequencing revealed that the patient carries a novel homozygous missense mutation that results in substitution of Thr211 with Pro in the activation peptide of FX. Thr211 is the site of an O-linked glycosylation in the activation peptide of FX. We postulated that the lack of this post-translational modification specifically impacts the activation of FX by intrinsic Xase, thereby impairing thrombin generation in the subject. To test this hypothesis, we expressed both wild-type FX and FX containing this mutation in mammalian cells and following the purification of the zymogens to homogeneity characterized their properties in both purified and plasma-based assay systems. Analysis of the results suggests that Thr211 to Pro substitution renders the FX mutant a poor substrate for both physiological activators, however, at physiological concentration of the substrate, the clotting defect manifest itself only in the intrinsic pathway, thus explaining the bleeding phenotype for the patient carrying this mutation.
Introduction
Human factor X (FX) is a multi-domain vitamin K-dependent zymogen in plasma which plays a central role in the coagulation cascade (1,2). It consists of a light chain with 139 and a heavy chain with 346 amino acid residues which are linked together by a disulphide bond (3). Following activation to FXa, the protease assembles into the prothrombinase complex (FXa, factor Va, negatively charged membrane surfaces, and Ca2+) to convert prothrombin to thrombin. FX can be activated to FXa by either one of its physiological activators, the tissue factor (TF)-factor FVIIa (FVIIa) complex in the extrinsic pathway or the factor IXa (FIXa)-factor VIIIa (FVIIIa) complex in the intrinsic pathway (1,2). FX can also be activated by a specific enzyme derived from Russell's viper venom (4). All three activators specifically cleave a single peptide bond, Arg15-Ile16, (chymotrypsin numbering) (5) and remove a 52-residue activation peptide located at the amino-terminus of the heavy chain of FX to generate an active enzyme (1,3). Efficient cleavage of this peptide bond by both of the physiological activators occurs only in the presence of cofactors since neither FVIIa nor FIXa alone can activate FX at a significant rate (1,2). Similarly, FXa by itself has poor catalytic activity toward its substrate, prothrombin, in the absence of a cofactor (1,2). It has been hypothesized that FXa can generate a small amount of thrombin during the initiation phase of the clotting cascade which is sufficient to activate factor V to active form (FVa) in order to facilitate the assembly of the prothrombinase complex, thereby rapidly activating prothrombin to thrombin during the propagation phase of the clotting cascade (6).
Hereditary FX deficiency is a very rare bleeding disorder inherited as an autosomal recessive trait (7–9). The FX gene (F10, OMIM 613872) is located on chromosome 13q34 and composed of 8 exons spanning a region of 25 kb (7). Molecular defects in the F10 gene are the main causes of FX deficiency and more than 100 mutations have been reported (7–9). The incidence of the disorder is about 1 per million and the carrier frequency is about 1:500 (7). Most of the identified mutations are located in either the N-terminus γ-carboxyglutamic acid (Gla) domain or the C-terminus catalytic domain of the heavy chain (9). We present here a novel mutation (Thr211Pro) on the activation peptide of FX which was identified through exome sequencing of the FX gene in a sib-pair with a similar bleeding disorder who exhibit a clotting defect only in the intrinsic pathway. Residue Thr211 is the site of an O-linked glycosylation in the activation peptide of FX (10–12). To understand the molecular basis of the clotting defect in the bleeding patient, we expressed the FX cDNA containing this mutation in mammalian cells. Following purification to homogeneity, we compared the biochemical properties of the FX variant to recombinant FX expressed in the same expression/purification system in both purified and plasma-based assay systems. Analysis of both clinical and biochemical data suggests that the loss of Thr211 glycosylation site is specifically associated with a poor recognition of the mutant substrate by FIXa in the intrinsic pathway.
Materials and methods
Patient
The proband was a 54-year-old Chinese female who was born from consanguineous parents (first-degree cousin marriage) and experienced her first bleeding episode in the left fossa iliac at the age of 18 following a fall. She was required to have multiple transfusions with whole blood due to persistent bleeding. The haemostatic screening assays demonstrated an isolated slightly prolonged activated partial thromboplastin time (aPTT). Other routine coagulation tests, including prothrombin time (PT), platelet count, fibrinogen (Fg) and thrombin time (TT), were all within the normal range. In addition, the simplified thromboplastin generation-test (STGT) as well as functional tests of the liver and kidneys were normal. According to the clinical symptoms and laboratory test results, an undiagnosed bleeding disorder was predicted. The use of Prothrombin Complex Concentrate (PCC) was more effective than fresh frozen plasma (FFP) for prevention and cessation of bleeding in the patient. After this initial bleeding event, she began to experience easy bruising and suffered from oral or uterine bleeding episodes occurring spontaneously or trauma-related several times per year.
Coagulation assays with patient's plasma
The study was approved by the Ethics Committee of Ruijin Hospital, Shanghai Jiaotong University School of Medicine. After written informed consent, venous blood samples from the proband and the family members were obtained and tested for the FX activity (FX:C) by PT and aPTT assays. The FX chromogenic activity assay was based on activation by Russell's viper venom (RVV-X) and conducted according to the manufacturer's instructions using a chromogenic assay kit (Hyphen Biomed, Neucille suroise, France). The FX antigen (FX:Ag) assay and western blot analysis of the plasma FX from the proband and her family members were performed as previously described (13).
Thromboelastograph (TEG) and thrombin generation test (TGT) analysis
The TEG test was performed by a thromboelastograph coagulation analyzer 5000 (Haemoscope) using citrated fresh whole blood obtained from the patient and the normal control according to manufacturer's instructions. Four TEG parameters were used to assess coagulation functions: reaction time (R-time), clot formation time (K-time), maximum amplitude (MA), and composite index (CI). R-time, K-time, and MA represent the activities of coagulation factors, the function of fibrinogen and platelets, and the absolute strength of the fibrin clot, respectively. The value of CI is an indicator of the overall coagulation state.
The thrombin generation test was conducted using the proband and normal control's PPP following manufacturer's protocol, reagents and equipment (Thrombinoscope BV, Maastricht, The Netherlands). PPP-Reagent Low with a mixture of 1 pM tissue factor and 4 μM phospholipids (final concentrations) was used. Five parameters, including lag time, time to peak (TTP), peak height (Peak, nM), area under the curve, referred to the endogenous thrombin potential (ETP, nM*min) and a derived parameter for the maximal thrombin generation velocity (Rate, nM/min) were used to assess TG dynamics and to evaluate the hypocoagulable state in the patient.
Genetic analysis
The genomic DNA of the proband and her family members was extracted from peripheral blood leukocytes using a standard protocol. Whole exome sequencing was performed for the proband as previously described (14).
Mutagenesis and expression of recombinant proteins
The expression and purification of wild-type FX in human embryonic kidney (HEK-293) cells has been described (12). The Thr211 to Pro (T211P) substitution mutant of FX (cDNA numbering) (1) was constructed by PCR methods and expressed using the same expression/purification vector system. The mutant was isolated from 40-L cell culture supernatants by a combination of immunoaffinity and ion exchange chromatography using HPC4 monoclonal antibody coupled to Affi-Gel 10 (Bio-Rad) and a Mono Q column, respectively, as described (12).
Human plasma proteins including FVIIa, FIXa and FX and RVV-X were purchased from Haematologic Technologies Inc. (Essex Junction, VT). Human FVIIIa was a generous gift from Dr. Philip Fay (University of Rochester, New York). Phospholipid vesicles containing 80% phosphatidylcholine and 20% phosphatidylserine (PC/PS) were prepared as described (15). TF lacking the cytoplasmic domain (dc-TF) was expressed and incorporated into PC/PS vesicles as described (16). The PT (Thrombomax with Ca2+) and aPTT (Alexin) reagents were purchased from Sigma (St. Louis, MO). Normal pooled plasma and FX deficient plasma were purchased from George King Bio-Medical, Inc. (Overland Park, KS). The chromogenic substrate Spectrozyme FXa (SpFXa) was purchased from American Diagnostica (Greenwich, CT).
Activation by the FVIIa-TF complex
The initial rate of the concentration-dependence of the activation of recombinant wild-type and mutant FX, expressed in the same expression/purification vector system, by FVIIa was evaluated both in the absence and presence of TF incorporated into PC/PS vesicles as described (12,16). Briefly, the activation of increasing concentrations of FX derivatives (5–600 nM) by FVIIa (100 nM in the absence of TF and 0.05 nM in the presence of TF) was monitored in the absence or presence of relipidated dc-TF (2 nM) in 0.1 M NaCl, 0.02 M Tris-HCl (pH 7.5) containing 0.1 mg/mL bovine serum albumin, 0.1% polyethylene glycol 8000 and 5 mM Ca2+ (TBS/Ca2+) for 2–20 min at room temperature in 30-μL reactions in 96-well assay plates. The activation reactions were terminated by an addition of 20 μL of EDTA to obtain a final concentration of 20 mM, and the rate of FXa generation was determined by an amidolytic activity assay using 50 μL of 0.2 mM SpFXa. Km and kcat values in the presence of TF were calculated from the Michaelis-Menten equation.
Activation by the FIXa-FVIIIa complex
The initial rate of activation of recombinant FX derivatives by FIXa was studied both in the absence and presence of FVIIIa on PC/PS vesicles as described (12). Briefly, the activation of increasing concentrations of FX derivatives (5–600 nM) by FIXa (100 nM in the absence of FVIIIa and 0.1 nM in the presence of the cofactor) on PC/PS vesicles (50 μM) was monitored in the absence or presence of a saturating concentration of FVIIIa (25 nM) in TBS/Ca2+ for 2–20 min at room temperature in 30 μL reactions in 96-well assay plates. Activation reactions were terminated by an addition of 20 μL EDTA and the concentration of FXa generated was determined from standard curves as described above.
Activation by RVV-X
The initial rate of the concentration-dependence of recombinant wild-type and mutant FX activation by RVV-X was studied as described (17). Briefly, the activation of increasing concentrations of FX derivatives (5–600 nM) by RVV-X (0.1 nM) was monitored in TBS/Ca2+ for 5 min at room temperature in 30-μL reactions in 96-well assay plates. Following termination of the reaction by EDTA, the rate of FXa generation was determined as described above.
Clotting activity
The clotting activities of both recombinant wild-type and the recombinant FX variant were evaluated by both PT and aPTT assays using a STart 4 fibrinometer (Diagnostica/Stago, Asnieres, France) as described (16). The clotting activities of samples in both assays were assessed at 4 different dilutions ranging from 0.2 to 1.6 μg/ml FX (final concentrations).
Results
Factor X activity assays
Clotting assays indicated that the proband's plasma has a prolonged aPTT (50.2 s), however, all other routinely checked haemostatic parameters were in the normal range (data not shown). The FX:C level, based on the PT assay, was 108.3% and the value of the FX chromogenic activity, based on the RVV-X assay, was 109.4% (Table 1). By contrast, the FX:C level, based on the aPTT assay, was 4.4%. The FX:Ag level was in the normal range in the proband and all family members. The patient was diagnosed as having moderate FX deficiency as characterized by a defect only in the intrinsic pathway. The FX:C levels in both of her parents and her sister (heterozygote) were in the normal range (Table 1) in both extrinsic and intrinsic pathways.
Table 1.
Analysis of FX antigen and activity levels in the proband and family memebers
| Pedigree | aPTT (s) | PT(s) | FX:C (%)a | FX:C (%)b | FX:C (%)c | FX:Ag (%) |
|---|---|---|---|---|---|---|
| Proband | 50.2 | 11.3 | 4.4 | 108.3 | 109.4 | 91.92 |
| Father | 30.1 | 10.9 | 110.3 | 129.3 | ND | ND |
| Mother | 28.9 | 12.0 | 115.6 | 134.6 | ND | ND |
| Brother | 53.6 | 11.4 | 3.1 | 96.1 | 91.1 | 90.7 |
| Sister | 29.5 | 10.7 | 101.6 | 143.2 | 143.7 | 123.5 |
| Normal range | 27.2–41.0 | 10.0–16.0 | 50–150 | 50–150 | 60–130 | 80–120 |
ND, not done;
aPTT based assay
PT based assay
chromogenic assay
Western-blot analysis under both reducing and non-reducing conditions confirmed that the concentrations of FX in the plasmas of the proband and her family members are similar to that observed in normal individuals (Fig.1A). Nevertheless, under reducing conditions, the band representing the heavy chain of FX (HC) in the proband and her brother migrated slightly faster than that of both her sister and the normal control (Fig.1B).
Figure 1.
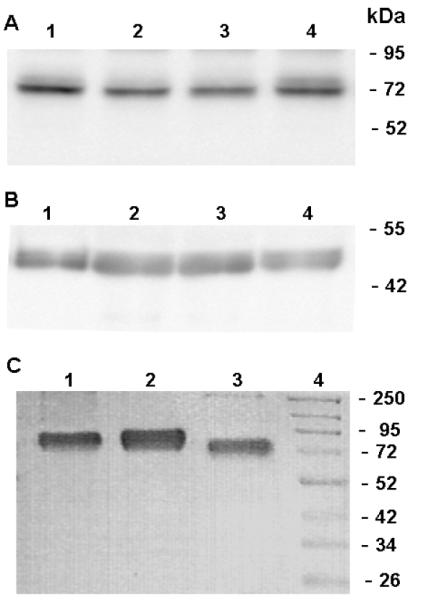
Western blot and SDS-PAGE analysis of FX. (A) Plasma proteins were fractionated on a 10% SDS-PAGE under non-reducing (A) and reducing (B) conditions. Lanes 1–4 in both panels represent plasma FX from the pedigree: lane 1, normal control; lane 2, proband; lane 3, brother; and lane 4, sister. (C) SDS-PAGE analysis of plasma-derived FX and recombinant FX derivatives under non-reducing conditions: Lane 1, plasma-derived FX; lane 2, recombinant FX; and lane 3, FX-Thr211Pro. Lane 4 represents molecular mass standards in kDa.
Assessment of hemostasis using TEG and TGT analysis
TEG provides a dynamic and global assessment of coagulation. In the proband, the value of R-time was increased to 18.2 min, while the level of CI was decreased to - 11.2 and the K value was prolonged to 5.1. These results indicated that the patient was in a hypocoagulable state (Fig. 2A). According to the TGT results (Fig. 2B), the ETP value (nM*min) in the proband's plasma was 364±36.0, which is markedly low when compared to the normal control (1086±12.0), indicating a decreased amount of thrombin generation in the proband's plasma. Both the Peak and Rate levels of the proband were much lower than those of the normal controls, suggesting a decrease in the maximal concentration of thrombin generation. Moreover, both the TTP and lag-time values of the proband were prolonged compared to those of the normal controls. These results indicated that the proband had a defect in thrombin generation.
Figure 2.
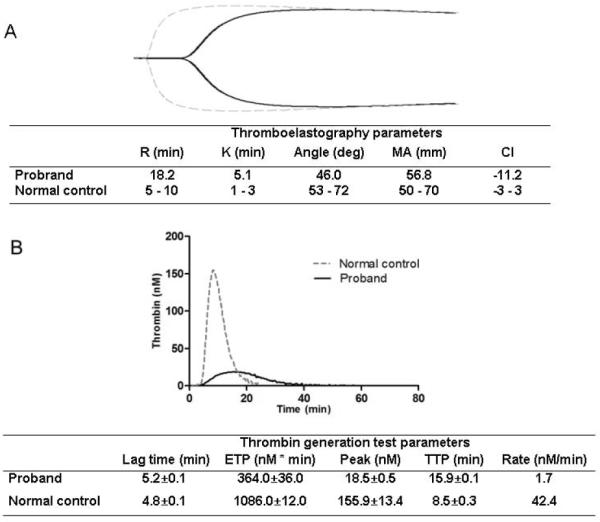
Assessment of hemostasis using TEG and TGT analysis. Panel A represents TEG results and panel B represents TGT results of the proband and the control. 1 pM, PPP-Reagent Low (contains 1 pM TF).
Genetic analysis
Based on the recessive model with a consanguineous relationship and clinical data (coagulation), a homozygous missense mutation (c.631A>C, p.T211P) in the F10 gene was identified as a candidate mutation responsible for the defect, which was subsequently confirmed by Sanger sequencing. Her brother was a homozygote and her sister was a heterozygote for this mutation (Fig. 3). Thr211 is located on the activation peptide of the FX heavy chain. It has been established that this residue is the site of an O-linked glycosylation on the activation peptide of FX (10–12). This finding explained the basis for the slightly lower molecular mass observed for FX derived from the plasma of the proband and her brother under reducing conditions (Fig. 1B) since their variant FX residue Pro211 cannot be glycosylated.
Figure 3.
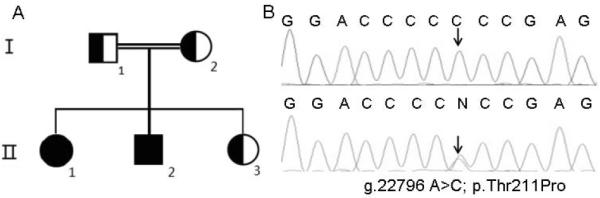
Genetic defect of the F10 gene in the pedigree. Panel A represents the pedigree and panel B represents the identified mutation in the pedigree.
Expression and purification of the FX-Thr211Pro variant
To characterize the molecular basis for the clotting defect in the proband, both wild-type FX and the Thr211Pro variant of FX were expressed in HEK-293 cells (using the same expression/purification vector system) and purified by immunoaffinity chromatography as described in “Materials and methods”. SDS-PAGE analysis under non-reducing conditions indicated that the recombinant FX variant has been purified to homogeneity and that it migrates with an apparent molecular mass of ~70 kDa which was slightly lower than that of recombinant wild-type FX (~75 kDa), indicating that residue Pro211 lacks the carbohydrate attachment as expected (Fig. 1C). It should be noted that recombinant FX produced in either the HPC4 epitope-tagged expression/purification vector system (as in this study) or in a vector system without the HPC4-tag have indistinguishable zymogenic and enzymatic activities that are also indistinguishable from the plasma-derived FX (data not presented). We have fully characterized and compared the zymogenic activity of the epitope-tagged FX to the plasma-derived FX in both purified system using the physiological activators and in plasma-based PT and APTT assays. Both recombinant and plasma-derived FX exhibit indistinguishable activities in all assays (18).
FX activation by extrinsic Xase
The concentration-dependence of the activation of recombinant wild-type and Thr211Pro variant of FX by either FVIIa alone or FVIIa in complex with TF in extrinsic Xase indicated that the Km for the activation of the variant FX by the protease in both the absence and presence of TF has been elevated (Fig. 4). While no Km parameter for the activation of the FX variant by FVIIa could be obtained in the absence of TF (Fig. 4A), the Km for the zymogen in the presence of the cofactor was determined to be elevated ~7.5-fold (Fig. 4B). Thus, in contrast to a Km of ~60 nM for the activation of FX by extrinsic Xase, this value was increased to ~450 nM for the FX variant (Fig. 4B). Nevertheless, kcat for the activation of FX by FVIIa was improved for the variant both in the absence and presence of the cofactor (Fig. 4). Thus, in contrast to a kcat of ~133 nM/min/nM for the activation of FX by extrinsic Xase, the corresponding value was increased to ~439 nM/min/nM for the FX variant, representing ~3.3-fold improvement in the rate of mutant zymogen activation by the activator complex (Fig. 4).
Figure 4.
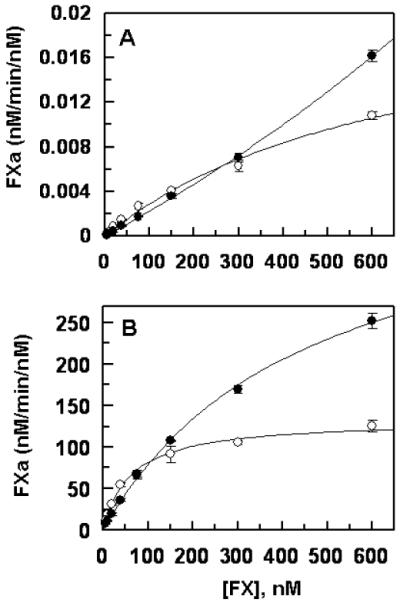
Comparisons of the activation rates of recombinant FX-WT and FX-Thr211Pro by FVIIa. (A) In the absence of TF, FVIIa (100 nM) was added to increasing concentrations of FX-WT (○) and FX-Thr211Pro (●) on PC/PS vesicles (50 μM) in TBS/Ca2+ at room temperature. After 20 min incubation, the reactions were terminated by EDTA and the rate of FXa generation was measured from the cleavage rate of SpFXa as described under “Materials and methods”. (B) The same as (A) except that FVIIa (0.05 nM) in complex with relipidated dcTF (2 nM) was incubated with FX derivatives for 2 min. Data are derived from three independent measurements (±SD).
FX activation by intrinsic Xase
Similar to activation by FVIIa, FIXa in the absence of FVIIIa exhibited elevated Km and kcat parameters toward the FX variant (Fig. 5A). However, FIXa in complex with FVIIIa in intrinsic Xase exhibited significantly reduced kcat toward the FX variant without a change in Km for the substrate (Fig. 5B). Thus, FIXa activated both recombinant FX and the variant with similar Km values of 140–160 nM in the presence of FVIIIa, however, kcat for the activation of the FX variant was decreased ~4-fold in the presence of the cofactor (Fig. 5B).
Figure 5.
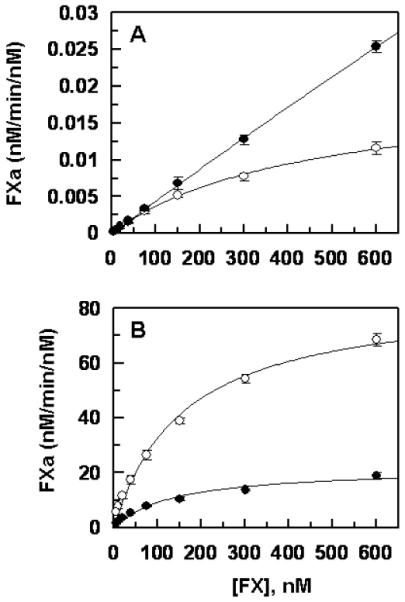
Comparisons of the activation rates of recombinant FX-WT and FX-Thr211Pro by FIXa. (A) In the absence of FVIIIa, FIXa (100 nM) was incubated with increasing concentrations of FX-WT (○) and FX-Thr211Pro (●) on PC/PS vesicles (50 μM) in TBS/Ca2+. After 20 min incubation at room temperature, the reactions were terminated by EDTA, and the rate of FXa generation was measured as described under “Materials and methods”. (B) The same as (A) except that FIXa (0.1 nM) in complex with FVIIIa ( 25 nM) was used for the activation of the zymogens for 2–5 min. Data are derived from three independent measurements (±SD).
FX activation by RVV-X
The RVV-X activation of both recombinant wild-type FX and the variant is shown in Fig. 6. RVV-X activated the FX variant with ~5-fold elevated Km, however, kcat for the activation of the variant was increased ~2-fold (Fig. 6 legend). Thus, the kinetic parameters for the activation of the FX variant by RVV-X resemble those of the extrinsic pathway (elevated Km and kcat values).
Figure 6.
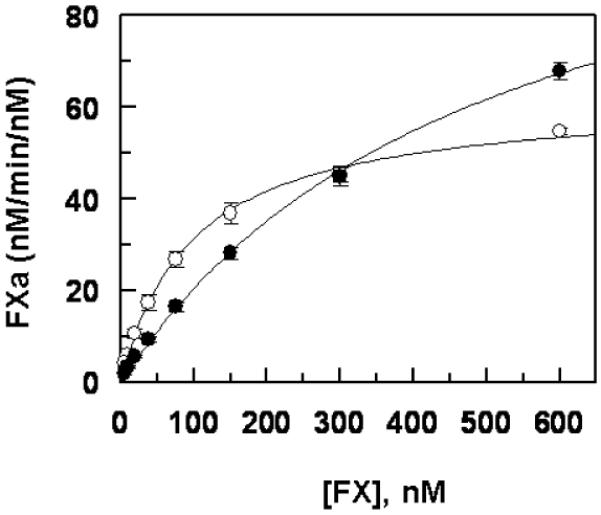
Comparisons of the activation rates of recombinant FX-WT and FX-Thr211Pro by RVV-X. RVV-X (0.1 nM) was incubated with increasing concentrations of FX-WT (○) and FX-Thr211Pro (●) in TBS/Ca2+. After 5 min incubation at room temperature, the reactions were terminated by EDTA, and the rate of FXa generation was measured as described under “Materials and methods”. Data are derived from three independent measurements (±SD).
Analysis of the clotting activity of recombinant FX zymogens
Both PT and aPTT assays were used to evaluate the clotting activities of recombinant FX derivatives. Both plasma-derived and recombinant wild-type FX (FX-WT) exhibited indistinguishable clotting activities in the PT assay (data not shown). In this assay, the zymogen concentration-dependence of the clotting activities indicated that at a low and sub-physiological concentration of FX (<1 μg/mL) the FX variant exhibits a lower clotting activity (Fig. 7A). However, at higher concentrations (>1.6 μg/mL) both FX-WT and FX-Thr211Pro showed comparable clotting activities (Fig. 7A). By contrast, the clotting activity of the FX variant was impaired in the aPTT assay independent of the concentration of the zymogen (Fig. 7B). These results are in agreement with the clotting data obtained from the patient's plasma which showed an impaired aPTT, but a normal PT. Thus, by increasing the concentration of the FX variant in the PT assay, it was possible to restore the defect in the clotting activity of this mutant (Fig. 7A), supporting the kinetic data that the defect in the activation of the variant by extrinsic Xase is due to a defect in Km for the substrate (Fig. 4B). By contrast, increasing the concentration of the FX variant in the aPTT assay did not restore the defect in the clotting activity of the variant (Fig. 7B), suggesting that the defect in the clotting activity of the FX variant is due to a defect in kcat for the activation of FX by intrinsic Xase that is in agreement with the kinetic data presented in Fig. 5B.
Figure 7.
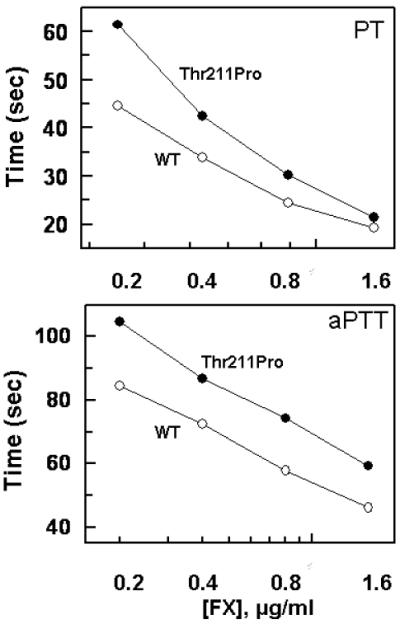
Plasma clotting activities of recombinant FX-WT and FX-Thr211Pro. The clotting activities of FX-WT (○) FX-Thr211Pro (●) were determined using FX-deficient plasma by both PT (panel A) and aPTT (panel B) assays as a function of different concentrations of the zymogens at 37 °C as described under “Materials and methods”. Data are derived from three independent measurements (±SD).
Discussion
In this study, exome sequencing identified a novel missense mutation (Thr211 Pro) in the FX sequence of a bleeding patient who has a clotting defect only in the intrinsic pathway. The plasma derived from the patient with this mutation had normal antigen and activity levels as determined by an ELISA and by a PT based assay, respectively. However, the patient's plasma exhibited a very low level of FX:C (4.4%) in the aPTT based assay, suggesting that the activation of the FX variant has been specifically impaired in the intrinsic pathway of the zymogen activation. This resembles the bleeding phenotype of both hemophilia A and B where the capacity of the extrinsic pathway to generate FXa is intact, but a defect in the amplification phase by intrinsic Xase causes bleeding diathesis in the patient. In addition, both global coagulation assays of TEG and TGT revealed a hypocoagulable status in the patient. This is a typical type II FX deficiency which is characterized by a low FX activity but a normal antigen level (7–9). Thr211 is the site of an O-linked glycosylation site in the activation peptide of FX and this post-translational modification appears to play a key role in the FX activity level in the intrinsic pathway. This is the first study to identify a FX deficiency due to a mutation involving a glycosylation site residue in the activation peptide of FX. Nevertheless, one deletion (Tyr163delAT), located in the activation peptide of FX, has also been reported in the database (9). The phenotypic features of type II FX deficiency can be heterogeneous. In the majority of patients, a clotting defect is observed in both the intrinsic and extrinsic pathways. However, in a few cases, clotting defects of FX deficiency involve either the intrinsic or the extrinsic system (19).
The activation peptide of human FX is heavily glycosylated with O-linked carbohydrates at Thr199 and Thr211 sites and N-linked carbohydrates at Asn221 and Asn231 sites (10,11). Recent results have indicated that the carbohydrate residues of the activation peptide play key roles in the recognition and efficient activation of FX by both of its physiological activators in the extrinsic and intrinsic pathways (12). However, the clinical data, showing that the bleeding patient has a clotting defect in only the intrinsic pathway, suggested that the absence of the O-linked glycosylation at position 211 due to Thr211Pro mutation specifically affects the activation of FX by intrinsic Xase. To test this hypothesis and to establish the molecular basis of the clotting defect in the bleeding patient, we expressed the Thr211 to Pro substitution mutant of the FX cDNA in mammalian cells and compared its zymogenic properties to recombinant FX (expressed by the same vector system) in both purified and plasma based assay systems. Analysis of the kinetic data indicated that both of the physiological activators of FX activate the FX variant with altered kinetic parameters. Thus, the activation of the FX variant by FVIIa in extrinsic Xase occurred with a significantly elevated Km, though the impairment in Km for the substrate was compensated by an improvement in kcat of the activation reaction. Thus, the net effect in the kinetic reaction was a comparable or improved activation of the FX variant by FVIIa in the extrinsic pathway at the near physiological concentration of the zymogen (~10 μg/mL, ~150 nM). As shown in Fig. 4B, extrinsic Xase generated a comparable or greater amount of FXa at FX concentration of above 100 nM when either recombinant wild-type or variant FX was used as the substrate. The plasma-based PT assay supported this finding since, similar to the purified system, the FX variant exhibited low clotting activity at the sub-physiological concentrations (<1.6 μg/mL) of the zymogen, but increasing the concentration of the FX variant to above 1.6 μg/mL corrected the clotting defect of the FX variant in the PT assay (Fig. 4). These results explain the normal FX:C level obtained with the patient's plasma using the PT-based assay (Table 1). Thus, the improvement in kcat largely compensates for the impaired Km for the FX variant in the patient's plasma, resulting in normal clotting in the extrinsic pathway.
In contrast to the extrinsic pathway, FIXa in the intrinsic complex exhibited a significantly reduced kcat for the activation of the FX variant without a significant change in Km for the substrate (Fig. 5B). The net effect was a markedly lower catalytic efficiency for the FIXa-VIIIa complex toward the FX variant in the intrinsic pathway independent of the concentration of the zymogen. The prolonged clotting time observed with the plasma-based aPTT assay supports the kinetic data since increasing the concentration of the FX variant did not restore the clotting defect caused by the mutation. Thus, a defect in kcat during activation by intrinsic Xase is responsible for the lower FX clotting activity in the plasma of the bleeding patient and her Pro211 homozygote brother. It was interesting to note that the other three family members with heterozygous Pro211 mutation had normal FX:C levels based on both PT and aPTT assays. This can be explained by previous findings that variations in the concentration of FX over the range of 50% to 150% do not alter the peak and/or the amount of total thrombin generation (20). Thus, the 50% normal FX zymogen in the heterozygote carriers is sufficient for maintaining normal hemostasis. Moreover, FIXa in the intrinsic Xase complex exhibits a Km of ~60 nM (Fig. 6) for FX that is nearly 2.5-fold lower than the plasma concentration of the zymogen in circulation (21). Such a low Km for the zymogen, which is primarily mediated by its assembly into negatively charged phospholipids in intrinsic Xase (20,21), readily compensates for a 2-fold drop in the concentration of the normal zymogen, explaining the lack of bleeding and normal FX:C levels in the intrinsic pathway in the heterozygote family members. Thus, a phospholipid-dependent Km effect, together with an improved kcat for the activation of the FX-Thr211Pro variant by the FVIIa-TF complex account for the normal clotting activity of the patient's plasma in the extrinsic pathway. The clotting defect in the homozygote patient relates to the inability of FVIIIa to effectively promote the kcat of FX activation by FIXa in the intrinsic Xase complex.
Finally, the observation that the kcat defect for the activation of the FX variant was specific for FIXa in the intrinsic Xase complex but not for FIXa alone suggests that the carbohydrate side chain of Thr211 on the activation peptide of FX constitutes a cofactor-dependent recognition site for the FIXa-FVIIIa complex in the intrinsic pathway. Thus, the interaction of the Thr211 carbohydrate with an exosite on intrinsic Xase plays a key role in the transition-state stabilization of the reaction intermediate in the intrinsic pathway. Structural and kinetic data have predicted a possible mechanistic role for the substrate-assisted catalysis in coagulation reactions (22,23). The interesting finding of this study now provides firm support for this hypothesis in an in vivo setting and demonstrates how substrate-assisted catalysis, mediated by a carbohydrate side chain, can contribute to efficient coagulation zymogen activation in the clotting cascade in general and FX activation by FIXa in the intrinsic pathway in particular.
Acknowledgements
The authors wish to thank the patient and her family members for their participation in this study and Audrey Rezaie for proofreading the manuscript. This research was partly supported by grants awarded by the National Heart, Lung, and Blood Institute of the National Institutes of Health HL 101917 and HL 62565 to ARR.
Footnotes
Disclosure of conflict of interests The authors declare no competing financial interest.
References
- 1.Leytus SP, Foster DC, Kurachi K, Davie EW. Gene for human factor X: A blood coagulation factor whose gene organization is essentially identical with that of factor IX and protein C. Biochemistry. 1986;25:5098–102. doi: 10.1021/bi00366a018. [DOI] [PubMed] [Google Scholar]
- 2.Mann KG, Nesheim ME, Church WR, Haley P, Krishnaswamy S. Surface-dependent reactions of the vitamin K-dependent enzyme complexes. Blood. 1990;76:1–16. [PubMed] [Google Scholar]
- 3.Stenflo J. Structure-function relationships of epidermal growth factor modules in vitamin K-dependent clotting factors. Blood. 1991;78:1637–51. [PubMed] [Google Scholar]
- 4.Furie BC, Furie B. Coagulant protein Russell's viper venom. Methods Enzymol. 1976;45:191–205. doi: 10.1016/s0076-6879(76)45019-x. [DOI] [PubMed] [Google Scholar]
- 5.Bode W, Mayr I, Baumann U, Huber R, Stone SR, Hofsteenge J. The refined 1.9 Å crystal structure of human a-thrombin: interaction with D-Phe-Pro-Arg chlorometheylketone and significance of the Tyr-Pro-Pro-Trp insertion segment. EMBO J. 1989;8:3467–75. doi: 10.1002/j.1460-2075.1989.tb08511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mann KG, Butenas S, Brummel K. The dynamics of thrombin generation. Arterioscler Thromb vasc Biol. 2003;23:17–25. doi: 10.1161/01.atv.0000046238.23903.fc. [DOI] [PubMed] [Google Scholar]
- 7.Peyvandi F, Duga S, Akhavan S, Mannucci PM. Rare coagulation deficiencies. Haemophilia. 2002;8:308–21. doi: 10.1046/j.1365-2516.2002.00633.x. [DOI] [PubMed] [Google Scholar]
- 8.Karimi M, Menegatti M, Afrasiabi A, Sarikhani S, Peyvandi F. Phenotype and genotype report on homozygous and heterozygous patients with congenital factor X deficiency. Haematologica. 2008;93:934–8. doi: 10.3324/haematol.12211. [DOI] [PubMed] [Google Scholar]
- 9.Herrmann FH, Auerswald G, Ruiz-Saez A, Navarrete M, Pollmann H, Lopaciuk S, Batorova A, Wulff K. For the Greifswald factor X deficiency study group. Factor X deficiency: clinical manifestation of 102 subjects from Europe and Latin America with mutations in the factor 10 gene. Haemophilia. 2006;12:479–89. doi: 10.1111/j.1365-2516.2006.01303.x. [DOI] [PubMed] [Google Scholar]
- 10.Inoue K, Morita T. Identification of O-linked oligosaccharide chains in the activation peptides of blood coagulation factor X: The role of the carbohydrate moieties in the activation of factor X. Eur J Biochem. 1993;218:153–63. doi: 10.1111/j.1432-1033.1993.tb18361.x. [DOI] [PubMed] [Google Scholar]
- 11.Sinha U, Wolf DL. Carbohydrate residues modulate the activation of coagulation factor X. J Biol Chem. 1993;268:3048–51. [PubMed] [Google Scholar]
- 12.Yang L, Manithody C, Rezaie AR. Functional role of O-linked and N-linked glycosylation sites present on the activation peptide of factor X. J Thromb Haemost. 2009;7:1696–702. doi: 10.1111/j.1538-7836.2009.03578.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang WB, Fu QH, Zhou RF, et al. Molecular characterization of two novel mutations causing factor X deficiency in a Chinese pedigree. Haemophilia. 2005;11:31–7. doi: 10.1111/j.1365-2516.2005.01063.x. [DOI] [PubMed] [Google Scholar]
- 14.Carmichael H, Shen Y, Nguyen T, Hirschhorn J, Dauber A. Whole exome sequencing in a patient with uniparental disomy of chromosome 2 and a complex phenotype. Clin Genet. 2012 Nov 21; doi: 10.1111/cge.12064. doi: 10.1111/cge.12064. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smirnov MD, Esmon CT. Phosphatidylethanolamine incorporation into vesicles selectively enhances factor Va inactivation by activated protein C. J Biol Chem. 1994;269:816–9. [PubMed] [Google Scholar]
- 16.Neuenschwander PF, Bianco-Fisher E, Rezaie AR, Morrissey JH. Phosphatidylethanolamine augments factor VIIa-tissue factor activity: enhancement of sensitivity to phosphatidylserine. Biochemistry. 1995;34:13988–93. doi: 10.1021/bi00043a004. [DOI] [PubMed] [Google Scholar]
- 17.Kittur FS, Manithody C, Rezaie AR. Role of the N-terminal epidermal growth factor-like domain of factor X/Xa. J Biol Chem. 2004;279:24189–96. doi: 10.1074/jbc.M402302200. [DOI] [PubMed] [Google Scholar]
- 18.Manithody C, Yang L, Rezaie AR. Role of basic residues of the autolysis loop in the catalytic function of factor Xa. Biochemistry. 2002;41:6780–8. doi: 10.1021/bi0255367. [DOI] [PubMed] [Google Scholar]
- 19.Girolami A, Scarparo P, Scandellari R, Allemand E. Congenital factor X deficiencies with a defect only or predominantly in the extrinsic or in the intrinsic system: a critical evaluation. Am J Hematol. 2008;83:668–71. doi: 10.1002/ajh.21207. [DOI] [PubMed] [Google Scholar]
- 20.Butenas S, van't Veer C, Mann KG. “Normal” thrombin generation. Blood. 1999;94:2169–78. [PubMed] [Google Scholar]
- 21.Hoyer LW. Hemophilia A. N Engl J Med. 1994;330:38–47. doi: 10.1056/NEJM199401063300108. [DOI] [PubMed] [Google Scholar]
- 22.Sichler K, Banner DW, D'Arcy A, et al. Crystal structures of uninhibited factor VIIa link its cofactor and substrate-assisted activation to specific interactions. J Mol Biol. 2002;322:591–603. doi: 10.1016/s0022-2836(02)00747-7. [DOI] [PubMed] [Google Scholar]
- 23.Neuenschwander PF. Exosite occupation by heparin enhances the reactivity of blood coagulation factor IXa. Biochemistry. 2004;43:2978–86. doi: 10.1021/bi035452d. [DOI] [PubMed] [Google Scholar]