

Abstract
Burn injury progression has not been well characterized at the cellular level. To define burn injury progression in terms of cell death, histopathologic spatiotemporal relationships of cellular necrosis and apoptosis were investigated in a validated porcine model of vertical burn injury progression. Cell necrosis was identified by High Mobility Group Box 1 protein and apoptosis by Caspase 3a staining of tissue samples taken 1h, 24h and 7 days post-burn. Level of endothelial cell necrosis at 1h was predictive of level of apoptosis at 24h (Pearson's r=0.87) and of level of tissue necrosis at 7 days (Pearson's r=0.87). Furthermore, endothelial cell necrosis was deeper than interstitial cell necrosis at 1h (p<0.001). Endothelial cell necrosis at 1h divided the zone of injury progression (Jackson's zone of stasis) into an upper subzone with necrotic endothelial cells and initially viable adnexal and interstitial cells at 1h that progressed to necrosis by 24h, and a lower zone with initially viable endothelial cells at 1h, but necrosis and apoptosis of all cell types by 24h. Importantly, this spatiotemporal series of events and rapid progression resembles myocardial infarction and stroke, and implicates mechanisms of these injuries, ischemia, ischemia reperfusion, and programmed cell death, in burn progression.
Keywords: Burn Injury Progression, HMGB1, Caspase, Zone of Stasis, Apoptosis
Introduction
Burn injury progression can convert initially partial thickness burns to full-thickness burns that heal slowly, require excision and grafting, and have increased complications like infection, wound contraction, and scarring.1 Despite seventy years of study, the pathobiology of burn injury progression remains unclear and most of what is known is established at the tissue rather than cellular level, hindering development of effective treatments. Pathologic signs of tissue injury progression in burns were first defined by Douglas Jackson.2 He described burns in terms of a central zone of immediate necrosis, an intermediate zone of stasis with diminished blood flow, and a viable surrounding zone of hyperemia. The zone of stasis is initially distinct from the central zone of necrosis, but undergoes necrosis in the days following injury until it can no longer be distinguished. This process has become known as burn injury progression. Jackson further described survival of epithelial adnexae (hair follicles, eccrine and apocrine glands, and their ducts) as the critical factor in early re-epithelialization, an observation recently confirmed in transgenic mice.3 Thus, necrosis of adnexal structures became the hallmark of full-thickness injury. This analytic framework led Jackson to later hypothesize that tissue in the zone of stasis may be salvageable.4 A fundamental challenge in burn pathobiology is to better understand cell death in the zone of stasis so that burn therapies can be developed to target the mechanisms of burn injury progression.
Burns are complex and dynamic injuries involving endothelial damage, vessel occlusion, thrombosis, cytokine release, white blood cell infiltration, apoptosis and progressive necrosis. Recently, Shupp et al. discussed commonly proposed mechanisms of burn injury progression, concluding that many processes may be at work.5 Determining causative pathologic features is challenging since histopathologic signs of apoptosis and necrosis evolve and expression of their associated biomarkers varies over time.6 Histopathologic analysis of burns has long implicated dermal ischemia as the mechanism of burn progression, with both densely packed erythrocytes7 and microthrombi observed in dermal vessels8. Recent work in our laboratory demonstrated strong correlation between endothelial cell necrosis at 1h post-burn and scar contraction at day 28.9It appears that early endothelial necrosis and microvascular occlusion results in ischemia that initiates further tissue injury beyond the initial zone of necrosis. Precise delineation of apoptosis and necrosis in the zone of stasis in a clinically relevant model is required to confirm these relationships.
We previously studied burn injury progression in a porcine horizontal burn injury progression model due to its common use in burn literature and relative ease of immunohistochemical-stain interpretation.6 We found horizontal progression of cellular necrosis across the epidermis between adjacent burns.6 It was clear to us that the next challenge was to build upon these studies in a vertical burn injury progression model that better mimics the important clinical issue of injury progression: burn conversion due to vertical burn progression. Using sequential biopsies of burn sites in pigs, we examined the relationship of endothelial cell necrosis at 1h post-burn to subsequent adnexal and interstitial cell necrosis at 24h, interstitial and adnexal apoptosis at 24h, and ultimate burn depth at 7 days.
Necrosis and apoptosis are distinct processes with specific detectable biomarkers. Necrosis is classically considered passive, but recent investigations have identified active, enzymatically mediated programmed necrosis (necroptosis) in myocardial infarctions and strokes.10,11 Signs of cellular necrosis may be subtle or delayed in routine tissue staining, but immunohistochemical staining for High Mobility Group Box 1 Protein (HMGB1) allows for rapid detection. HMGB1 is a small (∼30kD) nuclear protein that diffuses readily and early into cytoplasm and to extracellular environment during necrosis.12,13 Apoptosis, an active process, can be triggered by several pathways that often ultimately converge on Caspase 3a (CC3a) protein, a major executioner protein of apoptosis that was used to identify apoptosis in this study.14 We recently demonstrated in a horizontal burn injury progression model that immunohistochemical stains for these biomarkers, Caspase 3a and HMGB1, allow extension of past investigations of cutaneous burn injury progression through precise identification of cell death.6 Here we show a vertical progression of epithelial adnexal necrosis and interstitial cell necrosis over a 24h period post-burn and demonstrate that level of endothelial cell necrosis at 1h post-burn predicts level of necrosis of other skin cells by 24h, level of apoptotic cells at 24h, and level of tissue necrosis at day 7.
Materials and Methods
Study Design
This study is an immunohistochemical analysis of burn biopsies that were first presented in “Validation of a Vertical Progression Porcine Burn Model”.9 Furthermore, an additional experiment with two more animals was performed to validate observations reported here using the same animal protocol as reported in that paper.9 The complete animal protocol can be found in the previous paper. All studies were conducted with the approval of the Institutional Animal Care and Use Research Review Board. Tissue blocks were stained using immunohistochemical probes for HMGB1, H&E and CC3a as previously described.6
Experimental Procedure
Cutaneous burns from dorsum of three pigs were evaluated histomorphometrically. Study protocols were approved by Stony Brook University's Institutional Animal Care and Use Research Review Board and were in compliance with National Research Council Guidelines.15 Pigs were anesthetized and given analgesia.One pig had twenty burns inflicted using a hot aluminum bar heated to 70°C, 80°C, or 90°C for 20 or 30s. Two additional pigs each had 20 burns inflicted using the same procedure but with only 70°/30s, 80°/20s, and 80°/30s burn conditions. Two burns were excluded for insufficient burning based on gross assessment, leaving 58 wounds among 3 pigs for evaluation at each time-point. Full-thickness cutaneous biopsies were taken at 1h, 24h, and day 7 post-burning. Animals were euthanized after 28 days.Biopsies were fixed in formalin for 24h, dehydrated in alcohol, xylene-cleared, paraffin-embedded and cut into five-micron sections. Heat antigen retrieval was performed in Trilogy (Cell Marque, Rocklin, CA) at 92°C for 60 minutes. Slides to be stained for HMGB1 were blocked for alkaline phosphatase, while slides to be stained for CC3a were blocked for horseradish peroxidase. Tissue was stained for necrosis with anti-HMGB1 (Abcam 34269, Cambridge MA) incubated overnight at 4°C at a dilution of 1:200, detected with a polymer detection kit (Biocare M4U536L, Concord CA) using vulcan red (Biocare FR8055, Concord, CA). Apoptosis was detected with anti-CC3a antibodies (Cell Signaling Technology 9661, Danvers, MA) incubated overnight at 4°C at a dilution of 1:100 detected with goat anti-rabbit secondary antibodies conjugated to horse radish peroxidase (Vector Labs 4005). Standard Hematoxylin and Eosin slides were also prepared (Poly Scientific, Bay Shore, NY).
Outcome Measurements
Viable dermal thickness was measured from the interface of dermis with subcutaneous fat upwards (from bottom of the dermis to the top). This interface was defined by continuous dermal collagen above and continuous adipose cell layer below. Measurements were made using an Olympus Bx61 conventional microscope (Center Valley, PA) with a calibrated micrometer in one ocular and a calibrated reticle grid in the other ocular. Digital images were taken using an Olympus DP71 12 megapixel camera (Center Valley, PA).
Viable epithelial adnexa were assessed on each biopsy, and recorded as present or absent. Burns with complete necrosis of all adnexal structures were considered full-thickness. For all other parameters three measurements were made per slide. Thickness of viable dermis below the zone of interstitial cell necrosis was measured by ocular microscopy using a 20× field. Distance recorded was from dermal-subcutaneous interface upward to the 20× field where the majority of interstitial cells were necrotic. Level of endothelial cell necrosis was determined by measuring thickness of dermis from dermal-subcutaneous interface upward to the level where at least three quarters of endothelial cells were necrotic as judged by HMGB1 staining patterns.6 Apoptotic cells were identified by positive CC3a staining. Distance from dermal-subcutaneous interface upward to the lowest group of apoptotic cells was recorded as dermal thickness below the apoptotic zone. At day 7, viable dermis was clearly characterized by HMGB1 nuclear staining of dermal cells and was measured from dermal-subcutaneous interface to the lower bound of necrotic tissue where there was complete absence of HMGB1 staining. For measures where cell necrosis or apoptosis extended into subcutaneous tissue, dermal thickness was recorded as zero.
Statistical Analysis
Pearson correlation coefficients were computed between thickness of dermis below level of endothelial cell necrosis at 1h and thickness of dermis below apoptotic zone at 24h, between thickness of dermis below level of endothelial cell necrosis at 1h and thickness of viable dermis at day 7, and between thickness of dermis below apoptotic zone at 24h and thickness of dermis below necrotic dermis at day 7. Paired sign tests and paired, two-tailed Student's T-tests were used to test differences in means between thickness of dermis below the various measures of cellular death to show significant differences between levels of injury for different cell types and at different time points.
Results
Cell Necrosis and Apoptosis in Vertical Progression of Burn Injury
The range of time and temperature conditions used in these experiments created burns from superficial dermal (70°/20s) to immediate full-thickness burns (90°/20s). Most importantly, some burns retained viable epithelial structures at 1h, but progressed to full-thickness injury by 24h.70°/30s, 80°/20s and 80°/30s burns all showed progression, but they differed in frequency and time of conversion to full-thickness injury. Representative burns are shown in Figure 1. Only four of eleven 70°/30s burns converted to full thickness by 24h post-burn, burns created at 80°C for 20s were mostly partial thickness at 1h but nearly all full-thickness by 24h (14/15) and burns made at 80°/30s were mostly full-thickness at 1h (11/17) and all full-thickness by 24h. In 70°/30s burns, significant viable dermis remained at day 7, whereas dermal cells were entirely necrotic in many 80°/20s burns and in all 80°/30s burns.
Figure 1. Level of endothelial cell necrosis at 1h predicts further interstitial cell necrosis, apoptosis, and conversion at 24h and tissue necrosis at day 7.

HMGB1 at 1h (A, B) and 24h (C, D), Caspase 3a at 24h (E, F), and HMGB1 at day 7 (G, H) are shown for representative burns made at 70°/30s, 80°/20s, and 80°/30s at low power and high power. Boxes on 80°/20s burns are shown at high power. Red dots indicate level of endothelial necrosis, with viable endothelial cells below and necrotic cells above the dots, and black dots similarly define the interface of viable and necrotic epithelial adnexae. Green arrow heads bracket zone of apoptotic cells. In 80°/20s burn in panel A, the zone between black and red dots has necrotic endothelial cells and viable epithelial adnexal cells.
The 80°/20 burn in Figure 1 and Figure 2 illustrates progression of a mid-reticular burn to full-thickness dermal necrosis by day 7. This burn had viable adnexae (Figure 1B, 2A&D), but endothelial cell necrosis into deep dermis at 1h (Figure 1A, 2A, 3A). By 24h, no epithelial structures remained viable (Figure 1C&D, 2B&E) and a band of apoptotic cells appeared in the deep dermis (Figure 1E). Apoptotic epithelial cells, detached from an apocrine gland wall, as well as apoptotic interstitial cells were present in the apoptotic band at 24h (Figure 1F). At day 7, complete necrosis of all dermal cells and all epithelial adnexae confirmed that complete conversion to a full-thickness injury had occurred (Figure 1G&H, 2 C&F). The process of conversion is illustrated in Figure 1, and in greater detail in Figure 2. At 1h, viable adnexae were found in the deep dermis, confirmed by HMGB1 staining, but at his level necrotic vessels were evident from cytoplasmic rather than nuclear staining (Figure 1 and Figure 2 A&D, 3A&B). This area is demarcated by black and red dots in Figure 1A, and by blue dots in Figure 2A. At 24h, no viable adnexae (Figure 1 C, D, E, F and Figure 2 B&E) were present in this zone, and necrosis of adnexal structures was evident from necrotic epithelial cells detaching from gland walls and absence of viable nuclei in these structures. At day 7, much of the necrotic dermis was detaching, and all adnexal structures were necrotic, confirming conversion seen at 24h (Figure 1 G&H, Figure 2 C&F).
Figure 2. Conversion from partial to full-thickness of a representative 80°/20s burn in 24h, confirmed at day 7.

A. Low power H&E at 1h reveals viable (Green Arrow) at 1h flanked by necrotic blood vessels(red arrows, high power in Figure 3). The region of dermis with these initially viable adnexae that progress to necrosis by 24h, compromises the vertical component of the zone of stasis as defined by cellular necrosis. B. By24hnecrosis of all adnexae is apparent. C. Full-thickness injury is confirmed by complete necrosis of adnexae, infiltration of granulation tissue, and necrotic, sloughing dermis. D. High power nuclear staining HMGB1 stain of apocrine coil at 1h indicated by black arrow in A proves that these adnexal structures are not necrotic. E. Necrosis of apocrine coil indicated by black arrow in B is evident by absence of viable nuclei on coil wall and detachment of pyknotic epithelial calls. F. Completely necrotic hair follicle labeled with black arrow in C is surrounded by neutrophils and granulation tissue and has no viable epithelial cells.
Figure 3. High magnification reveals necrotic endothelial cells in deep dermal vessels labeled in Figure 2.
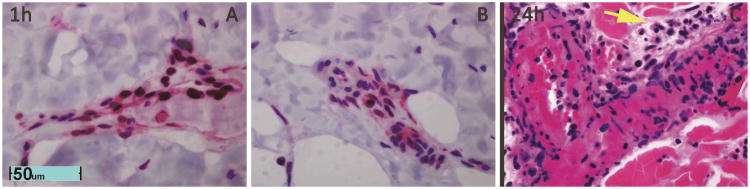
A and B. Loss of nuclear HMGB1 staining of endothelial cell cyptoplams in vessels labeled in zone stasis in Figure 2A indicate necrosis of endothelial cell of deep dermal vessels at 1h. C. By 24h necrosis on H&E pyknotic nuclei and hypereosinophilic vessel walls. Extravasated polymorphonuclear cells and monocytes are also evident (Yellow Arrow).
In most 80°C/30s burns, full-thickness endothelial and adnexal necrosis was evident at 1h (Figure 1A), and in many a band of apoptosis was seen below the dermis at 24h (Figure 1E). Complete dermal necrosis was universally observed at day 7 (Figure 1G). The entire dermis failed to stain for HMGB1 at day 7, and remnants of necrotic apocrine ducts and glands surrounded by granulation tissue extended from subcutaneous fat. Unlike the 80°/20s burn, this 80°/30s burn was clearly full-thickness at 1h post injury. Most 70°/30s at 1h (Figure 1A) demonstrated endothelial injury limited to mid-dermis, viable adnexae below the zone of apoptosis at 24h (Figure 1C and 1E), and viable apocrine glands and neo-epidermis at day 7 (Figure 1G). These wounds illustrate three important types of burn injuries: burns that progress without conversion to full-thickness (70°/30s), burns that progress with conversion to full-thickness (80°/20s), and burns that are initially full-thickness (80°/30s). At 1h, level of endothelial cell necrosis approximated, yet underestimated, level of necrosis at day 7. Conversely, level of apoptosis seemed to slightly overestimate level injury at day 7, with areas with significant apoptosis at 24h confirmed as viable at day 7 (Figures 1 E&G for 70°/20s burn and Figure 4 for an 80°/20s burn).
Figure 4. Zone of apoptosis at 24h approximates final level of injury.

A Caspase 3a staining of a non-progressing 80°/20s burn demonstrates a zone of apoptotic cells in the mid-recticular dermis. Location of the zone aproximates level of inflamatory cell infiltration and granulation tissue at day 7, but extends further. Complete necrosis at day 7 above this level is demonstrated by homogeneous pink staining due to complete absence of nuclei which contrasts sharply with intense blue hematoxlyin staining caused by inflammatory cells and granulation tissue directly below. B. At 24h, the region labeled by the black arrow in figure A is shown at high power, revealing apoptosis of many cells due to nuclear CC3a staining.
Quantification of Progression of Epithelial Adnexal and Interstitial Necrosis
In this porcine vertical burn injury progression model, “mid-dermal” burns may rapidly convert to full-thickness injury as judged by complete necrosis of epithelial adnexae (Figure 5). Most importantly, level of endothelial cell necrosis was predictive of burn conversion (Figure 5). 80°/20s burns had the highest rate of conversion from partial to full-thickness. In addition, progression occurred to and beyond the level of endothelial cell necrosis identified at 1h. 80°/20s burns that converted to full-thickness show a clear progression of interstitial cell necrosis from 1 to 24h also with a mean difference in dermal thickness with viable cells of 0.48 mm (p=.011, Table 1, i.e. interstitial cell necrosis was 0.48 mm deeper at 24h than at 1h). Importantly, endothelial cell necrosis was initially an average of 0.35 mm deeper than interstitial cell necrosis at 1h (p<0.001, Table 1) for the converting 80°/20s burns, but depth of endothelial cell and interstitial cell necrosis were comparable at 24h.
Figure 5. Progression of cell necrosis by HMGB1 in peri-burn tissue from 1 to 24h.

A. At 1h (grey histograms) endothelial cell necrosis is near full-thickness in 80°/20s, 80°/30s, and 90°/20s burns, and relatively more superficial in 70°/20s and 70°/30s burns. B. Level of endothelial cell necrosis predicts full-thickness injury at 24h (black histograms) with nearly all 80°/20s burns and all 80°/30s and 90°/20s ultimately full-thickness. 80°/20s burns were notable for progression from partial thickness to full-thickness between 1h and 24h post-burn judging by necrosis of hair follicles and apocrine glands.
Table 1. Endothelial Cell Necrosis at 1h divides the Zone of Stasis into an upper subzone with complete progression and a lower subzone with partial progression and variable recovery.
| Progressing 80°-20s Burns1 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Viable Dermis Below: | Mean A-B4 | SD5 | # of wounds:6 | p-values A>B7 | ||||
| A | B | (mm) | (mm) | A>B | A<B | Sign Test | T-test | |
| Zone of Stasis (ZOS) ICN1h less ICN24h | ICN2 1h | ICN 24h | 0.48 | 0.30 | 9 | 1 | 0.011 | <0.001 |
| Upper Subzone, ZOS ICN1h less ECN1h | ICN 1h | ECN3 1h | 0.35 | 0.18 | 10 | 0 | < 0.001 | <0.001 |
| Lower Subzone, ZOS ECN1h less A24h3 | ECN 1h | A 24h3 | 0.30 | 0.33 | 6 | 0 | 0.016 | 0.075 |
ICN = interstitial cell necrosis
ECN = endothelial cell necrosis
A = Apoptosis
Mean difference
Standard deviations
Number of wounds where A & B measurable
Statistical significance.
Zone of Apoptosis at 24h
By 24h, a thin zone of apoptosis was discerned in 35 of 58 burns at 24h, extending to or below the boundary of endothelial necrosis defined at 1h within the same burn (p<0.001). Fourteen of 15 70° burns had a clearly discernible zone of apoptosis, while only 21 of 43 of 80° and 90° burns had an identifiable apoptosis zones in the deep dermis or subcutaneous fat. Apoptosis was less apparent in subcutaneous fat due to the much lower density of cells and limited number of cell types compared to the dermis. In 70° burns, 10 /14 burns with discernible zones of apoptosis at 24h showed recovery of much of this zone by day 7. For example, recovery was apparent in the 70°/30s burn shown in Figure 1 andin the partial thickness 80°/20s burn in Figure 4. Thus, burn injury progression did not occur beyond the apoptotic zone, and sometimes a significant portion of it remained viable.
Endothelial Cell Necrosis Predicts Depth of Zone of Apoptosis and Ultimate Viable Dermis
There was excellent (>0.80) positive correlation between depth of endothelial cell necrosis at 1h and depth of dermal cell apoptosis at 24h, and between depth of endothelial cell necrosis at 1h and depth of dermal necrosis at day 7 (Table 2, Figure 6). Importantly, apoptosis and eventual necrosis at day 7 often extended beyond the level of endothelial cell necrosis at 1h. Correlation of depth of the apoptotic zone at 24h with dermal necrosis at day 7 was also excellent (Table 2, Figure 6). Relationships between endothelial cell necrosis at 1h, apoptosis at 24h, and depth of dermal necrosis at day 7 are illustrated in scatter plots in Figure 6. The relationship between endothelial cell necrosis at 1h and dermal necrosis at day 7 also had an excellent correlation. The second scatter plot shows the strong correlation between level of endothelial cell necrosis and apoptosis, but that apoptosis extended deeper into the dermis, with extension of the apoptotic zone into subcutaneous fat seen in 80°/20s, 80°/30s burns and 90°/20s burns. Survival of adnexae below the lower limit of the zone of apoptosis signifies partial-thickness injury, as no progression was observed beyond this level. Importantly, level of apoptosis may overestimate final level of burn depth. The red ellipse in Figure 6 illustrates several wounds where apoptosis extends into the deep dermis yet where ultimately greater than 1mm of viable dermis was evident at day 7.
Table 2. Pearson Correlation Coefficients show 1h endothelial cell necrosis level predicts 24h apoptosis level and day 7 tissue necrosis level, and 24h apoptosis level predicts day 7 tissue necrosis level.
| Thickness of dermis below apoptotic zone at 24h | Thickness of dermis below necrotic tissue at day 7 | |
|---|---|---|
| Thickness of dermis below EC necrosis at 1h | 0.87, N=33 burns | 0.87, N=55 burns |
| Thickness of dermis below apoptotic zone at 24h | -- | 0.82, N=35 burns |
Figure 6. Strong correlation between endothelial cell (EC) necrosis 1h, apoptotic zone (AZ) 24h, and dermal necrosis day 7.
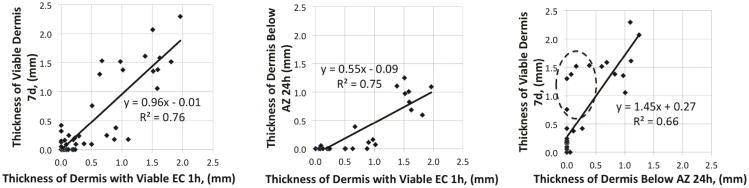
Level of endothelial cell necrosis at 1h post injury is strongly predictive of burn depth (A), and level of apoptosis at 1h, although level apoptosis extends significantly deeper (B). Level of apoptosis is predictive of level of dermal necrosis at day 7, but overestimates it, with several burns with apoptosis to the deep dermis show significant recovery at day 7 (dashed ellipse, C).
Discussion
Expansion of prior work on cell death in burn injury progression
These results identify a discrepancy between levels of endothelial, interstitial, and adnexae necrosis 1h after burns in pigs. Endothelial injury was initially deeper and more predictive of ultimate progression. These findings are hardly surprising given that endothelial cells are critical for perfusion, and when injured release inflammatory mediators and express adhesion molecules that recruit leukocytes,16increase vascular permeability, and promote coagulation. Previously, our lab studied necrosis and apoptosis in the ischemic zone of a porcine hot comb model of horizontal burn injury progression, finding necrosis to be the predominate form of cell death at 24h.17 In a subsequent study, a gradient of necrosis was observed to move across initially viable burn interspace over 24 hours, with an apoptotic zone found deep to necrotic cells at 24h post burn.6 Like the present study, this spatiotemporal pattern of progressive necrosis proximally and apoptosis distally to the initial injured site is similar to ischemic injury of the heart or brain.6 Our prior studies were limited by the burn model used. The hot comb model cannot compare an individual wound across multiple time-points, because the entire wound is consumed by biopsy. Furthermore and more significantly, it cannot model burn injury progression in the vertical dimension which is critical because burn conversion determines necessary treatment and clinical outcome.
To address these shortcomings, in our prior study a new vertical model of burn injury progression was developed that would model conversion of partial to full-thickness burns and allow each burn to be biopsied at every time-point of interest.9 Conversion was demonstrated by wounds with viable dermis evident on H&E staining at 1h and significant contraction at 28d, and endothelial cell necrosis at 1h was predictive of scar area (presumably secondary to wound contraction) at 28d post-burn. Like our previous study,6 apoptosis was observed deep to necrotic dermis at 24h, but was not significantly present at earlier time-points. In converting wounds, and only rare, sporadic apoptotic cells were seen at 48h and 7d.9
Several novel findings emerge extending our previous work. First, it establishes that endothelial cell necrosis is predictive of both level of apoptosis at 24h and final histologic level of injury at day 7. Second, it quantitatively defines spatiotemporal relationships between necrosis and apoptosis of various cells types, finding an endothelial cell necrosis to be initial more extensive than necrosis of interstitial and epithelial adnexal cells. Third, it finds that burn injury progression and conversion occurred within 24 hours following injury under these experimental conditions. This differs from previously published work that suggested extensive progression over several days following injury.18 Finally, previous results from our horizontal hot comb model of burn injury progression were confirmed in a more clinically relevant vertical model of burn injury progression that compares the same wounds across multiple time-points. The predictive power of endothelial cell necrosis and its appearance at levels deeper in the dermis 1h post-burn than necrosis of other cell types fits with an ischemic mechanism of burn injury progression. Extension of necrosis beyond 1h level of endothelial cell necrosis and delayed appearance of apoptosis implicate soluble factors inducing further cell death in ischemic tissue. Both mechanisms are consistent with rapid progression observed in the first 24 hours post-injury found in this study.
HMGB1 and Caspase 3a as biomarkers of cell death
Reliable biomarkers of necrosis and apoptosis are required for precise delineation of cell death. Nuclear loss of HMGB1 is an excellent early (1h – 24h) biomarker of cell necrosis,17,18 and here is shown to be a useful marker of viable at day 7 also. HMGB1 assesses viability of epithelial adnexal structures, determining burn severity. Vimentin is a useful stain for interstitial cell necrosis,19 but it cannot assess necrosis as early as HMGB1 nor can it evaluate epithelial structures, which may explain why burn injury progression was found occurring at later time points using vimentin.19 Caspase 3a identifies cells committed to apoptosis and is more specific for apoptosis than TUNNEL staining, a commonly used alternative.20 Although Caspase 3a has been identified in a number of settings aside from apoptosis,21 in burns its appearance is unlikely to be due to these roles. Nevertheless, no one or two biomarkers can be used to absolutely define apoptosis or any other type of cell death.22 Ultimately, in our opinion, probing each tissue specimen with multiple biomarkers for necrosis, apoptosis and autophagy may be needed to define tissue cell death processes in molecular terms.
Rethinking the Zone of Stasis
HMGB1 and Caspase 3a staining revealed progression of adnexal necrosis and apoptosis in 80°/20s burns over 24h following injury, not unlike Jackson's description of epithelial adnexae in the zone of stasis that eventually undergo necrosis.4 Jackson observed this change histologically at 3 days post injury, and inferred that these structures must have been killed immediately by thermal insult. HMGB1, however, reveals that this is not the case. Adnexae were still viable at 1h in 80°/20s burns, and progressively underwent necrosis over 24 hours following injury, showing that immediate thermal-induced necrosis or apoptosis was not responsible and that these structures may be salvageable.
We used depth of endothelial cell necrosis at 1h post-burn to divide the zone of stasis into distinct upper and lower subzones. The boundary between these subzones was defined by the limit of endothelial cell necrosis at 1h. Directly below this boundary, the lower subzone had seemingly viable endothelial cells as well as interstitial cells and adnexae at 1h, but demonstrated necrosis and apoptosis of all cell types at 24h post-injury. The upper subzone always progressed to complete necrosis while in many cases the lower subzone did not.
Distinct histopathologic findings in each subzone suggest different pathophysiologic processes of injury progression. Progressive necrosis of interstitial and epithelial adnexal cells in the upper subzone may be due to ischemia caused by the necrotic endothelium and subsequent vessel plugging. Both occlusion by densely packed erythrocytes and thrombosis have been proposed as causes of vascular stasis, and recent work by our group confirms both processes occur concurrently in burns.23 Therefore, it appears that necrotic endothelial cells allow erythrocyte aggregation and are a nidus for coagulation leading to ischemia and progressive necrosis of the upper subzone. This theory of burn progression is further supported by efficacy of therapies to reduced ischemia by increasing oxygen supply using anticoagulants,24 plasminogen activators,25 and hyperbaric oxygen therapy26 to limit burn progression in rodent models. However, an important difference between rodent burn studies and our burn studies in swine, is that microthrombi appeared within the first few hours in rodents whereas in porcine peri-burn tissue do not appear until 48 hours after burns in concert with studies by Moritz.27
In contrast, injury of the lower subzone may be a combination of ischemia and soluble factors released from ischemic, necrotic or inflammatory cells that induce either apoptosis or necroptosis, as previously observed in myocardial infarction28and stroke29,30.Although endothelial cells at 1h in the lower subzone were not predominately necrotic, there may be sufficient necrotic cells, sub-lethally injured cells, and dermal edema to reduce perfusion. Cytokines that ordinarily activate pro-survival pathways may induce apoptosis and necroptosis in an ischemic environment.31 Diffusion of cytokines and other soluble factors from the upper subzone, and invading inflammatory cells may then induce apoptosis and necroptosis in potentially viable cells in the lower subzone through cytokine release. Re-endothelialization of denuded vessels reestablishing sufficient perfusion may change the effect of these cytokines from pro-death to pro-survival. Viable endothelial cells have been shown in vivo to migrate to vessels denuded of endothelial cells and reestablish vascular tone and normal blood flow.32 Endothelial cells reaching the lower subzone may limit burn progression in that area. Salvaging the zone of stasis may depend on re-endothelializing blood vessels and reestablishing blood supply before surrounding fibroblasts and adnexal epithelial cells succumb to ischemia or programmed cell death. The role of apoptosis is clearly demonstrated, and we believe that the progression of necrosis seen in burns and similarity of burn progression to myocardial infarctions and strokes as described implicates necroptosis as well. Further work is required to determine the extent that it plays.
Inhibiting cell death will allow more time for ischemia to be corrected, and peptide inhibitors of pro-apoptotic protein c-Jun have been shown to speed burn healing in mice.33Inhibitors of necroptosis have not been used in burns but offer the promise of new therapies. Thus antagonizing these two forms of programmed cell death could potentially limit burn progression for the same reason it might improve outcomes in myocardial infarctions and strokes, by promoting cell survival while blood flow is being reestablished.
Conclusion
The level of endothelial cell necrosis at 1h correlates with level of interstitial and adnexal cell necrosis and apoptosis at 24h, and with depth of ultimate tissue necrosis at day 7 demonstrating a critical role for these cells in burn injury progression. HMGB1 and CC3a, biomarkers of cell necrosis and apoptosis, respectively, identified two subzones within the zone of stasis: an upper subzone characterized by endothelial necrosis in the midst of viable interstitial and epithelial adnexal cells, and a lower subzone characterized by viability of all cell types 1h after injury, but progressing to necrosis and apoptosis in the first 24 hours following injury. Without treatment, the upper zone always progressed to necrosis, while the lower zone progressed in some burns and recovered in others. This pattern of necrosis and apoptosis progressing rapidly over 24 hours suggests that burn progression is mediated by ischemia and programmed cell death. Therapies to reverse dermal ischemia and prevent apoptosis and/or necroptosis could limit burn injury progression and prevent conversion to full-thickness injury. Furthermore, these treatments may be transferable to progressive injury in other pathologic states.
Acknowledgments
The authors commend Joel Israel, Laurie Crawford, Fubao Lin PhD, Marcia Tonnesen MD, Catherine Silverstein, Nicole Steinhauff, Jean Rooney, and Thomas Zimmerman DVM, for their diligence on behalf of the study.
This research was supported by a National Institute of Health RC2 stimulus grant from NIAMS, ARO59384.
Footnotes
Conflicts of Interest: The authors state no conflicts of interest.
References
- 1.Singh V, Devgan L, Bhat S, Milner SM. The pathogenesis of burn wound conversion. Ann Plast Surg. 2007;59(1):109–15. doi: 10.1097/01.sap.0000252065.90759.e6. [DOI] [PubMed] [Google Scholar]
- 2.Jackson DM. Diagnosis of the Depth of Burning. Br J Surg. 1953:588–596. doi: 10.1002/bjs.18004016413. [DOI] [PubMed] [Google Scholar]
- 3.Langton AK, Herrick SE, Headon DJ. An extended epidermal response heals cutaneous wounds in the absence of a hair follicle stem cell contribution. J Invest Dermatol. 2008;128:1311–1318. doi: 10.1038/sj.jid.5701178. [DOI] [PubMed] [Google Scholar]
- 4.Jackson DM. Second Thoughts on the Burn Wound. J Trauma. 1969;9(10):839–62. doi: 10.1097/00005373-196910000-00003. [DOI] [PubMed] [Google Scholar]
- 5.Shupp JW, Nasabzadeh TJ, Rosenthal DS, Jordan MH, Filder P, Jeng JC. A Review of the Local Pathophysiologic Bases of Burn Wound Progression. J Burn Care Res. 2010;31(6):849–73. doi: 10.1097/BCR.0b013e3181f93571. [DOI] [PubMed] [Google Scholar]
- 6.Lanier ST, McClain SA, Lin F, Singer AJ, Clark RAF. Spatiotemporal progression of cell death in the zone of ischemia surrounding burns. Wound Repair Regen. 2011;19(5):622–632. doi: 10.1111/j.1524-475X.2011.00725.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sevitt S. Local Blood-Flow Changes in Experimental Burns. J Pathol Bacteriol. 1949;61(3):427–442. [Google Scholar]
- 8.Boykin JV, Erikson E, Pittman RN. Microcirculation of a scald burn: an in vivo experimental study of the hairless mouse ear. Burns. 1981;7(5):335–338. [Google Scholar]
- 9.Singer AJ, Hirth D, McClain SA, Crawford L, Lin F, Clark RAF. Validation of a Vertical Progression Porcine Burn Model. J Burn Care Res. 2011;32(6):638–46. doi: 10.1097/BCR.0b013e31822dc439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 2008;135:1311–1323. doi: 10.1016/j.cell.2008.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 12.Sun NK, Chao CC. The cytokine activity of HMGB1–extracellular escape of the nuclear protein. Chang Gung Med J. 2005;28:673–82. [PubMed] [Google Scholar]
- 13.Fiuza C, Bustin M, Talwar S, Tropea M, Gerstenberger E, Shelhamer JH, Suffredini AF. Inflammation promoting activity of HMGB1 on human microvascular endothelial cells. Blood. 2003;101:2652–2660. doi: 10.1182/blood-2002-05-1300. [DOI] [PubMed] [Google Scholar]
- 14.Fischer U, Janicke RU, Schulze-Osthoff K. Many cuts to ruin: a comprehensive update of Caspase substrates. Cell Death Differ. 2003;10:76–100. doi: 10.1038/sj.cdd.4401160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.National Research Council. Guide for the Care and Use of Laboratory Animals. Washington DC: National Academy Press; 1996. [Google Scholar]
- 16.Muller WA. Leukocyte-endothelial cell interactions in the inflammatory response. Lab Invest. 2002;82:521. doi: 10.1038/labinvest.3780446. [DOI] [PubMed] [Google Scholar]
- 17.Singer AJ, McClain SA, Taira BR. Apoptosis and necrosis in the ischemic zone adjacent to third degree burns. Acad Emerg Med. 2008;15(6):549–54. doi: 10.1111/j.1553-2712.2008.00115.x. [DOI] [PubMed] [Google Scholar]
- 18.Hirth D, Singer AJ, Clark RAF, McClain SA. Histopathologic Staining of Low Temperature Cutaneous Burns: Comparing Biomarkers of Epithelial and Vascular Injury Reveals Utility of HMGB1 and Hematoxylin Phloxine Saffron. Wound Repair Regen. 2012;20(6):918–27. doi: 10.1111/j.1524-475X.2012.00847.x. [DOI] [PubMed] [Google Scholar]
- 19.Nanney LB, Wenczak BA, Lynch JB. Progressive burn injury documented with vimentin immunostaining. J Burn Care Rehabil. 1996;17(3):191–8. doi: 10.1097/00004630-199605000-00003. [DOI] [PubMed] [Google Scholar]
- 20.Huerta S, Goulet EJ, Huerta-Yepez S, Livingston EH. Screening and detection of apoptosis. J Surg Res. 2007;139(1):143–56. doi: 10.1016/j.jss.2006.07.034. [DOI] [PubMed] [Google Scholar]
- 21.Launay S, Hermine O, Fontenay M, Kroemer G, Solary E, Garrido C. Vital functions for lethal caspases. Oncogene. 2005;24(33):5137–48. doi: 10.1038/sj.onc.1208524. [DOI] [PubMed] [Google Scholar]
- 22.Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. Nomenclature Committee on Cell Death 2009. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16(1):3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou W, Hirth D, McClain SA, Singer AJ, Clark RAF. Vascular Occlusion in Burn Injury: Erythrocyte aggregation or thrombus formation? J Burn Care Res. 2011;32(2)(Supplement):S112. Abstr. [Google Scholar]
- 24.Nisanci M, Eski M, Sahin I, Ilgan S, Isik S. Saving the zone of stasis in burns with activated protein C: an experimental study in rats. Burns. 2010;36(3):397–402. doi: 10.1016/j.burns.2009.06.208. [DOI] [PubMed] [Google Scholar]
- 25.Işik S, Sahin U, Ilgan S, Güler M, Günalp B, Selmanpakoğlu N. Saving the zone of stasis in burns with recombinant tissue-type plasminogen activator (r-tPA): an experimental study in rats. Burns. 1998;24(3):217–23. doi: 10.1016/s0305-4179(97)00116-2. [DOI] [PubMed] [Google Scholar]
- 26.Türkaslan T, Yogun N, Cimşit M, Solakoglu S, Ozdemir C, Ozsoy Z. Is HBOT treatment effective in recovering zone of stasis? An experimental immunohistochemical study. Burns. 2010;36(4):539–44. doi: 10.1016/j.burns.2009.06.210. [DOI] [PubMed] [Google Scholar]
- 27.Moritz AR. Studies of thermal injury III. Amer J Path. 1947;23:915–41. [PMC free article] [PubMed] [Google Scholar]
- 28.Smith CC, Davidson SM, Lim SY, Simpkin JC, Hothersall JS, Yellon DM. Necrostatin: a potentially novel cardioprotective agent? Cardiovasc Drugs Ther. 2007;21:227–233. doi: 10.1007/s10557-007-6035-1. [DOI] [PubMed] [Google Scholar]
- 29.Rosenbaum DM, Degterev A, David J, Rosenbaum PS, Roth S, Grotta JC, et al. Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. J Neurosci Res. 2010;88(7):1569–76. doi: 10.1002/jnr.22314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1(2):112–9. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 31.Christofferson DE, Yuan J. Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol. 2009;22:263–268. doi: 10.1016/j.ceb.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Itoh Y, Toriumi H, Yamada S, Hoshino H, Suzuki N. Resident endothelial cells surrounding damaged arterial endothelium reendothelialize the lesion. Arterioscler Thromb Vasc Biol. 2010;30(9):1725–32. doi: 10.1161/ATVBAHA.110.207365. [DOI] [PubMed] [Google Scholar]
- 33.Giles N, Rea S, Beer T, Wood FM, Fear MW. A peptide inhibitor of c-Jun promotes wound healing in a mouse full-thickness burn model. Wound Repair Regen. 2008;16:58–64. doi: 10.1111/j.1524-475X.2007.00331.x. [DOI] [PubMed] [Google Scholar]