

Abstract
To further understand the role of XIAP in acute myeloid leukemia (AML), we suppressed XIAP expression by antisense oligonucleotides and determined the effect on gene expression profiles and biological pathways. XIAP inhibition upregulated expression of proteasome genes in a manner similar to the proteasome inhibitor bortezomib or MG132; decreased 20S proteasome activity, an effect which was diminished in the presence of a pan-caspase inhibitor; and increased IκBα, Mcl-1, and HSP70in AML cells. In addition to multiple functions already described, XIAP contributes to increased proteasome activity in AML cells, and the antitumor effect of XIAP inhibition may be mediated in part through caspase-dependent proteasome inhibition.
Keywords: XIAP, proteasome, gene expression, AML, caspase, IκBα
Introduction
It is well established that X-linked inhibitor of apoptosis (XIAP), a member of the inhibitors of apoptosis (IAP) family, inhibits caspases and suppresses apoptosis mediated through both intrinsic and extrinsic pathways(1–6). XIAP is highly expressed in many types of cancer and is associated with chemoresistance. However, it has become evident in recent years that XIAP not only inhibits caspases and suppresses apoptosis, but also modulates multiple cellular functions.
XIAP was found to be critical in modulating reactive oxygen species levels via regulation of antioxidative genes, leading to inhibition of reactive oxygen species-induced apoptosis(7). Overexpression of XIAP in endothelial cells increases the level of the CDK inhibitor p21cip1/waf1 and decreases the rate of cell proliferation(8), while patients deficient in XIAP develop an X-linked lymphoproliferative syndrome(9). XIAP was also reported to play a role in mammary development(10). XIAP is involved in multiple signaling pathways. For example, XIAP is a component of the TGFβ family signaling pathway(11)and can regulate TGFβ-mediated JNK activation(12). It can also activate NF-κB(8). Interestingly, it was found recently that the activation of NF-κB by XIAP/survivin leads to autocrine production of fibronectin, which activates the cell motility kinase FAK and SRC via β1 integrin signaling, resulting in tumor cell invasion in vitro and metastasis in vivo(13). Clearly, in addition to regulating apoptosis, XIAP participates in many processes ranging from adaptive response to cellular stress, cell proliferation, differentiation, signaling, and motility.
To exploit XIAP as a therapeutic target, it is important to fully understand its function and participating pathways. To understand the function of XIAP in a more comprehensive way in AML, we downregulated XIAP expression by antisense oligonucleotide (ASO) in AML cell lines, determined the resulting changes in gene expression profiles, and analyzed pathways affected by XIAP downregulation in these cells. We report that inhibition of XIAP reduced proteasome activity in a caspase-dependent manner, leading to the activation of the proteasome recovery pathway. To our knowledge, this is the first report that XIAP plays a role in proteasome activity.
Materials and Methods
Cells and cell cultures
HL -60 (ATCC) and OCI-AML3 cells were cultured in RPMI 1640 medium, supplemented with 10% heat-inactivated fetal calf serum, 2 mM L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin. OCI-AML3 cells were kindly provided by Dr. M. Minden (Ontario Cancer Institute, Toronto, Ontario, Canada).
Treatment of cells
To inhibit the expression of XIAP by ASO, exponentially growing HL-60 and OCI-AML3 cells were electroporated as described previously(14) with XIAP ASO or a control oligonucleotide (both from ISIS Pharmaceuticals, Carlsbad, California) using Nucleofector solution T and program K-17 for HL-60 cells (3 μg oligonucleotide/106 cells) and program X-001 for OCI-AML3 cells (2 μg oligonucleotide/106 cells) according to the manufacturer’s instructions (Amaxa Biosystems, Cologne, Germany) for 24 and 48 hours. To inhibit proteasome activity, exponentially growing OCI-AML3 cells (0.4 ×106 cells/mL) were treated with proteasome inhibitor bortezomib (20 nM, LC Laboratories, Woburn, MA) or MG132 (1 μM). For cells treated with both oligonucleotide and pan-caspase inhibitor z-VAD (EMB Millipore Chemicals, Billerica, MA), cells were incubated with z-VAD (20 μM) for 1 hour prior to electroporation. For cells treated with both oligonucleotide and proteasome inhibitor bortezomib, bortezomib (10 nM) was added right after cells were electroporated.
Gene expression profiling
Total RNA was extracted using the RNAqueous kit (Ambion, Austin, TX). After confirmation of RNA quality using a Bioanalyzer 2100 instrument (Agilent), 300 ng of total RNA was amplified and biotin-labeled through an Eberwine procedure using an Illumina TotalPrep RNA Amplification kit (Ambion) and hybridized to Illumina HT12 version 4 human whole-genome arrays. Processing of bead-level data was by methods previously described(15). Significance testing for differentially-expressed probes was by the Wilcoxon rank-sum test applied to individual processed bead values, with false-discovery rate significance values (q) determined by the method of Benjamini and Hochberg(16). No fold-change threshold was applied, and significance was set at p≤0.01, FDR≤0.1. Hierarchical clustering and heat mapping used Cluster and Treeview software from Eisen et al(17). Gene set analysis applied gene set enrichment analysis(18) and the hypergeometric distribution test(19), using gene sets with 20–300 genes and FDR≤0.1, to gene sets from MSigDB and individual literature sources.
RT-PCR
RNAs were reverse-transcribed with random heximers (Roche Applied Science, Indianapolis, IN, USA) by superscript III reverse transcriptase (Invitrogen) at 50°C for 50 minutes and then 70°C for 15 minutes. The PCR amplification mixture (25μL) contained cDNA, primers and probe for Nrf1 (Hs00192316_m1), PSMB4 (Hs00160598_m1), PSMC2 (Hs00739800_m1), PSMC4 (Hs00197826_m1), or PSMD12 (Hs00356667_m1) and Taq-Man universal PCR master mix (Applied Biosystems, Foster City, CA, USA). Thermal cycle conditions included holding the reaction at 50°C for 2 minutes and at 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. Results were collected and analyzed by an ABI Prism 7900HT fast real-time PCR Sequence Detection System (Applied Biosystems). The abundance of each transcript relative to that of ABL1 was calculated using the 2−ΔCtmethod, where ΔCt is the mean Ct of the transcript of interest minus the mean Ct of the transcript for ABL(20). Results are expressed as folds of gene expression levels in treated cells over respective control cells.
Cell viability
Cell viability was determined by trypan blue exclusion using a Vi-Cell XR Cell Counter (Beckman Coulter, Fullerton, California). Apoptosis was estimated by flow cytometry measurements of phosphatidylserine externalization(21) with the Annexin V-Cy5 (BD Biosciences, San Jose, CA) using a FACSArray Bioanalyzer (BD Biosciences, San Jose, CA). Membrane integrity was assessed simultaneously by 7-amino-actinomycin D (7AAD) exclusion in the annexin V-stained cells.
Western blot analysis
XIAP, IκBα, Mcl-1, and HSP70 protein levels were determined by western blot analysis, as described previously(14). Antibodies against XIAP were purchased from BD-Transduction Laboratory (San Diego, CA), against IκBα from Imgenex (San Diego, CA), and against Mcl-1 and HSP70 from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA) and Cell Signaling Technology (Danvers, MA), respectively. Signals were detected using Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, Nebraska), and quantitated using Odyssey Software version 3.0 (LI-COR Biosciences). β-Actin was used as a loading control.
Proteasome activity assay
20S proteasome activity was determined in a fixed number of cells using a 20S Proteasome Assay Kit (Cayman Chemical, Ann Arbor, MI) following the manufactory’s instruction. Briefly, treated cells were pelleted from the 20S proteasome assay buffer and lysed in the 20S proteasome lysis buffer. The lysates were then mixed with a specific 20S substrate, SUC-LLVY-AMC which, upon cleavage by the active enzyme of 20S proteasome, generates a highly fluorescent product that can be measured using excitation and emission wavelength of 360 nm and 480 nm, respectively. Samples in duplicates or triplicates were read by Victor3 Multilabel Reader (PerkinElmer, Waltham, MA) using Ex 355nm/Em 460nm filters.
Statistical analysis
Experiments were carried out in triplicates and results are expressed as mean ± standard error. Statistical differences between groups were determined using paired Student’s t-test. P values of less than 0.05 were considered statistically significant (*).
Results
Inhibition of XIAP by ASO activates the proteasome recovery pathway in AML cells
To understand the function of XIAP in AML, we inhibited XIAP expression in HL-60 and OCI-AML3 cells using XIAP ASO (Figure 1A) and determined gene expression profiles in these cells. Each experiment was performed in duplicate or triplicate, using either ASO or a control oligonucleotide (NSO), for a duration of 24 or 48 hr. For each cell line and duration, differentially-expressed genes were identified by comparing one of two replicates of ASO treatment to one of two control replicates. In all 16 possible comparisons, analysis (by the hypergeometric distribution test) of the list of differentially-expressed genes was enriched for the Gene Ontology term “proteasomal protein catabolic process”. This result was confirmed by gene set enrichment analysis (data not shown). A subtracted heat map of “proteasomal protein catabolic process” genes, comparing values for ASO-treated cells to corresponding NSO-treated cells, showed a core of genes that were consistently upregulated by ASO treatment in both cell lines (Figure 1B), consisting predominantly of genes encoding proteasomal components.
Figure 1.

Inhibition of XIAP by ASO upregulates the expression of proteasome component genes in AML cells. A. Western blotting of HL60 and OCI-AML3 cells transfected with XIAP ASO or control oligonucleotide (NSO) confirms reduction of XIAP levels by ASO. B. A heat map shows hierarchically-clustered genes in the Gene Ontology term “proteasomal protein catabolic process” with consistent upregulation following XIAP inhibition by ASO in both HL-60 and OCI-AML3 cells. Each column represents a replicate of ASO treatment compared to a time- and cell line-matched replicate of NSO treatment. Multiple rows for the same gene indicate different probes for that gene. The color bar represents the relative fold-change in gene expression values. hr, hour.
Proteasome inhibition inducing new proteasome synthesis is an evolutionarily conserved “bounce-back” phenomenon, recently reported to be induced by proteasome inhibitors in human cancer cells via a proteasome recovery pathway mediated by the transcription factor Nrf1(22). To investigate whether upregulation of proteasomal component genes by XIAP inhibition was likely to indicate a bounce-back response, we assayed gene expression profiles in OCI-AML3 cells after chemical inhibition of proteasome activity, by bortezomib. Upregulation of genes in the larger Gene Ontology term “proteasome complex” induced by bortezomib indicated a bounce-back response and were similar to changes induced by ASO treatment (Figure 2), peaking at 9 to12 hours, strongly suggesting that XIAP downregulation inhibits proteasome function. This bounce back phenomenon was further confirmed by MG-132 (Figure 2A). At the concentration used, MG-132 was able to inhibit proteasome activity as measured by 20S proteasome activity assay (Figure 2A).
Figure 2.

Inhibition of XIAP expression and chemical inhibition of proteasome activity similarly upregulate the expression of proteasome component genes. For both A and B, each column corresponds to one replicate of an experimental condition (ASO, bortezomib, or MG132) as compared to a replicate of the appropriate control condition in the cell line indicated. For ASO, replicates are matched to NSO-treated controls at the same time point shown. For bortezomib and MG132, controls were at time zero, which for MG132 are the average of two array replicates. Numbers on sample labels correspond to particular replicates among multiple replicates. A. The heat map shows relative expression of consistently-upregulated hierarchically-clustered genes compiled from Gene Ontology terms for the 20S proteasome complex. Multiple rows for the same gene indicate different probes for that gene. The color bar represents the relative fold-change in gene expression values. 20S proteasome activity is also shown in OCI-AML3 cells treated with 1 μM MG-132. B. Relative gene expressions in ASO or bortezomib treated vs. control cells determined by Taq-Man RT-PCR. hr, hour.
To validate our findings, we determined the expression by Taq-Man RT-PCR of Nrf1 and several genes that are involved in the “bounce-back” phenomenon and whose expressions were induced by ASO and proteasome inhibitors using the same RNA samples as described in Figure 2A. Figure 2B showed that the expression levels of PSMB4, PSMC2, PSMC4, and PSMD12 were increased in both ASO and bortezomib treated cells compared to control cells, while only a moderate increase in Nrf1 expression was observed in ASO treated cells suggesting that in addition to Nrf1, other mediators may also regulate expression of proteasome genes.
Inhibition of XIAP decreases proteasome activity in a caspase-dependent manner in AML cells
We next used a proteasome activity assay to measure directly the effect of XIAP inhibition on proteasome function inHL -60 cells. The degradation rate by 20S proteasomes was decreased by XIAP inhibition, as well as by bortezomib treatment, and was further decreased when XIAP inhibition was combined with bortezomib (Figure 3A). Similar results were observed in OCI-AML3 cells (not shown). The assay was performed after 24 hours of treatment, when there was still minimal cell death produced by the treatments (Figure 3B). Using the levels of proteasome target proteins as a further measure of proteasome activity, we found that XIAP inhibition, as well as bortezomib treatment increased the IκBα, HSP70, and Mcl-1 levels, more so when combined with bortezomib (Figure 3C). Using the pan-caspase inhibitor z-VAD to determine whether the reduction in proteasome activity by XIAP inhibition involves caspase activation, we found that caspase inhibition abolished the reduction of proteasome activity by XIAP inhibition but had no effect on bortezomib-induced reduction of proteasome activity (Figure 4), indicating that the reduction of proteasome activity by XIAP inhibition is caspase-dependent in AML cells.
Figure 3.

Inhibition of XIAP decreases proteasome activity. A. 20S proteasome activity is decreased in HL-60 cells treated for 24 hours with ASO or bortezomib, and further decreased by their combination. Data are the result of 3 experiments. Bars indicate the standard error of the mean. Asterisks indicate significant (p < 0.05) differences from treatment with the NSO control plus DMSO. B. An apoptosis assay shows no differences between treatments at 24 hours and apoptosis induction with ASO at 48 hours. The comparisons were between treated groups with NSO control in cells positive for either Annexin V (AnnV) or 7-AAD. The degree of apoptosis seen in the control is likely attributable to the effects of electroporation. C. Western blotting shows increases in IκBα, HSP70, and Mcl-1 levels in HL-60 cells treated for 24 hours with ASO or bortezomib, and a further increase by their combination. Downregulation of XIAP by ASO is confirmed. hr, hour.
Figure 4.
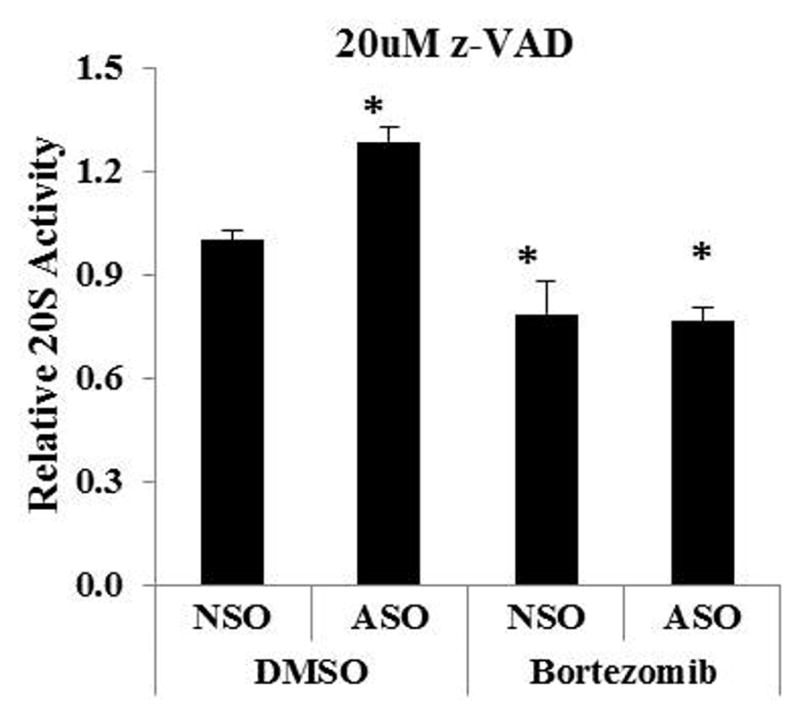
Inhibition of XIAP decreases proteasome activity in a caspase-dependent manner in HL-60 cells. HL-60 cells were pretreated for 1 hour with pan-caspase inhibitor z-VAD, electroporated with XIAP NSO/ASO, and then treated with bortezomib for 24 hours in the presence of z-VAD. The previously-seen reduction in 20S proteasome activity by ASO treatment was abolished, including its enhancement of the effect of bortezomib by the caspase inhibitor, but it had no effect on the inhibition of proteasome activity by bortezomib. Data are the result of 3 experiments. Bars indicate the standard error of the mean. Asterisks indicate significant (p < 0.05) differences from treatment with the NSO control plus DMSO.
Discussion
In this study, we sought to understand the biological function of XIAP in AML cells. We reduced XIAP levels by ASO, and first examined the effects by gene expression profiling. Genes encoding proteasomal components were consistently upregulated by XIAP inhibition, suggesting activation of a proteasome recovery pathway (mediated through transcription factor Nrf1(22) or other regulators) that occurs in mammalian cells following inhibition of proteasome activity. This suspicion was strengthened by the observation that treatment of AML cells with the small-molecule proteasome inhibitor bortezomib or MG132 produced similar upregulation of proteasomal genes, and confirmed by direct measurements of proteasome activity following ASO treatment. Furthermore, the decrease in proteasome activity following XIAP inhibition was caspase-dependent, as expected given the known caspase-inhibitory function of XIAP. We have therefore identified a new function of XIAP in AML cells: maintaining proteasome activity through suppression of caspase activity.
This study was carried out in HL-60 and OCI-AML3 cells. These two cell lines were derived from different patients and both have a complex aberrant karyotype. This newly identified function of XIAP likely applies to other AML cells. However, since AML is molecularly extremely heterogeneous, we cannot rule out the possibility that XIAP may function differently in other AML subtypes with a different genetic background.
Caspase-mediated apoptosis and proteasome system are two regulated proteolysis pathways that play important roles in cell biology and pathological conditions. Activation of caspases by targeting antiapoptotic proteins such as XIAP and inhibition of proteasome degradation by proteasome inhibitors such as bortezomib were explored as therapeutic strategies for cancer treatment. These two pathways also crosstalk and cooperate. The anticancer effect of proteasome inhibition is mediated mainly by activation of apoptosis(23). Activated caspases are substrates for proteasomal degradation and are stabilized by proteasome inhibitors(24). It has been reported that caspases not only attack the 26S proteasome and can cleave 20 of its 33 subunits(24) but also degrade specific subunits of the 19S regulator complex of proteasomes during apoptosis(25, 26), thereby inhibit the proteasomal degradation of substrates and enhance apoptosis. XIAP is a potent cellular caspase inhibitor. Our results that XIAP downregulation promotes caspase-dependent inhibition of proteasome activity in AML cells agree with negative regulation of proteasomal activity by caspases as reported in the literatures. Since these two protein degradation systems negatively regulate each other, there is therapeutic potential for a synergistic combination of proapoptotic drugs and proteasome inhibitors. Indeed, we observed that at 10 nM, bortezomib slightly enhanced cell death induction of XIAP ASO (Figure 3B, 48 hours) although it had no effect on viability of HL-60 cells at this dose by itself.
XIAP is known to activate NF-κB signaling, but the mechanism is not entirely known. Because we found that XIAP inhibition increases levels of IκBα, a principal inhibitor of NF-κB activity, it is plausible that high levels of XIAP, frequently observed in malignant cells, function to maintain high proteasome activity, suppressing IκBα levels and activating the NF-κB pathway. Consistent with this, bortezomib also decreased the mRNA level of XIAP, which is known to be a downstream target of NF-κB, further suggesting that inhibition of XIAP and/or the proteasome can disrupt the feed-forward NF-κB activation frequently found in malignant cells.
In summary, our findings demonstrate an additional function of XIAP in AML cells secondary to its suppression of caspase activity: maintaining the activities of the proteasome and the NF-κB signaling pathway. This provides a potential mechanism for the efficacy of XIAP inhibition in clinical trials of AML. In fact, we have demonstrated that an ASO directed against XIAP induced apoptosis preferentially in AML stem/progenitor cells in a clinical trial(27). NF-κB was reported to be highly active in primitive human AML cells(28). Currently, various SMAC mimetics that target multiple IAPs, including XIAP are entering clinical trials. Our findings provide additional support for the therapeutic targeting of XIAP in AML.
Acknowledgments
We thank Deanna A. Alexander for assisting with manuscript preparation.
This work was supported in part by grants from the National Institutes of Health (P01 CA055164 and MD Anderson’s Cancer Center Support Grant CA016672) and by the Paul and Mary Haas Chair in Genetics (M.A).
Footnotes
Conflict of interest: no conflict of interest.
Authors’ contributions: BZC conceptualized the study, analyzed the data, and wrote the paper, DHM, ZW, WM, and PYM did experiments and edited the paper, MA supported the study and edited the paper, and RED analyzed the data and wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Deveraux QL, Roy N, Stennicke HR, Van AT, Zhou Q, Srinivasula SM, et al. IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J. 1998;17:2215–23. doi: 10.1093/emboj/17.8.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shiozaki EN, Chai J, Rigotti DJ, Riedl SJ, Li P, Srinivasula SM, et al. Mechanism of XIAP-mediated inhibition of caspase-9. Mol Cell. 2003;11(2):519–27. doi: 10.1016/s1097-2765(03)00054-6. Epub 2003/03/07. [DOI] [PubMed] [Google Scholar]
- 3.Deveraux QL, Takahashi R, Salvesen GS, Reed JC. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 1997;388(6639):300–4. doi: 10.1038/40901. [DOI] [PubMed] [Google Scholar]
- 4.Chai J, Shiozaki E, Srinivasula SM, Wu Q, Datta P, Alnemri ES, et al. Structural basis of caspase-7 inhibition by XIAP. Cell. 2001;104(5):769–80. doi: 10.1016/s0092-8674(01)00272-0. Epub 2001/03/21. [DOI] [PubMed] [Google Scholar]
- 5.Riedl SJ, Renatus M, Schwarzenbacher R, Zhou Q, Sun C, Fesik SW, et al. Structural basis for the inhibition of caspase-3 by XIAP. Cell. 2001;104(5):791–800. doi: 10.1016/s0092-8674(01)00274-4. Epub 2001/03/21. [DOI] [PubMed] [Google Scholar]
- 6.Scott FL, Denault JB, Riedl SJ, Shin H, Renatus M, Salvesen GS. XIAP inhibits caspase-3 and -7 using two binding sites: evolutionarily conserved mechanism of IAPs. EMBO J. 2005;24(3):645–55. doi: 10.1038/sj.emboj.7600544. Epub 2005/01/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Resch U, Schichl YM, Sattler S, de Martin R. XIAP regulates intracellular ROS by enhancing antioxidant gene expression. Biochem Biophys Res Commun. 2008;375(1):156–61. doi: 10.1016/j.bbrc.2008.07.142. Epub 2008/08/12. [DOI] [PubMed] [Google Scholar]
- 8.Levkau B, Garton KJ, Ferri N, Kloke K, Nofer JR, Baba HA, et al. xIAP induces cell-cycle arrest and activates nuclear factor-kappaB: new survival pathways disabled by caspase-mediated cleavage during apoptosis of human endothelial cells. Circ Res. 2001;88(3):282–90. doi: 10.1161/01.res.88.3.282. Epub 2001/02/17. [DOI] [PubMed] [Google Scholar]
- 9.Rigaud S, Fondaneche MC, Lambert N, Pasquier B, Mateo V, Soulas P, et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature. 2006;444(7115):110–4. doi: 10.1038/nature05257. Epub 2006/11/03. [DOI] [PubMed] [Google Scholar]
- 10.Olayioye MA, Kaufmann H, Pakusch M, Vaux DL, Lindeman GJ, Visvader JE. XIAP-deficiency leads to delayed lobuloalveolar development in the mammary gland. Cell Death Differ. 2005;12:87–90. doi: 10.1038/sj.cdd.4401524. [DOI] [PubMed] [Google Scholar]
- 11.Birkey Reffey S, Wurthner JU, Parks WT, Roberts AB, Duckett CS. X-linked inhibitor of apoptosis protein functions as a cofactor in transforming growth factor-beta signaling. J Biol Chem. 2001;276(28):26542–9. doi: 10.1074/jbc.M100331200. Epub 2001/05/18. [DOI] [PubMed] [Google Scholar]
- 12.Kaur S, Wang F, Venkatraman M, Arsura M. X-linked inhibitor of apoptosis (XIAP) inhibits c-Jun N-terminal kinase 1 (JNK1) activation by transforming growth factor beta1 (TGF-beta1) through ubiquitin-mediated proteosomal degradation of the TGF-beta1-activated kinase 1 (TAK1) J Biol Chem. 2005;280(46):38599–608. doi: 10.1074/jbc.M505671200. Epub 2005/09/15. [DOI] [PubMed] [Google Scholar]
- 13.Mehrotra S, Languino LR, Raskett CM, Mercurio AM, Dohi T, Altieri DC. IAP regulation of metastasis. Cancer Cell. 2010;17(1):53–64. doi: 10.1016/j.ccr.2009.11.021. Epub 2010/02/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carter BZ, Milella M, Tsao T, McQueen T, Schober WD, Hu W, et al. Regulation and targeting of antiapoptotic XIAP in acute myeloid leukemia. Leukemia. 2003 doi: 10.1038/sj.leu.2403113. [DOI] [PubMed] [Google Scholar]
- 15.Ma W, Wang M, Wang ZQ, Sun L, Graber D, Matthews J, et al. Effect of long-term storage in TRIzol on microarray-based gene expression profiling. Cancer Epidemiol Biomarkers Prev. 2010;19(10):2445–52. doi: 10.1158/1055-9965.EPI-10-0565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: a Pactical and Powerful approach to Multiple testing. JR statist Soc B. 1995;57(1):289–300. [Google Scholar]
- 17.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95(25):14863–8. doi: 10.1073/pnas.95.25.14863. Epub 1998/12/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(43):15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jakt LM, Cao L, Cheah KS, Smith DK. Assessing clusters and motifs from gene expression data. Genome Res. 2001;11(1):112–23. doi: 10.1101/gr.148301. Epub 2001/01/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(−Delta Delta C) method. Methods. 2001;25(4):402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 21.Martin S, Reutelingsperger C, McGahon A, Rader J, van Schie R, LaFace D, et al. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and ABL. J Exp Med. 1995;182:1545–56. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Radhakrishnan SK, Lee CS, Young P, Beskow A, Chan JY, Deshaies RJ. Transcription Factor Nrf1 Mediates the Proteasome Recovery Pathway after Proteasome Inhibition in Mammalian Cells. Molecular Cell. 2010;38(1):17–28. doi: 10.1016/j.molcel.2010.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Orlowski RZ, Kuhn DJ. Proteasome inhibitors in cancer therapy: lessons from the first decade. Clin Cancer Res. 2008;14(6):1649–57. doi: 10.1158/1078-0432.CCR-07-2218. Epub 2008/03/19. [DOI] [PubMed] [Google Scholar]
- 24.Gray DC, Mahrus S, Wells JA. Activation of Specific Apoptotic Caspases with an Engineered Small-Molecule-Activated Protease. Cell. 2010;142(4):637–46. doi: 10.1016/j.cell.2010.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adrain C, Creagh E, Cullen S, Martin S. Caspase-dependent inactivation of proteasome function during programmed cell death in Drosophila and man. J Biol Chem. 2004;279(35):36923–30. doi: 10.1074/jbc.M402638200. Epub 2004/06/24. [DOI] [PubMed] [Google Scholar]
- 26.Sun XM, Butterworth M, MacFarlane M, Dubiel W, Ciechanover A, Cohen GM. Caspase activation inhibits proteasome function during apoptosis. Mol Cell. 2004;14(1):81–93. doi: 10.1016/s1097-2765(04)00156-x. Epub 2004/04/08. [DOI] [PubMed] [Google Scholar]
- 27.Carter BZ, Mak DH, Morris SJ, Borthakur G, Estey E, Byrd AL, et al. XIAP antisense oligonucleotide (AEG35156) achieves target knockdown and induces apoptosis preferentially in CD34+38− cells in a phase 1/2 study of patients with relapsed/refractory AML. Apoptosis: an international journal on programmed cell death. 2011;16(1):67–74. doi: 10.1007/s10495-010-0545-1. Epub 2010/10/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guzman ML, Neering SJ, Upchurch D, Grimes B, Howard DS, Rizzieri DA, et al. Nuclear factor-kappaB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood. 2001;98(8):2301–7. doi: 10.1182/blood.v98.8.2301. [DOI] [PubMed] [Google Scholar]