

Abstract
The vascular endothelium starts to age at the first heartbeat. There is no longer a need to demonstrate that an increased resting heart rate—above 70 b.p.m.—is associated with the onset of cardiovascular events and reduces lifespan in humans. Each cardiac cycle imposes a mechanical constraint on the arteries, and we would like to propose that this mechanical stress damages the vascular endothelium, its dysfunction being the prerequisite for atherogenesis. Consequently, reducing heart rate could protect the endothelium and slow the onset of atherosclerosis. The potential mechanisms by which reducing heart rate could be beneficial to the endothelium are likely a combination of a reduction in mechanical stress and tissue fatigue and a prolongation of the period of steady laminar flow, and thus sustained shear stress, between each systole. With age, irreparable damage accumulates in endothelial cells and leads to senescence, which is characterized by a pro-atherogenic phenotype. In the body, the highest mechanical stress occurs in the coronary vessels, where blood only flows during diastole and even reverses during systole; thus, coronary arteries are the prime site of atherosclerosis. All classical risk factors for cardiovascular diseases add up, to accelerate atherogenesis, but hypertension, which further raises mechanical stress, is likely the most damaging. By inducing flow through the arteries, the heart rate determines shear stress and its stability: mechanical stress and the associated damage induced by each systole are efficiently counteracted by the repair capacities of a healthy endothelium. The maintenance of a physiological, low heart rate may be key to prolonging the endothelial healthy lifespan and thus, vascular health.
Keywords: Endothelium, Resting heart rate, Mechanical stress, Cellular maintenance, Cellular repair, Atherosclerosis
1. Context
Resting heart rate is a basic and simple physiological parameter of the physical examination which has important prognostic implications: there is a remarkable negative correlation between resting heart rate and life expectancy, in individuals free of disease and in patients with cardiovascular diseases.1,2 Resting heart rate is a strong predictor of cardiovascular mortality,3,4 independently of other known risk factors such as hypertension, diabetes, or smoking. Heart rate is a key determinant of coronary blood flow and myocardial O2 consumption. Thus, an increased resting heart rate reduces the ischaemic threshold and cardiac performance. Because of myocardial mechanical and metabolic stimulation, heart rate likely imposes a stress not only on the myocardium but also on the vasculature, which could play an important role in determining cardiac and vascular damage to influence life expectancy.
High resting heart rate has been identified as a potential accelerator of atherosclerosis via its negative effects on the endothelium.5 More than five decades ago, Dr R. Altschul (1954) wrote: ‘It has been said that one is as old as one’s arteries. In view of the supreme importance of endothelium in arterial function, I should like to modify …this statement by saying that one is as old as one’s endothelium’.6 This was a visionary statement at a time when the endothelium was merely considered as an anti-coagulant sheet of cellophane. With age, the endothelium loses its ability to protect the arterial wall, promoting adhesion of platelets and infiltration of neutrophils, losing its smooth muscle cell anti-proliferative action, its dilatory function in favour of contraction, and expressing pro-inflammatory and oxidative markers.7,8 In this review, we will focus on trying understanding how heart rate reduction could be used as a means to slow the onset of atherosclerosis and thus to prolong vascular half life. The overall concept remains a hypothesis: unavoidable mechanical damage induced by each heartbeat starts in utero. It seems only logical that the addition of each risk factor for cardiovascular disease (including an elevated resting heart rate) will magnify the damage induced by each heartbeat and shorten vascular lifespan.
2. Healthy ageing and endothelial dysfunction or the natural history of atherosclerosis
With ageing, physiological functions decline.9 This is also true for endothelial functions.10 In short, endothelial ageing is first characterized by reduced endothelium-dependent dilation both in vivo and in vitro in all vascular bed tested.11–13 To put it simply, this dysfunction (as we will refer to throughout this review) is due to a reduced production and release of nitric oxide (NO) in response to flow and agonists following an increase in inflammation and oxidative stress.11 The consequences are not only functional, but also structural: the endothelium influences the structure of its underlying arterial wall, which becomes stiffer with age.14 An increase in arterial wall rigidity augments systolic and pulse pressure, and in turn strongly increases the risk of atherosclerosis, hypertension, myocardial infarction, and stroke.15
The mechanisms leading to the progressive decline in endothelial function remain uncertain. During our life, damage to the endothelium will activate maintenance repair systems. A working hypothesis is that this damage stems from the daily pounding of the cycling pressure, a classical risk factor for cardiovascular diseases, but also viral infections, autoimmune diseases, and inflammatory diseases in general. If the damage is minimal or maintenance system efficient (as in young subjects), endothelial cells will likely correct the defect and keep going. If the damage is irreversible, the damaged cells senesce and are eventually eliminated by a mechanism yet to be discovered, while a sister endothelial cell will divide and fill the gap. It is also proposed that circulating progenitor endothelial cells assume some repair function,16 although this has not yet been clearly demonstrated in vivo.17 Cell division for maintenance is likely the main response of an injured endothelium. In the long term, however, the regenerated endothelium is dysfunctional18 and endothelial cells can only undergo a finite number of divisions, after which they enter senescence and stop dividing.19,20 Senescence is a state of quiescence in which cells are still metabolically active and, most importantly, express a pro-inflammatory, pro-oxidative, and pro-atherogenic phenotype.21 The gradual accumulation of senescent endothelial cells leads to a progressive decline in endothelial function.22 Thus, while endothelial cell division contributes to repair and maintenance of endothelial function, this leads ultimately to senescence, and thus, endothelial dysfunction (Figure 1). Consequently, any condition that increases damage will accelerate endothelial cell division and so hasten dysfunction.23–25
Figure 1.

A model for the mechanisms of age-dependent endothelial dysfunction. Physiological levels of stress on the endothelium are generated by heartbeats and metabolic oxidative stress. Upon addition of a risk factor such as hypertension, damage increases, augmenting endothelial cell turnover and thus senescence.25 Exponential accumulation of senescent endothelial cells is pro-atherogenic.
3. Risk factors for cardiovascular diseases accelerate endothelial dysfunction
At exhaustion of the endothelial repair capacities, atherosclerosis develops according to a process that has been very well reviewed.26,27 Briefly, atherosclerotic plaques associated with excessive expansive remodelling evolve to high-risk plaques in areas of low endothelial shear stress, a condition that promotes continued local lipid accumulation, inflammation, oxidative stress, matrix breakdown, and eventually further plaque progression and excessive expansive remodelling. In the endothelium of patients with severe coronary artery disease (CAD), and exposed to multiple risk factors, we observed that while the sum of all risk factors for cardiovascular diseases accelerates endothelial senescence in plaque-free arteries, hypertension is sufficiently damaging per se to accelerate endothelial cell turnover and lead to premature senescence (Figure 1), which is associated with atherosclerosis.25 Hypertension likely magnifies the mechanical stress of each heartbeat by increasing peripheral resistance. In addition, hypertension-dependent endothelial dysfunction will accelerate large artery stiffening (see in what follows), thus magnifying further the stress of each heartbeat, and will increase pulse pressure, a risk factor for stroke, to name only one consequence. Therefore, elevated resting heart rate and high blood pressure have synergistic effects on cardiovascular mortality.1
4. Causes for elevated resting heart rate
Tachycardia is a well-defined medical condition with resting heart rate above 100 b.p.m. Such condition can be the consequence of fever (18 beats per degree Celsius), sympathetic activation of the heart (shock or semi-shock, loss of blood), and toxic conditions to the heart such as weakening of the heart that elicits a sympathetic reflex to increase the heart rate.28 In contrast, the origin of a higher-than normal resting heart rate, which is directly relevant to this review, is not clearly defined. Mental stress increases sympathetic drive in humans and increases resting heart rate.29–31 Obesity is associated with an increase in sympathetic drive in humans and can be at the basis of the associated hypertension and elevated resting heart rate.32 Interestingly, however, a recent 20 year prospective study demonstrates that an increase in resting heart rate may predispose to obesity and diabetes mellitus,33 suggesting that an early rise in sympathetic drive may promote these metabolic changes. Nagae et al.34 demonstrated the importance of central oxidative stress in the regulation of blood pressure and heart rate and how obesity increases the sympathetic drive (also see the section on the regulation of the baroreflex sensitivity).
These observations lead us to consider the role of NO as a regulator of the sympathetic outflow, since a rise in reactive oxygen species (ROS) production favours the degradation of NO. Nitric oxide is known to depress the sympathetic activity, likely by increasing parasympathetic tone both centrally35 and peripherally (for review, see Danson et al.36). A rise in oxidative stress down-regulates this pathway and favours the sympathetic drive as shown in diabetes32 and hypertension.36 In spontaneously hypertensive rats, L-arginine supplementation corrects local cardiac noradrenergic hyperactivity, probably via increased pre-synaptic substrate availability of the NOS pathway and reduced tyrosine hydroxylase levels, limiting noradrenaline production.35 There is therefore a strong inhibitory effect of NO on the sympathetic drive, and thus the regulation of heart rate.
Finally, and to our knowledge, there are no genome wide association studies that tested the genetic association with resting heart rate. Overall, the central hypothesis for an increased resting heart rate below tachycardia but above normal range (>70 b.p.m.) is an increased sympathetic drive either directly or through a reduction in parasympathetic tone. Alternative hypotheses have yet to be defined but increased ROS production is emerging as an important mechanism activating the sympathetic drive.
5. High heart rate and the potential link with endothelial dysfunction
Cardiac contraction induces a pulsatile pressure and flow: it is accepted that the pulsatile increase in pressure leads to fatigue failure of the vascular wall in the long term, while the pulsatile rise in blood flow is beneficial to the endothelium.26,27 At normal low heart rate (between 60 and 70 b.p.m., based on the impact of resting heart rate on event rate;37 Figure 2), mechanical stress-induced damage is likely efficiently counterbalanced by the protective function of the normal endothelium under physiological shear stress conditions.
Figure 2.
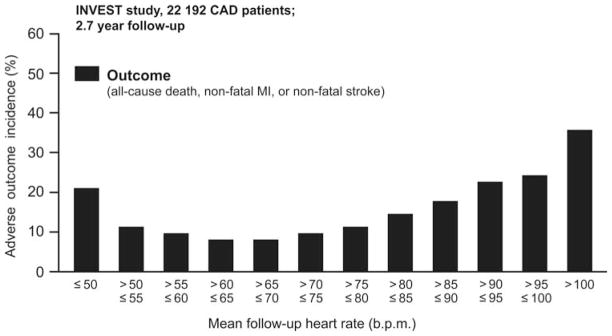
Relationship between follow-up resting heart rate for all patients and incidence of adverse outcomes. Among all patients, the nadir for follow-up resting heart rate was 59 b.p.m. Adapted from Kolloch et al. Impact of resting heart rate on outcomes in hypertensive patients with coronary artery disease: findings from the INternational VErapamil-SR/trandolapril STudy (INVEST). Eur Heart J 2008;29:1327–1334.37
5.1 The pulsatile change in pressure
The pulsatile increase in pressure generated by cardiac contraction is the main cause of vascular stress. The conduit arteries are elastic because the heart is a reciprocating pump. Thus, during systole, a bolus of blood is ejected into the aorta and the pressure stretches the aortic wall to accommodate the bolus. At the end of systole, the aortic valve closes, the blood starts to flow through the systemic circulation driven by the elastic energy stored in the aortic wall, which returns to its end-diastolic diameter. This mechanical stress is therefore largely absorbed by the compliance of the conduit artery wall. If the aorta and other conduit arteries were not rubber-like but copper-like, the required systolic pressure would have to be greater to permit flow throughout the entire vascular tree and the heart rate would need to be much higher than 70 b.p.m. Hypertension, which increases peripheral resistance, imposes a greater pressure. This no doubt magnifies the mechanical stress and accelerates the damage induced by repeated cardiac cycles.38 Likewise, while conceptualizing a bridge, an engineer takes into account material strength and fatigue capacity, and thus resistance to stresses such as high winds, waves, and earthquakes. Any of these stresses, if higher than normal, will shorten the half-life of the construction.
5.2 The pulsatile change in flow
Before presenting the effects of heart rate on shear stress—the frictional (tangential) force per unit area created by flow of viscous blood over the inner vessel wall and the luminal surface of the endothelium—and endothelial function, we would like to summarize briefly the mechanisms by which flow mediates dilation and regulates eNOS expression.
A rise in flow induces a transient rise in intracellular Ca2+ necessary for calmodulin to interact with caveolin, which maintains eNOS inactive in the basal state. Following calmodulin activation, heat shock protein (Hsp) 90 binds to eNOS39 favouring the recruitment of Akt leading to the phosphorylation of eNOS on ser-1177, its sustained enzymatic activation, production of NO, and dilation.40,41
Haemodynamic shear stress also regulates eNOS expression in the endothelium. Recent data clearly point to a role for H2O2 in this process, and thus to the primary involvement of superoxide ( ).42,43 In agreement with the concept that oxidative stress up-regulates eNOS expression is the demonstration that exercise increases the production of ROS together with eNOS.44
The cardiac contraction is responsible for pulsatile flow and thus for haemodynamic shear stress. The latter is characterized by Poiseuille’s law, which states that shear stress is proportional to blood flow and its viscosity, and inversely proportional to the third power of the inner diameter. Shear stress is low in the veins (1–6 dynes/cm2; 10 dynes/cm2 = 1 Pa = 1 N/m2), high and pulsatile in conduit arteries (10–70 dynes/cm2), and averages 15 dynes/cm2 in arterioles. Overall, in the arterial tree, shear stress averages 15–20 dynes/cm.2,26 Shear stress is important for three reasons. First, it is a strong endothelial stimulus. A physiological increase in shear stress stimulates the production of NO—and to a lesser extent prostacyclin—which immediately dilates brachial arteries in humans,13 except large conduit arteries, which are essentially elastic. Second, sustained shear stress maintains the expression of the NO-producing enzyme, which maintains basal release of NO, which is known to reduce platelet aggregation, neutrophil adhesion, inflammation, constricting factors, and free radical production.26,27 Third, conversely, a reduction in flow constricts small arteries, increases resistance, and thus further reduces flow locally. The response to shear stress of the endothelium therefore permits fine-tuning of nutrient delivery to accommodate metabolic demand.8
There are, however, two extreme conditions where shear stress no longer plays a regulatory role, i.e. low and even negative shear stress (−4 to +4 dynes/cm2) on the one hand, and extremely high shear stress (>70–100 dynes/cm2) on the other hand. The former occurs in areas of the coronary circulation at each cardiac cycle, but also in zones of recirculation of blood flow such as behind bifurcations or a plaque. Extremely high shear stress occurs in the aortic valve, but also at complex atherosclerotic plaques and stents, both of which are rigid and therefore do not dilate in response to systolic pressure or to shear stress. Thus, if one compares shear stress in veins and arteries, it may not be its physiological level that contributes to the maintenance of a healthy endothelium, but rather its stability.
Blood flows in the coronary arteries during diastole and reverses during systole as the contracting myocardium squeezes the subendocardial coronary arteries.45 Nowhere else in the body is such mechanical stress imposed on an artery. Shear stress in coronary arteries is uneven, and its mean value is well below 10 dynes/cm2. The consequence is a lower NO production and thus reduced protection against stresses. Not surprisingly the coronary circulation is the prime site for early endothelial dysfunction,46 followed by atherosclerosis.47 Conversely, cerebral arteries are better protected from such extreme mechanical stress and so are less prone to early atherosclerosis than the coronaries.48 In cerebral arteries, shear stress is more stable, and pulsatility is quickly reduced by myogenic responses—the reduction in diameter induced by a rise in intraluminal pressure—leading to faster dampening of pulse pressure.49 Therefore, a high resting heart rate shortens the diastolic coronary perfusion phase,45 favours bi-directional changes in flow, and reduces the average shear stress, and thus the release of protective NO. Finally, this likely promotes endothelial dysfunction.
6. Heart rate and atherosclerosis
The first evidence of the prognostic importance of heart rate was presented by Levy et al.50 in 1945. In this report, the authors showed that the rate of developing hypertension with age was more than doubled in the presence of transient tachycardia (Figure 3). Numerous epidemiological data now show that an increase in resting heart rate accelerates the atherosclerotic process.3 Accordingly, the reduction in resting heart rate by metoprolol slows endothelial cell replication (a marker of dysfunction/activation) in psychologically stressed monkeys,30 while the reduction in resting heart rate by surgical ablation of the sinoatrial node slows the atherosclerotic process in monkeys fed a high fat diet.51,52 Regular exercise, which decreases resting heart rate, is beneficial to the cardiovascular system.53 It increases the number of healthy life years,54 slows the progression of atherosclerosis,55 and a year of regular exercise that lowers resting heart rate from 71 to 65 b.p.m. even reduces the recurrence of cardiovascular events post-angioplasty.56 Hence, lowering resting heart rate through exercise is beneficial even in secondary prevention. The benefits gained from regular exercise are, however, by far not limited to the reduction in heart rate but also due to the improvement in the endothelial function, the reduction in blood pressure and the better metabolic profile.57
Figure 3.
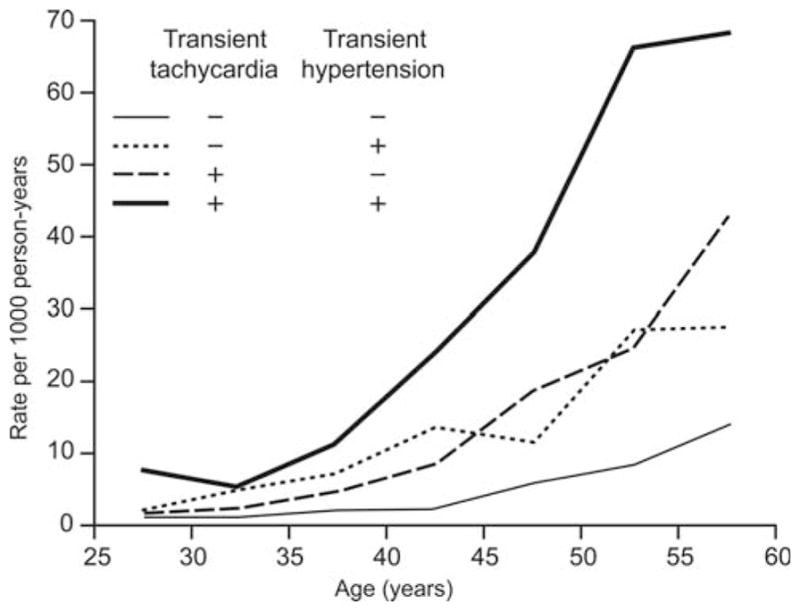
Rate of developing sustained hypertension by age according to the presence or absence of transient tachycardia and transient hypertension. Adapted from Levy et al. Transient tachycardia: pronostic significance alone and in association with transient hypertension. JAMA 1945;129:585–588.50
The discovery and clinical development of new drugs that selectively reduce heart rate without concomitant negative effects on myocardial contractility, relaxation, and conduction constitute a significant advance for the medical treatment of angina. To date, only the bencyclobutane derivative ivabradine is available in medical practice in Europe.58 This small molecule selectively inhibits the activity of the depolarizing pacemaker current If and thus slows heart rate without affecting blood pressure or cardiac contractility.58 In other words, ivabradine slows heart rate by reducing the ‘steepness’ of the If current slope of diastolic depolarization in the sinoatrial nodes and thus increases diastolic duration. Ivabradine not only protects endothelial function,59 but also slows atherosclerosis in apolipoprotein E-deficient mice,60 which is in complete agreement with the clinical data cited earlier. However, besides the reduction in resting heart rate, the underlying vascular protective mechanisms of ivabradine remain to be elucidated.
6.1 Clinical benefits of ivabradine
The pathophysiological basis and experimental data support the potential of heart rate reduction as an intervention to improve endothelial function, to attenuate progression of atherosclerosis and ultimately to prevent coronary events. Whether lowering heart rate with ivabradine in patients with stable CAD receiving optimal background therapy could result in reduction of cardiovascular events prompted the BEAUTIFUL (morBidity-mortality EvAlUaTion of the If inhibitor ivabradine in patients with coronary disease and left ventricULar dysfunction) study, the results of which have been recently reported.61 BEAUTIFUL is a large, international, randomized, placebo-controlled morbidity–mortality trial in a high-risk population of patients with CAD and left ventricular systolic dysfunction (mean left ventricular ejection fraction was 32.4%), conducted between December 2004 and December 2006. The study enrolled 10 917 eligible patients; 5479 patients were randomized to receive ivabradine on top of conventional cardiovascular treatments as recommended by guidelines, and 5438 patients were randomly assigned to the placebo group. The mean heart rate at baseline was 71.6 b.p.m.
Although the primary endpoint of the study (composite of cardiovascular mortality, admission to hospital for acute myocardial infarction, and admission to hospital for new-onset or worsening heart failure) was not met in the overall population, in the pre-specified subgroup of patients with resting heart rate ≥70 b.p.m., ivabradine reduced significantly the risk of coronary events by 22%, fatal and non-fatal myocardial infarction by 36%, and reduced coronary revascularization by 30%. These benefits were observed despite the fact that 84% of these patients received beta-blockers.
The BEAUTIFUL evidence has extended current knowledge of the role of heart rate as a risk factor in CAD by also conducting a prospective analysis of the data from the placebo arm of the BEAUTIFUL study to assess the association of heart rate with different clinical outcomes.4 Patients with a heart rate of 70 b.p.m. or more had significantly increased risks of cardiovascular death (34%), admission to hospital for heart failure (53%), admission to hospital for myocardial infarction (46%), and coronary revascularization (38%) after adjustment for other predictors of outcome.
Ongoing and future clinical studies will provide further evidence on the extent of the cardioprotective benefits of heart rate lowering with ivabradine in cardiovascular disease. The SHIFT (Systolic Heart failure with the If inhibitor ivabradine Trial) study, which is currently investigating the effects of ivabradine in congestive heart failure, will provide important information concerning importance of heart rate reduction with ivabradine in the management of heart failure. This large-scale, randomized, placebo-controlled trial has recruited over 7000 patients with moderate to severe heart failure and resting heart rate ≥70 b.p.m. Throughout the study, all patients have continued their current standard treatments for heart failure and any other cardiovascular conditions. The results of these studies will extend current knowledge of the role of heart rate as a risk factor in heart failure.
7. By what mechanism pure heart rate reduction could slow age-related endothelial dysfunction?
Two possible mechanisms can be put forward, keeping in mind that first irregular shear stress and second mechanical stress are damaging. The first mechanism is well described in a recent review:62 by reducing heart rate, the interval of sustained blood flow during diastole increases. The second mechanism, also well described in a recent review,14 is only mechanical and is a direct consequence of a slower heart rate, which slows tissue fatigue. In addition to these mechanisms, it is also important to consider that a low heart rate has a positive regulatory consequence: the baroreflex sensitivity increases.38
7.1 Shear stress
Exercise and ivabradine improve endothelial function and slow the onset and progression of atherosclerosis. But how do we know that shear stress is important in the response to heart rate? To answer this question specifically, one needs to study a vascular bed where the cycling mechanical stress imposed by each heartbeat is limited. In small, 100 μm diameter, mouse resistance cerebral arteries, the pulsatile nature of the flow is not eliminated, but is largely dampened by the myogenic contraction of upstream arteries. Consequently, blood pressure is lowered to 60 mmHg, compared with 100 mmHg in same size peripheral gracilis arteries.63 Ivabradine was tested in mice with dyslipidaemia, in which endothelial function declines from the age of 3 to 6 months.12,59 A treatment with ivabradine, from the age of 3 to 6 months, reduced heart rate by 10% and prevented the decline in cerebrovascular endothelial function.59 Since ivabradine did not affect blood pressure, only a slower heart rate accounted for this effect. By slowing heart rate, the diastolic perfusion time increases, and thus flow is more constant.45,62 A more constant shear stress in the physiological range, i.e. over 15 and below 70 dynes/cm2, likely explains part of the benefit of a lower resting heart rate. The stability of shear stress is so important that the initial growth of an atherosclerotic plaque will promote an endothelium-dependent expansion of the vascular lumen to maintain shear stress within normal values, a response known as the ‘Glagov phenomenon’.64 This response is genetically programmed and demonstrates how a constant shear stress is fundamental to the overall regulation of arterial function.
7.2 Mechanical stress
Ageing is associated with a change in the structure of the wall, which becomes stiffer.14 The most damaging effect of the hypertensive phenotype is likely a magnification of the mechanical stress imposed on the wall by each heartbeat, which is reminiscent of an accelerated ageing process.65 This has two consequences: the heart and the large conduit arteries hypertrophy to compensate for the increased resistance pressure. In the long term, cardiac hypertrophy turns into heart failure, while loss of arterial elasticity,66 by magnifying mechanical stress, accelerates endothelial cell turnover,67 promoting premature senescence of the endothelium24 and atherosclerosis.68 Not only does the endothelium lose its protective capacities, but very high shear stresses (>70 dynes/cm2) generated by the lack of elasticity during the systole activate platelets and favour thrombosis.23,26,27
We observed the ‘age-retarding’ effect of a lower heart rate on conduit arteries in dyslipidaemic mice using ivabradine.59 After 3 month treatment with ivabradine, the renal arteries of dyslipidaemic mice dilated to the same extent as healthy arteries. In these large conduit arteries, pulse pressure is at its maximum, as is the mechanical stress. Ivabradine, which slowed heart rate by around 10% from the age of 3 to 6 months, prevented the decline in endothelium-dependent dilations. Although we have no definite demonstration, it is possible that the reduction in heart rate minimized the damaging effect of the mechanical stress and consequently attenuated the deleterious effect of dyslipidaemia. In hypercholesterolaemic mice, ivabradine slowed the progression of atherosclerosis,60 in agreement with the concept that each heartbeat generates damage. It may come to a surprise, however, that in the study by Custodis et al., 60 high-fat fed ApoE−/− mice had a lower resting heart rate compared with wild-type mice; yet, lowering further heart rate by 13% prevented the decline in endothelial function and development of atherosclerotic plaques. It was known that resting heart rate differed from one strain to another,64 but there is also the possibility that high-cholesterol limits somehow heart function/rate. It would therefore have been informative to have a group of ApoE−/− mice fed a normal diet. Finally, it has been suggested that ivabradine may have pleiotropic effects on cardiac infarct size and reperfusion injury.45,69 The mechanisms by which ivabradine could protect the heart post-ischaemia remain to be discovered. In isolated arteries, however, ivabradine has no direct acute effect on either contractions or dilations.59
7.3 The contrasting effect of β-adrenergic receptor blockade
While β-adrenergic receptor antagonists reduce heart rate, atenolol and most of the selective β-blockers impair endothelial function, decrease insulin sensitivity, and increase lipid levels,70 conditions that increase the burden of cardiovascular risks. New generation of ‘vasodilating’ β-blockers (such nebivolol and carvedilol)71 have been reported to improve metabolism and endothelial function.72 It is clear that this new generation of agents are better at reducing central aortic blood pressure,73 which may explain their apparent better efficacy, but large clinical trials are needed to validate these early findings.
The endothelium expresses β-adrenergic receptors and their activation by circulating catecholamines has been proposed to activate a survival pathway.74 Although this remains an hypothesis, this may explain why the LIFE (Losartan Intervention For Endpoint) study showed that for an equal resting heart rate, there was a 28% greater cardiovascular risk associated with pulse pressure in atenolol-treated patients with hypertension and left ventricular hypertrophy than in patients treated with the angiotensin II receptor antagonist losartan. Most importantly, an even greater risk (84%) of stroke was reported in patients treated with atenolol compared with losartan.75,76 In the MORE (Multicentre Olmesartan atherosclerosis Regression Evaluation) study, when the baseline plaque-volume was greater than 33.7 μL, atenolol did not promote its regression in contrast to the angiotensin II receptor antagonist olmesartan.77 In this latter study, heart rate was not reported but the two receptor antagonists reduced blood pressure similarly. Meta-analyses have shown that despite lowering blood pressure and heart rate, β-adrenergic receptor blockage was not effective in reducing cardiovascular events when compared with either placebo or other antihypertensive agents.78
Activation of endothelial β-adrenergic receptors stimulates NO release,79 and a reduced bioavailability of NO is linked to increased arterial stiffness.80 Arterial stiffness is so damaging to the cardiovascular system that restoring arterial elasticity would be of great therapeutic benefit.81 In our study,59 pure heart rate reduction by ivabradine protected both cerebral and renal endothelial function. We also tested the effects of metoprolol, a β-adrenergic receptor antagonist, which reduced heart rate similarly to ivabradine without reducing blood pressure. Metoprolol partially protected renal but not cerebral arterial dilation. This suggests that a reduction in resting heart rate limits mechanical stress in conduit renal arteries, a beneficial effect that may be partially counterbalanced by inhibition of the protective endothelial β-adrenergic receptor survival pathway. Mechanical stress being of less impact in cerebral arteries, inhibition of endothelial β-adrenergic receptors likely negated the beneficial effects of an improved shear stress profile. As mentioned previously, this remains however, a working hypothesis to be tested.
7.4 The baroreflex
By reducing heart rate, the sensitivity of the baroreflex increases.38,82 This is confirmed in humans using β-adrenergic receptor antagonists83,84 but not with all of them.85 The autonomic nervous system is perhaps the most important defender of homeostasis. Thus, by reducing heart rate and increasing baroreflex sensitivity, there is a better match between blood pressure and sympathetic activity, favouring greater parasympathetic tone, as observed with the non-selective β-adrenergic receptor antagonist carvedilol in heart failure patients.84 This also means better long-term regulation of the rennin–angiotensin–aldosterone system, as suggested by a reduced level of circulating angiotensin-II in ivabradine-treated rats.86 Parasympathetic tone, however, decreases with age and this is associated with a reduction in resting heart rate variability between night and day and a lower beat-to-beat adaptation to physiological haemodynamic stimuli.87
Long-term regulation of blood pressure is highly dependent on the kidney via blood volume regulation.88,89 Blood pressure increases with age, but this is directly proportional to daily salt consumption: in societies where salt intake is high, blood pressure rises by 1 mmHg per year after the age of 30.90 In this context, what could be the role of the baroreflex? The relationship between short-term baroreflex, sympathetic activity, heart rate, and blood pressure is obvious, but the link with hypertension is not, because it is generally accepted that the baroreflex is not an important regulator of blood pressure in the long term. Baro-denervation in animals does not change blood pressure over a 24 h period.91 Total peripheral resistance and cardiac output do not change, but mean blood pressure variability increases enormously between 50 and 140 mmHg compared with 80 and 125 mmHg in controls. This therefore supports the accepted idea that the baroreflex is a short-term regulator of blood pressure. There are, however, indications to the contrary. In baro-denervated animals, a high-salt diet increases blood pressure dramatically.92 In addition, chronic activation of the baroreflex causes sustained reduction in blood pressure in normotensive animals, with little evidence that there is activation of renal mechanisms to increase blood volume and thus, normalize pressure.93 It has been shown that sympathetic activity increases with age and obesity, which raises blood pressure.94–96 Importantly, NO synthesis blockade impairs the cardiovascular autonomic adaptations induced by physical training in rats that might be, at least in part, ascribed to a decreased baroreflex sensitivity,97 a mechanisms that has been confirmed in rabbits with congestive heart failure due to an augmentation in the vagal tone.98 In fact, NO has more impact on the regulation by the baroreflex of heart rate than on the renal sympathetic nerve activity.99 In leptin-deficient mice, which are diabetic, hypertensive, and with an elevated resting heart rate, the rennin–angiotensin system is activated and could be responsible for the greater sympathetic and decreased parasympathetic tone in these mice.100 These observations have important implications for prematurely ageing and obese western populations: since ageing, hypertension, and obesity increase arterial stiffness and reduce baroreflex sensitivity, any way to increase this sensitivity is likely to be beneficial for the cardiovascular system and its longevity. A reduction in heart rate does so, and a better baroreflex control doubtless accounts for the long-term beneficial effects of exercise on the endothelium, the vascular wall, and thus cardiovascular longevity.
8. Concluding remarks
In this review, we have described evidences suggesting that resting heart rate, if elevated, is deleterious to the cardiovascular system. Our working hypothesis is that each heartbeat imposes a mechanical stress on the endothelium, and clearly over time, billions of them can induce damage. Mechanical stress is unavoidable, but its magnitude varies from one individual to another. We propose that this mechanical stress is magnified by additional stress factors, such as hypertension, as well as smoking, dyslipidaemia, and diabetes. If this hypothesis was validated, it would be essential to detect and treat cardiovascular risk factors—elevated resting heart rate being one of them—as early as during childhood.101 Stress and repair start in utero102 and atherosclerosis develops soon after childhood, as discovered in young US soldiers killed during the Korean War. Early control of risk factors will therefore benefit the patient. Reducing an abnormal elevated resting heart rate would be a way of increasing the number of healthy years of life by minimizing the primary stress imposed on the vascular endothelium and wall.
Acknowledgments
Funding
This work is supported by the Canadian Institutes for Health Research (MOP14496; MOP87388; MOP89733), the Heart & Stroke Foundation of Canada, and the Foundation of the Montreal Heart Institute.
Footnotes
Conflict of interest: E.T. received research grants and an honorarium from Servier.
References
- 1.Palatini P, Benetos A, Julius S. Impact of increased heart rate on clinical outcomes in hypertension: implications for antihypertensive drug therapy. Drugs. 2006;66:133–144. doi: 10.2165/00003495-200666020-00001. [DOI] [PubMed] [Google Scholar]
- 2.Cook S, Togni M, Schaub MC, Wenaweser P, Hess OM. High heart rate: a cardiovascular risk factor? Eur Heart J. 2006;27:2387–2393. doi: 10.1093/eurheartj/ehl259. [DOI] [PubMed] [Google Scholar]
- 3.Diaz A, Bourassa MG, Guertin MC, Tardif JC. Long-term prognostic value of resting heart rate in patients with suspected or proven coronary artery disease. Eur Heart J. 2005;26:967–974. doi: 10.1093/eurheartj/ehi190. [DOI] [PubMed] [Google Scholar]
- 4.Fox K, Ford I, Steg PG, Tendera M, Robertson M, Ferrari R. Heart rate as a prognostic risk factor in patients with coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): a subgroup analysis of a randomised controlled trial. Lancet. 2008;372:817–821. doi: 10.1016/S0140-6736(08)61171-X. [DOI] [PubMed] [Google Scholar]
- 5.Bohm M, Reil JC. Perspectives of I(f) inhibition by ivabradine in cardiology. Drugs. 2007;67(Suppl 2):43–49. doi: 10.2165/00003495-200767002-00006. [DOI] [PubMed] [Google Scholar]
- 6.Altschul R. Endothelium, its Development, Morphology, Function, and Pathology. New York: MacMillan; 1954. p. vii. [Google Scholar]
- 7.Libby P. Inflammatory mechanisms: the molecular basis of inflammation and disease. Nutr Rev. 2007;65:S140–S146. doi: 10.1111/j.1753-4887.2007.tb00352.x. [DOI] [PubMed] [Google Scholar]
- 8.Liu Y, Gutterman DD. Vascular control in humans: focus on the coronary microcirculation. Basic Res Cardiol. 2009;104:211–227. doi: 10.1007/s00395-009-0775-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holliday R. Aging is no longer an unsolved problem in biology. Ann NY Acad Sci. 2006;1067:1–9. doi: 10.1196/annals.1354.002. [DOI] [PubMed] [Google Scholar]
- 10.Brandes RP, Fleming I, Busse R. Endothelial aging. Cardiovasc Res. 2005;66:286–294. doi: 10.1016/j.cardiores.2004.12.027. [DOI] [PubMed] [Google Scholar]
- 11.Csiszar A, Ungvari Z, Edwards JG, Kaminski P, Wolin MS, Koller A, et al. Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ Res. 2002;90:1159–1166. doi: 10.1161/01.res.0000020401.61826.ea. [DOI] [PubMed] [Google Scholar]
- 12.Gendron ME, Thorin-Trescases N, Villeneuve L, Thorin E. Aging associated with mild dyslipidemia reveals that COX-2 preserves dilation despite endothelial dysfunction. Am J Physiol. 2007;292:H451–H458. doi: 10.1152/ajpheart.00551.2006. [DOI] [PubMed] [Google Scholar]
- 13.Taddei S, Virdis A, Mattei P, Ghiadoni L, Gennari A, Fasolo CB, et al. Aging and endothelial function in normotensive subjects and patients with essential hypertension. Circulation. 1995;91:1981–1987. doi: 10.1161/01.cir.91.7.1981. [DOI] [PubMed] [Google Scholar]
- 14.Greenwald SE. Ageing of the conduit arteries. J Pathol. 2007;211:157–172. doi: 10.1002/path.2268. [DOI] [PubMed] [Google Scholar]
- 15.Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part I: aging arteries: a ‘set up’ for vascular disease. Circulation. 2003;107:139–146. doi: 10.1161/01.cir.0000048892.83521.58. [DOI] [PubMed] [Google Scholar]
- 16.Foteinos G, Hu Y, Xiao Q, Metzler B, Xu Q. Rapid endothelial turnover in atherosclerosis-prone areas coincides with stem cell repair in apolipoprotein E-deficient mice. Circulation. 2008;117:1856–1863. doi: 10.1161/CIRCULATIONAHA.107.746008. [DOI] [PubMed] [Google Scholar]
- 17.Hill JM, Zalos G, Halcox JP, Schenke WH, Waclawiw MA, Quyyumi AA, et al. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348:593–600. doi: 10.1056/NEJMoa022287. [DOI] [PubMed] [Google Scholar]
- 18.Shimokawa H, Aarhus LL, Vanhoutte PM. Porcine coronary arteries with regenerated endothelium have a reduced endothelium-dependent responsiveness to aggregating platelets and serotonin. Circ Res. 1987;61:256–270. doi: 10.1161/01.res.61.2.256. [DOI] [PubMed] [Google Scholar]
- 19.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 20.Chang E, Harley CB. Telomere length and replicative aging in human vascular tissues. Proc Natl Acad Sci USA. 1995;92:11190–11194. doi: 10.1073/pnas.92.24.11190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Voghel G, Thorin-Trescases N, Farhat N, Mamarbachi AM, Villeneuve L, Fortier A, et al. Chronic treatment with N-acetyl-cystein delays cellular senescence in endothelial cells isolated from a subgroup of atherosclerotic patients. Mech Ageing Dev. 2008;129:261–270. doi: 10.1016/j.mad.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burrig KF. The endothelium of advanced arteriosclerotic plaques in humans. Arterioscler Thromb. 1991;11:1678–1689. [PubMed] [Google Scholar]
- 23.Chen J, Goligorsky MS. Premature senescence of endothelial cells: Methusaleh’s dilemma. Am J Physiol. 2006;290:H1729–H1739. doi: 10.1152/ajpheart.01103.2005. [DOI] [PubMed] [Google Scholar]
- 24.Minamino T, Komuro I. Vascular cell senescence: contribution to atherosclerosis. Circ Res. 2007;100:15–26. doi: 10.1161/01.RES.0000256837.40544.4a. [DOI] [PubMed] [Google Scholar]
- 25.Voghel G, Thorin-Trescases N, Farhat N, Nguyen A, Villeneuve L, Mamarbachi AM, et al. Cellular senescence in endothelial cells from atherosclerotic patients is accelerated by oxidative stress associated with cardiovascular risk factors. Mech Ageing Dev. 2007;128:662–671. doi: 10.1016/j.mad.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 26.Malek AM, Alper SL, Izumo S. Hemodynamic shear stress and its role in atherosclerosis. JAMA. 1999;282:2035–2042. doi: 10.1001/jama.282.21.2035. [DOI] [PubMed] [Google Scholar]
- 27.Chatzizisis YS, Coskun AU, Jonas M, Edelman ER, Feldman CL, Stone PH. Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodeling: molecular, cellular, and vascular behavior. J Am Coll Cardiol. 2007;49:2379–2393. doi: 10.1016/j.jacc.2007.02.059. [DOI] [PubMed] [Google Scholar]
- 28.Guyton AC, Hall JE. Cardiac arrhythmias and their electrocardiographic interpretation. In: Hall G, editor. Textbook of medical physiology. 9. Philadelphia: W.B. Saunders company; 1996. pp. 149–150. [Google Scholar]
- 29.Esler M, Alvarenga M, Pier C, Richards J, El-Osta A, Barton D, et al. The neuronal noradrenaline transporter, anxiety and cardiovascular disease. J Psychopharmacol. 2006;20(Suppl 4):60–66. doi: 10.1177/1359786806066055. [DOI] [PubMed] [Google Scholar]
- 30.Strawn WB, Bondjers G, Kaplan JR, Manuck SB, Schwenke DC, Hansson GK, et al. Endothelial dysfunction in response to psychosocial stress in monkeys. Circ Res. 1991;68:1270–1279. doi: 10.1161/01.res.68.5.1270. [DOI] [PubMed] [Google Scholar]
- 31.Kaplan JR, Manuck SB, Adams MR, Weingand KW, Clarkson TB. Inhibition of coronary atherosclerosis by propranolol in behaviorally predisposed monkeys fed an atherogenic diet. Circulation. 1987;76:1364–1372. doi: 10.1161/01.cir.76.6.1364. [DOI] [PubMed] [Google Scholar]
- 32.Esler M, Straznicky N, Eikelis N, Masuo K, Lambert G, Lambert E. Mechanisms of sympathetic activation in obesity-related hypertension. Hypertension. 2006;48:787–796. doi: 10.1161/01.HYP.0000242642.42177.49. [DOI] [PubMed] [Google Scholar]
- 33.Shigetoh Y, Adachi H, Yamagishi S, Enomoto M, Fukami A, Otsuka M, et al. Higher heart rate may predispose to obesity and diabetes mellitus: 20-year prospective study in a general population. Am J Hypertens. 2009;22:151–155. doi: 10.1038/ajh.2008.331. [DOI] [PubMed] [Google Scholar]
- 34.Nagae A, Fujita M, Kawarazaki H, Matsui H, Ando K, Fujita T. Sympathoexcitation by oxidative stress in the brain mediates arterial pressure elevation in obesity-induced hypertension. Circulation. 2009;119:978–986. doi: 10.1161/CIRCULATIONAHA.108.824730. [DOI] [PubMed] [Google Scholar]
- 35.Lee CW, Li D, Channon KM, Paterson DJ. L-arginine supplementation reduces cardiac noradrenergic neurotransmission in spontaneously hypertensive rats. J Mol Cell Cardiol. 2009;47:149–155. doi: 10.1016/j.yjmcc.2009.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Danson EJ, Li D, Wang L, Dawson TA, Paterson DJ. Targeting cardiac sympathovagal imbalance using gene transfer of nitric oxide synthase. J Mol Cell Cardiol. 2009;46:482–489. doi: 10.1016/j.yjmcc.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 37.Kolloch R, Legler UF, Champion A, Cooper-Dehoff RM, Handberg E, Zhou Q, et al. Impact of resting heart rate on outcomes in hypertensive patients with coronary artery disease: findings from the INternational VErapamil-SR/trandolapril STudy (INVEST) Eur Heart J. 2008;29:1327–1334. doi: 10.1093/eurheartj/ehn123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sa Cunha R, Pannier B, Benetos A, Siche JP, London GM, Mallion JM, et al. Association between high heart rate and high arterial rigidity in normotensive and hypertensive subjects. J Hypertens. 1997;15:1423–1430. doi: 10.1097/00004872-199715120-00009. [DOI] [PubMed] [Google Scholar]
- 39.Vequaud P, Thorin E. Endothelial G protein beta-subunits trigger nitric oxide-but not endothelium-derived hyperpolarizing factor-dependent dilation in rabbit resistance arteries. Circ Res. 2001;89:716–722. doi: 10.1161/hh2001.097783. [DOI] [PubMed] [Google Scholar]
- 40.Balligand JL, Feron O, Dessy C. eNOS activation by physical forces: from short-term regulation of contraction to chronic remodeling of cardiovascular tissues. Physiol Rev. 2009;89:481–534. doi: 10.1152/physrev.00042.2007. [DOI] [PubMed] [Google Scholar]
- 41.Drouin A, Thorin E. Flow-induced dilation is mediated by Akt-dependent activation of endothelial nitric oxide synthase-derived hydrogen peroxide in mouse cerebral arteries. Stroke. 2009;40:1827–1833. doi: 10.1161/STROKEAHA.108.536805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cai H, Davis ME, Drummond GR, Harrison DG. Induction of endothelial NO synthase by hydrogen peroxide via a Ca(2+)/calmodulin-dependent protein kinase II/janus kinase 2-dependent pathway. Arterioscler Thromb Vasc Biol. 2001;21:1571–1576. doi: 10.1161/hq1001.097028. [DOI] [PubMed] [Google Scholar]
- 43.Cai H, McNally JS, Weber M, Harrison DG. Oscillatory shear stress upregulation of endothelial nitric oxide synthase requires intracellular hydrogen peroxide and CaMKII. J Mol Cell Cardiol. 2004;37:121–125. doi: 10.1016/j.yjmcc.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 44.Ji LL. Antioxidants and oxidative stress in exercise. Proc Soc Exp Biol Med. 1999;222:283–292. doi: 10.1046/j.1525-1373.1999.d01-145.x. [DOI] [PubMed] [Google Scholar]
- 45.Heusch G. Heart rate in the pathophysiology of coronary blood flow and myocardial ischaemia: benefit from selective bradycardic agents. Br J Pharmacol. 2008;153:1589–1601. doi: 10.1038/sj.bjp.0707673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ludmer PL, Selwyn AP, Shook TL, Wayne RR, Mudge GH, Alexander RW, et al. Paradoxical vasoconstriction induced by acetylcholine in atherosclerotic coronary arteries. N Engl J Med. 1986;315:1046–1051. doi: 10.1056/NEJM198610233151702. [DOI] [PubMed] [Google Scholar]
- 47.Quyyumi AA, Dakak N, Andrews NP, Husain S, Arora S, Gilligan DM, et al. Nitric oxide activity in the human coronary circulation. Impact of risk factors for coronary atherosclerosis. J Clin Invest. 1995;95:1747–1755. doi: 10.1172/JCI117852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paciaroni M, Hennerici M, Agnelli G, Bogousslavsky J. Statins and stroke prevention. Cerebrovasc Dis. 2007;24:170–182. doi: 10.1159/000104474. [DOI] [PubMed] [Google Scholar]
- 49.Thorin-Trescases N, Bartolotta T, Hyman N, Penar PL, Walters CL, Bevan RD, et al. Diameter dependence of myogenic tone of human pial arteries.Possible relation to distensibility. Stroke. 1997;28:2486–2492. doi: 10.1161/01.str.28.12.2486. [DOI] [PubMed] [Google Scholar]
- 50.Levy RL, White PD, Stroud WD. Transient tachycardia: pronostic significance alone and in association with transient hypertension. JAMA. 1945;129:585–588. [PubMed] [Google Scholar]
- 51.Beere PA, Glagov S, Zarins CK. Experimental atherosclerosis at the carotid bifurcation of the cynomolgus monkey. Localization, compensatory enlargement, and the sparing effect of lowered heart rate. Arterioscler Thromb. 1992;12:1245–1253. doi: 10.1161/01.atv.12.11.1245. [DOI] [PubMed] [Google Scholar]
- 52.Beere PA, Glagov S, Zarins CK. Retarding effect of lowered heart rate on coronary atherosclerosis. Science. 1984;226:180–182. doi: 10.1126/science.6484569. [DOI] [PubMed] [Google Scholar]
- 53.Green DJ, O’Driscoll G, Joyner MJ, Cable NT. Exercise and cardiovascular risk reduction: time to update the rationale for exercise? J Appl Physiol. 2008;105:766–768. doi: 10.1152/japplphysiol.01028.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chakravarty EF, Hubert HB, Lingala VB, Fries JF. Reduced disability and mortality among aging runners: a 21-year longitudinal study. Arch Intern Med. 2008;168:1638–1646. doi: 10.1001/archinte.168.15.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Niebauer J, Hambrecht R, Velich T, Hauer K, Marburger C, Kalberer B, et al. Attenuated progression of coronary artery disease after 6 years of multifactorial risk intervention: role of physical exercise. Circulation. 1997;96:2534–2541. doi: 10.1161/01.cir.96.8.2534. [DOI] [PubMed] [Google Scholar]
- 56.Hambrecht R, Walther C, Mobius-Winkler S, Gielen S, Linke A, Conradi K, et al. Percutaneous coronary angioplasty compared with exercise training in patients with stable coronary artery disease: a randomized trial. Circulation. 2004;109:1371–1378. doi: 10.1161/01.CIR.0000121360.31954.1F. [DOI] [PubMed] [Google Scholar]
- 57.Ristow M, Zarse K, Oberbach A, Kloting N, Birringer M, Kiehntopf M, et al. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc Natl Acad Sci USA. 2009;106:8665–8670. doi: 10.1073/pnas.0903485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vilaine JP. The discovery of the selective I(f) current inhibitor ivabradine.A new therapeutic approach to ischemic heart disease. Pharmacol Res. 2006;53:424–434. doi: 10.1016/j.phrs.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 59.Drouin A, Gendron ME, Thorin E, Gillis MA, Mahlberg-Gaudin F, Tardif JC. Chronic heart rate reduction by ivabradine prevents endothelial dysfunction in dyslipidaemic mice. Br J Pharmacol. 2008;154:749–757. doi: 10.1038/bjp.2008.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Custodis F, Baumhakel M, Schlimmer N, List F, Gensch C, Bohm M, et al. Heart rate reduction by ivabradine reduces oxidative stress, improves endothelial function, and prevents atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2008;117:2377–2387. doi: 10.1161/CIRCULATIONAHA.107.746537. [DOI] [PubMed] [Google Scholar]
- 61.Fox K, Ford I, Steg PG, Tendera M, Ferrari R. Ivabradine for patients with stable coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): a randomised, double-blind, placebo-controlled trial. Lancet. 2008;372:807–816. doi: 10.1016/S0140-6736(08)61170-8. [DOI] [PubMed] [Google Scholar]
- 62.Heusch G, Skyschally A, Gres P, van Caster P, Schilawa D, Schulz R. Improvement of regional myocardial blood flow and function and reduction of infarct size with ivabradine: protection beyond heart rate reduction. Eur Heart J. 2008;29:2265–2275. doi: 10.1093/eurheartj/ehn337. [DOI] [PubMed] [Google Scholar]
- 63.Heistad DD. What’s new in the cerebral microcirculation? Landis Award lecture. Microcirculation. 2001;8:365–375. doi: 10.1038/sj/mn/7800109. [DOI] [PubMed] [Google Scholar]
- 64.Korshunov VA, Berk BC. Strain-dependent vascular remodeling: the ‘Glagov phenomenon’ is genetically determined. Circulation. 2004;110:220–226. doi: 10.1161/01.CIR.0000134958.88379.2E. [DOI] [PubMed] [Google Scholar]
- 65.Taddei S, Virdis A, Mattei P, Ghiadoni L, Fasolo CB, Sudano I, et al. Hypertension causes premature aging of endothelial function in humans. Hypertension. 1997;29:736–743. doi: 10.1161/01.hyp.29.3.736. [DOI] [PubMed] [Google Scholar]
- 66.Wallace SM, Yasmin, McEniery CM, Maki-Petaja KM, Booth AD, Cockcroft JR, et al. Isolated systolic hypertension is characterized by increased aortic stiffness and endothelial dysfunction. Hypertension. 2007;50:228–233. doi: 10.1161/HYPERTENSIONAHA.107.089391. [DOI] [PubMed] [Google Scholar]
- 67.Schwartz SM, Benditt EP. Aortic endothelial cell replication. I. Effects of age and hypertension in the rat. Circ Res. 1977;41:248–255. doi: 10.1161/01.res.41.2.248. [DOI] [PubMed] [Google Scholar]
- 68.Schwartz SM. Role of endothelial integrity in atherosclerosis. Artery. 1980;8:305–314. [PubMed] [Google Scholar]
- 69.Heusch G. Pleiotropic action(s) of the bradycardic agent ivabradine: cardiovascular protection beyond heart rate reduction. Br J Pharmacol. 2008;155:970–971. doi: 10.1038/bjp.2008.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lithell HO. Effect of antihypertensive drugs on insulin, glucose, and lipid metabolism. Diabetes Care. 1991;14:203–209. doi: 10.2337/diacare.14.3.203. [DOI] [PubMed] [Google Scholar]
- 71.Pedersen ME, Cockcroft JR. The vasodilatory beta-blockers. Curr Hypertens Rep. 2007;9:269–277. doi: 10.1007/s11906-007-0050-2. [DOI] [PubMed] [Google Scholar]
- 72.Celik T, Iyisoy A, Kursaklioglu H, Kardesoglu E, Kilic S, Turhan H, et al. Comparative effects of nebivolol and metoprolol on oxidative stress, insulin resistance, plasma adiponectin and soluble P-selectin levels in hypertensive patients. J Hypertens. 2006;24:591–596. doi: 10.1097/01.hjh.0000209993.26057.de. [DOI] [PubMed] [Google Scholar]
- 73.Dhakam Z, Yasmin, McEniery CM, Burton T, Brown MJ, Wilkinson IB. A comparison of atenolol and nebivolol in isolated systolic hypertension. J Hypertens. 2008;26:351–356. doi: 10.1097/HJH.0b013e3282f283c9. [DOI] [PubMed] [Google Scholar]
- 74.Cockcroft J. A review of the safety and efficacy of nebivolol in the mildly hypertensive patient. Vasc Health Risk Manag. 2007;3:909–917. [PMC free article] [PubMed] [Google Scholar]
- 75.Dahlof B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, de Faire U, et al. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet. 2002;359:995–1003. doi: 10.1016/S0140-6736(02)08089-3. [DOI] [PubMed] [Google Scholar]
- 76.Fyhrquist F, Dahlof B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, et al. Pulse pressure and effects of losartan or atenolol in patients with hypertension and left ventricular hypertrophy. Hypertension. 2005;45:580–585. doi: 10.1161/01.HYP.0000161186.55933.6b. [DOI] [PubMed] [Google Scholar]
- 77.Stumpe KO, Agabiti-Rosei E, Zielinski T, Schremmer D, Scholze J, Laeis P, et al. Carotid intima-media thickness and plaque volume changes following 2-year angiotensin II-receptor blockade. The Multicentre Olmesartan atherosclerosis Regression Evaluation (MORE) study. Ther Adv Cardiovasc Dis. 2007;1:97–106. doi: 10.1177/1753944707085982. [DOI] [PubMed] [Google Scholar]
- 78.Lindholm LH, Carlberg B, Samuelsson O. Should beta blockers remain first choice in the treatment of primary hypertension? A meta-analysis. Lancet. 2005;366:1545–1553. doi: 10.1016/S0140-6736(05)67573-3. [DOI] [PubMed] [Google Scholar]
- 79.Ciccarelli M, Cipolletta E, Santulli G, Campanile A, Pumiglia K, Cervero P, et al. Endothelial beta2 adrenergic signaling to AKT: role of Gi and SRC. Cell Signal. 2007;19:1949–1955. doi: 10.1016/j.cellsig.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 80.Wilkinson IB, Franklin SS, Cockcroft JR. Nitric oxide and the regulation of large artery stiffness: from physiology to pharmacology. Hypertension. 2004;44:112–116. doi: 10.1161/01.HYP.0000138068.03893.40. [DOI] [PubMed] [Google Scholar]
- 81.Hadley EC, Lakatta EG, Morrison-Bogorad M, Warner HR, Hodes RJ. The future of aging therapies. Cell. 2005;120:557–567. doi: 10.1016/j.cell.2005.01.030. [DOI] [PubMed] [Google Scholar]
- 82.Ramirez-Marrero FA, Charkoudian N, Hart EC, Schroeder DR, Zhong L, Eisenach JH, et al. Cardiovascular dynamics in healthy subjects with differing heart rate responses to tilt. J Appl Physiol. 2008;105:1448–1453. doi: 10.1152/japplphysiol.90796.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Melenovsky V, Simek J, Sperl M, Malik J, Wichterle D. Relation between actual heart rate and autonomic effects of beta blockade in healthy men. Am J Cardiol. 2005;95:999–1002. doi: 10.1016/j.amjcard.2004.12.048. [DOI] [PubMed] [Google Scholar]
- 84.Mortara A, La Rovere MT, Pinna GD, Maestri R, Capomolla S, Cobelli F. Nonselective beta-adrenergic blocking agent, carvedilol, improves arterial baroflex gain and heart rate variability in patients with stable chronic heart failure. J Am Coll Cardiol. 2000;36:1612–1618. doi: 10.1016/s0735-1097(00)00900-1. [DOI] [PubMed] [Google Scholar]
- 85.Floras JS, Jones JV, Hassan MO, Sleight P. Effects of acute and chronic beta-adrenoceptor blockade on baroreflex sensitivity in humans. J Auton Nerv Syst. 1988;25:87–94. doi: 10.1016/0165-1838(88)90013-6. [DOI] [PubMed] [Google Scholar]
- 86.Dedkov EI, Zheng W, Christensen LP, Weiss RM, Mahlberg-Gaudin F, Tomanek RJ. Preservation of coronary reserve by ivabradine-induced reduction in heart rate in infarcted rats is associated with decrease in perivascular collagen. Am J Physiol. 2007;293:H590–H598. doi: 10.1152/ajpheart.00047.2007. [DOI] [PubMed] [Google Scholar]
- 87.Bonnemeier H, Richardt G, Potratz J, Wiegand UK, Brandes A, Kluge N, et al. Circadian profile of cardiac autonomic nervous modulation in healthy subjects: differing effects of aging and gender on heart rate variability. J Cardiovasc Electrophysiol. 2003;14:791–799. doi: 10.1046/j.1540-8167.2003.03078.x. [DOI] [PubMed] [Google Scholar]
- 88.Rettig R, Folberth C, Stauss H, Kopf D, Waldherr R, Unger T. Role of the kidney in primary hypertension: a renal transplantation study in rats. Am J Physiol. 1990;258:F606–F611. doi: 10.1152/ajprenal.1990.258.3.F606. [DOI] [PubMed] [Google Scholar]
- 89.Romero JC, Reckelhoff JF. State-of-the-Art lecture. Role of angiotensin and oxidative stress in essential hypertension. Hypertension. 1999;34:943–949. doi: 10.1161/01.hyp.34.4.943. [DOI] [PubMed] [Google Scholar]
- 90.Meneton P, Jeunemaitre X, de Wardener HE, MacGregor GA. Links between dietary salt intake, renal salt handling, blood pressure, and cardiovascular diseases. Physiol Rev. 2005;85:679–715. doi: 10.1152/physrev.00056.2003. [DOI] [PubMed] [Google Scholar]
- 91.Cowley AW, Jr, Liard JF, Guyton AC. Role of baroreceptor reflex in daily control of arterial blood pressure and other variables in dogs. Circ Res. 1973;32:564–576. doi: 10.1161/01.res.32.5.564. [DOI] [PubMed] [Google Scholar]
- 92.Osborn JW, Hornfeldt BJ. Arterial baroreceptor denervation impairs long-term regulation of arterial pressure during dietary salt loading. Am J Physiol. 1998;275:H1558–H1566. doi: 10.1152/ajpheart.1998.275.5.H1558. [DOI] [PubMed] [Google Scholar]
- 93.Lohmeier TE, Dwyer TM, Hildebrandt DA, Irwin ED, Rossing MA, Serdar DJ, et al. Influence of prolonged baroreflex activation on arterial pressure in angiotensin hypertension. Hypertension. 2005;46:1194–1200. doi: 10.1161/01.HYP.0000187011.44201.2e. [DOI] [PubMed] [Google Scholar]
- 94.Seals DR, Bell C. Chronic sympathetic activation: consequence and cause of age-associated obesity? Diabetes. 2004;53:276–284. doi: 10.2337/diabetes.53.2.276. [DOI] [PubMed] [Google Scholar]
- 95.Shibao C, Gamboa A, Diedrich A, Ertl AC, Chen KY, Byrne DW, et al. Autonomic contribution to blood pressure and metabolism in obesity. Hypertension. 2007;49:27–33. doi: 10.1161/01.HYP.0000251679.87348.05. [DOI] [PubMed] [Google Scholar]
- 96.Sundlof G, Wallin BG. Human muscle nerve sympathetic activity at rest. Relationship to blood pressure and age. J Physiol. 1978;274:621–637. doi: 10.1113/jphysiol.1978.sp012170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Souza HCD, De Araújo JE, Martins-Pinge MC, Cozza IC, Martins-Diasa DP. Nitric oxide synthesis blockade reduced the baroreflex sensitivity in trained rats. Auton Neurosci. 2009 doi: 10.1016/j.autneu.2009.04.007. (in press) [DOI] [PubMed] [Google Scholar]
- 98.Liu JL, Kulakofsky J, Zucker IH. Exercise training enhances baroreflex control of heart rate by a vagal mechanism in rabbits with heart failure. J Appl Physiol. 2002;92:2403–2408. doi: 10.1152/japplphysiol.00039.2002. [DOI] [PubMed] [Google Scholar]
- 99.Liu JL, Murakami H, Zucker IH. Effects of NO on baroreflex control of heart rate and renal nerve activity in conscious rabbits. Am J Physiol. 1996;270:R1361–R1370. doi: 10.1152/ajpregu.1996.270.6.R1361. [DOI] [PubMed] [Google Scholar]
- 100.Goncalves AC, Tank J, Diedrich A, Hilzendeger A, Plehm R, Bader M, et al. Diabetic hypertensive leptin receptor-deficient db/db mice develop cardioregulatory autonomic dysfunction. Hypertension. 2009;53:387–392. doi: 10.1161/HYPERTENSIONAHA.108.124776. [DOI] [PubMed] [Google Scholar]
- 101.Gidding SS. Measuring children’s blood pressure matters. Circulation. 2008;117:3163–3164. doi: 10.1161/CIRCULATIONAHA.108.787168. [DOI] [PubMed] [Google Scholar]
- 102.Tarry-Adkins JL, Martin-Gronert MS, Chen JH, Cripps RL, Ozanne SE. Maternal diet influences DNA damage, aortic telomere length, oxidative stress, and antioxidant defense capacity in rats. FASEB J. 2008;22:2037–2044. doi: 10.1096/fj.07-099523. [DOI] [PubMed] [Google Scholar]