

Abstract
Background
Remogliflozin etabonate (RE) is the prodrug of remogliflozin, a selective inhibitor of the renal sodium-dependent glucose transporter 2 (SGLT2), which could increase urine glucose excretion (UGE) and lower plasma glucose in humans.
Methods
This double-blind, randomized, placebo-controlled, single-dose, dose-escalation, crossover study is the first human trial designed to evaluate safety, tolerability, pharmacokinetics (PK) and pharmacodynamics of RE. All subjects received single oral doses of either RE or placebo separated by approximately 2 week intervals. In Part A, 10 healthy subjects participated in 5 dosing periods where they received RE (20 mg, 50 mg, 150 mg, 500 mg, or 1000 mg) or placebo (4:1 active to placebo ratio per treatment period). In Part B, 6 subjects with type 2 diabetes mellitus (T2DM) participated in 3 dose periods where they received RE (50 mg and 500 mg) or placebo (2:1 active to placebo per treatment period). The study protocol was registered with the NIH clinical trials data base with identifier NCT01571661.
Results
RE was generally well-tolerated; there were no serious adverse events. In both populations, RE was rapidly absorbed and converted to remogliflozin (time to maximum plasma concentration [Cmax;Tmax] approximately 1 h). Generally, exposure to remogliflozin was proportional to the administered dose. RE was rapidly eliminated (mean T½ of ~25 min; mean plasma T½ for remogliflozin was 120 min) and was independent of dose. All subjects showed dose-dependent increases in 24-hour UGE, which plateaued at approximately 200 to 250 mmol glucose with RE doses ≥150 mg. In T2DM subjects, increased plasma glucose following OGTT was attenuated by RE in a drug-dependent fashion, but there were no clear trends in plasma insulin. There were no apparent effects of treatment on plasma or urine electrolytes.
Conclusions
The results support progression of RE as a potential treatment for T2DM.
Trial registration
ClinicalTrials.gov NCT01571661
Keywords: Remogliflozin etabonate, Sodium-dependent glucose transporter 2 inhibitor, Pharmacokinetics, Pharmacodynamics, Type 2 diabetes mellitus
Background
Type 2 diabetes mellitus (T2DM) is characterized by abnormalities of glucose and lipid homeostasis, which drive secondary micro- and macrovascular complications. Clinical evidence indicates that maintaining glycemic control and reducing postprandial glucose excursions can lower the risk of diabetic complications, e.g. reduce the risk of myocardial infarction, renal disease and retinopathy [1,2]. Despite the availability of multiple classes and combinations of antidiabetic agents, the clinical management of T2DM remains challenging, with the majority of patients failing to achieve and maintain target glycemic levels in practice [3]. There is a continued need for novel therapeutic approaches, particularly those with complementary modes of action that will enable further improvement of glycemic control.
Glucose homeostasis is a complex process controlled by gastrointestinal absorption, tissue utilization, hepatic/renal gluconeogenesis and renal filtration/reabsorption/excretion. Under normal physiological conditions when the glomerular filtrate reaches the proximal tubule, glucose is primarily reabsorbed through the active sodium-dependent glucose transporter 2 (SGLT2) located on the apical or luminal membrane of the epithelial cell in the S1 segment [4-6].
SGLT1 is a high-affinity, low-capacity glucose/galactose co-transporter primarily expressed in the intestine and in the kidney [7,8]. In contrast, SGLT2 is a low-affinity, high-capacity glucose transporter selectively expressed in the kidney. Together, SGLT1 and SGLT2 are responsible for the active reabsorption of glucose across the renal luminal membrane [9,10]. Once reabsorbed by the renal epithelial cell, glucose is transported to the blood by facilitated diffusion via the sodium-independent glucose transporter 2 (GLUT-2). The uptake of glucose in the proximal tubules by SGLT1 and SGLT2 is highly efficient, resulting in complete reabsorption of glucose. In humans, genetic alterations in SGLT2 increase renal glucose excretion (up to 200 g/day) with no apparent adverse effects on renal function or carbohydrate metabolism [11].
SGLT2 is currently the focus of interest as a potential therapeutic target for reducing hyperglycemia in T2DM, and several selective SGLT2 inhibitors have been developed [12-16]. In diabetic animal models, pharmacological inhibition of SGLT2 leads to glucosuria, and improvement of plasma glucose levels, followed by a reduction of insulin resistance [17-19].
SGLT2 inhibitors have the potential to offer distinct advantages over currently available diabetic treatments. Because SGLT2 inhibitors work by an insulin-independent mechanism, this class of compounds may be of benefit as adjunctive therapy in patients whose pancreatic function is diminished or in patients who have insulin resistance. Thus, treatment with SGLT2 inhibitors may be appropriate in all stages of T2DM, provided the patient still has adequate renal function to deliver the drug to the site of action in the kidney. Another advantage is that SGLT2 inhibitors cause calorie wasting by loss of glucose in the urine, thus offering the potential for promoting weight loss, whereas some other anti-diabetic treatments such as sulfonylureas and insulin promote weight gain.
Remogliflozin etabonate is the ester prodrug of remogliflozin [20], which is the active entity that selectively inhibits SGLT2. Remogliflozin undergoes further transformation to GSK279782, an active metabolite. The structures of remogliflozin etabonate, remogliflozin and GSK279782 are presented in Figure 1.
Figure 1.
Structures of remogliflozin etabonate, remogliflozin, and GSK279782. Structures of (A) remogliflozin etabonate, (B) remogliflozin and (C) GSK279782).
Remogliflozin etabonate causes a concentration-dependent increase in urinary glucose excretion in mice and rats [20,21]. Unlike earlier SGLT inhibitors, such as phlorizin and T-1095, remogliflozin displays a high level of selectivity for SGLT2 over SGLT1 [22]. This single-dose evaluation was the first study to be conducted with remogliflozin etabonate in humans and was designed to provide safety, tolerability, PK and pharmacodynamic information.
Methods
This single center study was conducted at Profil Institute for Clinical Research (Chula Vista, CA, USA) and was conducted in accordance with Good Clinical Practice and the principles of the Declaration of Helsinki. The study protocol and subject information were reviewed and approved by Biomedical Research Institute of America investigational reviewer board (San Diego, CA, USA) and all subjects provided written, informed consent prior to start of study-related procedures.
Subjects
Ten healthy male and female subjects followed by a separate group of 6 subjects with T2DM were enrolled in this study. Enrollment of women was restricted to those who were postmenopausal or surgically sterile. All subjects gave written informed consent prior to participation in any study-related procedures. Healthy subjects were required to be 18–55 years of age, and have a body mass index (BMI) of 19.0 to 30.0 kg/m2 inclusive. Subjects with T2DM were required to be 30–60 years of age, have a BMI of 22–35 kg/m2, to be healthy other than having been diagnosed with T2DM at least 6 months prior to entry in the study, and to have been maintained on a stable treatment regimen for at least 3 months. Diabetic subjects were required to have hemoglobin A1c (HbA1c) ≤10% and fasting plasma glucose <280 mg/dL at screening. Participants with diabetes were required to be on a stable treatment regimen using a single oral antidiabetic agent (either sulfonylureas, rosiglitazone, metformin or acarbose) or management by diet and exercise. All T2DM subjects also had to be willing and medically able to discontinue their diabetes medications for up to 72 h during each treatment period. Subjects were excluded if they had been taking diuretics, corticosteroids or other medications that might result in electrolyte depletion; had required insulin during the last 3 months; had significant renal disease; or if their participation would have resulted in donation of blood in excess of 550 mL within an 8-week period.
Study design
This double-blind, randomized, placebo-controlled, single escalating-dose crossover study was conducted in two parts: Part A consisted of a randomized, dose-escalation in healthy subjects; Part B was of similar design, but conducted in subjects with T2DM and included an evaluation of pharmacodynamics using a 50 g oral glucose tolerance test (OGTT).
Part A
Ten healthy subjects were evaluated in 5 study sessions, each separated by approximately 2 weeks. At each study session, subjects received either an oral dose of remogliflozin etabonate or placebo after an overnight fast. Remogliflozin etabonate doses were 20 mg, 50 mg, 150 mg, 500 mg and 1000 mg. Over the course of participation in the study, each subject received 4 of the 5 active remogliflozin etabonate doses and 1 dose of placebo (4:1 active to placebo ratio per treatment period). The available safety and PK results from each dosing period were evaluated before proceeding to the next dose level.
Part B
Six subjects with T2DM received two doses of remogliflozin etabonate and a placebo dose, in a randomized, dose escalating, crossover design, along with an oral glucose load on three study sessions separated by 7–14 days. Full PK and safety profiles were measured on Day 1 of each dosing period. The doses selected for this portion of the study, 50 mg and 500 mg, were based on data obtained in Part A. In each dosing period, subjects were assigned to active vs placebo in a 2:1 ratio.
All subjects were admitted to the unit two nights prior to receiving study drug to establish baseline safety parameters and fluid intake levels over a 36 h period. Subjects remained in the unit for at least 24 h after doses were administered for monitoring of clinical laboratory parameters exploratory biomarkers, vital signs, ECGs and adverse events. While confined to the clinical research unit, all subjects received meals standardized with respect to calories, fat, protein, carbohydrate, and sodium content; however, detailed dietary information was not captured in this study. In Part A, subjects were dosed following an overnight fast; lunch and dinner were provided at 4 and 10 h after dosing, respectively. Fifteen minutes after dosing in each treatment period in Part B, a fasting OGTT was performed using 50 g glucose (administered as 50 g Glucola™). A 50 g glucose load was chosen since the OGTT was being performed in subjects already known to have diabetes. The glucose drink was consumed by subjects within approximately 5 minutes. Blood samples for the measurement of glucose, insulin, and intact glucagon-like peptide 1 (GLP-1) were collected for 24 hours following dose administration. Provided that there were no safety or tolerability concerns, subjects were released from the clinic on day 2 of each treatment period until their return for the next treatment or follow-up period. Each subject was involved in the study for approximately 8 weeks (from screening to follow-up).
Pharmacokinetic assessments
Blood collections and analysis
On each dosing day, a series of 2.0 mL blood samples were collected at pre-dose and 10, 20, 30 and 45 min, and 1, 1.25, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 16, and 24 h post-dose for the determination of remogliflozin etabonate, remogliflozin and GSK279782 in plasma by using high-performance liquid chromatography with tandem mass spectrometry (MS/MS) as described [23].
Pharmacokinetic calculations
Non-compartmental PK analysis of plasma concentration–time data was performed using WinNonlin Version 4.1 (Pharsight Corporation, Mountainview, CA, USA). The Cmax and Tmax were obtained directly from the data. Areas under the plasma concentration–time curves from time zero to the last quantifiable time point (AUC[0–last]) and extrapolated to infinity (AUC[0–∞]) were calculated using the log-linear trapezoidal method. The terminal plasma elimination rate-constant (λz) was estimated from log-linear regression analysis of the terminal phase of the plasma concentration–time curve, and the T½ was calculated as T½ = ln2/λz. Ratios of AUC(0–∞) remogliflozin to AUC(0–last) remogliflozin etabonate were calculated including molecular weight corrections.
Pharmacodynamic assessments
Plasma pharmacodynamics
Blood samples for glucose were taken at 0 (pre-dose), and 1, 2, 4, 8 and 12 h after dosing in each treatment period (Part A). For Part B, blood samples for glucose, insulin and GLP-1 were taken at check-in on day -2, and at 0 h (pre-dose), and 0.5, 1, 1.5, 2, 4 h (prior to lunch), 4.5, 5, 6, 8, 10, 12 and 24 h after dosing on day 1 of each treatment period.
Glucose and insulin sample handling
For glucose, plasma was analyzed using a YSI 2300 Glucose Analyzer (Yellow Springs International Life Sciences, Yellow Springs, OH, USA). For insulin, plasma was rapidly prepared and frozen at -70°C until analyzed by LabCorp (San Diego, CA, USA) using a chemiluminescent immunometric assay method (Siemen’s Immulite 2000 analyzer with Immulite Insulin Kit L2KIN2).
GLP-1 sample handling
For assay of intact GLP-1, blood was collected into a chilled EDTA tube and protease inhibitors (DPP4 inhibitor obtained from EMD Millipore, St. Charles, MO) were immediately added. Samples were then spun down and plasma split into two separate tubes and frozen at -70°C until analyzed by Pathway Diagnostics (Malibu, CA, USA) by ELISA (kit # EGLP-35 K, EMD Millipore). The lowest level of intact GLP-1 this assay can detect is 2 pM (with a minimum plasma sample size of 0.4 mL).
Urine pharmacodynamics
Sample collection
Urine samples were collected at pre-dose, and over a series of intervals (0–2, 2–4, 4–6, 6–8, 8–12 and 12–24 h post-dose) for the analysis of creatinine, glucose and electrolytes (Na, K and Cl). All fluid intake was recorded, as well as urine volume, over the 24 hours before and after dosing in each treatment period. Urine was tested for protein on the first morning void of day -1 and 24 h after dosing.
Calculations
Creatinine clearance (CLCR) was calculated as the amount of creatinine excreted in 24 h (Ae0–24 h) divided by the mean of the pre-dose and 24-h post-dose plasma creatinine levels. The percentage of filtered glucose excreted in the urine for each individual time period was calculated as the amount of glucose excreted during that time period divided by (CLCR × PG × time interval length), where CLCR is the creatinine clearance for the time interval, PG is the plasma glucose concentration closest to the midpoint of the time interval, and the time interval is the period (min) of urine collection (CLCR × PG represents the glucose filtered load). For the 24-h period, the percentage of filtered glucose excreted was calculated as the amount of glucose excreted over 24 h divided by the sum over the individual time intervals of (CLCR × PG × time interval length).
Statistical analysis
Safety and pharmacodynamic data were summarized using descriptive statistics. This was a small exploratory study and no formal hypothesis-testing was conducted. Dose proportionality with respect to Cmax, and AUC was assessed using the power model y = α doseβ, where y = Cmax or AUC, and α denotes a random subject effect. The exponent β in the power model will be estimated by regressing the loge-transformed PK parameters on loge dose, i.e. ln(PK parameter) = ln(α) + β * ln(dose). Dose proportionality implied that β = 1 and was assessed by estimating β and its corresponding 90% confidence interval. The power model was fitted by restricted maximum likelihood using SAS Proc Mixed.
Results
Subject demographics
In Part A, 10 subjects (8 males, 2 females, mean age of 39 years, mean BMI of 24.5 mg/kg2, and mean baseline fasting plasma glucose 4.7 mmol/L [range 4.2 to 5.1 mmol/L, SD 0.29 mmol/L]) were randomized; 9 completed the study (1 subject participated in all the study visits but did not return for the follow up visit). In Part B, 6 T2DM subjects (2 males, 4 females, mean age of 53 years, mean BMI of 30.5 mg/kg2, and mean baseline fasting plasma glucose 8.9 mmol/L [range 5.81 to 12.1 mmol/L, SD 2.47 mmol/L]) were randomized and all completed the dosing period.
Safety and tolerability
Remogliflozin etabonate was generally well-tolerated by all subjects. There were no obvious patterns suggesting an effect of remogliflozin etabonate on clinical laboratory results, urine electrolytes, vital signs or ECGs. There were no deaths, serious adverse events or adverse events (AEs) leading to withdrawal. The most frequently reported AE was headache. AEs are summarized in Table 1.
Table 1.
Summary of adverse events
| |
|
Remogliflozin etabonate dose |
|
||||
|---|---|---|---|---|---|---|---|
| Placebo | 20 mg | 50 mg | 150 mg | 500 mg | 1000 mg | Total | |
|
Healthy Subjects |
n = 10 |
n = 8 |
n = 8 |
n = 8 |
n = 8 |
n = 8 |
n = 10 |
| |
n (%) |
n (%) |
n (%) |
n (%) |
n (%) |
n (%) |
n (%) |
| At least one adverse event |
4 (40) |
2 (25) |
3 (38) |
1 (13) |
3 (38) |
1 (13) |
8 (80) |
|
Adverse events reported by >1 subject in total | |||||||
| Headache |
1 (10) |
1 (13) |
1 (13) |
0 |
2 (25) |
1 (13) |
4 (40) |
| Blood creatine phosphokinase increased |
1 (10) |
0 |
0 |
0 |
1 (13) |
0 |
2 (20) |
|
Drug-related adverse events | |||||||
| Headache |
1 (10) |
1 (13) |
1 (13) |
0 |
0 |
1 (13) |
3 (30) |
| Dizziness |
0 |
0 |
0 |
0 |
0 |
1 (13) |
1 (10) |
| Diarrhea |
0 |
1 (13) |
0 |
0 |
0 |
0 |
1 (10) |
| Hot flush |
0 |
0 |
0 |
0 |
0 |
1 (13) |
1 (10) |
|
T2DM subjects |
n = 6 |
|
n = 6 |
|
n = 6 |
|
n = 6 |
|
n (%) |
n (%) |
n (%) |
n (%) |
||||
| At least one adverse event |
3 (50) |
1 (17) |
2 (33) |
4 (67) |
|||
|
Adverse events reported by >1 subject in total | |||||||
| Muscle cramp |
1 (17) |
|
1 (17) |
|
0 |
|
2 (33) |
| β2-microglobulin increased |
0 |
|
0 |
|
2 (33) |
|
2 (33) |
|
Drug-related adverse events | |||||||
| Nausea |
0 |
|
0 |
|
1 (17) |
|
1 (17) |
| β2-microglobulin increased |
0 |
|
0 |
|
1 (17) |
|
1 (17) |
| Pain in extremity | 0 | 0 | 1 (17) | 1 (17) | |||
n = number of subjects reporting event.
% = percentage of subjects reporting event.
Urine beta-2 microglobulin levels, an exploratory biomarker that was measured as a potential early indicator of renal toxicity, were within the normal range at both baseline and after treatment for all subjects except for two subjects with diabetes.
One of these subjects had normal beta-2 microglobulin values at 1 and 4 days after dosing with 500 mg remogliflozin etabonate. However, 11 days after dosing, this subject returned to the clinic for the Day -2 visit of the 3rd treatment period. At this time, the subject’s beta-2 microglobulin levels were elevated to 2.5 μg/mL. The values returned to normal (<0.3 μg/mL) within 2 days. The investigator attributed the elevated pre-placebo levels to other concomitant disease. A second subject, however, did have what was considered by the investigator to be a drug-related elevation of beta-2 microglobulin of 1.62 μg/mL on day 1 after dosing with 500 mg remogliflozin etabonate. The value returned to normal levels within 4 days. No associated changes in serum creatinine and urea or urine microalbumin were observed.
Pharmacokinetics
Healthy subjects
PK parameters are summarized in Table 2 (remogliflozin etabonate), and Table 3 (remogliflozin and GSK279782). Remogliflozin etabonate was rapidly absorbed and extensively hydrolyzed to the active entity in all dose groups. The median Tmax estimates for the prodrug ranged from 0.52 to 1.25 h and median T½ estimates ranged from 0.26 to 0.71 h. The concentration–time profiles are shown for the 50 mg dose in Figure 2 illustrating low circulating plasma concentrations of prodrug relative to active entity.
Table 2.
Summary of plasma remoglifozin etabonate pharmacokinetic parameters in healthy subjectsa,b
| Remogliflozin etabonate dose | 20 mg | 50 mg | 150 mg | 500 mg | 1000 mg |
|---|---|---|---|---|---|
|
AUC(0–∞) (ng•h/mL) |
NQc |
3.70 (46)d |
9.77 (48)d |
36.8 (67)e |
126 (44)f |
|
AUC(0–t) (ng.h/mL) |
1.61 (67) g |
3.56 (56) |
9.51 (42) |
35.4 (62) |
107 (53) |
|
Cmax (ng/mL) |
1.89 (78) |
4.98 (61) |
17.6 (48) |
41.6 (81) |
144 (59) |
|
Tmax (h) |
0.625 |
0.625 |
0.515 |
1.25 |
0.625 |
| 0.33-2.03 |
0.17-1.50 |
0.17-1.50 |
0.33-2.50 |
0.33-2.50 |
|
| T½ (h) | NQc | 0.353 (56)d | 0.256 (35)d | 0.263e (27) | 0.707f (56) |
a. Geometric mean and CV% except for Tmax (median and range).
b. n = 8 unless otherwise specified.
c. NQ, not quantifiable because concentrations at later time points were below the limit of quantification.
d. n = 6.
e. n = 4.
f. n = 7.
g. n = 5.
Table 3.
Summary of plasma remogliflozin and GSK279782 PK parameters in healthy subjectsa
| Remogliflozin etabonate dose | 20 mg | 50 mg | 150 mg | 500 mg | 1000 mg | |||||
|---|---|---|---|---|---|---|---|---|---|---|
|
Analyte |
Rb |
279782 |
Rb |
279782 |
Rb |
279782 |
Rb |
279782 |
Rb |
279782 |
|
AUCc(0-∞) (ng•h/mL) |
|
|
|
|
|
|
|
|
|
|
| Geometric mean (CV%) |
133 (45) |
51.8 (48) |
324 (29) |
145 (48) |
991 (25) |
447 (44) |
3721 (29) |
1523 (40) |
10257 (17) |
3995 (22) |
|
Cmax (ng/mL) |
|
|
|
|
|
|
|
|
|
|
| Geometric mean (CV%) |
61 (54) |
17.5 (72) |
158 (44) |
50.2 (62) |
515 (37) |
155 (50) |
1703 (45) |
498 (44) |
4822 (37) |
1286 (28) |
|
Tmax (h) |
|
|
|
|
|
|
|
|
|
|
| Median |
0.89 |
1.26 |
1.14 |
1.38 |
0.66 |
1.00 |
1.50 |
1.50 |
1.25 |
1.25 |
| (range) |
(0.50–1.5) |
(0.75–2.5) |
(0.50–1.5) |
(1.00–2.0) |
(0.33–2.0) |
(0.75–2.0) |
(0.50–3.0) |
(1.00–4.0) |
(0.50–3.0) |
(0.75–3.0) |
|
T½ (h) |
|
|
|
|
|
|
|
|
|
|
| Geometric mean (CV%) |
1.38 (21) |
1.54 (11) |
1.47 (15) |
2.19 (17) |
1.59 (13) |
2.28(13) |
2.57 (29) |
3.07 (13) |
2.86 (17) |
3.50 (12) |
|
AUC ratio (remogliflozin/remogliflozin etabonate) |
|
|
|
|
|
|
|
|
|
|
| Geometric mean |
84d |
— |
81 |
— |
102 |
— |
105 |
— |
95 |
— |
| CV% | (39) | (34) | (53) | (86) | (69) | |||||
a. n = 8 unless otherwise specified.
b. R = remogliflozin.
c. The median percentage of AUC(0-∞) extrapolated for R was low ranging from 0.07% to 2.1% across all doses and for 279782 ranging from 0.29% to 4.51%.
d. n = 5.
Figure 2.
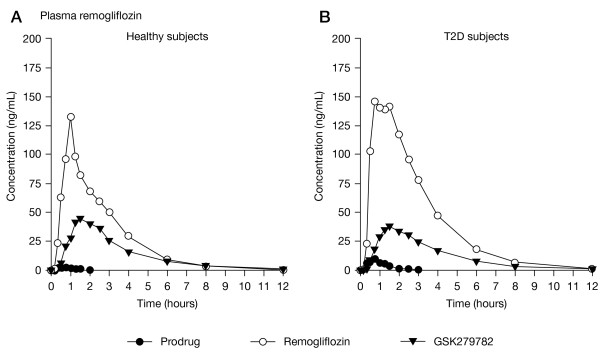
Mean plasma concentration-time profiles for remogliflozin etabonate (prodrug), remogliflozin (active entity), and GSK279782 (metabolite) following a 50 mg dose of remogliflozin etabonate to subjects.
Remogliflozin appeared in plasma at relatively high concentrations within 10 min of remogliflozin etabonate dosing. Mean remogliflozin Cmax of the active entity occurred within 1.5 h of prodrug dosing and was eliminated from plasma more slowly than prodrug. The mean T½ estimates ranged from 1.38 to 2.86 h. The AUC(0-∞) ratios of remogliflozin to prodrug (or AUC(0-t) when AUC(0-∞) not available for prodrug), ranged from 81 to 105 indicating extensive conversion to active entity. The ratios were consistent across all doses.
An active metabolite of remogliflozin, GSK279782, was monitored in this study and found to be present in relatively high concentrations in plasma. An estimated mean AUC(0-∞) ratio of GSK279782 to remogliflozin was 40 to 45%. The median Tmax estimates for GSK279782 ranged from 1.0 to 1.5 h and median T½ estimates ranged from 1.54 to 3.50 h. The concentration profiles of GSK279782 followed a similar time course to those of remogliflozin with only a small delay in appearance of Cmax and slightly longer T½.
The statistical analysis showed AUC(0-∞) and Cmax for all three analytes increased nearly dose proportionally over the 50-fold dose range of 20 to 1000 mg in healthy subjects. For remogliflozin etabonate, the mean slopes and 90% confidence intervals (CI) for AUC(0-∞) and Cmax were 1.17 (1.04, 1.30) and 1.04 (0.94, 1.14). For remogliflozin, the mean slopes and 90% CIs for AUC(0-∞) and Cmax were 1.09 (1.06, 1.12) and 1.08 (1.02, 1.14). For GSK279782, the mean slopes and 90% CIs for AUC(0-∞) and Cmax were 1.08 (1.05, 1.12 and 1.07 (1.01, 1.12).
Type 2 diabetes subjects
The PK parameters for remogliflozin etabonate, remogliflozin, and GSK279782 in subjects with T2DM are shown in Table 4. The prodrug was rapidly absorbed and extensively hydrolyzed to the active entity in patients as well as non-diabetic subjects. There were no discernable differences in mean Tmax or T½ values of remogliflozin or GSK279782 between patients and healthy subjects. The apparent differences in AUCs for remogliflozin and AUC ratios between populations are potentially the result of small sample size and variability rather than differences in absorption or metabolism.
Table 4.
Summary of plasma remogliflozin etabonate, remogliflozin, and GSK279782 PK parameters in T2DM subjectsa
| |
Remogliflozin etabonate |
Remogliflozin |
GSK279782 |
|||
|---|---|---|---|---|---|---|
| Remogliflozin etabonate dose | 50 mg | 500 mg | 50 mg | 500 mg | 50 mg | 500 mg |
|
AUC(0–∞)c(ng•h/mL) |
|
|
|
|
|
|
| Geometric mean (CV%) |
8.91 (58) |
91.9 (46) b |
523 (38) |
5176 (44) |
130 (50) |
1293 (45) |
|
Cmax (ng/mL) |
|
|
|
|
|
|
| Geometric mean (CV%) |
9.56 (39) |
83.9 (89) |
195 (46) |
1891 (49) |
34.6 (39) |
314 (39) |
|
Tmax (h) |
|
|
|
|
|
|
| Median |
0.58 |
0.75 |
1.46 |
2.50 |
1.74 |
2.75 |
| (range) |
(0.33–0.78) |
(0.33–2.50) |
(0.33–2.00) |
(0.33–4.00) |
(0.75–4.00) |
(0.75–4.00) |
|
T½ (h) |
|
|
|
|
|
|
| Geometric mean (CV%) |
0.61 (32) |
0.82 (69) b |
1.59 (27) |
3.93 (25) |
2.05 (24) |
3.28 (23) |
|
AUC(0–∞) ratio |
|
|
|
|
|
|
| (Remogliflozin/remogliflozin etabonate) Geometric mean (CV%) | — | — | 59 (62) | 54 (38) | — | — |
a. n = 6 unless otherwise specified.
b. n = 2.
c. The median percentage of AUC(0-∞) extrapolated for remogliflozin etabonate ranged from 10% to 11% and for remogliflozin was low ranging from 0.14% to 0.42% and for GSK279782 was 0.47% to 2.0%.
Pharmacodynamics
Urine glucose
In both populations, the total amount of glucose excreted in urine from 0–24 h increased in a dose-dependent manner; however, urine glucose excretion increased less than proportionally with increasing doses of remogliflozin etabonate (Table 5), suggesting a plateau of effect. The urine glucose excretion was higher in the T2DM subjects due to higher plasma glucose concentrations. When urine glucose excretion was corrected for circulating plasma glucose concentrations and CLCR (to provide an estimate of percentage filtered glucose load or FGL%), the FGL% was similar between the populations. Figure 3 illustrates the similarity between populations and the saturation of urine glucose excretion with increasing doses. The saturation is related to maximal SGLT2 transporter inhibition. Figure 4 shows the cumulative mean values for the amount of glucose excreted in healthy subjects over time for each dose.
Table 5.
24-h urinary glucose and electrolyte excretion, after single-dose administration of remogliflozin etabonate (20–1000 mg) in healthy volunteers (n = 8 per remogliflozin etabonate group and n = 10 for placebo) and subjects with T2DM (n = 6)
| Parameter | Placebo |
Remogliflozin etabonate dose, mg |
||||
|---|---|---|---|---|---|---|
| 20 | 50 | 150 | 500 | 1000 | ||
|
Healthy subjects | ||||||
| Urinary glucose excretion (mmol) |
6.5 (18.6)a |
67.1 (17.9) |
96.7 (17.1)b |
168 (49.4)b |
223 (49.5) |
304 (137) |
| Filtered glucose excreted in urine (%) |
0.9 (2.4)a |
9.0 (2.2) |
12.7 (3.7)b |
25.5 (7.8)b |
34.2 (5.0) |
26.4 (11.7) |
| Urinary sodium excretion (mmol) |
162 (49.4)a |
148 (64.0) |
212 (67.6)b |
176 (38.7)b |
143 (55.0) |
207 (62.9) |
| Urinary chloride excretion (mmol) |
141 (39.8)a |
136 (56.6) |
189 (51.4)b |
179 (49.6)b |
126 (55) |
201 (61.5) |
| Urinary potassium excretion (mmol) |
62.2 (15.7)a |
66.6 (27.7) |
75.2 (21.3)b |
59.7 (20.3)b |
55.8 (17.2) |
89.1 (24.7) |
|
T2DM subjects | ||||||
| Urinary glucose excretion (mmol) |
40.4 (62.4)c |
|
384 (210) |
|
642 (256) |
|
| Filtered glucose excreted in urine (%) |
2.3 (3.6)c |
|
15.9 (5.9)c |
|
21.6 (9.1) |
|
| Urinary sodium excretion (mmol) |
196 (39.2)c |
|
173 (35.5)c |
|
301 (128) |
|
| Urinary chloride excretion (mmol) |
181 (48.3)c |
|
168 (23.9)c |
|
287 (147) |
|
| Urinary potassium excretion (mmol) | 71.7 (12.1)c | 65.8 (4.5)c | 108 (65.8) | |||
Results are expressed as mean (SD).
a. n = 9.
b. n = 7.
c. n = 5.
Figure 3.

The filtered glucose load (%) vs dose in healthy volunteers (filled circles) and subjects with T2DM (open triangles).
Figure 4.

Mean cumulative 24-h urine glucose excretion following single-dose administration of remogliflozin etabonate (20 to 1000 mg) in healthy subjects.
Urine electrolytes
Urine electrolytes are also summarized in Table 5. Urine excretion of electrolytes was highly variable, and no treatment-related changes were observed. Much of the variability in the 500 mg dose period can be attributed to one subject (#12) whose values for all three electrolytes were roughly 2-fold higher than those of the other participants during that period.
Plasma glucose and insulin
For healthy subjects, AUC values for plasma glucose were very similar between placebo and remogliflozin etabonate periods at both 0–4 h (mean of approximately 18 mmol•h/L) and 0–12 h (mean of approximately 55–59 mmol•h/L) following study drug administration.
For T2DM subjects, the increases in plasma glucose following an OGTT were clearly attenuated between placebo and 50 mg RE; the effect on glucose was altered little by increasing the dose from 50 mg to 500 mg RE. There were no clear trends in plasma insulin following the glucose load. The baseline adjusted AUC(0–4) values for plasma glucose and insulin following an OGTT, are depicted in Figure 5.
Figure 5.
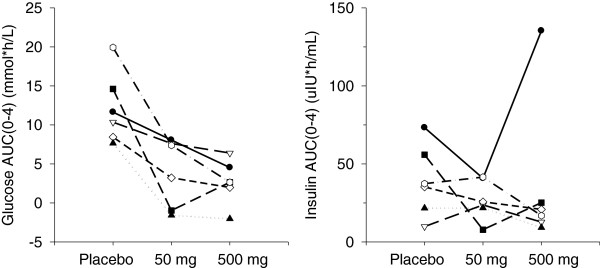
Plasma glucose and insulin AUC0-4 h following glucose challenge in subjects with T2DM.
Plasma intact GLP-1
In T2DM subjects, the median baseline adjusted AUC(0–4) for plasma GLP-1 following an OGTT was 6.36 pM*h (range, 0 to 16.6) for placebo, -8.85 pM*h (range, -46.7 to 33.4) for remogliflozin etabonate 50 mg, and -1.95 pM*h (range, -11.6 to 7.0) for remogliflozin etabonate 500 mg. These data are difficult to interpret because the ranges for placebo, 50 mg, and 500 mg groups contain zero, and also because the analytical method likely did not include extraction of the samples, which can confound the analysis of intact GLP-1 [24,25].
Fluid balance
Fluid balance data for both healthy and T2DM subjects are shown in Table 6.
Table 6.
Summary of fluid balance data (mL)
| |
|
Remogliflozin etabonate dose |
||||
|---|---|---|---|---|---|---|
| Placebo | 20 mg | 50 mg | 150 mg | 500 mg | 1000 mg | |
|
Healthy Subjects |
(n = 10) |
(n = 8) |
(n = 8) |
(n = 8) |
(n = 8) |
(n = 8) |
| -24 h to pre-dose |
-462 (605) |
-460 (1311) |
-338 (525) |
-512 (731) |
-278 (1250) |
-126 (798) |
| 0–12 h post-dose |
163 (668) |
243 (1161) |
-570 (582) |
-86 (909) |
-159 (531) |
-273 (331) |
| 12–24 h post-dose |
-739 (423) |
-581 (387) |
-551 (530) |
-564 (478) |
-424 (334) |
-495(367) |
|
T2DM subjects |
n = 6 |
|
n = 6 |
|
n = 6 |
|
| -24 h to pre-dose |
-312 (457) |
|
-504 (294) |
|
188 (1879) |
|
| 0–12 h post-dose |
569 (400) |
|
-78(433) |
|
-29 (749) |
|
| 12–24 h post-dose | -908 (524) | -803 (658) | -1088(679) | |||
Fluid balance = total fluid intake minus total urine volume.
Results are expressed as mean (SD).
On the day prior to dosing in healthy subjects, fluid balance (total fluid intake minus total urine volume) was negative (mean volumes in the range of -126 to -512 mL). In the 0–12 h interval after dosing, fluid balance shifted to positive for the placebo (+163 mL) and 20 mg remogliflozin etabonate (+242 mL) periods, while the 50 to 1000 mg remogliflozin etabonate treatment periods remained negative. For the 12 to 24 h post-dose interval, all regimens had a fluid balance that was negative. There was no evidence of a clear dose–response.
In T2DM subjects, fluid balance was variable in the 24 h prior to dosing; T2DM subjects assigned to placebo and remogliflozin etabonate 50 mg had a negative fluid balance (mean volumes ranging from -312 mL to -504 mL), while those assigned to remogliflozin etabonate 500 mg had a positive fluid balance (mean volume +188 mL). From 0–12 h after dosing, fluid balance was positive for the placebo regimen and negative for the remogliflozin etabonate 50 mg and 500 mg regimens. For the 12–24 h interval, all regimens had negative fluid balance (mean volumes ranging from -802 mL to -1087 mL). As with the healthy subjects, there was no clear dose–response.
Discussion
By promoting urinary glucose excretion, SGLT2 inhibitors offer a novel mechanism of antidiabetic action that is complementary to currently available classes of drugs which reduce hepatic gluconeogenesis (e.g. metformin), increase glucose flux into muscle and fat (e.g. insulin sensitizers such as thiazolidinediones) or stimulate β-cell insulin secretion (e.g. GLP-1-based therapies).
In both healthy and T2DM subject populations, the prodrug, remogliflozin etabonate was extensively converted to its active entity, remogliflozin, and to an active metabolite, GSK279782, which is as potent as remogliflozin in inhibiting SGLT-2 in vitro [23]. The PK parameters (Cmax and AUC) of all three analytes (remogliflozin etabonate, remogliflozin, and GSK279782) increased nearly dose proportionally over the 50-fold dose range, 20 to 1000 mg, of remogliflozin etabonate in healthy subjects. Although limited data are available, there appeared to be dose proportionality in subjects with T2DM between 50 and 500 mg doses as well. The PK parameters were similar between the healthy subjects and T2DM subjects.
Evidence of the desired pharmacological effect was seen in the dose-dependent increase in urinary glucose excretion in healthy subjects and in T2DM subjects following administration of remogliflozin etabonate. The total amount of glucose excreted in the 24 h after dosing increased less than proportional to the dose and exposure to remogliflozin in healthy subjects. The plateau in glucose excretion was observed between exposures associated with single doses of 150 mg and 500 mg, suggesting maximal inhibition of the SGLT2 transporter at this dose range. Subjects with T2DM showed increased glucose excretion over 24 hours with increasing doses of remogliflozin etabonate, but the percentage of filtered glucose excreted was similar between the two study groups. This amount of urine glucose excretion has been predicted to provide clinically meaningful reduction in plasma glucose [26]. For subjects with T2DM, the increase in plasma glucose following an oral glucose load appeared to be blunted in a drug-dependent fashion following dosing with remogliflozin etabonate, suggesting that SGLT2 inhibition is an appropriate mechanism for glucose lowering in these patients.
Conclusions
This single-dose evaluation is the first clinical study to be conducted with remogliflozin etabonate, and it provides safety, tolerability, pharmacokinetic and pharmacodynamic information in both healthy subjects and those with T2DM. In this study, single oral doses of remogliflozin etabonate (20 mg to 1000 mg for healthy subjects; 50 mg and 500 mg for subjects with T2DM) were generally safe and well-tolerated. Clinically significant fluid and electrolyte imbalances were not seen in this study, but longer repeat dose studies will be required to establish the safety profile. Remogliflozin etabonate increased urinary glucose excretion in a dose dependent fashion and also blunted increases in plasma glucose following an oral glucose load, which suggests that remogliflozin etabonate could be useful as a treatment for T2DM.
Competing interests
AK, ROCS, EKH, RLD, WT, GAS, JWP, CJ, and DN are/were all employees of GlaxoSmithKline at the time of the study; IM is an employee of Kissei Pharmaceutical Company.
Authors’ contributions
AK, ROCS, EKH, RLD, WT, GAS, JWP, CJ, and DN participated in the design of the study, its co-ordination and performed the statistical analysis. IM contributed to study design and the analysis, and MH contributed to study conduct. All authors have been involved in critically revising the drafts of the manuscript and read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Contributor Information
Anita Kapur, Email: anita.x.kapur@gsk.com.
Robin O’Connor-Semmes, Email: robin.l.o.connor-semmes@gsk.com.
Elizabeth K Hussey, Email: elizabeth.k.hussey@gsk.com.
Robert L Dobbins, Email: robert.l.dobbins@gsk.com.
Wenli Tao, Email: wenli.x.tao@gsk.com.
Marcus Hompesch, Email: marcus.hompesch@profilinstitute.com.
Glenn A Smith, Email: glenn.a.smith@gsk.com.
Joseph W Polli, Email: joseph.w.polli@gsk.com.
Charles D James Jr, Email: jamesc3@labcorp.com.
Imao Mikoshiba, Email: imao_mikoshiba@pharm.kissei.co.jp.
Derek J Nunez, Email: derek.j.nunez@gsk.com.
Acknowledgements
The authors thank Laurene Wang-Smith for her assistance with PK analyses.
Editorial assistance in the preparation of this manuscript was provided by Katie Green, International Medical Press, funded by GlaxoSmithKline.
References
- Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med. 2008;359:1577–1589. doi: 10.1056/NEJMoa0806470. [DOI] [PubMed] [Google Scholar]
- UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34) Lancet. 1998;352:854–865. [PubMed] [Google Scholar]
- Giugliano D, Standl E, Vilsboll T, Betteridge J, Bonadonna R, Campbell I, Schernthaner G, Staels B, Trichopoulou A, Farinaro E. Is the current therapeutic armamentarium in diabetes enough to control the epidemic and its consequences? What are the current shortcomings? Acta Diabetol. 2009;46:173–181. doi: 10.1007/s00592-009-0134-3. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Lee WS, You G, Brown D, Hediger MA. The human kidney low affinity Na+/glucose cotransporter SGLT2. Delineation of the major renal reabsorptive mechanism for D-glucose. J Clin Invest. 1994;93:397–404. doi: 10.1172/JCI116972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells RG, Pajor AM, Kanai Y, Turk E, Wright EM, Hediger MA. Cloning of a human kidney cDNA with similarity to the sodium-glucose cotransporter. Am J Physiol. 1992;263:F459–F465. doi: 10.1152/ajprenal.1992.263.3.F459. [DOI] [PubMed] [Google Scholar]
- You G, Lee WS, Barros EJ, Kanai Y, Huo T, Khawaja S, Wells R, Nigam S, Hediger M. Molecular characteristics of Na(+)-coupled glucose transporters in adult and embryonic rat kidney. J Biol Chem. 1995;270:29365–29371. doi: 10.1074/jbc.270.49.29365. [DOI] [PubMed] [Google Scholar]
- Bakris GL, Fonseca VA, Sharma K, Wright EM. Renal sodium-glucose transport: role in diabetes mellitus and potential clinical implications. Kidney Int. 2009;75:1272–1277. doi: 10.1038/ki.2009.87. [DOI] [PubMed] [Google Scholar]
- Nishimura M, Naito S. Tissue-specific mRNA expression profiles of human ATP-binding cassette and solute carrier transporter superfamilies. Drug Metab Pharmacokinet. 2005;20:452–477. doi: 10.2133/dmpk.20.452. [DOI] [PubMed] [Google Scholar]
- Idris I, Donnelly R. Sodium-glucose co-transporter-2 inhibitors: an emerging new class of oral antidiabetic drug. Diabetes Obes Metab. 2009;11:79–88. doi: 10.1111/j.1463-1326.2008.00982.x. [DOI] [PubMed] [Google Scholar]
- Wright EM, Turk E. The sodium/glucose cotransport family SLC5. Pflugers Arch. 2004;447:510–518. doi: 10.1007/s00424-003-1063-6. [DOI] [PubMed] [Google Scholar]
- Santer R, Kinner M, Lassen CL, Schneppenheim R, Eggert P, Bald M, Brodehl J, Daschner M, Ehrich J, Kemper M, Li Volti S, Neuhaus T, Skovby F, Swift P, Schaub J, Klaerke D. Molecular analysis of the SGLT2 gene in patients with renal glucosuria. J Am Soc Nephrol. 2003;14:2873–2882. doi: 10.1097/01.asn.0000092790.89332.d2. [DOI] [PubMed] [Google Scholar]
- Handlon AL. Sodium glucose co-transporter 2 (SGLT2) inhibitors as potential antidiabetic agents. Expert Opin Ther Pat. 2005;13:1531–1540. [Google Scholar]
- Hussey E, Clark R, Amin D, Kipnes M, O’Connor-Semmes R, O’Driscoll E, Leong L, Murray S, Dobbins R, Layko D, Nunez D. The single-dose pharmacokinetics and pharmacodynamics of sergliflozin etabonate, a novel inhibitor of glucose reabsorption, in healthy volunteers and subjects with type 2 diabetes mellitus. J Clin Pharmacol. 2010;50:623–635. doi: 10.1177/0091270009351879. [DOI] [PubMed] [Google Scholar]
- Isaji M. Sodium-glucose cotransporter inhibitors for diabetes. Curr Opin Investig Drugs. 2007;8:285–292. [PubMed] [Google Scholar]
- Komoroski B, Vachharajani N, Feng Y, Li L, Kornhauser D, Pfister M. Dapagliflozin, a novel, selective SGLT2 inhibitor, improved glycemic control over 2 weeks in patients with type 2 diabetes mellitus. Clin Pharmacol Ther. 2009;85:513–519. doi: 10.1038/clpt.2008.250. [DOI] [PubMed] [Google Scholar]
- Komoroski B, Vachharajani N, Boulton D, Kornhauser D, Geraldes M, Li L, Pfister M. Dapagliflozin, a novel SGLT2 inhibitor, induces dose-dependent glucosuria in healthy subjects. Clin Pharmacol Ther. 2009;85:520–526. doi: 10.1038/clpt.2008.251. [DOI] [PubMed] [Google Scholar]
- Asano T, Ogihara T, Katagiri H, Sakoda H, Ono H, Fujishiro M, Anai M, Kurihara H, Uchijima Y. Glucose transporter and Na+/glucose cotransporter as molecular targets of anti-diabetic drugs. Curr Med Chem. 2004;11:2717–2724. doi: 10.2174/0929867043364360. [DOI] [PubMed] [Google Scholar]
- Ehrenkranz JR, Lewis NG, Kahn CR, Roth J. Phlorizin: a review. Diabetes Metab Res Rev. 2005;21:31–38. doi: 10.1002/dmrr.532. [DOI] [PubMed] [Google Scholar]
- Katsuno K, Fujimori Y, Takemura Y. Sergliflozin, a novel selective inhibitor of low-affinity sodium glucose cotransporter (SGLT2), validates the critical role of SGLT2 in renal glucose reabsorption and modulates plasma glucose level. J Pharmacol Exp Ther. 2007;320:323–330. doi: 10.1124/jpet.106.110296. [DOI] [PubMed] [Google Scholar]
- Fujimori Y, Katsuno K, Nakashima I, Ishikawa-Takemura Y, Fujikura H, Isaji M. Remogliflozin etabonate, in a novel category of selective low-affinity sodium glucose cotransporter (SGLT2) inhibitors, exhibits antidiabetic efficacy in rodent models. J Pharmacol Exp Ther. 2008;327:268–276. doi: 10.1124/jpet.108.140210. [DOI] [PubMed] [Google Scholar]
- Harrington WW, Milliken NO, Binz JG. Remogliflozin etabonate, a potent and selective sodium-dependent glucose transporter 2 antagonist, produced sustained metabolic effects in zucker diabetic fatty rats. Diabetes. 2008;58:529-P. [Google Scholar]
- Wright EM, Turk E, Martin MG. Molecular basis for glucose-galactose malabsorption. Cell Biochem Biophys. 2002;36:115–121. doi: 10.1385/CBB:36:2-3:115. [DOI] [PubMed] [Google Scholar]
- Sigafoos J, Bowers G, Castellino S, Culp AG, Wagner DS, Reese JM, Humphreys JE, Hussey EK, O’Connor-Semmes RL, Kapur A, Tao W, Dobbins RL, Polli JW. Assessment of the drug interaction risk for remogliflozin etabonate, a sodium-dependent glucose cotransporter-2 inhibitor: evidence from in vitro, human mass balance, and ketoconazole interaction studies. Drug Metab Dispos. 2012;40:1–13. doi: 10.1124/dmd.112.047258. [DOI] [PubMed] [Google Scholar]
- Yabe D, Watanabe K, Sugawara K, Kuwata H, Kitamoto Y, Sugizaki K, Fujiwara S, Hishizawa M, Hyo T, Kuwabara K, Yokota K, Iwasaki M, Kitatani N, Kurose T, Inagaki N, Seino Y. Comparison of incretin immunoassays with or without plasma extraction: Incretin secretion in Japanese patients with type 2 diabetes. J Diabetes Investig. 2012;3:70–79. doi: 10.1111/j.2040-1124.2011.00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon CF, Holst JJ, Sugawara K. Immunoassays for the incretin hormones GIP and GLP-1. Best Pract Res Clin Endocrinol Metab. 2009;23(4):425–462. doi: 10.1016/j.beem.2009.03.006. ISSN 1521-690X, 10.1016/j.beem.2009.03.006. [ http://www.sciencedirect.com/science/article/pii/S1521690X09000256] [DOI] [PubMed] [Google Scholar]
- O’Connor-Semmes R, Hussey EK, Dobbins RL, Nunez DJ. Simulation and prediction of long-term clinical efficacy in type 2 diabetes resulting from inhibition of the renal sodium-glucose cotransporter 2 (SGLT2) Diabetes. 2008;57(2132-PO):A589. [Google Scholar]