

Abstract
The cardiac cycle imposes a mechanical stress that dilates elastic carotid arteries, while shear stress largely contributes to the endothelium-dependent dilation of downstream cerebral arteries. In the presence of dyslipidemia, carotid arteries stiffen while the endothelial function declines. We reasoned that stiffening of carotid arteries would be prevented by reducing resting heart rate (HR), while improving the endothelial function would regulate cerebral artery compliance and function. Thus we treated or not 3-mo-old male atherosclerotic mice (ATX; LDLr−/−: hApoB+/+) for 3 mo with the sinoatrial pacemaker current inhibitor ivabradine (IVA), the β-blocker metoprolol (METO), or subjected mice to voluntary physical training (PT). Arterial (carotid and cerebral artery) compliance and endothelium-dependent flow-mediated cerebral dilation were measured in isolated pressurized arteries. IVA and METO similarly reduced (P < 0.05) 24-h HR by ≈15%, while PT had no impact. As expected, carotid artery stiffness increased (P < 0.05) in ATX mice compared with wild-type mice, while cerebral artery stiffness decreased (P < 0.05); this paradoxical increase in cerebrovascular compliance was associated with endothelial dysfunction and an augmented metalloproteinase-9 (MMP-9) activity (P < 0.05), without changing the lipid composition of the wall. Reducing HR (IVA and METO) limited carotid artery stiffening, but plaque progression was prevented by IVA only. In contrast, IVA maintained and PT improved cerebral endothelial nitric oxide synthase-dependent flow-mediated dilation and wall compliance, and both interventions reduced MMP-9 activity (P < 0.05); METO worsened endothelial dysfunction and compliance and did not reduce MMP-9 activity. In conclusion, HR-dependent mechanical stress contributes to carotid artery wall stiffening in severely dyslipidemic mice while cerebrovascular compliance is mostly regulated by the endothelium.
Keywords: cerebrovascular compliance, endothelial function, resting heart rate
Large elastic artery stiffness and endothelial dysfunction are hallmarks of aging and premature atherosclerosis (51, 58). It is postulated that these changes are, at least partly, the consequence of the accumulation of damage associated with the repeated mechanical stress imposed by the heart rate (HR), a process magnified by risk factors for cardiovascular diseases (19, 51, 58). Although never directly tested, endothelial dysfunction and arterial stiffening have been suggested to be closely associated in peripheral elastic arteries (46, 50, 57, 60) and to feed each other’s deterioration in a vicious cycle (14, 56). Because of the cyclic mechanical stress imposed by each heartbeat on the vasculature (19, 58), elevated resting HR >80 beats/min in humans is a known risk factor for cardiovascular events and premature death (8, 19, 21, 28). Accordingly, the stiffening of the carotid artery is an excellent predictor of cardiovascular (1, 36), peripheral, and also cerebral vascular diseases (42, 52, 61). On the other hand, regular exercise is beneficial to the cardiovascular system and is associated with an augmented life expectancy by reducing all causes of mortality (8, 33, 39). Both a reduction in resting HR and an improved endothelium-dependent endothelial nitric oxide synthase (eNOS) function are associated with these beneficial effects (26, 29, 33, 38).
The effects of a reduction in resting HR and exercise on the cerebral circulation are, however, unknown. Severe dyslipidemia induces endothelial dysfunction in cerebral arteries (22), but its effects on cerebral artery wall compliance are unknown. Few studies have shown that in rat and mouse models of hypertension (4, 5), of inhibition of peroxisome proliferator-activated receptor-γ signaling (13, 32), and of ischemia/reperfusion (35), cerebral endothelial dysfunction is consistently associated with a paradoxical increase in cerebral arterial and arteriolar compliance, suggestive of cerebral wall weakening rather than wall stiffening. Thus the mechanisms by which chronic mechanical stress alters arterial wall compliance may be different in carotid and cerebral arteries. We reasoned that if the compliance of elastic carotid arteries is directly affected by the pulsatile mechanical forces, the compliance of muscular cerebral arteries is essentially controlled by the endothelium. To validate this hypothesis, we modulated HR for 3 mo in atherosclerotic mice displaying endothelial dysfunction (31), using three different indirect approaches, all relevant to the clinical setting and in prevention. First, mice were treated with the sinoatrial pacemaker current inhibitor ivabradine (IVA); it selectively reduces HR without affecting cardiac function (43) and consequently decreases the chronic mechanical stress and has been shown to maintain the endothelial function (17, 18, 23). Second, mice were treated with metoprolol (METO) that reduces HR but does not preserve the cerebral endothelial function, likely by interfering with endothelial β-adrenergic receptor (AR)-nitric oxide (NO) production (12, 18, 23). The third approach was nonpharmacological, using voluntary physical training (PT), proven to improve endothelial function in both humans and animals (33, 48). While exercise training reduces resting HR in humans, its effects in mice are not known, although forced exercise has been shown to decrease HR at rest (9). Our data demonstrate that, in contrast to the carotid arteries, cerebral artery compliance increases with severe dyslipidemia and that, in agreement with our hypothesis, the repeated mechanical stress imposed by the cardiac cycle directly regulates carotid artery stiffening while the endothelium is central to maintain compliance of cerebral arteries. In the cerebral circulation, the mechanical stress only amplifies endothelial dysfunction, favoring functional remodeling.
METHODS
The procedures and protocols were approved by the Montréal Heart Institute Animal Ethical Committee and performed in accordance with the Guide for the Care and Use of Laboratory Animals of Canada and the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996).
Experiments were conducted on cerebral arteries from the circle of Willis and carotid arteries isolated from 3- and 6-mo-old (mo) male C57Bl/6 control wild-type and atherosclerotic (ATX) mice (22, 31). These ATX mice are severely dyslipidemic (Table 1) and develop, under a normal diet, atherosclerotic plaque in large conductance arteries such as the aorta, the renal arteries, and the carotids but not in the cerebral arteries. ATX mice are knockout for the LDL receptor but express the human apolipoprotein B-100 (LDLr−/−:hApoB+/+) (31). ATX mice were randomly assigned to receive a 3-mo treatment (from 3 to 6 mo) with ivabradine (18, 23) (15 mg·kg−1·day−1; ATX + IVA; n = 11) or metoprolol (23) (80 mg·kg−1·day−1; ATX + METO; n = 11) in the drinking water. In another set of experiments, ATX mice (n = 9) were subjected to 3 mo of voluntary training and compared with sedentary ATX and WT mice. Each mouse was kept in a cage with free access to a running wheel (Lafayette Instruments, Lafayette, IN), without reward. The mice ran for 9.0 ± 0.7 h/24 h, mostly during the night (6,160 ± 619 m/day at an average speed of 13 ± 1 m/min; Fig. 1).
Table 1.
Evolution of body weight, plasma lipids and glucose levels, HR, SBP, and pulse pressure in mice
| Weight, g | Total cholesterol, mmol/l | LDL cholesterolmmol/l | HDL cholesterol, mmol/l | Triglycerides, mmol/l | Glucose, mmol/l | Average 24-h HR, beats/min | SBP, mmHg | Pulse, mmHg | |
|---|---|---|---|---|---|---|---|---|---|
| 3-mo WT | 27 ± 1 (12) | 2.1 ± 0.2 (4) | 0.1 ± 0.1 (4) | 1.6 ± 0.1 (4) | 1.2 ± 0.2 (4) | 18.1 ± 2.7 (4) | 543 ± 15 (6) | 137 ± 6 (6) | 31.0 ± 1.7 (6) |
| 3-mo ATX | 27 ± 1 (15) | 16.2 ± 1.1† (4) | 9.8 ± 0.5† (4) | 1.9 ± 0.1 (4) | 7.8 ± 0.6† (4) | 16.2 ± 1.7 (4) | 522 ± 10 (10) | 147 ± 2† (23) | 31.6 ± 0.8 (23) |
| 6-mo WT | 38 ± 1† (20) | 2.3 ± 0.2 (9) | 0.7 ± 0.2 (9) | 1.2 ± 0.1 (9) | 0.8 ± 0.1 (4) | 24.0 ± 2.4 (9) | 532 ± 12 (7) | 127 ± 3 (7) | 30.3 ± 1.9 (6) |
| 6-mo ATX | 31 ± 1*‡ (20) | 16.1 ± 1.0* (9) | 10.1 ± 0.8* (9) | 1.6 ± 0.1* (9) | 7.4 ± 0.8* (9) | 13.8 ± 2.2* (9) | 504 ± 15 (7) | 149 ± 4* (11) | 32.9 ± 1.0 (6) |
| 6-mo ATX + IVA | 33 ± 1 (11) | 16.1 ± 1.9 (4) | 10.3 ± 1.3 (4) | 1.9 ± 0.1 (4) | 7.1 ± 0.6 (4) | 9.6 ± 1.3 (4) | 434 ± 10§ (6) | 149 ± 6 (6) | 33.7 ± 1.0 (11) |
| 6-mo ATX + METO | 32 ± 1 (11) | 20.2 ± 1.0§ (4) | 13.5 ± 0.5§ (9) | 1.7 ± 0.1 (4) | 9.0 ± 0.6 (4) | 11.0 ± 1.6 (4) | 439 ± 7§ (6) | 150 ± 5 (6) | 37.1 ± 2.1 (6) |
| 6-mo ATX + PT | 31 ± 1 (6) | 17.8 ± 2.1 (5) | 11.4 ± 2.0 (5) | 1.6 ± 0.1 (5) | 7.5 ± 0.8 (5) | 15.4 ± 3.0 (5) | 516 ± 30 (3) | 159 ± 4 (6) | 33.0 ± 1.2 (6) |
Results are means ± SE of (n) mice. HR, heart rate (telemetry); SBP, systolic blood pressure (tail-cuff); Pulse, pulse pressure (tail-cuff); WT: wild-type mice; ATX: LDLr−/−:hApoB+/+ mice; ATX + IVA: LDLr−/−:hApoB+/+ mice treated with ivabradine (15 mg · kg−1 · day−1); ATX + METO: LDLr−/−:hApoB+/+ mice treated with metoprolol (80 mg · kg−1 · day−1); ATX + PT: LDLr−/−:hApoB+/+ mice subjected to 3 mo of voluntary physical training.
P < 0.05 vs. 3-mo WT;
P < 0.05 vs. 6-mo WT;
P < 0.05 vs. 3-mo ATX;
P < 0.05 vs. 6-mo ATX.
Fig. 1.

Time during which exercise occurred and changes in heart rate induced by training. A: recording of the running activity of 3 different LDLr−/−:hApoB+/+ (ATX) mice housed in a cage with free access to a running wheel. Cumulative duration of the training, which occurred mostly during the night, was 9 ± 1 h. B: example of the simultaneous recording of the speed of running and of the changes in heart rate (HR) monitored by telemetry in an ATX mouse. C: schematization of the heart rate in sedentary, ivabradine (IVA)-, or metoprolol (METO)-treated ATX mice and in mice exposed to exercise. Heart rate is expressed in beats per minute (bpm) or in beats per 24 h. Difference in heart rate (ΔHR) between IVA- or METO-treated ATX mice and trained ATX mice is indicated.
Heart rate was monitored from 3 to 6 mo after implantation of open-heart radiofrequency transmitters under isoflurane (2.5% in O2, 0.5 l/min for induction and 1.5% for maintenance) anesthesia (Data Sciences International, Arden Hills, MN) (11, 23). Analgesia was maintained by buprenorphine (0.05 mg/kg before surgery and every 8 h for 48 h). Electrocardiogram leads were placed in lead II position. Data were analyzed with ECG Auto (version 2.4.6; EMKA Technologies, Paris, France). In voluntary trained mice, HR was monitored by telemetry for a week before death (n = 3) to avoid physical interferences between the implantable telemetry probe and the exercise capacities of the mice. Telemetry data were confirmed by a weekly monitoring of HR using a tail-cuff device during the 3-mo training protocol. At 3 and 6 mo, mice were anesthetized (44 mg/kg ketamine and 2.2 mg/kg xylazine) and blood was collected; the plasma was frozen at −80°C. The thoracic aorta was dissected and frozen at −80°C. The brain was removed from the cranial cavity and frozen at −80°C or placed in ice-cold physiological salt solution (PSS, pH 7.4, mmol/l: 130 NaCl, 4.7 KCl, 1.18 KH2PO4, 1.17 MgSO4, 14.9 NaHCO3, 1.6 CaCl2, 0.023 EDTA, and 10 glucose) to isolate cerebral arteries for reactivity (24) and compliance studies. The left carotid arteries were removed and placed in ice-cold PSS for compliance studies.
Isolated arteries were cannulated at both ends and pressurized as previously described (24). Flow-mediated dilations (FMD) were induced on phenyleprine precontracted cerebral arteries (24). A single cumulative curve (from 0 to 25 μl/min, with 2 μl/min steps between 0 and 10 μl/min, followed by three 5 μl/min steps, at constant pressure of 60 mmHg) was performed on each segment. Shear stress (t; dyn/cm2) was calculated according to [τ = 4ηQ/πr3], where represents the viscosity (0.009 P), Q the flow rate through the lumen (ml/s), and r the inside radius (cm). The applied calculated shear stress was in the physiological range (≈0 –25 dyn/cm2; Refs. 47, 53). Data are presented as the percentage of dilatation for a given average value of shear stress. The average shear stress represents the average of each shear stress obtained for each data set presented on the same graph that was in a ±1.0 dyn/cm2 interval.
Reactivity and compliance studies were performed on different arterial segments. Passive pressure-diameter curves were conducted in a Ca2+-free PSS containing 1 mM of EGTA to abolish myogenic tone and to assess the mechanical properties of the arteries. Internal and external diameter changes were measured after each increment of intraluminal pressure (from 10 –120 mmHg with a first 10-mmHg step followed by 20-mmHg steps for cerebral arteries and from 60 to 180 mmHg with 20-mmHg steps for carotid arteries), to calculate mechanical parameters. The circumferential wall strain (Strain, %) was calculated according to [(D − Dinitial mmHg)/Dinitial mmHg], where D is the internal diameter at a given pressure and Dinitial mmHg is the initial diameter at the initial pressure (10 mmHg for cerebral arteries and 60 mmHg for carotid arteries). The incremental distensibility (ID; %/mmHg), which represents the percentage of change of the arterial internal diameter for each mmHg change in intraluminal pressure, was calculated according to [(D1 −D0)/(D1 * ΔP) * 100], where D0 is the internal diameter before the pressure increment, D1 is the internal diameter after the pressure increment, and ΔP is the change in pressure (10 or 20 mmHg).
Plasma parameters
Total cholesterol, LDL, HDL, triglycerides, and glucose levels were measured at the Montreal Heart Institute Biochemistry Laboratory (Montreal, QC, Canada).
Staining of atherosclerotic lesions and quantification of plaque areas
Thoracic aortas still attached to the heart were removed and dissected from surrounding tissues. The aortic arch and the aorta were opened longitudinally from the aortic valve through the end of the thoracic aorta, pinned on a Petri dish, fixed in 0.01 M PBS (pH 7.4) and 10% paraformaldehyde, and stained for 1 h with 1–2-propandiol 0.5% Oil Red O. Stained aortas were photographed and digitalized. Plaque areas were quantified using Gimp 2.6 software (www.gimp.org) and expressed in percentage of total aorta area.
Histological Verhoeff-Van Gieson staining
Cerebral arteries were fixed in 10% formalin PBS-buffered solution and processed for standard histological procedures and embedded in paraffin. Sections of 6 μm were obtained by microtome, deparaffinized in xylene, and hydrated in ethanol. All of the slides obtained were stained with Verhoeff’s elastic stain to visualize the elastic fibers and counter-stained with Van Gieson’s solution to visualize collagen.
Isolation of cerebral vessels and Western blot
Whole brain vessels were isolated as previously described (45). For Western blot analysis, 30 μg (for collagen type I/III) or 50 μg (for elastin) of cerebral vessels proteins were mixed with a discontinuous Laemmli buffer and loaded on a 10% acrylamide SDS-PAGE gels. After 45 min of migration at 200 V, gels were transferred on nitrocellulose. Membranes were incubated with an anti-collagen I/III (1:10 000; Calbiochem, Darmstadt, Germany) or with an anti-elastin (1: 6,000; Abcam, Cambridge, MA). To enhance elastin detection sensitivity, after primary antibody incubation, membranes were incubated with a biotin-conjugated anti-mouse IgG (1:10,000) followed by incubation with a horseradish peroxidase-conjugated with streptavidin (1:10,000). α-Actin was used as a loading control (anti-α-actin 1:100,000; Sigma-Aldrich Canada, Oakville, ON, Canada). Quantification was made by densitometric analysis using Quantity One software.
Gelatin zymography
Zymography using gelatin-containing gels was performed as described previously (37). Briefly, a modified Laemmli buffer without mercaptoethanol was added to the vessel proteins samples, without heating, on a 6% SDS-polyacrylamide gel containing 1% gelatin. Migration was conducted at 4°C and 10 mA for 1 h, followed by a 90 V migration for 2 h. After migration, SDS was removed from the gel by washing 5 × 10 min with 2.5% (v/v) Triton X-100 at room temperature. Gels were incubated in a zymography buffer (38 mM Tris·HCl pH 7.4, 13 mM CaCl2, 10 μM ZnCl2, 0.02% NaN3, and 0.03% Brij 35) at 37°C for 72 h and then stained with Coomassie brilliant blue. Gelatinolytic activity was visualized as clear bands against the dark background. Samples of purified human pro-MMP-9 (Chemicon International) and pro-MMP-2 (Enzo Life Sciences, Plymouth Meeting, PA) were used as a positive control. Quantification was made by densitometric analysis using Gimp 2.6 software. When sample quantity allowed it, experiments were made in duplicate.
Cholesterol composition of cerebral vessels
Lipids extraction was performed by adding 200 μl of chloroform and 1% Triton X-100 to 20 mg of isolated cerebrals vessels. Samples were sonicated for 20 min and mixed by inversion at 4°C overnight. Samples were filtrated through a gaze to remove suspended particles and collected in clean tubes in order be air dried and vacuum dried at 50°C for 30 min. Dry lipids were redissolved in 200 μl cholesterol reaction buffer from a cholesterol/cholesteryl ester quantitation kit (Calbiochem), and the total cholesterol concentration was calculated according to the manufacturer’s instructions.
Fatty acids composition of cerebral vessels
Lipids extraction method was adapted from Ruiz et al. (55). One milliliter of chloroform:methanol (2:1) was added to 20 mg of isolated cerebral vessels. Samples were sonicated 20 min and mixed by inversion at 4°C overnight. Samples were then filtrated through a gaze to remove suspended particles and collected in glass tubes. A N2 evaporation was used to concentrate fatty acids that were redissolved in 200 μl of hexane:chloroform:methanol (95:3:2) for the column separation step. Dissolved fatty acids were loaded on the 0.5-g NH2 bond columns (Varian, Lake Forest, CA). Triglycerides were eluted with 4 ml of chloroform, and free fatty acids and phospholipids were eluted in the same fraction by 2.5 ml of methanol:chloroform (6:1) followed by 2.5 ml of methanol:chloroform (6:1) 0.05 M sodium acetate. Free fatty acid concentration was negligible and thus not detectable by gas chromatography coupled with flame ionisation detection (GC-FID). An external standard of nonadecanoic acid (C19:0) was added in each fraction for relative quantification, and the solvent was evaporated by N2. Fatty acids were redissolved in 2 ml of hexane:methanol (1:4) 10% acetyl chloride and heated for 1 h at 80°C under agitation for esterification. Then, tubes were cooled down and 5 ml of K2CO3 6% were added to neutralize the excess of acetyl chloride. The tubes were agitated by inversion and centrifuged 10 min at high speed to accelerate the phase of separation between hexane and the aqueous solution. The hexane phase was transferred in vials for analysis by GC-FID (Agilent Technologies, Santa Clara, CA). The system is coupled with an Agilent DB-WAX capillary column (60 m × 0.25 mm id; 0.25-μm film thickness). The carrier gas was hydrogen at a flow rate of 1.9 ml/min. Two microliters of each sample were injected at 40°C in the column. Initially, the oven is held at 40°C for 7 min. Then, the temperature is increased to 120°C with a 10°C/min rate, to 180°C with a 1°C/min rate, and finally, 218°C is reached with a rate of 0.5°C/min. Fatty acid concentrations in the phospholipids fraction were calculated based on the concentration of the external standard (C19:0).
Statistics
The n refers to the number of animals used in each protocol. Results are presented as means ± SE. Unpaired Student’s t-test and one-way ANOVA were used, when adequate. A value of P < 0.05 was considered statistically significant.
Materials
METO and phenyleprine were purchased from Sigma-Aldrich Canada. IVA was provided by Institut de Recherches Inter-nationales Servier (Courbevoie, France).
RESULTS
Phenotype of WT and ATX mice
Plasma total cholesterol, LDL cholesterol, and triglycerides levels were higher in ATX mice compared with WT mice (Table 1). ATX mice (at 6 mo of age) were hypertensive, and blood pressure was not normalized by IVA, METO, or PT. IVA and METO similarly reduced HR (Table 1). In contrast, PT had no impact on 24-h HR compared with sedentary ATX mice (Table 1). Trained mice were indeed active for a cumulative time of 9 ± 1 h daily (during night time, Fig. 1), running 6.2 ± 0.6 km/day at a speed of 11 ± 1 m/min with an average running HR of 623 ± 47 beats/min, which is significantly higher than sedentary 6-mo ATX mice (504 ± 15 beats/min, P < 0.05); when mice were not running, HR was 452 ± 20 beats/min, which tended to be lower than sedentary 6-mo ATX mice (P = 0.086) and which is not significantly different from IVA- and METO-treated mice. In the group of trained mice, the daily (9 h of running and 15 h of rest) HR, i.e., the mean cumulative HR at rest and during running, was therefore not significantly different from sedentary 6-mo ATX mice (Table 1 and Fig. 1). Weekly HR acquisition by tail-cuff used in parallel during the protocol of PT reinforces data obtained by telemetry revealing a 10% reduction in HR at rest in 6-mo-trained (612 ± 18 beats/min; n = 6; P < 0.05) compared with sedentary ATX mice (678 ± 16 beats/min; n = 6).
Carotid artery compliance
Compared with 6-mo WT mice, carotid arteries from 6-mo ATX mice were less compliant, as illustrated by a decrease of the circumferential strain from 120 to 180 mmHg (Fig. 2A). IVA and METO, but not PT, equally reduced carotid stiffness (Fig. 2B).
Fig. 2.
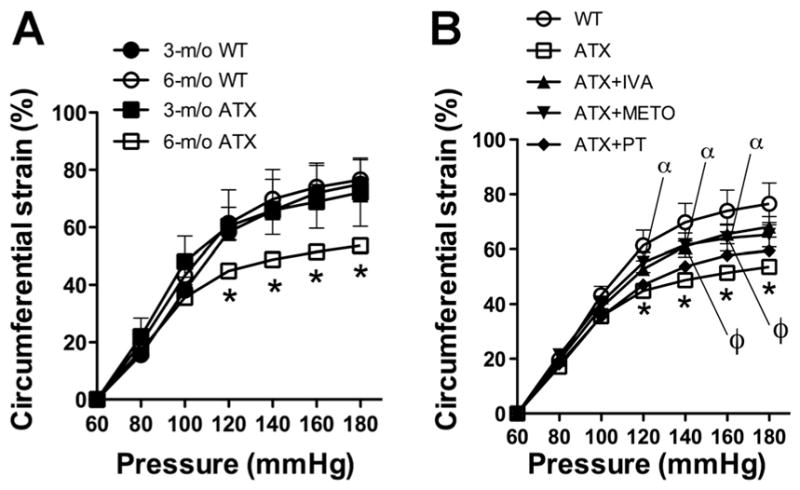
Effect of aging and atherosclerosis on the compliance of peripheral carotid arteries expressed as strain (%) from 3- and 6-mo wild-type (WT) mice and ATX mice (A) and following 3-mo exposure to voluntary physical training (PT) or treatment with either IVA or METO (B). Results are means ± SE of 7–16 mice. *P < 0.05 vs. 6-mo WT; ϕP < 0.05 IVA vs. ATX; αP < 0.05 METO vs. ATX.
Cerebral artery compliance and endothelial function
Cerebral artery compliance increased with age in WT mice, a change magnified by atherosclerosis in ATX mice (Fig. 3A). The circumferential strain of cerebral arteries from 3-mo ATX mice was similar to that measured in 6-mo WT mice (Fig. 3A). The increase in strain of cerebral arteries of ATX mice was associated with an increase in ID between 20 and 60 mmHg (Fig. 3B). In parallel, endothelium-dependent flow-mediated FMD was significantly reduced in cerebral arteries from 3- and 6-mo ATX mice at 6.7 ± 0.4 and 12.1 ± 0.5 dyn/cm2, compared with age-matched WT mice (Fig. 3C). The effect of Nω-nitro-L-arginine (L-NNA; 10 μM), an inhibitor of eNOS, was tested on the responses to flow: as previously reported (22), in 3- and 6-mo WT mice, FMD is clearly eNOS dependent and almost abolished by L-NNA (Fig. 3, D and E). In 3-mo ATX mice, FMD are reduced compared with WT mice, but the response is still sensitive to L-NNA (Fig. 3D). In 6-mo ATX mice, FMD are very low, in the presence or absence of L-NNA, suggesting major eNOS dysfunction (Fig. 3E).
Fig. 3.

Age and atherosclerosis increase cerebral compliance and alter endothelial function. Cerebrovascular compliance expressed as circumferential strain (%; A), distensibility expressed as incremental distensibility (ID; %/mmHg; B), and flow-mediated endothelium-dependent dilation (C) of pressurized cerebral arteries from 3- and 6-mo WT mice and ATX mice. Effects of endothelial nitric oxide synthase inhibition by Nω-nitro-L-arginine (L-NNA) on flow-mediated dilation of cerebral arteries isolated from 3-mo WT and ATX mice (D) and 6-mo WT and ATX mice (E). Results are means ± SE of 7 mice. **P < 0.05 vs. 3-mo WT mice; *P < 0.05 vs. 6-mo WT mice; ωP < 0.05 vs. 3-mo ATX mice.
IVA normalized the strain of cerebral arteries at physiological pressures from 60 to 80 mmHg (Fig. 4A), attributable to the maintenance of the distensibility between 20 and 60 mmHg. IVA also prevented further decline in endothelial dysfunction in ATX mice, illustrated by a greater FMD at 16.3 ± 0.6 dyn/cm2 compared with cerebral arteries isolated from 6-mo untreated ATX mice (Fig. 4A) but a similar endothelial dysfunction compared with 3-mo ATX sedentary mice (Fig. 3C). In contrast, METO tended to further impair strain and ID and significantly worsened FMD (Fig. 4B).
Fig. 4.

Effect of a chronic treatment of 6-mo ATX mice with IVA (A) and METO (B) and exposure to voluntary PT (C) on the cerebral compliance expressed as circumferential strain (%), the distensibility expressed as ID (%/mmHg), and flow-mediated endothelium-dependent dilation of pressurized cerebral arteries. Results are means ± SE of 6 to 10 mice. *P < 0.05 vs. 6-mo WT mice; φP < 0.05 vs. 6-mo ATX mice.
After 3 mo of voluntary exercise (PT), the strain and ID were completely normalized to WT values (Fig. 4C) and thus improved compared with 3-mo ATX sedentary mice (Fig. 3C). In cerebral arteries from trained ATX mice, FMD induced by shear stresses from 3.2 ± 0.2 to 17.0 ± 0.1 dyn/cm2 was increased compared with arteries from 6-mo sedentary ATX (Fig. 4C), and endothelial function was even normalized to WT values from 7.1 ± 0.4 to 11.6 ± 0.4 dyn/cm2 (Fig. 4C), demonstrating that PT not only stopped but also reversed the endothelial dysfunction in ATX mice.
When all groups are considered, there is a negative correlation (P < 0.05; r = 0.824) between cerebral FMD obtained at 12.0 ± 0.5 dyn/cm2 (the only physiological shear stress shared between the different groups of mice) and the cerebral circumferential strain measured at the physiological intraluminal pressure of 60 mmHg (Fig. 5).
Fig. 5.
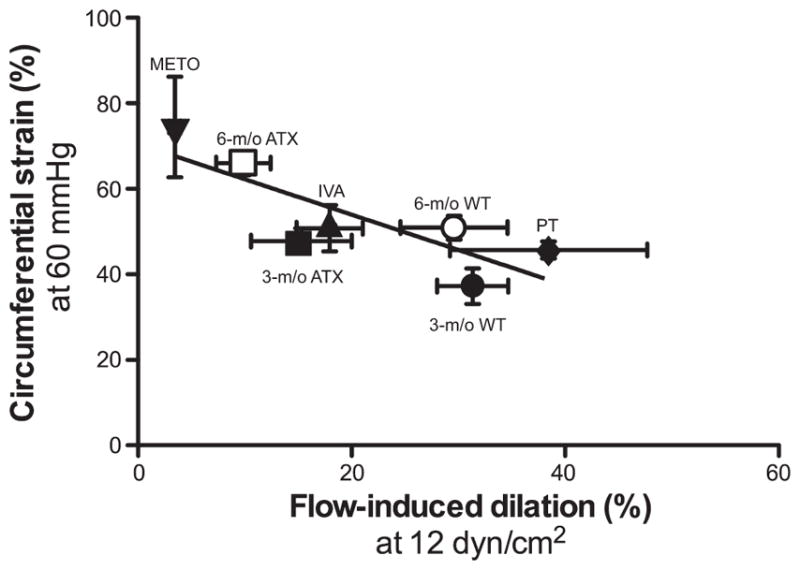
Correlation between endothelial flow-mediated dilation (%) observed at 12.0 ± 0.5 dyn/cm2 and the circumferential strain (%) observed at an intraluminal pressure of 60 mmHg, in cerebral arteries isolated from 3- and 6-mo WT mice and ATX mice, and in 6-mo ATX mice treated for 3 mo with either IVA or METO, or ATX mice exposed for 3 mo to voluntary PT. There is a negative and significant correlation between both parameters (P = 0.0227; r = 0.824; n = 7). Results are means ± SE.
Factors modulating cerebrovascular compliance
We tested whether the lipid composition of cerebral vessels could underlie the increase in compliance by altering the membrane fluidity of the cells. The content of fatty acids and cholesterol in cerebral vessels was, however, similar between WT and severely dyslipidemic ATX mice (Table 2).
Table 2.
Vessel wall lipid composition of total cerebral vessel from WT and ATX mice
| 3-mo WT | 3-mo ATX | 6-mo WT | 6-mo ATX | |
|---|---|---|---|---|
| Fatty acids, % | ||||
| Palmitic acid (C:16:0) | 18.9 ± 0.8 | 18.8 ± 0.7 | 18.5 ± 0.4 | 18.0 ± 1.0 |
| Palmitoleic acid (C:16:1) | 0 | 0 | 0 | 0 |
| Stearic acid (C:18:0) | 19.3 ± 0.7 | 19.2 ± 0.2 | 19.7 ± 0.3 | 19.3 ± 0.4 |
| Oleic acid Ω 9 (C18:1n9) | 31.2 ± 0.9 | 32.2 ± 0.8 | 31.7 ± 0.9 | 32.3 ± 1.4 |
| Oleic acid Ω 7 (C18:1n7) | 5.2 ± 0.3 | 5.2 ± 0.1 | 4.9 ± 0.2 | 5.1 ± 0.2 |
| Linoleic acid (C18:2) | 1.7 ± 0.5 | 2.2 ± 0.3 | 1.8 ± 0.5 | 2.2 ± 0.3 |
| Linolenic acid Ω 6 (C18:3n6) | 0 | 0 | 0 | 0 |
| Linolenic acid Ω 3 (C18:3n3) | 0 | 0 | 0 | 0 |
| Homo-γ-linoleic acid (C20:3n6) | 0 | 0 | 0 | 0 |
| Arachidonic acid (C20:4) | 9.9 ± 0.3 | 9.7 ± 0.3 | 9.9 ± 0.2 | 9.9 ± 0.2 |
| Eicosapentaneoic acid (C20:5) | 0 | 0 | 0 | 0 |
| Decosahexanoic acid (C22:6) | 13.7 ± 0.5 | 12.8 ± 0.7 | 13.5 ± 0.4 | 13.2 ± 0.2 |
| Total cholesterol, μg/mg | 7.0 ± 0.3 | 7.3 ± 0.6 | NA | NA |
Results are means ± SE for n = 4. Fatty acid composition in phospholipids is expressed as the percentage of total fatty acids detected by gas chromatography coupled with flame ionisation detection. Total free cholesterol was quantified by colorimetric detection (μg/mg). NA, not available.
We also tested whether differences in pulse pressure could contribute to the changes in compliance: the pulse pressure, calculated from the systolic and diastolic blood pressure measured by tail-cuff, was similar between the groups of mice (Table 1).
Changes in the hemodynamic environment associated with atherosclerosis, such as oxidative stress and NO bioavaibility, are known to trigger MMP-dependent remodeling (30). Such remodeling could affect cerebrovascular compliance. Accordingly, MMP-9 activity increased in cerebral vessels from 3 to 6-mo in WT and ATX mice (Fig. 6); MMP-9 activity was, however, higher in 6-mo ATX compared with age-matched WT mice, and this was prevented by IVA and PT but not by METO (Fig. 6). MMP-2 activity was very low and similar between all groups (data not shown).
Fig. 6.

A: representative example of zymography illustrating metalloproteinase-9 (MMP-9) activities of cerebral vessels from WT and ATX mice. B: effect of a chronic treatment of 6-mo ATX mice with IVA or METO and after 3-mo exposure to voluntary PT on the gelatinase activity of cerebral vessels. Each graph represents the %changes to respective controls. Results are means ± SE of 6 mice. **P < 0.05 vs. 3-mo WT mice; *P < 0.05 vs. 6-mo WT mice; ωP < 0.05 vs. 3-mo ATX mice; ϕP < 0.05 vs. 6-mo ATX mice.
We finally tested whether the collagen type I or III content of cerebral blood vessels could be modified by severe dyslipidemia and modulated by IVA, METO, and PT. Cerebrovascular Col I/III protein expression was similar between WT and ATX mice and was not altered by treatments or by PT (Fig. 7, A and B). Collagen type IV and V were not detected. Histological analysis of collagen expression (Verhoeff-Van Gieson staining; Fig. 7E) did not enable to reveal differences between cerebral arteries isolated from 6-mo WT and ATX mice (Fig. 6). Elastin expression was similar between 6-mo WT and ATX mice (Fig. 7, C and D).
Fig. 7.

A: representative example of Western blot illustrating collagen I and III (Col I/III) protein expression of total cerebral vessels from WT and ATX mice. B: effect of a chronic treatment of 6-mo ATX mice with IVA or METO and after 3-mo exposure to voluntary exercise (PT) on Col I/III protein expression of total cerebral vessels. Each graph represents the protein abundancy normalized to its respective internal standard after adjustment for loading with Ponceau red. Results are means ΔSE of 4 mice. C: representative example of Western blot illustrating elastin expression of total cerebral vessels from 6-mo WT and ATX mice. D: bar graph of the elastin abundancy normalized to α-actin. E: representative example of cerebral arteries stained with Verhoeff-Van Gieson to reveal elastic fibers in black and collagen in red (×100). No differences were detected between cerebral arteries isolated from 6-mo WT and ATX mice.
Atherosclerotic plaque progression
ATX mice spontaneously develop atherosclerotic plaque in the aorta and the carotid arteries. The lesions, hardly noticeable at 3 mo, covered 7.1 ± 1.6% of the total surface of the thoracic aorta at 6 mo, including the aortic arch (Fig. 8). IVA significantly slowed plaque progession; METO tended to decrease it as well, while PT did not limit plaque progression. No atherosclerotic plaques were observed in cerebral arteries.
Fig. 8.
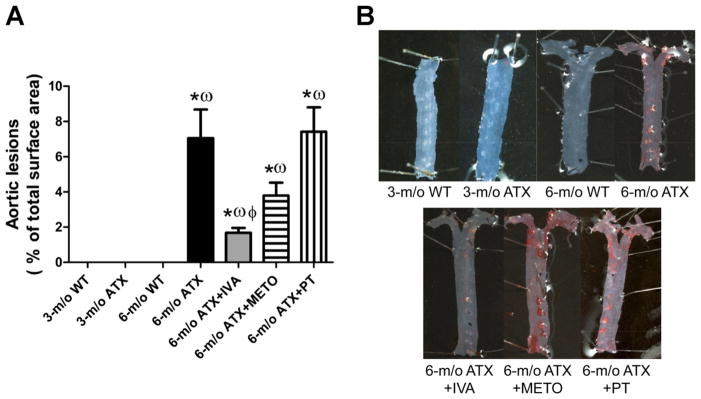
Effect of 3-mo chronic treatement of ATX mice with IVA or METO and effect of 3-mo exposure to voluntary PT on the the progression of atheroclerotic lesions in the aortic arch and thoracic aorta. Aortic lesions quantification (A) and representative picture of Oil Red O-stained aorta (B). Results are means ± SE of 9 –20 mice. *P < 0.05 vs. 6-mo WT mice; ωP < 0.05 vs. 3-mo ATX mice; ϕP < 0.05 vs. 6-mo ATX mice.
DISCUSSION
The results of this study demonstrate for the first time that, in mice, severe dyslipidemia is associated with a paradoxical increase in cerebrovascular compliance and that while chronic HR-associated mechanical stress directly contributes to carotid artery stiffening, cerebrovascular compliance is essentially regulated by the endothelium. These conclusions are indirectly validated through the use of three experimental approaches: 1) IVA that reduced HR and thus mechanical stress and maintained endothelial function and cerebrovascular compliance, 2) METO that also reduced HR but worsened endothelial function and did not maintain the compliance, and (3) PT that normalized the endothelial function and cerebrovascular compliance without reducing mechanical stress.
Previous studies (3, 7, 15, 59) demonstrated that dyslipidemia has a significant impact on conduit artery stiffness. Accordingly, we found that carotid arteries stiffened from the age of 3 to 6 mo in ATX but not in WT mice. In contrast to the carotids, cerebral arteries isolated from ATX mice were more compliant, suggestive of a weakening of the wall. Although similar increased cerebrovascular compliance has been previously reported in animal models of hypertension (4, 5), PPARγ inhibition (13, 32), and ischemia/reperfusion (35), this is the first study to report such a paradoxical cerebrovascular wall weakening in a mouse model of severe dyslipidemia and atherosclerosis in the large peripheral conductance arteries.
The beneficial effect of voluntary PT on cerebrovascular compliance indirectly demonstrates that the endothelium, but not the mechanical stress, tightly controls wall biomechanics. Voluntary running, which did not reduce HR over a 24-h period (Table 1), not only maintained but also improved endothelial function (eNOS-dependent FMD; Fig. 4C) and normalized both cerebrovascular compliance (Fig. 4C) and MMP-9 activity (Fig. 6). Exercise induces acute increases in blood flow and thus shear stress, a stimulus known to improve the endothelial function by upregulating eNOS activity (25, 63). There is, however, an apparent disconnect between the beneficial effect of PT on the cerebral arteries (Fig. 4C) and its lack of effect on both carotid stiffness (Fig. 2) and aortic plaque size (Fig. 8). In contrast to PT, HR reduction by IVA and METO (Table 1) equally reduced carotid stiffness (Fig. 2), demonstrating that the cyclic mechanical stress is critical for determining carotid elasticity, in accordance with previous reports in animal models of atherosclerosis (3, 7). Although exercise reduced resting HR 15 h per day, it has been at the expense of a significant increase in HR during exercise for a cumulative time of 9 h (Fig. 1): therefore, over a 24-h period, HR averaged 516 beats/min in trained ATX mice (Table 1), which is not different from sedentary mice. In contrast, HR averaged 435 beats/min in ATX mice treated with IVA or METO. This represents a reduction of 80 beats/min in treated mice compared with PT mice, i.e., a reduction of 115,200 heartbeats per 24 h (Fig. 1). Therefore, because of the supreme importance of HR and accumulated cyclic stress on large elastic stiffness, the lack of effect of PT observed on large elastic artery stiffness and plaque formation could be explained. In monkeys (62) and humans (34), moderate exercise (<3 h per week) was associated with a small reduction in resting HR without preventing plaque progression despite improving the endothelial function (62). Only intensive training (>5 h per wk) led to a reduction in plaque size in humans (34). Furthermore, it has been reported that 1 h of daily forced exercise in high-fat fed LDLr−/− mice slowed plaque progression (48): although in the latter study no hemodynamic data were reported, this experimental approach decreases resting HR in rats (9). Hence, voluntary exercise in mice may represent a low to moderate exercise activity in humans. Another discrepancy is that METO did not prevent plaque progression as well as IVA (Fig. 8) despite equal HR reduction; this may be dependent on the deleterious effect of METO on the endothelial function (Fig. 4B). This would therefore suggest that if HR is essential for plaque progression, the endothelium could modulate its growth. We (31) previously observed that plaque progresses, although not as rapidly, under an endothelium with a perfectly normal function in ATX mice treated with the polyphenol catechin, which does not reduce HR.
We (23) previously reported that IVA has no direct vascular effects. Therefore, the beneficial effects of pure HR reduction by IVA on the cerebral endothelial function in ATX mice (Fig. 4A) also demonstrate that the cardiac cycle magnifies the endothelial damage induced by severe dyslipidemia. While IVA has been previously shown to improve carotid distensibility in WKY rats and wall stress in SHR (2), IVA also prevented the decline in aorta endothelial relaxant function in dyslipidemic mice and increased brain capillary density (17, 18). Because IVA is highly specific for its molecular target, these data suggest that in addition to inducing elastic artery fatigue and stiffness, cyclic mechanical stress is responsible for magnifying the endothelial damage induced by severe dyslipidemia, which favors cerebral artery wall weakening (19, 58).
The molecular mechanism by which a reduction in HR is beneficial to the cardiovascular system remains, however, unclear. In large elastic arteries, it is accepted that age-dependent stiffness is dependent on wall fatigue (51) and that this process is magnified by risk factors for cardiovascular diseases (41). Our data suggest that in muscular cerebral arteries a reduction in the mechanical stress will likely tip the balance towards maintenance mechanisms allowing for slowing the damages that target the endothelium. The deleterious effects of METO on the endothelial function and cerebral compliance (Fig. 4B) suggest that β1-adrenergic receptor (β1-AR), expressed on the endothelium (20), could contribute to the maintenance of vessel integrity (23). It has been postulated that catecholamines act as endothelial cell survival factors (16, 27, 40). Recently, β-AR stimulation during voluntary exercise has been shown to be instrumental to the cardiac protection following ischemia in mice (12). By extension, circulating catecholamines could protect the cerebral arteries during exercise as well as at rest. Our data support this concept, since antagonism of cerebrovascular β-AR with METO fully counteracted the beneficial effects of HR lowering that were evidenced by IVA in our study (Fig. 4, A and B) and in others (18).
In addition to the catecholamine hypothesis proposed above, MMP-2 and -9 have been widely studied for their implication in vascular remodling since they cleave the basal membrane, collagens (type III, IV, and V), elastin, and laminin (49). We observed that MMP-9 activity was augmented in cerebral vessels from 6-mo ATX mice (Fig. 6). Such an increase in MMP-9 activity could reduce arterial stiffness by disturbing the cell-to-cell connections and their matrix; this would likely be sufficient to weaken the wall since, like in humans, mouse cerebral arteries lack external elastic lamina and adventitia (44). In addition, this increase in MMP-9 activity is in accordance with the magnified endothelial dysfunction observed in ATX mice (Fig. 3C), since oxidative stress and nitric oxide (NO) bioavaibility are known to trigger MMP-dependent remodeling (30); likewise, in eNOS−/− mice, cerebral artery walls are weakened and this is independent of pulse pressure (6). Exercise and IVA, but not METO, strongly reduced MMP-9 activity compared with sedentary and untreated ATX mice (Fig. 6), which is associated with better eNOS-dependent flow-mediated dilation and the normalization of cerebrovascular compliance (Fig. 4). On the other hand, the direct impact of MMP activity on the endothelial function remains to be studied. Of interest, MMP-9 has been reported to inactivate endothelial β2-AR by cleavage of their extracellular domain, reducing endothelium-dependent relaxation (54), providing a mechanism by which MMPs could also reduce endothelial function independently of a reduced eNOS function.
Baumbach et al. (5) proposed that the increase in cerebral arteriolar compliance associated with hypertension is linked to a reduction in the stiff collagen vs. the compliant elastin contained in the arteriolar wall extracellular matrix. Collagen I and III are the most prominent types in the vasculature, 60 and 30%, respectively, of total collagen content (10). We did not find a significant difference in collagen I and III expression among the different groups of mice (Fig. 7, A and B). At 6 mo, elastin content was not altered by severe dyslipidemia (Fig. 7, C–E). We also tested whether the lipid composition of cerebral vessels could underlie the increase in compliance by altering the membrane fluidity of the cells as well as cell mass, but we found that the lipid composition was identical in WT and ATX mice (Table 2). This is, however, the first report of vascular wall lipid composition of total mouse cerebral vessels. Therefore, neither collagen and elastin expression per se nor lipid composition of the arterial wall seems to account for the changes in cerebral compliance associated with severe dyslipidemia.
Changes in cerebrovascular wall thickness, such as an increase in vascular smooth muscle, one of the most compliant components of the vascular wall, could account for the increase in cerebrovascular compliance observed in ATX mice (Fig. 3A). Vessel hypertrophy was not evaluated in the present study, but compared with WT mice, lumen diameter decreased in cerebral arteries from 6-mo ATX mice, wall thickness increased, and as a result, the wall-to-lumen ratio increased significantly (data not shown). However, while IVA, METO, and exercise significantly increased the inner diameter in cerebral arteries from ATX mice (data not shown), only IVA and exercise prevented the rise in compliance (Fig. 4). Therefore, changes in passive structure (wall thickness, inner diameter, and wall-to-lumen ratio) may contribute to the observed alterations in the vascular compliance, while other pathways, such the rise in MMP-9, may be more important.
Limits of the study
While cerebrovascular compliance and endothelial function were assessed in arteries from the circle of Willis, all the molecular biology studies (MMPs activity, collagen, and elastin protein expression) were conducted in blood vessels extracted from the whole brain (including the circle of Willis). Since the cerebral arteries and the whole brain blood vessels are of different origin and exposed to different hemodynamic factors, conclusions should be made with caution. Another limit of the study is that active MMP-2 activity is also revealed while evaluating MMP-9 activity. However, the signal concerning MMP-2 was low compared with that of MMP-9. Finally, the conclusion that changes in compliance in cerebral arteries in ATX mice were regulated by endothelial function is based on indirect evidence. In the literature, although never directly tested, endothelial function and arterial stiffness of large peripheral elastic arteries have been suggested to be closely associated. To the best of our knowledge, direct endothelium-dependent regulation of cerebrovascular compliance has never been reported, and we are the first to suggest a link between the two parameters.
In summary, the stiffening of the carotid arteries observed in atherosclerotic mice is predominantly related to HR-associated cyclic stress and independent of the endothelial function. In contrast, cerebral arteries from ATX mice are characterized by an increased compliance suggestive of a weakening of the wall, a change that can be prevented by maintaining or improving cerebral endothelial function by a chronic treatment with IVA or by voluntary exercise, respectively. By reducing HR and thus by decreasing the mechanical strain stress directly applied onto the cerebral endothelium, IVA maintained cerebral endothelial function without improving it, reduced MMP-9 activity, and in turn was able to prevent cerebral arterial wall functional remodling. Voluntary exercise training maximized these benefits likely through mechanisms involving physiological transient increases in shear stress and sympathetic tone. This study has marked clinical relevance since cerebral endothelial function and compliance are critical for the ability of cerebral arterioles to maintain their autoregulatory functions, which are known to decline with age and prematurely in the presence of risk factors for cardiovascular diseases, leading to impaired cognitive functions and dementia.
Acknowledgments
GRANTS
This work was supported in part by the Montreal Heart Institute Foundation, Heart and Stroke Foundation of Quebec, and Canadian Institutes of Health Research (MOP89733) and by an unrestricted educational grant from Servier (France). A. Drouin holds the Frederick Banting and Charles Best Canada Graduate Scholarships-Doctoral Award in association with the Canadian Institutes for Health Research.
Footnotes
AUTHOR CONTRIBUTIONS
V. B., A. D., C. D.R., J. -C. T., and E. T. conception and design of research; V. B., A. D., M. -A. G., N. D., and I. F. -R. performed experiments; V. B., A. D., M. -A. G., N. T. -T., I. F. -R., C. D. R., and E. T. analyzed data; V. B., N. T. -T., C. D. R., and E. T. interpreted results of experiments; V. B. and N. T. -T. prepared figures; V.B. drafted manuscript; V. B., A. D., M. -A. G., N. D., N. T. -T., I. F. -R., C. D. R., J. -C. T., and E. T. approved final version of manuscript; N. T. -T., C. D. R., J. -C. T., and E. T. edited and revised manuscript.
DISCLOSURES
J. -C. Tardif has recieved honoraria from Servier Pharmaceuticals that provided Ivabradine and partly supported the present study. This study was also supported in part by an unrestricted educational grant from Servier (to C. Des Rosiers, J.-C. Tardif, and E. Thorin).
References
- 1.Aatola H, Koivistoinen T, Hutri-Kahonen N, Juonala M, Mikkila V, Lehtimaki T, Viikari JS, Raitakari OT, Kahonen M. Lifetime fruit and vegetable consumption and arterial pulse wave velocity in adulthood: the Cardiovascular Risk in Young Finns Study. Circulation. 2010;122:2521–2528. doi: 10.1161/CIRCULATIONAHA.110.969279. [DOI] [PubMed] [Google Scholar]
- 2.Albaladejo P, Carusi A, Apartian A, Lacolley P, Safar ME, Benetos A. Effect of chronic heart rate reduction with ivabradine on carotid and aortic structure and function in normotensive and hypertensive rats. J Vasc Res. 2003;40:320–328. doi: 10.1159/000072696. [DOI] [PubMed] [Google Scholar]
- 3.Bassiouny HS, Zarins CK, Kadowaki MH, Glagov S. Hemodynamic stress and experimental aortoiliac atherosclerosis. J Vasc Surg. 1994;19:426–434. doi: 10.1016/s0741-5214(94)70069-9. [DOI] [PubMed] [Google Scholar]
- 4.Baumbach GL, Heistad DD. Remodeling of cerebral arterioles in chronic hypertension. Hypertension. 1989;13:968–972. doi: 10.1161/01.hyp.13.6.968. [DOI] [PubMed] [Google Scholar]
- 5.Baumbach GL, Sigmund CD, Faraci FM. Cerebral arteriolar structure in mice overexpressing human renin and angiotensinogen. Hypertension. 2003;41:50–55. doi: 10.1161/01.hyp.0000042427.05390.5c. [DOI] [PubMed] [Google Scholar]
- 6.Baumbach GL, Sigmund CD, Faraci FM. Structure of cerebral arterioles in mice deficient in expression of the gene for endothelial nitric oxide synthase. Circ Res. 2004;95:822–829. doi: 10.1161/01.RES.0000146279.11923.14. [DOI] [PubMed] [Google Scholar]
- 7.Beere PA, Glagov S, Zarins CK. Retarding effect of lowered heart rate on coronary atherosclerosis. Science. 1984;226:180–182. doi: 10.1126/science.6484569. [DOI] [PubMed] [Google Scholar]
- 8.Benetos A, Thomas F, Bean KE, Pannier B, Guize L. Role of modifiable risk factors in life expectancy in the elderly. J Hypertens. 2005;23:1803–1808. doi: 10.1097/01.hjh.0000179512.71018.40. [DOI] [PubMed] [Google Scholar]
- 9.Benito B, Gay-Jordi G, Serrano-Mollar A, Guasch E, Shi Y, Tardif JC, Brugada J, Nattel S, Mont L. Cardiac arrhythmogenic remodeling in a rat model of long-term intensive exercise training. Circulation. 2011;123:13–22. doi: 10.1161/CIRCULATIONAHA.110.938282. [DOI] [PubMed] [Google Scholar]
- 10.Borel JP, Bellon G. Vascular collagens. General review. Pathol Biol (Paris) 1985;33:254–260. [PubMed] [Google Scholar]
- 11.Brouillette J, Grandy SA, Jolicoeur P, Fiset C. Cardiac repolarization is prolonged in CD4C/HIV transgenic mice. J Mol Cell Cardiol. 2007;43:159–167. doi: 10.1016/j.yjmcc.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 12.Calvert JW, Elston M, Pablo Aragon J, Nicholson CK, Moody BF, Hood RL, Sindler A, Gundewar S, Seals DR, Barouch LA, Lefer DJ. Exercise protects against myocardial ischemia-reperfusion injury via stimulation of β3-adrenergic receptors and increased nitric oxide signaling: role of nitrite and nitrosothiols. Circ Res. 2011 doi: 10.1161/CIRCRESAHA.111.241117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan SL, Chapman AC, Sweet JG, Gokina NI, Cipolla MJ. Effect of PPARgamma inhibition during pregnancy on posterior cerebral artery function and structure. Front Physiol. 2010;1:130. doi: 10.3389/fphys.2010.00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chatzizisis YS, Giannoglou GD. Coronary hemodynamics and atherosclerotic wall stiffness: a vicious cycle. Med Hypotheses. 2007;69:349–355. doi: 10.1016/j.mehy.2006.11.053. [DOI] [PubMed] [Google Scholar]
- 15.Cheng HM, Ye ZX, Chiou KR, Lin SJ, Charng MJ. Vascular stiffness in familial hypercholesterolaemia is associated with C-reactive protein and cholesterol burden. Eur J Clin Invest. 2007;37:197–206. doi: 10.1111/j.1365-2362.2007.01772.x. [DOI] [PubMed] [Google Scholar]
- 16.Ciccarelli M, Cipolletta E, Santulli G, Campanile A, Pumiglia K, Cervero P, Pastore L, Astone D, Trimarco B, Iaccarino G. Endothelial beta2 adrenergic signaling to AKT: role of Gi and SRC. Cell Signal. 2007;19:1949–1955. doi: 10.1016/j.cellsig.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 17.Custodis F, Baumhakel M, Schlimmer N, List F, Gensch C, Bohm M, Laufs U. Heart rate reduction by ivabradine reduces oxidative stress, improves endothelial function, and prevents atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2008;117:2377–2387. doi: 10.1161/CIRCULATIONAHA.107.746537. [DOI] [PubMed] [Google Scholar]
- 18.Custodis F, Gertz K, Balkaya M, Prinz V, Mathar I, Stamm C, Kronenberg G, Kazakov A, Freichel M, Bohm M, Endres M, Laufs U. Heart rate contributes to the vascular effects of chronic mental stress: effects on endothelial function and ischemic brain injury in mice. Stroke. 2011;42:1742–1749. doi: 10.1161/STROKEAHA.110.598607. [DOI] [PubMed] [Google Scholar]
- 19.Custodis F, Schirmer SH, Baumhakel M, Heusch G, Bohm M, Laufs U. Vascular pathophysiology in response to increased heart rate. J Am Coll Cardiol. 2010;56:1973–1983. doi: 10.1016/j.jacc.2010.09.014. [DOI] [PubMed] [Google Scholar]
- 20.Daly CJ, Ross RA, Whyte J, Henstridge CM, Irving AJ, McGrath JC. Fluorescent ligand binding reveals heterogeneous distribution of adrenoceptors and ‘cannabinoid-like’ receptors in small arteries. Br J Pharmacol. 2010;159:787–796. doi: 10.1111/j.1476-5381.2009.00608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diaz A, Bourassa MG, Guertin MC, Tardif JC. Long-term prognostic value of resting heart rate in patients with suspected or proven coronary artery disease. Eur Heart J. 2005;26:967–974. doi: 10.1093/eurheartj/ehi190. [DOI] [PubMed] [Google Scholar]
- 22.Drouin A, Bolduc V, Thorin-Trescases N, Belanger E, Fernandes P, Baraghis E, Lesage F, Gillis MA, Villeneuve L, Hamel E, Ferland G, Thorin E. Catechin treatment improves cerebrovascular flow-mediated dilation and learning abilities in atherosclerotic mice. Am J Physiol Heart Circ Physiol. 2011;300:H1032–H1043. doi: 10.1152/ajpheart.00410.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drouin A, Gendron ME, Thorin E, Gillis MA, Mahlberg-Gaudin F, Tardif JC. Chronic heart rate reduction by ivabradine prevents endothelial dysfunction in dyslipidaemic mice. Br J Pharmacol. 2008;154:749–757. doi: 10.1038/bjp.2008.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drouin A, Thorin E. Flow-induced dilation is mediated by Akt-dependent activation of endothelial nitric oxide synthase-derived hydrogen peroxide in mouse cerebral arteries. Stroke. 2009;40:1827–1833. doi: 10.1161/STROKEAHA.108.536805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duncker DJ, Bache RJ. Regulation of coronary blood flow during exercise. Physiol Rev. 2008;88:1009–1086. doi: 10.1152/physrev.00045.2006. [DOI] [PubMed] [Google Scholar]
- 26.Durrant JR, Seals DR, Connell ML, Russell MJ, Lawson BR, Folian BJ, Donato AJ, Lesniewski LA. Voluntary wheel running restores endothelial function in conduit arteries of old mice: direct evidence for reduced oxidative stress, increased superoxide dismutase activity and down-regulation of NADPH oxidase. J Physiol. 2009;587:3271–3285. doi: 10.1113/jphysiol.2009.169771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Figueroa XF, Poblete I, Fernandez R, Pedemonte C, Cortes V, Huidobro-Toro JP. NO production and eNOS phosphorylation induced by epinephrine through the activation of beta-adrenoceptors. Am J Physiol Heart Circ Physiol. 2009;297:H134–H143. doi: 10.1152/ajpheart.00023.2009. [DOI] [PubMed] [Google Scholar]
- 28.Fox K, Ford I, Steg PG, Tendera M, Robertson M, Ferrari R. Heart rate as a prognostic risk factor in patients with coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): a subgroup analysis of a randomised controlled trial. Lancet. 2008;372:817–821. doi: 10.1016/S0140-6736(08)61171-X. [DOI] [PubMed] [Google Scholar]
- 29.Fujimoto N, Prasad A, Hastings JL, Arbab-Zadeh A, Bhella PS, Shibata S, Palmer D, Levine BD. Cardiovascular effects of 1 year of progressive and vigorous exercise training in previously sedentary individuals older than 65 years of age. Circulation. 2010;122:1797–1805. doi: 10.1161/CIRCULATIONAHA.110.973784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Galis ZS, Khatri JJ. Matrix metalloproteinases in vascular remodeling and atherogenesis: the good, the bad, and the ugly. Circ Res. 2002;90:251–262. [PubMed] [Google Scholar]
- 31.Gendron ME, Theoret JF, Mamarbachi AM, Drouin A, Nguyen A, Bolduc V, Thorin-Trescases N, Merhi Y, Thorin E. Late chronic catechin antioxidant treatment is deleterious to the endothelial function in aging mice with established atherosclerosis. Am J Physiol Heart Circ Physiol. 2010;298:H2062–H2070. doi: 10.1152/ajpheart.00532.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Halabi CM, Beyer AM, de Lange WJ, Keen HL, Baumbach GL, Faraci FM, Sigmund CD. Interference with PPAR gamma function in smooth muscle causes vascular dysfunction and hypertension. Cell Metab. 2008;7:215–226. doi: 10.1016/j.cmet.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hambrecht R, Adams V, Erbs S, Linke A, Krankel N, Shu Y, Baither Y, Gielen S, Thiele H, Gummert JF, Mohr FW, Schuler G. Regular physical activity improves endothelial function in patients with coronary artery disease by increasing phosphorylation of endothelial nitric oxide synthase. Circulation. 2003;107:3152–3158. doi: 10.1161/01.CIR.0000074229.93804.5C. [DOI] [PubMed] [Google Scholar]
- 34.Hambrecht R, Niebauer J, Marburger C, Grunze M, Kalberer B, Hauer K, Schlierf G, Kubler W, Schuler G. Various intensities of leisure time physical activity in patients with coronary artery disease: effects on cardiorespiratory fitness and progression of coronary atherosclerotic lesions. J Am Coll Cardiol. 1993;22:468–477. doi: 10.1016/0735-1097(93)90051-2. [DOI] [PubMed] [Google Scholar]
- 35.Jimenez-Altayo F, Martin A, Rojas S, Justicia C, Briones AM, Giraldo J, Planas AM, Vila E. Transient middle cerebral artery occlusion causes different structural, mechanical, and myogenic alterations in normotensive and hypertensive rats. Am J Physiol Heart Circ Physiol. 2007;293:H628–H635. doi: 10.1152/ajpheart.00165.2007. [DOI] [PubMed] [Google Scholar]
- 36.Juonala M, Magnussen CG, Venn A, Dwyer T, Burns TL, Davis PH, Chen W, Srinivasan SR, Daniels SR, Kahonen M, Laitinen T, Taittonen L, Berenson GS, Viikari JS, Raitakari OT. Influence of age on associations between childhood risk factors and carotid intima-media thickness in adulthood: the Cardiovascular Risk in Young Finns Study, the Childhood Determinants of Adult Health Study, the Bogalusa Heart Study, and the Muscatine Study for the International Childhood Cardiovascular Cohort (i3C) Consortium. Circulation. 2010;122:2514–2520. doi: 10.1161/CIRCULATIONAHA.110.966465. [DOI] [PubMed] [Google Scholar]
- 37.Kleiner DE, Stetler-Stevenson WG. Quantitative zymography: detection of picogram quantities of gelatinases. Anal Biochem. 1994;218:325–329. doi: 10.1006/abio.1994.1186. [DOI] [PubMed] [Google Scholar]
- 38.Kojda G, Cheng YC, Burchfield J, Harrison DG. Dysfunctional regulation of endothelial nitric oxide synthase (eNOS) expression in response to exercise in mice lacking one eNOS gene. Circulation. 2001;103:2839–2844. doi: 10.1161/01.cir.103.23.2839. [DOI] [PubMed] [Google Scholar]
- 39.Kokkinos P, Myers J, Faselis C, Panagiotakos DB, Doumas M, Pittaras A, Manolis A, Kokkinos JP, Karasik P, Greenberg M, Papademetriou V, Fletcher R. Exercise capacity and mortality in older men: a 20-year follow-up study. Circulation. 2010;122:790–797. doi: 10.1161/CIRCULATIONAHA.110.938852. [DOI] [PubMed] [Google Scholar]
- 40.Kou R, Michel T. Epinephrine regulation of the endothelial nitric-oxide synthase: roles of RAC1 and beta3-adrenergic receptors in endothelial NO signaling. J Biol Chem. 2007;282:32719–32729. doi: 10.1074/jbc.M706815200. [DOI] [PubMed] [Google Scholar]
- 41.Kovacic JC, Moreno P, Hachinski V, Nabel EG, Fuster V. Cellular senescence, vascular disease, and aging: part 1 of a 2-part review. Circulation. 2011;123:1650–1660. doi: 10.1161/CIRCULATIONAHA.110.007021. [DOI] [PubMed] [Google Scholar]
- 42.Laurent S, Boutouyrie P. Arterial stiffness and stroke in hypertension: therapeutic implications for stroke prevention. CNS Drugs. 2005;19:1–11. doi: 10.2165/00023210-200519010-00001. [DOI] [PubMed] [Google Scholar]
- 43.Lauzier B, Vaillant F, Gelinas R, Bouchard B, Brownsey R, Thorin E, Tardif JC, Des Rosiers C. Ivabradine reduces heart rate while preserving metabolic fluxes and energy status of healthy normoxic working hearts. Am J Physiol Heart Circ Physiol. 2011;300:H845–H852. doi: 10.1152/ajpheart.01034.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee HY, Oh BH. Aging and arterial stiffness. Circ J. 2010;74:2257–2262. doi: 10.1253/circj.cj-10-0910. [DOI] [PubMed] [Google Scholar]
- 45.Li X, Geary GG, Gonzales RJ, Krause DN, Duckles SP. Effect of estrogen on cerebrovascular prostaglandins is amplified in mice with dysfunctional NOS. Am J Physiol Heart Circ Physiol. 2004;287:H588–H594. doi: 10.1152/ajpheart.01176.2003. [DOI] [PubMed] [Google Scholar]
- 46.McEniery CM, Wallace S, Mackenzie IS, McDonnell B, Yasmin Newby DE, Cockcroft JR, Wilkinson IB. Endothelial function is associated with pulse pressure, pulse wave velocity, and augmentation index in healthy humans. Hypertension. 2006;48:602–608. doi: 10.1161/01.HYP.0000239206.64270.5f. [DOI] [PubMed] [Google Scholar]
- 47.Muller JM, Davis MJ, Chilian WM. Integrated regulation of pressure and flow in the coronary microcirculation. Cardiovasc Res. 1996;32:668–678. [PubMed] [Google Scholar]
- 48.Napoli C, Williams-Ignarro S, De Nigris F, Lerman LO, Rossi L, Guarino C, Mansueto G, Di Tuoro F, Pignalosa O, De Rosa G, Sica V, Ignarro LJ. Long-term combined beneficial effects of physical training and metabolic treatment on atherosclerosis in hypercholesterolemic mice. Proc Natl Acad Sci USA. 2004;101:8797–8802. doi: 10.1073/pnas.0402734101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Newby AC. Metalloproteinases and vulnerable atherosclerotic plaques. Trends Cardiovasc Med. 2007;17:253–258. doi: 10.1016/j.tcm.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nigam A, Mitchell GF, Lambert J, Tardif JC. Relation between conduit vessel stiffness (assessed by tonometry) and endothelial function (assessed by flow-mediated dilatation) in patients with and without coronary heart disease. Am J Cardiol. 2003;92:395–399. doi: 10.1016/s0002-9149(03)00656-8. [DOI] [PubMed] [Google Scholar]
- 51.O’Rourke MF, Hashimoto J. Mechanical factors in arterial aging: a clinical perspective. J Am Coll Cardiol. 2007;50:1–13. doi: 10.1016/j.jacc.2006.12.050. [DOI] [PubMed] [Google Scholar]
- 52.Ochi N, Kohara K, Tabara Y, Nagai T, Kido T, Uetani E, Ochi M, Igase M, Miki T. Association of central systolic blood pressure with intracerebral small vessel disease in Japanese. Am J Hypertens. 2010;23:889–894. doi: 10.1038/ajh.2010.60. [DOI] [PubMed] [Google Scholar]
- 53.Papaioannou TG, Karatzis EN, Vavuranakis M, Lekakis JP, Stefanadis C. Assessment of vascular wall shear stress and implications for atherosclerotic disease. Int J Cardiol. 2006;113:12–18. doi: 10.1016/j.ijcard.2006.03.035. [DOI] [PubMed] [Google Scholar]
- 54.Rodrigues SF, Tran ED, Fortes ZB, Schmid-Schonbein GW. Matrix metalloproteinases cleave the beta2-adrenergic receptor in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2010;299:H25–H35. doi: 10.1152/ajpheart.00620.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ruiz J, Antequera T, Andres AI, Petron MJ, Muriel E. Improvement of a solid phase extraction method for analysis of lipid fractions in muscle foods. Analytica Chimica Acta. 2004;520:201–205. [Google Scholar]
- 56.Sugawara J, Komine H, Hayashi K, Yoshizawa M, Yokoi T, Otsuki T, Shimojo N, Miyauchi T, Maeda S, Tanaka H. Effect of systemic nitric oxide synthase inhibition on arterial stiffness in humans. Hypertens Res. 2007;30:411–415. doi: 10.1291/hypres.30.411. [DOI] [PubMed] [Google Scholar]
- 57.Takumi T, Yang EH, Mathew V, Rihal CS, Gulati R, Lerman LO, Lerman A. Coronary endothelial dysfunction is associated with a reduction in coronary artery compliance and an increase in wall shear stress. Heart. 2010;96:773–778. doi: 10.1136/hrt.2009.187898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thorin E, Thorin-Trescases N. Vascular endothelial ageing, heartbeat after heartbeat. Cardiovasc Res. 2009;84:24–32. doi: 10.1093/cvr/cvp236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tonstad S, Joakimsen O, Stensland-Bugge E, Ose L, Bonaa KH, Leren TP. Carotid intima-media thickness and plaque in patients with familial hypercholesterolaemia mutations and control subjects. Eur J Clin Invest. 1998;28:971–979. doi: 10.1046/j.1365-2362.1998.00399.x. [DOI] [PubMed] [Google Scholar]
- 60.Tounian P, Aggoun Y, Dubern B, Varille V, Guy-Grand B, Sidi D, Girardet JP, Bonnet D. Presence of increased stiffness of the common carotid artery and endothelial dysfunction in severely obese children: a prospective study. Lancet. 2001;358:1400–1404. doi: 10.1016/S0140-6736(01)06525-4. [DOI] [PubMed] [Google Scholar]
- 61.Tsuchikura S, Shoji T, Kimoto E, Shinohara K, Hatsuda S, Koyama H, Emoto M, Nishizawa Y. Central versus peripheral arterial stiffness in association with coronary, cerebral and peripheral arterial disease. Atherosclerosis. 2010;211:480–485. doi: 10.1016/j.atherosclerosis.2010.03.037. [DOI] [PubMed] [Google Scholar]
- 62.Williams JK, Kaplan JR, Suparto IH, Fox JL, Manuck SB. Effects of exercise on cardiovascular outcomes in monkeys with risk factors for coronary heart disease. Arterioscler Thromb Vasc Biol. 2003;23:864–871. doi: 10.1161/01.ATV.0000067934.12783.6A. [DOI] [PubMed] [Google Scholar]
- 63.Zhang QJ, McMillin SL, Tanner JM, Palionyte M, Abel ED, Symons JD. Endothelial nitric oxide synthase phosphorylation in treadmill-running mice: role of vascular signaling kinases. J Physiol. 2009;587:3911–3920. doi: 10.1113/jphysiol.2009.172916. [DOI] [PMC free article] [PubMed] [Google Scholar]