

Abstract
Multiple lines of evidence indicate that regional brain eicosanoid signaling is important in initiation and progression of neurodegenerative conditions that have a neuroinflammatory pathologic component, such as AD. We hypothesized that PGE2 receptor subtype 1 (EP1) signaling (linked to intracellular Ca2+ release) regulates Aβ peptide neurotoxicity and tested this in two complementary in vitro models: a human neuroblastoma cell line (MC65) producing Aβ1-40 through conditional expression of the APP C-terminal portion, and murine primary cortical neuron cultures exposed to Aβ1-42. In MC65 cells, EP1 receptor antagonist SC-51089 reduced Aβ neurotoxicity ~50% without altering high molecular weight Aβ immunoreactive species formation. Inositol-3-phosphate receptor antagonist 2-aminoethoxy-diphenyl borate offered similar protection. SC-51089 largely protected the neuron cultures from synthetic Aβ1-42 neurotoxicity. Nimodipine, a Ca2+ channel blocker, was completely neuroprotective in both models. Based on these data, we conclude that suppressing neuronal EP1 signaling may represent a promising therapeutic approach to ameliorate Aβ peptide neurotoxicity.
INTRODUCTION
Amyloid (A) β peptides are pleiotropic neurotoxins that accumulate in multiple soluble and insoluble forms in Alzheimer’s disease (AD) and are potent stimulators of innate immune response. Multiple lines of evidence, including observational data from large epidemiologic cohorts, autopsy series, cerebrospinal fluid biomarker profiles, and genome-wide association studies, as well as experimental data from multiple in vitro and in vivo models, have highlighted a potentially important role for regional brain innate immune activation and signaling though the eicosanoid products of cyclooxygenase (COX) isozymes in the metabolism of Aβ peptides and in the initiation and progression of AD. (Montine et al. 1999; Lim et al. 2000; Lim et al. 2001; Liang et al. 2005; Morihara et al. 2005; Combrinck et al. 2006; Hoshino et al. 2007). These data have motivated treatment trials in different stages of symptomatic AD and even an AD prevention trial with non-steroidal anti-inflammatory drugs (NSAIDs) that inhibit COX activity; the treatment trials failed and the prevention trial was terminated due to concerns over toxicity that were mostly related to “prothrombotic” events (Aisen et al. 2003; Szekely et al. 2007; Vlad et al. 2008). Despite these setbacks for NSAIDs as a therapeutic approach, the observational and experimental data compel investigation of specific sub-pathways of COX-dependent signaling as a potential avenue for disease modification of AD. Indeed, a current goal is to focus on the potentially therapeutic aspects of COX-dependent signaling while avoiding those that contribute to toxicity (Figure 1).
Figure 1. Inhibiting innate immunity as a therapeutic strategy for neurodegenerative diseases.
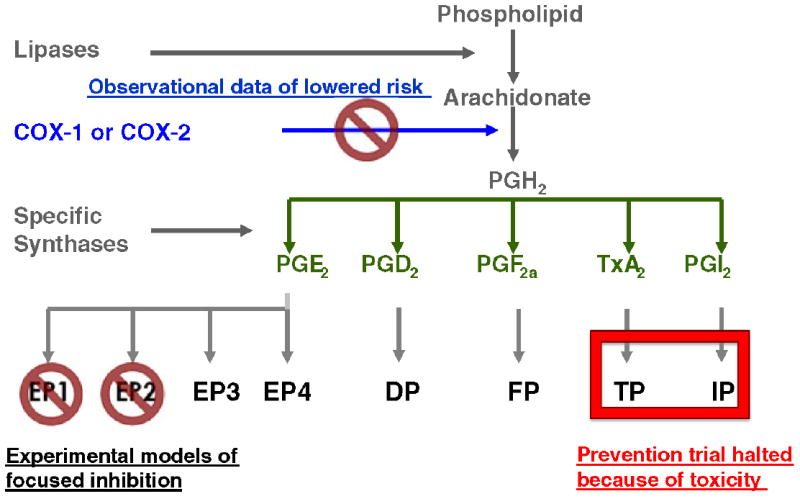
COX-dependent signaling involves a complex cascade that begins with catalysis by COX isozymes (constitutive COX1 and inducible COX2) of free arachidonic acid to PGH2, which serves as the substrate for multiple other enzymes that catalyze the conversion of PGH2 to PGD2, PGE2, PGF2a, PGI2a or thromboxane (Tx) A2. These six eicosanoid products of COX exert biological activity through diverse G protein-couple receptors (Hata et al. 2004).
Importantly, it is likely that the majority of the toxic effects observed with NSAIDs are related to alterations in the concentrations of PGI2 and TxA2 (Montine et al. 2010). We and others have highlighted beneficial effects in pre-clinical models of AD and other neurodegenerative diseases from the selective suppression of signaling through specific receptor subtypes for PGE2 that are called EP1, EP2, EP3, and EP4 (Shie et al. 2005; Shie et al. 2005; Shie et al. 2005; Kawano et al. 2006; Carrasco et al. 2007; Keene et al. 2009).
EP2 signaling is linked to Gs and increased intracellular cAMP and mediates various aspects of innate immune response in brain including neurotoxicity resulting from microglial activation. In addition, EP2 signaling suppresses microglia and macrophage non-Fc-mediated phagocytosis of multiple substrates in culture, including Aβ peptides, and decreases cerebral Aβ accumulation in a mouse model of AD, at least in part, through microglia-mediated mechanisms (Liang et al. 2005; Shie et al. 2005; Nagano et al.). These studies suggest an EP2 antagonist would be an effective therapeutic option for AD, since such a drug would be expected to limit immune-mediated neurotoxicity and enhance Aβ phagocytosis. However, EP2 receptor signaling is also important for synaptic plasticity (Yang et al. 2009) and thus other targets with even more specificity are needed.
EP1 activation is linked to release of intracellular Ca2+. We have shown that EP1 signaling also supports specific aspects of microglial activation that contribute to immune-mediated neurotoxicity (Li et al. 2011). In contrast to EP2 (Shie et al. 2005; Shie et al. 2005), EP1 signaling does not appear to significantly modulate microglial phagocytosis (unpublished data). These data again are encouraging for an EP1 antagonist as a potential approach to modulating microglial activation, but EP1 also is expressed widely on neurons in brain. Others have shown that blockage of EP1 signaling is partially protective in models of cerebral ischemic injury and suppresses Aβ accumulation in a mouse model of cerebral β amyloidogenesis, although it is not clear if these effects derive from suppression of EP1 signaling on neurons, glia, other cells in brain or blood vessels, or some combination (Kawano et al. 2006; Zhen et al. 2011). Here we addressed this gap in our knowledge by determining whether EP1 receptor antagonism mitigates direct neurotoxic effects of Aβ peptides in two different cell culture models.
MATERIALS AND METHODS
Materials
DMEM (4.5g/L D-glucose, L-glutamine), Neurobasal medium, B27 supplement, Sodium Pyruvate, MEM non-essential amino acids (NEAA), Penicillin/Streptomycin, Opti-MEM and Trypsin-EDTA were purchased from Invitrogen (Carlsbad, CA). Fetal Bovine Serum (FBS) was from Hyclone Laboratories (Logan, UT). α-Tocopherol, Nimodipine and Poly-D-Lysine were purchased from Sigma-Aldrich (St. Louis, MO). SC-51089 was from Cayman Chemical Company (Ann Arbor, MI). 2-aminoethoxy-diphenyl borate (2-APB) was from Tocris Bioscience (Ellisville, MO). Tetracycline was from Calbiochem (La Jolla, CA). Papain and DNase I were from Worthington Biochemical (Lakewood, NJ). Aβ1-42 peptide was from Bachem Americas, Inc (Torrance, CA).
Cell culture
Experiments used approximately 10,000 cells/well in 96 well plate for MTT experiments and 300,000 cells/well in 6 well plates for Western blots. MC65 is a human neuroblastoma cell line that conditionally expresses carboxy-terminal fragments of the amyloid precursor protein by withdrawal of tetracycline from the culture medium (Sopher et al. 1994; Woltjer et al. 2003) that was kindly provided by Dr. George Martin, University of Washington. MC65 cells were cultured in DMEM supplemented with sodium pyruvate, MEM non-essential amino acids, penicillin/streptomycin, 10% FBS and 1 μg/ml tetracycline. Primary neuron cultures were prepared from cerebral cortex obtained from postnatal (P1) C57BL/6 mice according to IACUC-approved protocol. After removing meninges, the cortex was incubated for 20 min at 37°C in Neurobasal medium containing 15 U/ml papain, 0.5 mM EDTA and 100 μg/ml DNase I. The mixture was spun at 1500 rpm for 5 min. The pellet was triturated with a flame-polished Pasteur pipette and passed through a 40 micron Nylon strainer. The cell suspension was plated on poly-D-lysine coated plates in culture medium (Neurobasal medium, B27 supplement and 0.5 mM glutamine).
MTT assay
Cells were seeded into 96-well plates for MTT assay. After treatments, culture medium was removed and 60 μl of MTT solution (0.25 mg/ml) was added followed by incubation for 2 h at 37°C. MTT formazan was solubilized by addition of 120 μl acid isopropanol (40 mM hydrocholoric acid in isopropanol), followed by shaking at room temperature for 10 min. The plates were read at 570 nm absorbance and blanks (MTT solution with acid isopropanol in wells with no cells) were subtracted from each well.
Western Blot analysis
Cell lysates were prepared in RIPA buffer. 6 μg of total protein was loaded and separated by 4-12% Bis-Tris gel (Invitrogen) followed by transfer to PVDF membrane. The membrane was then blotted with 6E10 monoclonal antibody for Aβ peptides (Covance, Emeryville, CA). The protein of interest was detected with ECL reagents (Thermo Scientific, Rockford, IL).
Statistical analyses
Data are summarized as mean of measured values or measured values normalized to control ± S.E.M of 5 to 12 separate cultures. Group comparisons using one- or two-way analysis of variance (ANOVA) and Bonferroni-corrected posttests were performed using GraphPad Prism 5 (San Diego, CA). Significance was set as P < 0.05.
RESULTS
We first tested our hypothesis using an immortalized human neuroblastoma cell line (MC65) conditionally expressing the C-terminal 99 amino acids of amyloid precursor protein (Sopher et al. 1994). Figure 2A shows the expected toxicity resulting from generation of C99 following removal of tetracycline suppression, its proteolysis to Aβ40 among other products, with aggregation to form high molecular weight (HMW) species (Figure 2B) (Woltjer et al. 2007). Toxicity in the MC65 cell model is closely associated with of HMW aggregate formation; both can be completely blocked by antioxidants such as α-tocopherol (Woltjer et al. 2007). Ca2+ channel blockers also can suppress MC65 toxicity but without apparent effect on HMW aggregate formation (Anekonda et al. 2011). We have replicated these results with nimodipine (Figures 2B and 3A), an L-type voltage-gated Ca2+ channel blocker.
Figure 2.
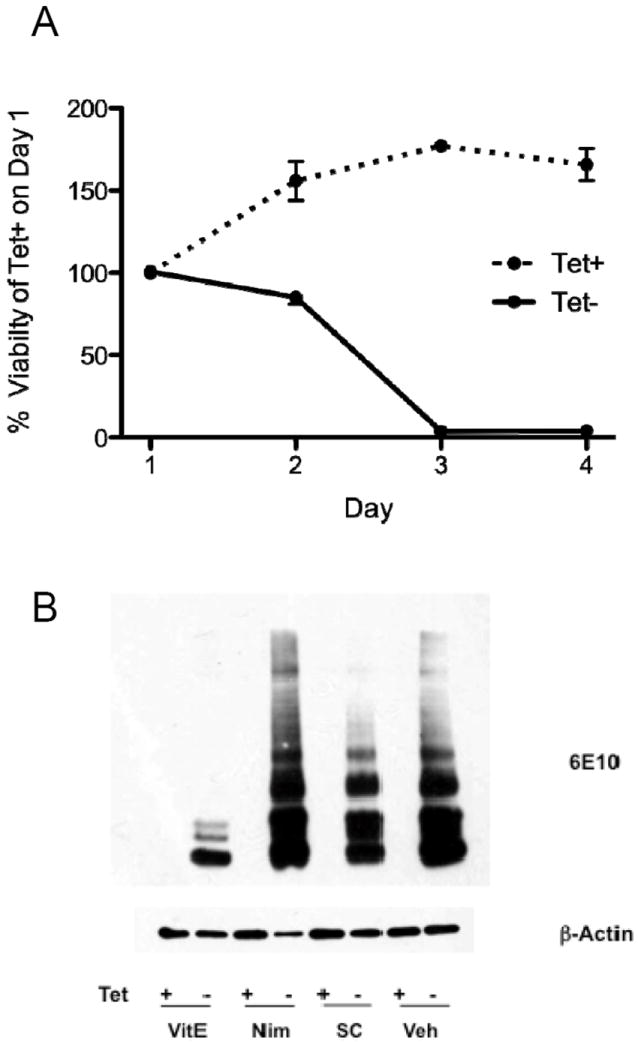
A. MC65 cells were plated on Day 0 in medium with (Tet+) or without (Tet-) tetracycline. Cell viability was assessed with the MTT assay and data presented as % (SEM) of Tet+ cultures on Day 1. Tet+ cultures continued to expand over the next 2 days before reaching a plateau. In contrast, Tet- cultures showed delayed but complete toxicity that developed over Days 1 to 3. B. Cultures of MC65 cells were incubated with (Tet+) or without (Tet-) tetracylcline (1 μg/ml) in the presence of the reagents for 48 h and then harvested in RIPA buffer, proteins separated by SDS-PAGE, and then probed with antibody 6E10 that recognizes an epitope in amino acids 1–16 of Aβ peptides. All Tet+ cultures failed to display any detectable 6E10-immunorective bands. Cultures exposed to drug vehicle (Veh) show the expected multiple species of 6E10-immunoreactive bands. Incubation with α-tocopherol (VitE, 100 μM) suppressed HMW aggregates of 6E10-immunoreative species, as previously described. In contrast, incubation with nimodipine (Nim, 25 μM) or SC-51089 (SC, 50 μM) did not block the formation of HMW species. Western blot for β-actin is loading control.
Figure 3.
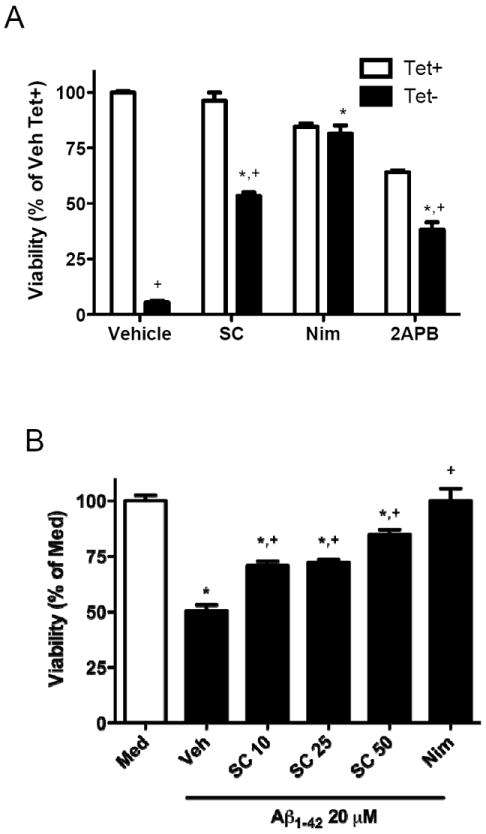
A. MC65 cells were seeded in 96-well plate in mediumwith (Tet+) or without (Tet-) tetracycline in the presence of the inhibitors at Day 0. The cells were cultured for an additional 72 h and then cell viability was assessed with an MTT assay with data presented as % (SEM) of Tet+ cultures with vehicle. Inhibitors were SC-51089 (SC, 20 μM), nimodipine (Nim, 25 μM), or 2-aminoethoxy-diphenyl borate (2APB, 20 μM). Two-way ANOVA had P<0.0001 for Tet+ vs. Tet-, for agents, and for interaction between these two terms. Bonferroni-corrected post-tests Tet- with Vehicle, SC, or 2APB. B. Murine primary cerebral cortical neurons were incubated with fresh medium (Med, clear bar) or medium that contained 20 μMAβ1–42 (filled bars) and the inhibitors for 48 h. Cell viability was assessed with an MTT assay. SC-51089 protected neurons from Aβ1–42 toxicity at all concentrations used (P<0.001 compared to vehicle for all concentrations of SC-51089) with the greatest protection provided at 50 μM; however, protection by SC-51089 was partial and significantly less than nimodipine (P<0.01 for SC-51089 at 10 or 25 μM, and P<0.05 for SC-51089 at 50 μM). One-way ANOVA had P<0.0001 with Bonferroni-corrected multiple paired comparisons having *P<0.001 compared to Med and +P<0.001 compared to Veh
We next determined whether the EP1 antagonist SC-51089 had effects similar as nimodipine. SC-51089 had little impact on the HMW aggregate formation relative to α-tocopherol (Figure 2B). Indeed, the amount of HMW Aβ-immunoreactive bands relative to α-tocopherol increased 7-fold for SC-51089 and 9-fold for nimodipine-exposed cultures. SC-51089 partially protected MC65 cells from cytotoxicity caused by C99 over-expression (Figure 3A). Nimodipine displayed modest toxicity with 84.5 ± 1.5 % viability in Tet+ cultures, but also fully protected from C99-caused toxicity. In contrast, SC-51089 showed no evidence of toxicity to MC65 cells (96.3 ± 4.0 % viability) but afforded only about half protection (53.3 ± 1.6 % viability) from C99-caused toxicity. We assesed the mechanism of SC-51089 by using IP3R inhibitor 2-aminoethoxydiphenyl borate (2APB), which also suppresses intracellular Ca2+ liberation. After normalizing to actin, the relative amount of HMW Aβ-immunoreactive bands in 2APB-exposed cultures was 94% of vehicle-exposed cultures. Incubation with 2APB was toxic with viability reduced to 64.1 ± 0.9 % viability; however, 2APB also protected about half of Tet- cells with 38.2 ± 3.3 % viability. These results validated that suppression of intracellular Ca2+ release can afford approximately 50% protection from the toxic effects of C99 overexpression.
Next, primary murine cerebral cortical neurons were exposed to synthetic Aβ1-42. This model of Aβ-mediated neurotoxicity differs from MC65 cells in two important ways beyond whether or not they are immortalized. First, MC65 cells model intracellular toxicity from C99 and its proteolytic products, including Aβ (Sopher et al. 1994), while primary cerebral cortical neurons exposed to synthetic peptide serves model extracellular Aβ-mediated toxicity. Second, MC65 cells proteolyze C99 to generate Aβ1-40 without detectable Aβ1-42, while the synthetic peptide used with primary neurons was Aβ1-42. Starting material of 20 μM Aβ1-42 was aggregated and then incubated with primary cerebral cortical neurons for 48 hr to yield 50.4 ± 2.7 % viability compared to cultures incubated with medium alone. This well-described neurotoxic effect of extracellular Aβ1-42 has been shown by others to be reduced by Ca2+ channel blockers (Ueda et al. 1997; Fu et al. 2006) such as nimodipine, a result that we corroborated (Figure 3B). Indeed, nimodipine afforded complete protection from extracellular exposure to Aβ1-42. In contrast, SC-51089 partially protected primary cerebral cortical neurons from Aβ1-42. Each concentration of SC-51089 significantly partially protected primary cerebral cortical neurons from Aβ1-42 (P < 0.001 for each concentration) (Figure 3B). Indeed, viability with SC-51089 at 10 (70.8 ± 2.1 %) or 20 (72.1 ± 1.3 %) μM remained significantly less than medium controls (P < 0.001 for all), was not significantly different, and was significantly lower than cultures exposed to nimodidine (P < 0.01 for both). Higher concentration of SC-51089 improved neuroprotection to 84.8 ± 2.3 % viability, only marginally less than nimodipine (P < 0.05).
DISCUSSION
Exposure of cultured neuroblastoma cells (Bate et al. 2003; Hanson et al. 2003; Jang et al. 2005), glia (Szaingurten-Solodkin et al. 2009 ; Blanco et al.), and primary murine cortical neurons (Bate et al. 2003; Bate et al. 2008) to Aβ peptides increases expression of enzymes necessary for production of PGE2 as well as secretion of PGE2. Aβ sterotaxic injection increases murine hippocampal COX2 expression and PGE2 production (Dargahi et al.), and hippocampal slice cultures from Ab-accumulating transgenic mice also show increased COX2 expression and PGE2 secretion (Quadros et al. 2003). Moreover, cerebrospinal fluid from patients with AD has increased levels of PGE2 (Montine et al. 1999). These data have led to an investigation of a potential role for PGE2 receptor subtype signaling in the pathogenesis of AD. Here we demonstrated that intracellular and extracellular Aβ-mediated neurotoxicity in vitro was at least partially EP1 receptor-dependent and that this pathway is conserved between mice and human cells. Indeed, these two cell culture models are so different, especially since one is an immortalized neuroblastoma cell line and the other primary neurons, that it is best to view them as complementary rather than validating. Using these models, we demonstrated partial efficacy of EP1 antagonism on both intracellular Aβ1-40 aggregate cytotoxicity, and extracellular Aβ1-42 neurotoxicity. Thus, while previous studies demonstrated beneficial effects in MC65 cells through α-tocopherol–mediated inhibition of Aβ aggregation (Woltjer et al. 2007), the current study revealed an alternative, aggregation-independent mechanism of protection related to EP1 signaling. While aggregation state alters the neurotoxicity of Aβ species in vitro (Resende et al. 2008), our data from MC65 cells suggest that EP1-dependent mechanisms of neuroprotection likely are distal to aggregation of Aβ peptides.
AD is a syndrome. Increased production of Aβ peptides, and perhaps other C terminal fragments of APP, appears to be sufficient to replicate the full spectrum of illness in individuals who inherit disease-causing autosomal dominant mutations. In some respects both the MC65 cell line used here and transgenic mice with cerebral β amyloidogenesis model this form of AD by using Aβ over-production to drive neuron cell stress, damage, and death. Thus, MC65 cells represent a crude model of the cellular consequences Aβ over-production. However, the more common form of AD, indeed the looming public health disaster that is AD, has accumulation of Aβ peptides as a necessary but perhaps not sufficient feature. It is less clear that “sporadic” AD is caused by Aβ over-production; in fact, recent evidence suggests that decreased clearance of Aβ is a more likely mechanism (Mawuenyega et al. 2010). From this perspective, addition of Aβ1-42 to cultures of primary cortical neurons is a crude model of over-accumulation rather than over-production of Aβ1-42. Our results from murine primary cortical neurons also suggest that EP1-dependent mechanisms are a component of the neurotoxic cascade following extracellular exposure to Aβ1-42.
NSAIDs block COX activity and thereby suppress both the cytotoxic and beneficial aspects of PG and TxA2 signaling. While extensive observation and epidemiologic literature support NSAIDs as potential therapeutics for preventing clinical expression of AD, NSAIDs appear to be too toxic to be used in this context (Szekely et al. 2007; Vlad et al. 2008). To circumvent this toxicity, we and several other groups have targeted specific PG pathways or eicosanoid receptors, including EP1, to retain therapeutic benefits while limiting untoward effects.
EP1 is one of a family of four functionally antagonistic G protein coupled receptors that are activated by PGE2 (Sugimoto et al. 2007). EP1 is noteworthy among PGE2 receptor subtypes in that it has the highest Kd for PGE2, suggesting that it will be the last to be activated in an environment of increasing PGE2 concentration. In this regard, EP1 may be an especially appealing therapeutic target because, at least in theory, the other functions of PGE2 that are mediated by EP2, EP3, and EP4 would be unaltered by an antagonist of EP1.
Not only does EP1 have the highest Kd for PGE2, but it also is unique among PGE2 receptor subtypes in that its activation is linked to liberation of intracellular Ca2+. Although not directly demonstrated by our experiments, we speculate that suppression of EP1 signaling was partially neuroprotective by limiting the rise in intracellular Ca2+. Indeed, previous work in MC65 cells and primary neuron cultures have shown that L-type voltage-gated Ca2+ blockers, such as nimodipine, can fully protect neurons from the toxic effects of Aβ peptides (Ueda et al. 1997; Fu et al. 2006; Anekonda et al. 2011). We replicated these results with nimodipine in our two cell culture models. Interestingly, EP1 provided partial protection in these same models, suggesting that PGE2-mediated activation of EP1 is a component of neurotoxicity from Aβ. These results provide a potential mechanistic explanation of earlier experiments that demonstrated neuroprotection from Aβ by inhibitors of COX.
While Aβ peptide neurotoxicity appears to be a key element in all forms of AD, a large body of observational and experimental data have identified pro-inflammatory and pro-oxidative states in diseased regions of brain in AD (Keene et al. 2011). Indeed, these three events appear to be linked mechanistically, as Aβ peptides can activate an innate immune response from glia, and innate immune response can promote production of oxidative metabolites and alter APP production and metabolism. We have shown previously that suppression of EP1 signaling in glia, both by genetic ablation and use of antagonists, suppresses the neurotoxic phenotype in microglia by selectively reducing secretion of cytokines IL-6 and TNF-α following innate immune activation (Li et al. 2011). In this regard, interpretation of recent results that show reduced Aβ peptide accumulation in EP1 deficient mice is somewhat limited because this outcome can be achieved by suppressing neuron production or metabolism, or glial immune activation (Zhen et al. 2011). Regardless, these cell culture and in vivo studies underscore multiple potentially beneficial effects of EP1 antagonists in models of AD, and underscore the possibility of EP1 antagonists as therapeutic agents for AD.
Acknowledgments
This work was supported by ES016754, AG005136, NS062684 and the Nancy and Buster Alvord Endowment. We thank Ms. Amy Look for administrative support and Ms. Carol Arnold for managerial support.
Footnotes
The authors declare that they have no conflict of interest.
References
- Aisen PS, Schafer KA, Grundman M, Pfeiffer E, Sano M, Davis KL, Farlow MR, Jin S, Thomas RG, Thal LJ. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA. 2003;289:2819–26. doi: 10.1001/jama.289.21.2819. [DOI] [PubMed] [Google Scholar]
- Anekonda TS, Quinn JF, Harris C, Frahler K, Wadsworth TL, Woltjer RL. L-type voltage-gated calcium channel blockade with isradipine as a therapeutic strategy for Alzheimer’s disease. Neurobiol Dis. 2011;41:62–70. doi: 10.1016/j.nbd.2010.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bate C, Marshall V, Colombo L, Diomede L, Salmona M, Williams A. Docosahexaenoic and eicosapentaenoic acids increase neuronal death in response to HuPrP82-146 and Abeta 1-42. Neuropharmacology. 2008;54:934–43. doi: 10.1016/j.neuropharm.2008.02.003. [DOI] [PubMed] [Google Scholar]
- Bate C, Veerhuis R, Eikelenboom P, Williams A. Neurones treated with cyclo-oxygenase-1 inhibitors are resistant to amyloid-beta1-42. Neuroreport. 2003;14:2099–103. doi: 10.1097/00001756-200311140-00018. [DOI] [PubMed] [Google Scholar]
- Blanco A, Alvarez S, Fresno M, Munoz-Fernandez MA. Amyloid-beta induces cyclooxygenase-2 and PGE2 release in human astrocytes in NF-kappa B dependent manner. J Alzheimers Dis. 2010;22:493–505. doi: 10.3233/JAD-2010-100309. [DOI] [PubMed] [Google Scholar]
- Carrasco E, Casper D, Werner P. PGE(2) receptor EP1 renders dopaminergic neurons selectively vulnerable to low-level oxidative stress and direct PGE(2) neurotoxicity. J Neurosci Res. 2007;85:3109–17. doi: 10.1002/jnr.21425. [DOI] [PubMed] [Google Scholar]
- Combrinck M, Williams J, De Berardinis MA, Warden D, Puopolo M, Smith AD, Minghetti L. Levels of CSF prostaglandin E2, cognitive decline, and survival in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2006;77:85–8. doi: 10.1136/jnnp.2005.063131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dargahi L, Nasiraei-Moghadam S, Abdi A, Khalaj L, Moradi F, Ahmadiani A. Cyclooxygenase (COX)-1 activity precedes the COX-2 induction in Abeta-induced neuroinflammation. J Mol Neurosci. 2011;45:10–21. doi: 10.1007/s12031-010-9401-6. [DOI] [PubMed] [Google Scholar]
- Fu H, Li W, Lao Y, Luo J, Lee NT, Kan KK, Tsang HW, Tsim KW, Pang Y, Li Z, Chang DC, Li M, Han Y. Bis(7)-tacrine attenuates beta amyloid-induced neuronal apoptosis by regulating L-type calcium channels. J Neurochem. 2006;98:1400–10. doi: 10.1111/j.1471-4159.2006.03960.x. [DOI] [PubMed] [Google Scholar]
- Hanson AJ, Prasad JE, Nahreini P, Andreatta C, Kumar B, Yan XD, Prasad KN. Overexpression of amyloid precursor protein is associated with degeneration, decreased viability, and increased damage caused by neurotoxins (prostaglandins A1 and E2, hydrogen peroxide, and nitric oxide) in differentiated neuroblastoma cells. J Neurosci Res. 2003;74:148–59. doi: 10.1002/jnr.10726. [DOI] [PubMed] [Google Scholar]
- Hata AN, Breyer RM. Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol Ther. 2004;103:147–66. doi: 10.1016/j.pharmthera.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Hoshino T, Nakaya T, Homan T, Tanaka K, Sugimoto Y, Araki W, Narita M, Narumiya S, Suzuki T, Mizushima T. Involvement of prostaglandin E2 in production of amyloid-beta peptides both in vitro and in vivo. J Biol Chem. 2007;282:32676–88. doi: 10.1074/jbc.M703087200. [DOI] [PubMed] [Google Scholar]
- Jang JH, Surh YJ. Beta-amyloid-induced apoptosis is associated with cyclooxygenase-2 up-regulation via the mitogen-activated protein kinase-NF-kappaB signaling pathway. Free Radic Biol Med. 2005;38:1604–13. doi: 10.1016/j.freeradbiomed.2005.02.023. [DOI] [PubMed] [Google Scholar]
- Kawano T, Anrather J, Zhou P, Park L, Wang G, Frys KA, Kunz A, Cho S, Orio M, Iadecola C. Prostaglandin E2 EP1 receptors: downstream effectors of COX-2 neurotoxicity. Nat Med. 2006;12:225–9. doi: 10.1038/nm1362. [DOI] [PubMed] [Google Scholar]
- Keene CD, Chang R, Stephen C, Nivison M, Nutt SE, Look A, Breyer RM, Horner PJ, Hevner R, Montine TJ. Protection of hippocampal neurogenesis from toll-like receptor 4-dependent innate immune activation by ablation of prostaglandin E2 receptor subtype EP1 or EP2. Am J Pathol. 2009;174:2300–9. doi: 10.2353/ajpath.2009.081153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keene CD, Cudaback E, Li X, Montine KS, Montine TJ. Apolipoprotein E isoforms and regulation of the innate immune response in brain of patients with Alzheimer’s disease. Curr Opin Neurobiol. 2011 doi: 10.1016/j.conb.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Cudaback E, Keene CD, Breyer RM, Montine TJ. Suppressed microglial E prostanoid receptor 1 signaling selectively reduces tumor necrosis factor alpha and interleukin 6 secretion from toll-like receptor 3 activation. Glia. 2011;59:569–76. doi: 10.1002/glia.21125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Wang Q, Hand T, Wu L, Breyer RM, Montine TJ, Andreasson K. Deletion of the prostaglandin E2 EP2 receptor reduces oxidative damage and amyloid burden in a model of Alzheimer’s disease. J Neurosci. 2005;25:10180–7. doi: 10.1523/JNEUROSCI.3591-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, Tran T, Ubeda O, Ashe KH, Frautschy SA, Cole GM. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer’s disease. J Neurosci. 2000;20:5709–14. doi: 10.1523/JNEUROSCI.20-15-05709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim GP, Yang F, Chu T, Gahtan E, Ubeda O, Beech W, Overmier JB, Hsiao-Ashec K, Frautschy SA, Cole GM. Ibuprofen effects on Alzheimer pathology and open field activity in APPsw transgenic mice. Neurobiol Aging. 2001;22:983–91. doi: 10.1016/s0197-4580(01)00299-8. [DOI] [PubMed] [Google Scholar]
- Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science. 2010;330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montine TJ, Sidell KR, Crews BC, Markesbery WR, Marnett LJ, Roberts LJ, 2nd, Morrow JD. Elevated CSF prostaglandin E2 levels in patients with probable AD. Neurology. 1999;53:1495–8. doi: 10.1212/wnl.53.7.1495. [DOI] [PubMed] [Google Scholar]
- Montine TJ, Sonnen JA, Milne G, Baker LD, Breitner JC. Elevated ratio of urinary metabolites of thromboxane and prostacyclin is associated with adverse cardiovascular events in ADAPT. PLoS One. 2010;5:e9340. doi: 10.1371/journal.pone.0009340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morihara T, Teter B, Yang F, Lim GP, Boudinot S, Boudinot FD, Frautschy SA, Cole GM. Ibuprofen suppresses interleukin-1beta induction of pro-amyloidogenic alpha1-antichymotrypsin to ameliorate beta-amyloid (Abeta) pathology in Alzheimer’s models. Neuropsychopharmacology. 2005;30:1111–20. doi: 10.1038/sj.npp.1300668. [DOI] [PubMed] [Google Scholar]
- Nagano T, Kimura SH, Takemura M. Prostaglandin E2 reduces amyloid beta-induced phagocytosis in cultured rat microglia. Brain Res. 2010;1323:11–7. doi: 10.1016/j.brainres.2010.01.086. [DOI] [PubMed] [Google Scholar]
- Quadros A, Patel N, Crescentini R, Crawford F, Paris D, Mullan M. Increased TNFalpha production and Cox-2 activity in organotypic brain slice cultures from APPsw transgenic mice. Neurosci Lett. 2003;353:66–8. doi: 10.1016/j.neulet.2003.08.076. [DOI] [PubMed] [Google Scholar]
- Resende R, Ferreiro E, Pereira C, Resende de Oliveira C. Neurotoxic effect of oligomeric and fibrillar species of amyloid-beta peptide 1-42: involvement of endoplasmic reticulum calcium release in oligomer-induced cell death. Neuroscience. 2008;155:725–37. doi: 10.1016/j.neuroscience.2008.06.036. [DOI] [PubMed] [Google Scholar]
- Shie FS, Breyer RM, Montine TJ. Microglia lacking E Prostanoid Receptor subtype 2 have enhanced Abeta phagocytosis yet lack Abeta-activated neurotoxicity. Am J Pathol. 2005;166:1163–72. doi: 10.1016/s0002-9440(10)62336-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shie FS, Montine KS, Breyer RM, Montine TJ. Microglial EP2 as a new target to increase amyloid beta phagocytosis and decrease amyloid beta-induced damage to neurons. Brain Pathol. 2005;15:134–8. doi: 10.1111/j.1750-3639.2005.tb00509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shie FS, Montine KS, Breyer RM, Montine TJ. Microglial EP2 is critical to neurotoxicity from activated cerebral innate immunity. Glia. 2005;52:70–7. doi: 10.1002/glia.20220. [DOI] [PubMed] [Google Scholar]
- Sopher BL, Fukuchi K, Smith AC, Leppig KA, Furlong CE, Martin GM. Cytotoxicity mediated by conditional expression of a carboxyl-terminal derivative of the beta-amyloid precursor protein. Brain Res Mol Brain Res. 1994;26:207–17. doi: 10.1016/0169-328x(94)90092-2. [DOI] [PubMed] [Google Scholar]
- Sugimoto Y, Narumiya S. Prostaglandin E receptors. J Biol Chem. 2007;282:11613–7. doi: 10.1074/jbc.R600038200. [DOI] [PubMed] [Google Scholar]
- Szaingurten-Solodkin I, Hadad N, Levy R. Regulatory role of cytosolic phospholipase A2alpha in NADPH oxidase activity and in inducible nitric oxide synthase induction by aggregated Abeta1-42 in microglia. Glia. 2009;57:1727–40. doi: 10.1002/glia.20886. [DOI] [PubMed] [Google Scholar]
- Szekely CA, Breitner JC, Zandi PP. Prevention of Alzheimer’s disease. Int Rev Psychiatry. 2007;19:693–706. doi: 10.1080/09540260701797944. [DOI] [PubMed] [Google Scholar]
- Ueda K, Shinohara S, Yagami T, Asakura K, Kawasaki K. Amyloid beta protein potentiates Ca2+ influx through L-type voltage-sensitive Ca2+ channels: a possible involvement of free radicals. J Neurochem. 1997;68:265–71. doi: 10.1046/j.1471-4159.1997.68010265.x. [DOI] [PubMed] [Google Scholar]
- Vlad SC, Miller DR, Kowall NW, Felson DT. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology. 2008;70:1672–7. doi: 10.1212/01.wnl.0000311269.57716.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woltjer RL, Maezawa I, Ou JJ, Montine KS, Montine TJ. Advanced glycation endproduct precursor alters intracellular amyloid-beta/A beta PP carboxy-terminal fragment aggregation and cytotoxicity. J Alzheimers Dis. 2003;5:467–76. doi: 10.3233/jad-2003-5607. [DOI] [PubMed] [Google Scholar]
- Woltjer RL, McMahan W, Milatovic D, Kjerulf JD, Shie FS, Rung LG, Montine KS, Montine TJ. Effects of chemical chaperones on oxidative stress and detergent-insoluble species formation following conditional expression of amyloid precursor protein carboxy-terminal fragment. Neurobiol Dis. 2007;25:427–37. doi: 10.1016/j.nbd.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Yang H, Zhang J, Breyer RM, Chen C. Altered hippocampal long-term synaptic plasticity in mice deficient in the PGE2 EP2 receptor. J Neurochem. 2009;108:295–304. doi: 10.1111/j.1471-4159.2008.05766.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen G, Kim YT, Li RC, Yocum J, Kapoor N, Langer J, Dobrowolski P, Maruyama T, Narumiya S, Dore S. PGE(2) EP1 receptor exacerbated neurotoxicity in a mouse model of cerebral ischemia and Alzheimer’s disease. Neurobiol Aging. 2011 doi: 10.1016/j.neurobiolaging.2011.09.017. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]