

Abstract
Introduction
With aging, oxidative stress accelerates vascular endothelial cell (EC) telomere shortening-induced senescence, and may promote atherosclerosis in humans. Our aim was to investigate whether an antioxidant treatment combined with telomerase (hTERT) over-expression would prevent senescence of EC isolated from patients with severe atherosclerosis.
Methods
Cells were isolated from internal mammary arteries (n = 11 donors), cultured until senescence with or without N-acetylcystein (NAC) and infected, or not, with a lentivirus over-expressing hTERT.
Results
Compared to control EC, hTERT-NAC cells had increased telomerase activity, longer telomeres and underwent more cell divisions. According to the donor, hTERT-NAC either delayed (n = 5) or prevented (n = 4) EC senescence, the latter leading to cell immortalization. Lack of cell immortalization by hTERT-NAC was accompanied by an absence of beneficial effect of NAC alone in paired EC. Accordingly, lack of EC immortalization by hTERT-NAC was associated with high endogenous susceptibility to oxidation. In EC where hTERT-NAC did not immortalize EC, p53, p21 and p16 expression increased with senescence, while oxidative-dependent DNA damage associated with senescence was not prevented.
Conclusion
Our data suggest that irreversible oxidative stress-dependent damages associated with cardiovascular risk factors are responsible for senescence of EC from atherosclerotic patients.
Keywords: Telomerase, Endothelium, Oxidative stress, Cellular senescence, Coronary artery disease
1. Introduction
Cellular senescence is a state of permanent growth arrest in which cells remain alive and metabolically active for months, but refractory to mitogenic stimuli (Ben-Porath and Weinberg, 2005; Chen and Goligorsky, 2006). Telomere attrition provided the first molecular explanation for the growth arrest observed in most of somatic cells cultured in vitro (Harley et al., 1990; Levy et al., 1992). Due to the end-replication problem, telomeres shorten at each cell division down to a threshold where it is recognized as DNA damage, and thus, initiates replicative senescence (Allsopp et al., 1995; Brown et al., 1997; Lou and Chen, 2006). Telomerase, a ribonucleo-protein responsible for telomere maintenance, is minimally active in somatic cells (Allsopp et al., 1995; Kim et al., 1994). On the other hand, senescence is referred to accelerated replicative senescence, when mild repeated oxidative stress induces telomeric DNA damage (Erusalimsky and Kurz, 2005; Houben et al., 2008; Kawanishi and Oikawa, 2004; Passos and Von Zglinicki, 2006; von Zglinicki, 2002; von Zglinicki et al., 2005). Senescence can also be telomere-independent, the so-called stress-induced senescence (SIS), when it is triggered prematurely by sub-lethal oxidative stress inducing non-telomeric DNA damages and growth arrest signals (Chen et al., 2007; Farhat et al., 2008; Klimova et al., 2009; Medrano et al., 1995; Shi et al., 2007). Cellular senescence is now recognized to play an active role in vivo as a tumor suppressor pathway, in the aging process, in the loss of regenerative potential in aging tissues and in the pathogenesis of cardiovascular diseases (Campisi and d’Adda di Fagagna, 2007; Cichowski and Hahn, 2008; Jeyapalan and Sedivy, 2008).
We recently reported that hypertension accelerates the biological aging of vascular EC isolated and cultured from patients with severe coronary artery disease (CAD) exposed for years to various risk factors (Voghel et al., 2007). We observed a significant interplay between replicative senescence and SIS. Chronic antioxidant treatment with N-acetylcystein (NAC) of EC isolated from CAD patients, temporarily delayed senescence by limiting SIS and by reducing telomere shortening via endogenous telomerase activation, unless EC had undergone irreversible oxidative stress-induced cellular and molecular damages (Voghel et al., 2008). In contrast, immortalization has been reported after ectopic expression of the catalytic subunit of telomerase (hTERT) of healthy human skin fibroblasts (Forsyth et al., 2003; Naka et al., 2004; Vaziri and Benchimol, 1998), adrenocortical cells (Yang et al., 2001), retinal pigment epithelial cells (Bodnar et al., 1998) and EC (Yang et al., 1999). Addition of exogenous H2O2 was shown, however, to induce senescence of fibroblasts despite hTERT over-expression (Gorbunova et al., 2002; Naka et al., 2004). Similarly, under hyperoxia, hTERT over-expression in fibroblasts delayed but did not prevent cellular senescence (Ahmed et al., 2008). To better understand the contribution of each pathway and the role played by telomere shortening in EC isolated from CAD patients, we examined whether over-expression of hTERT, in the absence and presence of an antioxidant, can protect from SIS. Our prediction was that if telomere shortening contributes to stress-induced senescence, over-expression of hTERT should be protective; in addition, if oxidative stress plays a role in replicative senescence, antioxidant treatment should be preventive. We found that hTERT over-expression by it-self could not immortalize EC, suggesting that telomere shortening and replicative senescence only play a limited role in the pathway leading to senescence in EC isolated from patients with CAD. We believe that accumulation of cellular damage associated with life-long exposure to cardiovascular risk factor bypasses telomere shortening. It is therefore the extent of the stress-dependent damage that determines EC survival.
2. Methods
2.1. Ethics statement
The study was approved by the ethical committee for clinical research of the Montreal Heart Institute and has therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki. In addition, all the donors gave their informed written consent prior to their inclusion in the study.
2.2. Cell culture
Segments of human distal internal mammary artery (n = 11, Table 1) discarded during primary coronary artery bypass were harvested. We cultured EC as previously described (Voghel et al., 2007). From passage 2 (referred as “initial passage”; cumulative population doubling level ΣPDL = 5.5 ± 0.4, n = 11), EC from each donor were chronically treated in the presence or absence of the antioxidant NAC (10 μM, according to (Voghel et al., 2008). At passage 3 (≈60% confluence), EC from each donor were divided in six groups: cells were non-infected (control), infected with the empty lentivirus FG12-CMV or infected with the lentivirus expressing the catalytic subunit of telomerase FG12-CMV-hTERT, in the absence or presence of NAC. Then, cells were serially passaged until senescence (referred as “final passage”; ΣPDL = 21.5 ± 1.5, n = 11 in control EC).
Table 1.
Clinical profile of patients undergoing coronary artery bypass graft.
| Men (n = 8), Women (n = 3) | |
|---|---|
| Age (years) | 67 ± 5 |
| Body mass index (kg/m2) | 27 ± 2 |
| History of disease (years) | 5 ± 2 |
| Left ventricular ejection fraction, % | 50 ± 5 |
| Dyslipidemia, % | 82 (9/11) |
| Hypertension, % | 73 (8/11) |
| Diabetes, % | 46 (5/11) |
| Nicotine use, % | 73 (8/11) |
Patients were treated with similar medications including aspirin, angiotensin-converting enzyme inhibitors, β-blockers, calcium channel blockers, statins and nitrates.
2.3. hTERT lentiviral infection of EC
The EcoRI/SalI hTERT gene obtained from pBabe-hTERT was inserted into the EcoRI/XhoI sites of the lentiviral vector FG12-CMV (kindly provided by Dr. Maria S. Soengas, University of Michigan, Ann Arbor, MI).
Virus stocks were prepared using transfected 293FT cells with 28 μg of FG12-CMV or FG12-CMV-hTERT with the packaging plasmids expressing Gag-Pol (34.5 μg), Rev (3.6 μg), Tat (3.6 μg) and VSV-G (3.6 μg). Culture supernatant containing viruses produced by packaging 293FT cells were collected, filtered (0.45 μm) and centrifuged at 87,000 × g for 2 h using a SW 28 rotor (Beckman) at 44 h, 52 h and 60 h post-transfection. After decantation, viruses harvested were resuspended in 200 μl of PBS for 2 h at 4 °C. Viruses collected were pooled together and 3 infections of human EC (0 h, 3 h and 7 h) were performed using 750 μl of viruses/infection supplemented with 4 μg/ml polybrene.
2.4. Telomerase activity
Telomere repeat amplification protocol (TRAP) assay was performed to quantify nuclear activity of telomerase as previously described (Kim et al., 1994), with minor modifications. Briefly, for each sample, a PCR amplification of telomerase extension products was performed using 2 ng of nuclear EC protein, 1× TRAP buffer (Tris HCl 200 mM pH 8.3, MgCl2 15 mM, KCl 680 mM, Tween 20 0.5%, EGTA 10 mM), dNTP 50 μM, [32P]-labelled M2-TS forward primer [5′-AATCCGTCGAGCAGAGTT-3′] 1.8 ng/μl, ACX reverse primer [5′-GCGCGG(CTTACC)3CTAACC-3′] 1.8 ng/μl and Taq polymerase 2U. The extension mixture was then loaded and run onto a 10% acrylamide/0.5× TBE gel. Gel was finally fixed 1 h in 5% acetic acid/5% methanol/10% glycerol and exposed on an autoradiogram film. All samples were run in duplicate and either lysis buffer or heat-inactivated samples were used as a negative control.
We also used the sensitive real-time PCR-TRAP assay to measure low nuclear hTERT activity in EC, as previously described (Voghel et al., 2008). Telomerase activity was quantified at the beginning of the cell culture (initial passage) and at final passage (senescence or when cell culture was stopped in immortalized cells) in control non-infected EC, CMV and hTERT infected cells, in the presence or absence of NAC.
2.5. Telomere length measurement
EC were grown in 75 cm2 flasks at early and subsequent passages until senescence. DNA was extracted from EC and telomeric restriction fragments lengths (TRF) were quantified using a Southern blot technique (Voghel et al., 2007; Zhang et al., 2000).
2.6. Senescence-associated β-galactosidase staining
Senescence-associated β-galactosidase (SA-β-Gal) was quantified as previously described (Dimri et al., 1995; Voghel et al., 2007).
2.7. Western blot
Nuclear protein levels of p16 (rabbit anti-p16, 1:200, Santa Cruz) and p21 (mouse anti-p21, 1:200, Santa Cruz) were quantified by Western blot. Protein levels were normalized by GAPDH (1:100000, Ambion).
2.8. Immunofluorescence
Immunostaining was used to assess the expression and sub-cellular localization of a marker of lipid peroxidation 4-hydroxy-nonenal (HNE (Voghel et al., 2008)) (rabbit anti-HNE, 1:200, Alpha Diagnostics), of the oxidative stress marker DHE (Molecular Probes), of a marker of SIS caveolin-1 (mouse anti-caveolin-1, 1:400, BD Transduction Laboratories), of a marker of DNA damage ATM (rabbit anti-ATM, 1:100, Santa Cruz), of cyclin-dependent kinase inhibitors p16 (rabbit anti-p16, 1:50, Santa Cruz), p21 (mouse anti-p21, 1:25, Santa Cruz), and tumor suppressor p53 (mouse anti-p53, 1:200, Upstate) at different levels of senescence. A similar protocol was followed for every antibody:
EC were grown on a glass coverslip and were fixed with paraformaldehyde (2%, 20 min). Cells were then washed twice with PBS for 5 min, blocked and permeabilized for 1 h, at room temperature with normal serum (2%) containing Triton X-100 (0.2%). Cells were incubated overnight, at 4 °C, with the primary antibody diluted in PBS containing 0.05% of Triton X-100. This was followed by 3 steps of washing of the cells with PBS, and incubation with the adequate secondary antibody TRITC- or FITC-conjugated (Sigma). DNA counterstaining was performed by finally incubating the cells with TOPRO-3 (2 μM; Molecular Probe). Negative controls were performed by omission of primary antibodies during the protocol.
Cells were visualized using a confocal microscope (LSM 510 Zeiss microscope). Five images from the same coverslip were recorded and analyzed using the semi-quantitative analysis software LSM 510 (Zeiss): it allows the selective measurement of the fluorescent signal in the nucleus, or the cytosol of each cell. Each image was analyzed and values of nuclear or cytosolic fluorescence (arbitrary units, a.u.), corrected by the number of the cells per image, were quantified. The average value of the 5 images from the same coverslip was used for statistics. In order to decrease the variability inherent to the technique and to compare the protein expressions between groups, the immunoassay was performed simultaneously in Control, NAC, hTERT and hTERT-NAC cells, at initial and final passages, in n = 6–11 patients.
Immunostaining could not be performed in CMV-infected EC because the green fluorescent protein GFP signal, inherent to the infection, interferes with the fluorescence of the secondary antibodies.
2.9. Estimation of intrinsic susceptibility to oxidation of EC
The fluorescent dye 5-(and -6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester (CM-H2DCFDA, Molecular Probes) was used as a non-specific marker of ROS and indirectly as an index of intrinsic susceptibility to oxidation. At passage 2, prior to infection and NAC treatment, control living cells (from n = 11 patients) were loaded with 5 μM of CM-H2DCFDA in DMEM without phenol red, for 30 min at 37 °C, washed and let to recover for 10–15 min at 37 °C. The fluorescence was then observed at 488 nm under a confocal microscope (LSM 510 Zeiss microscope). Negative control consisted in unstained cells (autofluorescence). Compared to healthy porcine aortic EC (PAEC) or mouse aortic EC, human cells used in this study – although at an early passage – displayed a very unusual unstable basal level of CM-H2DCFDA-fluorescence that significantly increased over time. This suggests that human EC from coronary patients display abnormal high levels of ROS and/or are very susceptible to oxidation. Human EC were exposed to the pro-oxidative laser light (0.05%; in PAEC, to detect a stable signal we used 5% laser) until the fluorescent signal saturated (Fig. 4A, B). The time course of each saturation profile was divided in 3 phases: the initiation, the linear and the termination phase (Fig. 4B). Accordingly, the lag time (delay before signal starts to increase), the time needed to obtain 50% of maximal signal (T50) and the termination time (when the signal plateaued) were respectively calculated. These parameters were used as an index of the susceptibility to oxidation, the shorter the times the higher the susceptibility to oxidation. Identical optimal parameters were used for all measurements of ROS in the different EC (n = 11 donors) (objective 63 × 1.4 plan-Apochromat oil, laser Argon 488 nm, 0.1% transmission; dichroïc mirror HFT 488 with filter LP505, pinhole size 1.7 Airy units). The frame size of the images was 512 × 300 pixels and the scan speed was 2.56 μs/pixel.
Fig. 4. High susceptibility to oxidation prevents EC immortalization by hTERT over-expression in the presence of NAC.

The endogenous susceptibility to oxidation was estimated in living cells loaded with CM-H2DCFDA in non-infected EC (n = 11) at initial passage, prior to NAC treatment or hTERT infection. Cells were exposed to a pro-oxidant laser light until fluorescence signal saturated (A, B). Typical profiles of fluorescence from 3 different EC donors are shown in (B). For comparison, live porcine aortic EC (PAEC) were used. In PAEC, the CM-H2DCFDA signal was stable and, to be detectable, the laser light had to be increased (from 0.05% in human cells to 5% in porcine cells). PAEC only behaved like hIMA EC if the superoxide dismutase was inhibited by DETC (100 μM). The time course of each saturation profile (n = 11) was divided in 3 phases: the initiation phase, the linear phase and the termination phase (B and C). The lag time (s), the time needed to obtain 50% of maximal signal (T50, s) and the termination time (s) were respectively calculated. The parameters T50, lag time and termination time were used as an index of the endogenous susceptibility to oxidation: the shorter the times, the higher the susceptibility. Endogenous susceptibility to oxidation was higher in EC that will senesce (C) and in EC isolated from smokers (D). *: p < 0.05 compared to EC that will not be immortalized by the combination hTERT-NAC (C) or compared to EC from smokers (D). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
2.10. Real-time RT-PCR
Total RNA was isolated using RNeasy kit (Qiagen) at early and subsequent passages until senescence. Real-time PCR was carried out on diluted RT products using the DNA-binding dye SYBR Green I for the detection of PCR products (Mx3005P system, Stratagene) according to the manufacturer’s instruction. Serial dilutions (100 ng to 1 pg) of human aortic EC (Cambrex) total RNA were used as standard. The following primers designed by primer express (Version 2.0) were used in order to quantify gene expression of Caveolin-1, p16, p21, p53 and Sirtuin-1:
| Primer | (5′-3′) Forward | (5′-3′) Reverse |
|---|---|---|
| Caveolin-1 | GCTGAGCGAGAAGCAAGTGT | TGGTGAAGCTGGCCTTCCAA |
| p16 | CATAGATGCCGCGGAAGGT | TGTAGGACCTTCGGTGACTG |
| p21 | GGACCTGTCACTGTCTTGTA | CCTCTTGGAGAAGATCAGCCG |
| p53 | TGAGGTTGGCTCTGACTGTA | TTCTCTTCCTCTGTGCGCCG |
| Sirtuin-1 | GAACAGGTTGCGGGAATCCA | CCTGATTAAAAATATCTCCTCGTA |
| GAPDH | CTGCACCACCAACTGCTTAGC | ACTGTGGTCATGAGTCCTTCCA |
RNA levels in each sample were calculated relative to GAPDH. Values are expressed as 2ΔΔCt where ΔΔCt is the difference between the ΔCt (sample-calibrator) for a specified gene and the correspondent ΔCt (sample-calibrator) for GAPDH (arbitrary units, a.u.).
2.11. Statistical analysis of the data
The Gaussian distribution of the parameters was verified using the D’Agostino and Pearson normality test. Logarithmic transformation was performed for DHE, HNE, ATM and p16/p21 data in order to have a normal distribution. Data are presented as mean ± SEM for continuous variables, with “n” indicating the number of patients. Paired t-tests or ANOVA (followed by Bonferroni post hoc test) were used when appropriate to compare data obtained in EC early in culture (initial passage) and at senescence (final passage) and to compare data obtained in control, EC-CMV and EC-hTERT, treated or not with NAC.
3. Results
3.1. Lack of effect of NAC on cellular senescence prevents cell immortalization by hTERT over-expression in the presence of the antioxidant
For each donor and for each condition, a time course of senescence was constructed (% of SA-β-Gal = f (replication potential, ΣPDL)), and the number of days needed to reach 50% of senescent EC (Daysβ-Gal 50%) was evaluated in order to measure the propensity to develop senescence. NAC, according to the donor, displayed a dichotomic response, i.e. the onset of senescence was either i) non-affected (Fig. 1A) or ii) slightly delayed (Fig. 1D). Similarly, the combination of hTERT and NAC either delayed (Fig. 1B, C) or prevented EC senescence, leading to immortalization (Fig. 1E, F). The response to NAC predicted the response to hTERT-NAC (Chi square test, p = 0.0079): in EC where NAC had no effect (similar Daysβ-Gal 50% in control and NAC-treated EC, 93 ± 5 and 90 ± 3 days, respectively, p > 0.05), a concomitant hTERT over-expression temporarily delayed senescence in matched EC (Daysβ-Gal 50% = 116 ± 11 days, p = 0.048) (Fig. 1A–C). In contrast, in EC where NAC alone delayed senescence (longer Daysβ-Gal 50% in NAC-treated EC versus control, 119 ± 10 days versus 91 ± 3 days, p = 0.0131), the combination hTERT-NAC immortalized the cells (Fig. 1D–F); these cultures were deliberately stopped after ≈ one year, as cells continued to proliferate (ΣPDL > 50) with a very low level of senescence.
Fig. 1. Lack of effect of NAC on senescence prevents immortalization by hTERT over-expression in the presence of NAC.
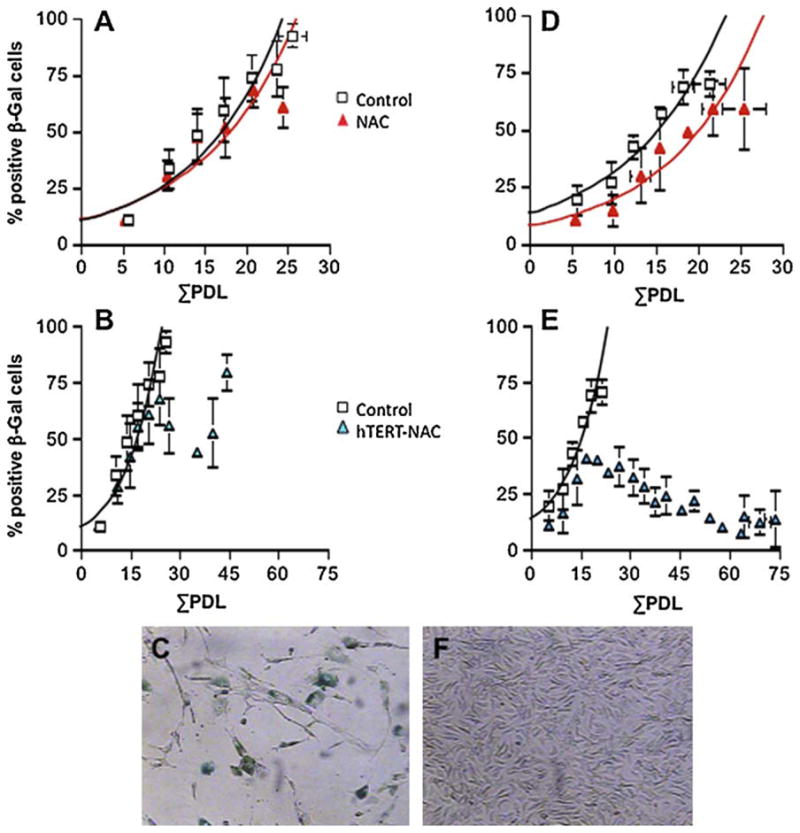
Senescence was quantified by cytochemical detection of β-galactosidase (β-Gal, in blue in the photographs) at pH 6. (A) Average (mean ± SEM, n = 5) senescence time course in non-infected and NAC-treated EC. When NAC had no beneficial effect, senescence was only delayed by hTERT infection combined with NAC treatment (hTERT-NAC) (B). (D) Average (n = 4) senescence time course in non-infected and NAC-treated EC. When NAC treatment delayed senescence, hTERT-NAC combination led to EC immortalization (E). Contrast phase photographs typical of a senescence-associated β-galactosidase staining in hTERT-NAC EC at final passages (× 400 magnification), when senescence was delayed (C) or prevented (F). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
Compared to control non-infected EC, infection with hTERT alone delayed the onset of senescence in all EC cultures (longer Daysβ-Gal 50% = 124 ± 16 days versus 96 ± 4 days, p = 0.045, n = 11), independently of the response to NAC (Fig. S1, online supplement). Time courses of EC senescence infected with the empty lentiviral vector FG12-CMV were super-imposable to those of non-infected EC (data not shown).
Ectopic hTERT over-expression increased significantly the replication potential (ΣPDL) of the cells by 35% (ΣPDL: 21 ± 2 versus 28 ± 4, Control versus hTERT, p < 0.05). Combined to NAC, hTERT over-expression increased the replication potential (ΣPDL) from 21 ± 2 to 34 ± 7 (in EC where senescence was delayed) and from 21 ± 2 to 73 ± 7 (n = 4) (in immortalized EC).
3.2. hTERT over-expression increases telomerase activity
In non-infected control cells nuclear hTERT activity was only detectable with the sensitive real-time TRAP assay (Fig. 2), and decreased between the beginning and the end of the culture (from 16.0 ± 1.5 to 9.1 ± 1.5, initial and final passages, p < 0.05, n = 11, Fig. 2B). In the presence of NAC, when compared to control untreated EC, nuclear hTERT activity slightly increased at initial passages (Control: 16.0 ± 1.5 versus NAC: 24.5 ± 3.1, p < 0.05, n = 11, data not shown) and at final passages (Control: 9.1 ± 1.5 versus NAC: 15.9 ± 2.1, p < 0.05, n = 11, data not shown). Telomerase over-expression in the absence (from 44.0 ± 4.8 to 66.6 ± 8.0, initial and final passages, p < 0.05, n = 11, data not shown) and presence of NAC (from 53.5 ± 4.8 to 81.1 ± 7.4, initial and final passages, p < 0.05, n = 11, Fig. 2), led to a significant increase in hTERT activity comparable to that of HEK293 cells (Fig. 2A), which further increased between the beginning and the end of the culture. Initial and final nuclear telomerase activities were similar in EC where senescence was delayed or prevented (data not shown).
Fig. 2. hTERT over-expression increases telomerase activity.

Nuclear telomerase activity (arbitrary units) was quantified at the beginning and the end of the cultures (at initial and final passages) in non-infected EC and in hTERT-NAC-EC, with both the conventional TRAP assay (A) and the very sensitive real-time TRAP assay (B). Two μg of nuclear protein were loaded for each sample (n = 10) and tumoral HEK293 cells were used as standard. For each sample, heat-inactivated (hi) proteins were used as negative control. *: p < 0.05 compared to initial passage, †: p < 0.05 compared to control EC.
3.3. hTERT over-expression maintains telomere length
Whereas there was no difference in initial telomere length between the groups (control: 9031 ± 308, NAC: 8679 ± 360, hTERT: 9274 ± 359, hTERT-NAC: 8612 ± 338 bp, n = 9–11 p > 0.05), final telomere lengths were longer in hTERT-NAC compared to their respective matched non-infected cells (control: 8761 ± 479, NAC: 8456 ± 375, hTERT-NAC: 9464 ± 609 bp, n = 9–11 p < 0.05, Fig. 3). Accordingly, Fig. 3C illustrates significant telomere shortening in non-infected or CMV-infected cells, but telomere stabilization in hTERT-infected EC. There was no difference in telomere length in hTERT-NAC EC where senescence was delayed or prevented (data not shown). Similarly, the telomere shortening rate in control EC was not an indicator of the capability of hTERT-NAC to immortalize matched EC: telomere shortening was not different between EC that will be immortalized (ΔRFL/PDL = −72.8 ± 19.2 bp/PDL) and EC that will senesce (ΔRFL/PDL = −30.4 ± 6.2 bp/PDL).
Fig. 3. hTERT over-expression stabilizes telomere length.
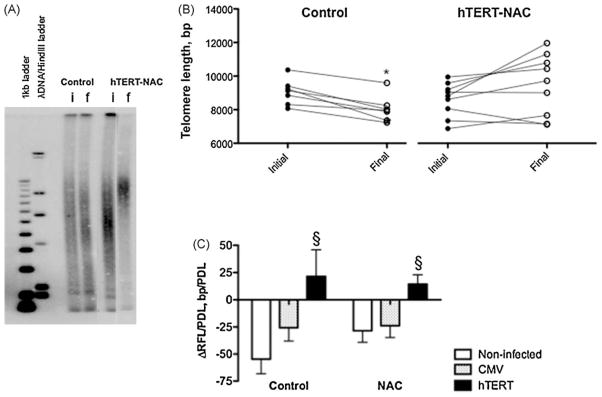
Telomere restriction fragment length (RFL, bp, n = 9) measured by Southern blot (A) at initial (i) and final (f) passage in non-infected EC and in hTERT-NAC EC (A, B). Ten μg of DNA were loaded for each sample. The rate of telomere shortening (ΔRFL/PDL, bp/PDL) was calculated in control and NAC-treated cells, in non-infected, CMV- or hTERT-infected EC (C). *: p < 0.05 compared to initial passage, §: p < 0.05 compared to non-infected EC.
3.4. High susceptibility to oxidation prevents cell immortalization by hTERT over-expression in the presence of the antioxidant
The endogenous global antioxidant capacities were estimated in non-infected living cells loaded with CM-H2DCFDA (n = 11) at initial passage, prior to NAC treatment or hTERT infection. Unlike healthy porcine aortic EC (PAEC), human internal mammary artery EC were very sensitive when exposed to a pro-oxidant laser light (Fig. 4A, B). The kinetic parameters of saturation (T50, lag time and termination time) were quantified and were significantly shorter in EC that will not be immortalized later in culture by the combination hTERT-NAC, suggesting higher susceptibility to oxidation (Fig. 4C). Similarly, T50, lag time and termination time were significantly lower in EC isolated from smokers, a condition associated with high oxidative stress and low antioxidant capacities (Farhat et al., 2008) (Fig. 4D). In contrast, neither basal nor maximal fluorescence intensities of CM-H2DCFDA differed between EC that will be immortalized or not (data not shown), suggesting that the levels of ROS were similar. PAEC exhibited a similar pattern of oxidative susceptibility when they were pre-treated with DETC, an inhibitor of the superoxide dismutase (Fig. 4B).
In addition, initial antioxidant capacities of the cells were positively associated with the beneficial effect of hTERT-NAC on cell proliferation (positive correlations between ΣPDL and i) T50: p = 0.0059, r = 0.862, n = 8; ii) lag time: p = 0.010, r = 0.869, n = 7; and iii) termination time: p = 0.0082, r = 0.809, n = 9).
3.5. Low endogenous sirtuin-1 gene expression is associated with the lack of immortalization by hTERT over-expression in the presence of the antioxidant
At initial passage, non-infected and non-treated EC that will be later immortalized by the combination hTERT-NAC, have higher initial sirtuin-1 mRNA level compared to cells that will respond to hTERT-NAC by a delayed senescence (0.896 ± 0.187 a.u. versus 0.494 ± 0.059 a.u., p < 0.05). At the end of the culture, in hTERT-NAC EC, the expression of sirtuin-1 was similar whether or not senescence was delayed (0.449 ± 0.292 a.u.) or prevented (0.859 ± 0.300 a.u.).
3.6. Combination of hTERT over-expression and NAC decreases oxidative stress
Senescence was associated with a rise in DHE that was prevented by NAC (Fig. 5). Over-expression of hTERT alone did not affect the increase in DHE associated with senescence, but the combination of NAC with hTERT over-expression prevented it (Fig. 5). The combination hTERT-NAC was more effective at preventing the rise in DHE in immortalized cells (Fig. 5C). Very similar data were obtained using the marker of lipid peroxidation HNE (Fig. S2, online supplement): senescence was associated with a rise in HNE that was lowered by NAC, prevented by the combination hTERT-NAC but not by hTERT alone. The combination hTERT-NAC was more effective to prevent the rise in HNE than NAC alone (p = 0.0211).
Fig. 5. Combination of NAC and hTERT over-expression decreased of oxidative stress in immortalized EC.

(A) Confocal images of DHE (in red; nuclear counterstaining in blue) at final passage in non-infected EC and in hTERT-NAC EC where senescence was delayed or prevented. (B and C) Nuclear fluorescence intensity (arbitrary units, a.u., corrected by cell number) of DHE, in non-infected EC, NAC-treated, hTERT-infected and hTERT-NAC EC, at initial and final passages (n = 6). (C) Nuclear DHE-fluorescence intensity in non-infected EC and hTERT-NAC EC where senescence was delayed or prevented, at initial and final passages. Data are mean ± SEM of logarithmic transformed values. *: p < 0.05 compared to initial passage; †: p < 0.05 compared to Control; ‡: p < 0.05 compared to hTERT-NAC EC or as indicated. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
3.7. Combination of hTERT over-expression and NAC decreases caveolin-1 expression
Caveolin-1 increased with senescence (Fig. 6). This cytosolic rise in caveolin-1 protein was not significantly affected by NAC or hTERT over-expression (data not shown), but was prevented by the combination hTERT-NAC in immortalized EC (Fig. 6B). The prevention of senescence-induced rise in caveolin-1 by the combination hTERT-NAC was also observed at the gene level (mRNA expression in Control EC at initial passage = 0.913 ± 0.122 versus Control EC at final passage = 1.164 ± 0.179, n = 11, p = 0.054; hTERT-NAC EC at final passage = 0.078 ± 0.019, p < 0.001 versus Control EC at final passage).
Fig. 6. Combination of NAC treatment and hTERT over-expression decreased ROS-induced caveolin-1 in immortalized EC.
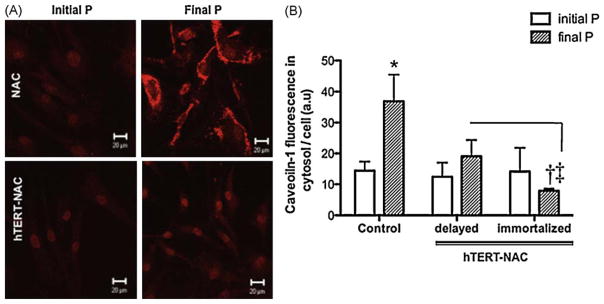
(A) Confocal images of caveolin-1 in NAC-treated EC at initial and final passage in non-infected EC and in hTERT-NAC EC. (B) Quantification of caveolin-1-fluorescence intensity in the cytosol (arbitrary units, a.u., corrected by cell number) in non-infected EC and hTERT-NAC EC where senescence was delayed or prevented, at initial and final passages (n = 10). *: p < 0.05 compared to initial passage; †: p < 0.05 compared to Control EC; ‡: p < 0.05 compared to hTERT-NAC-EC where senescence was delayed. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
3.8. hTERT-NAC abolishes p16, p21 and p53 expression in immortalized cells
In control untreated and non-infected cells, senescence led to an increase in p16 and p21 protein expressions (data not shown). This rise in p16 and p21 protein levels was abolished by the combination hTERT-NAC in immortalized EC (Fig. S3, online supplement). Similarly, while in non-infected cells, gene expression of p16 and p21 increased between the initial and final passages, this rise was prevented in immortalized hTERT-NAC EC, but not in hTERT-NAC EC where senescence was delayed (data not shown). In addition, p53 protein expression increased with senescence in non-infected cells (initial versus final passage in control: 0.28 ± 0.02 a.u. versus 0.91 ± 0.15 a.u., n = 11, p = 0.0026). This rise in p53 was lowered by hTERT-NAC in EC where senescence was delayed (final passage in hTERT-NAC: 0.17 ± 0.03 a.u., p < 0.05) and abolished in immortalized EC (final passage in hTERT-NAC: 0.05 ± 0.01 a.u., p = 0.0004).
3.9. Combination of hTERT over-expression and NAC decreases DNA damage
ATM protein expression was significantly decreased in hTERT-NAC immortalized EC (Fig. 7).
Fig. 7. Combination of NAC treatment and hTERT over-expression decreased ATM in immortalized EC.
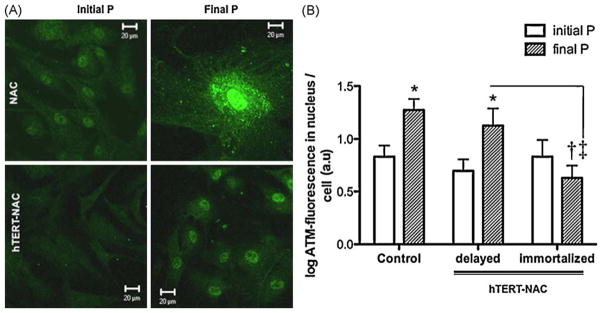
Immunostaining of ATM, a marker of DNA damage. (A) Confocal images of EC at initial and final passage in non-infected EC and in hTERT-NAC EC. (B) Quantification of ATM-fluorescence intensity in the nucleus (arbitrary units, a.u., corrected by cell number) in non-infected EC and hTERT-NAC EC where senescence was delayed or prevented, at initial and final passages (n = 10). Data are mean ± SEM of logarithmic transformed values. *: p < 0.05 compared to initial passage; †: p < 0.05 compared to Control EC; ‡: p < 0.05 compared to NAC-hTERT EC where senescence was delayed. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
Initial DNA damage levels were similar in all conditions and therefore did not predict if EC would be later immortalized or not by hTERT-NAC.
3.10. Impact of the clinical parameters on EC outcome
No particular clinical parameter (Table 1) could predict the response to the combination hTERT-NAC. We observed, however, that EC from active smoker donors have a lower probability to be immortalized by hTERT-NAC (Chi squared test, p = 0.0476). This is in line with the fact that EC from smokers display higher susceptibilities to oxidation (Fig. 4D). Although half of the donors suffered from heart failure (LVEF of 39 ± 6% compared to 62 ± 3% in donors with normal left ventricular function), this had no impact on EC immortalization (Chi squared test, p = 0.4652). Similarly, gender was not predictive of the response to hTERT-NAC (Chi squared test, p = 0.6210).
4. Discussion
The accumulation of senescent EC has been associated with endothelial dysfunction, a premise to atherosclerosis; accordingly, senescent EC are predominant in human atherosclerotic plaques (Samani et al., 2001). Two major pathways lead to senescence, telomere shortening and oxidative stress-mediated cellular damage, and a combination of both is involved in senescent EC of mammary arteries isolated from patients with CAD (Voghel et al., 2007). If internal mammary arteries do not develop atherosclerotic lesions, they have been exposed, however, for years to decades to the risk factors for CVD. Our data from this study suggest that telomerase activation is necessary to bypass replicative senescence, but not sufficient to maintain cellular proliferation and to prevent oxidative damages and SIS. Hence, the level of oxidative stress-dependent damage is the key determinant of EC survival in patients with CAD.
Different studies have shown that telomere shortening of human white blood cells can be a biomarker of accelerated cellular senescence in cardiovascular diseases such as myocardial infarction (Brouilette et al., 2003), heart failure (van der Harst et al., 2007), diabetes (Fitzpatrick et al., 2007) and CAD (Ogami et al., 2004). Thus, there is a strong apparent association between telomere length and diseases in which oxidative stress and inflammation play a role (Erusalimsky and Kurz, 2005). It has been reported, mainly in fibroblasts, that oxidative stress could cause telomere shortening due to i) the high and very susceptible guanine content of the telomeres, and ii) to the deficiency of telomeric DNA in repair of single-stranded breaks (Houben et al., 2008; Kawanishi and Oikawa, 2004; Passos and Von Zglinicki, 2006; von Zglinicki, 2002). Thus, the concept that chronic mild oxidative stress accelerates telomere shortening has emerged. However, more intense ROS-generating stressors can induce senescence via generation of telomere-independent DNA damage (Britt-Compton et al., 2009; Klimova et al., 2009). Furthermore, in epithelial cells, premature senescence is observed before telomeres become short enough to trigger replicative senescence and is associated to oxidative stress-induced DNA damage in non-telomeric regions (Ksiazek et al., 2009, 2007; Wang et al., 2009). In fact, senescence of human peritoneal mesothelial cells in culture was characterized by longer telomeres despite an increase in oxidative stress (Ksiazek et al., 2007). In EC, more relevant to our study, it has been reported that in baboons, a high fat diet augmented the prevalence of endothelial senescence independently of telomere length (Shi et al., 2007). In contrast, in HUVEC exposed to exogenous oxidative stress, an acceleration of telomere shortening was measured (Kurz et al., 2004). In EC isolated and cultured from patients with CAD, we believe that on the contrary to HUVEC or fibroblasts, endogenous oxidative stress bypasses telomere shortening and that EC enter senescence before telomeres shorten to their threshold length, via telomere-independent pathways. Accordingly, we found that telomere shortening rate is lower in hIMA EC with the higher endogenous oxidative stress: in the parental strains (control EC), ΔRFL/PDL was negatively correlated with T50-ROS (ΔRFL/PDL = 10.624–1.362 × T50-ROS, r2 = 0.742, p = 0.0275), Lag-ROS (ΔRFL/PDL = 0.235–0.267 × Lag-ROS, r2 = 0.956, p = 0.0040) and Termination-ROS (ΔRFL/PDL = 42.266–2.351 × Termination-ROS, r2 = 0.746, p = 0.0122), suggesting that small telomere shortening was associated with high susceptibility to stress (i.e. low ROS-parameters). Consequently, in our cellular model, telomere shortening rate in the parental strains is not an indicator of the capability of hTERT to immortalize the strains. Thus, the connection between ROS and senescence is complex and it is likely that both replicative and stress-induced senescence occur in vivo and contribute to aging and cardiovascular diseases.
The absence of immortalization by NAC in hTERT-infected EC is likely related to the failure of EC to inactivate ROS. The non-immortalized EC were characterized by three responses: 1) a failure of NAC alone to delay senescence, 2) a high initial susceptibility to oxidation, and 3) a low initial endogenous expression of the longevity gene Sirtuin-1 known, among others, to confer stress resistance (Langley et al., 2002; Rajendrasozhan et al., 2008).
In the present study, in vivo exposure to elevated oxidative stress prior to isolation of the cells was evidenced by high initial endogenous susceptibility to oxidation (Fig. 4), a defect exacerbated in smokers, as well as by a positive correlation (p = 0.0036, r = 0.81, n = 11; data not shown) between initial levels of the lipid peroxidation marker HNE and the initial gene expression of caveolin-1, a protein known to induce SIS (Chretien et al., 2008; Dasari et al., 2006; Voghel et al., 2008, 2007). Thereafter, culture-induced senescence was associated with a further rise in oxidative stress (DHE, HNE and caveolin-1). NAC, a direct scavenger of ROS, also known to be physiologically converted to metabolites that will stimulate glutathione synthesis, an important source of endogenous antioxidant (De Flora et al., 2001; Zafarullah et al., 2003), has the ability to temporarily abrogate the oxidative DNA damages (Tanaka et al., 2007). Using a chronic treatment at a low concentration of NAC (Haendeler et al., 2004), we recently demonstrated that only 50% of NAC-treated EC could be temporarily rescued from senescence, conditional to a reversible decrease in oxidative stress markers and damages (Voghel et al., 2008). In the present study, the dichotomic response to NAC was confirmed. We also observed that hTERT over-expression alone could not prevent senescence despite preserving telomere length. Because of the interplay between replicative senescence and SIS, hTERT over-expression per se was unable to counteract the damages induced by oxidative stress, and cells eventually cease to divide, confirming previous results showing that the life span of human aortic EC could only be temporarily extended by hTERT over-expression (Minamino et al., 2002). Similarly, hTERT-dependent immortalization of normal EC was prevented by exogenous H2O2 (Gorbunova et al., 2002).
Both oxidative stress and caveolin-1 negatively regulate eNOS (Cho and Park, 2005; Frank et al., 2004; Lin et al., 2006), which function is crucial for endothelial survival. On the other hand, NO activates hTERT and improves endothelial survival and function (Grasselli et al., 2008; Vasa et al., 2000). In a pro-atherogenic context, we reported that eNOS expression is dramatically decreased (Voghel et al., 2007) and therefore it is very unlikely that eNOS could play a role in the regulation of the expression and function of hTERT. Matsuchita et al. (Matsushita et al., 2001) also reported that expression of hTERT reversed the deficit in eNOS expression observed with increasing age. In contrast, despite hTERT over-expression, eNOS expression was not increased in our cellular model (data not shown). Because following hTERT infection, in the presence or absence of NAC, telomerase activity and telomere length were similar in delayed and immortalized EC, it is the reduction of damage facilitated by NAC exposure that permitted unlimited proliferation of the cells. This translates by a lower expression levels of oxidative stress markers (Figs. 5 and 6; Fig. S2 online supplement) and to the abolition of the rise in cell cycle inhibitors (p16, p21) triggering senescence (Fig. S3, online supplement) in the presence of NAC in immortalized hTERT-infected cells. Likewise, accumulation of molecular damages (ATM) was only prevented in hTERT-NAC immortalized EC (Fig. 7). Altogether, these data suggest that the potential effect of NAC to delay senescence by reducing oxidative stress-induced damage is a prerequisite for immortalization by hTERT-NAC.
The ability of NAC to permit hTERT-dependent immortalization of EC was directly related to the endogenous susceptibility to oxidation of these cells. Risk factors for CAD have been shown to increase oxidative stress. We hypothesized that the rise in ROS production by EC was associated with an augmented sensitivity to oxidative stimulus, and thus a greater vulnerability to oxidative stress, so that oxidative damage may trigger premature senescence. Our data demonstrate that EC with the lowest susceptibility to oxidation (Fig. 4) underwent more cell divisions and could be immortalized. In addition, we observed that nuclear (Lagtime = −12.998 + 0.299 × nuclear hTERT activity, r = 0.764, n = 9, p = 0.0166) and cytosolic (Lagtime = −4.26 + 0.288 × cytosolic hTERT activity, r = 0.657, n = 9, p = 0.0544) telomerase activity in hTERT-NAC infected cells was positively correlated with Lag time-ROS, suggesting that a high initial susceptibility to oxidative stress (low Lag time-ROS) is associated with a low global hTERT activity. In EC infected with hTERT, in the absence of NAC, we observed that cellular senescence was significantly delayed, but not prevented (Fig. S1), while nuclear telomerase activity was high and telomere length was maintained (Fig. 3). Therefore, the lack of immortalization by hTERT over-expression is unlikely due to the export of hTERT from the nucleus to the mitochondria, as reported in fibroblasts cultivated under hyperoxia, where over-expression of hTERT did not maintain telomere length and only delayed cellular senescence (Ahmed et al., 2008).
Finally, we found that initial sirtuin1 gene expression was higher in EC that could be immortalized by hTERT-NAC. Sirtuins (silencing information regulator) are nuclear nicotinamide adenine nucleotide-dependent histone deacetylases. In mammalian cells, sirtuin1 appears to control the cellular response to stress by regulating the FOXO family of Forkhead transcription factors (Brunet et al., 2004). Because FOXO transcription factors trans-activate a series of target genes that have critical roles in the cellular response to stress stimuli, endogenous sirtuin1 may potentiate FOXO’s ability to detoxify ROS and to repair damaged DNA (Michan and Sinclair, 2007; Westphal et al., 2007). It has been reported that sirtuin1 levels were reduced in macrophages and lungs of smokers and patients with chronic obstructive pulmonary disease due to its post-translational modifications by cigarette smoke-derived reactive components (Rajendrasozhan et al., 2008). This is in line with the fact that EC isolated from smoker patients have a lower probability to be immortalized by hTERT-NAC and that lower endogenous sirtuin1 levels were found in EC that would not be immortalized. Furthermore, sirtuin1 can also inhibit growth arrest by deacetylating and repressing p53-dependent PML-induced (Langley et al., 2002) and H2O2-induced cellular senescence (Furukawa et al., 2007; Ota et al., 2007). Another important function of sirtuin1 relevant to our study is that acetylation status of histones is implicated in DNA repair (Kruszewski and Szumiel, 2005; Michan and Sinclair, 2007). The exact role of sirtuin1 remains therefore to be elucidated in our model.
There are potential limitations to this study. The cells have been isolated from patients with various clinical backgrounds. Nonetheless, the damages induced by risk factors for CVD have been reproducible. In addition, this study does not include cells from healthy control for comparison. We have no access to control human internal mammary artery EC, but since we have two populations of cells (immortalized or not), we can draw some conclusions. It could be argued that the culture process isolated clones of EC prone to become immortalized. This however, is unlikely because of the frequency of occurrence (50%) of this phenomenon. Because of the small samples, multivariant analyses were not performed, although clinically relevant. We found, however, that smoking results in a lower probability for EC to be immortalized by hTERT-NAC, and that EC from smokers had a higher susceptibility to oxidative stress. Another limit, is while our approach globally estimates the capacities of EC to react against a pro-oxidant stimulus, it does not directly quantify the intrinsic antioxidant capacities. The observation that hTERT infection did not permit to discriminate between the two types of donor cells suggests that whether the oxidative damage is irreversible or not, hTERT activation alone cannot overcome this damage. Finally, because EC from different vascular regions are exposed to different levels and forms of shear stress and may have different expression levels of pro- and antioxidant enzymes, survival factors and cell cycle regulators, our data obtained in EC from human internal mammary artery should not be extrapolated to EC in general.
In conclusion, hTERT over-expression in EC from atherosclerotic patients delays replicative senescence by maintaining telomere integrity but cannot bypass CVD-related oxidative damage and the onset of SIS. Thus, in a pro-oxidative environment such as EC exposed in vivo to risk factors for CVD, efficient DNA repair mechanisms and control of ROS production are key factors allowing for the prevention of senescence.
Acknowledgments
We thank Guy Charron (Montreal Heart Institute) for fruitful discussions and technical advises, and the biological tissue bank (RETEB) of the Fonds de la Recherche en Santé du Québec for technical support.
Funding sources
This work was supported by the Canadian Institutes for Health Research (MOP 14496) and the Foundation of the Montreal Heart Institute.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.mad.2010.04.004.
Footnotes
Conflict of interest
None declared.
References
- Ahmed S, Passos JF, Birket MJ, Beckmann T, Brings S, Peters H, Birch-Machin MA, von Zglinicki T, Saretzki G. Telomerase does not counteract telomere shortening but protects mitochondrial function under oxidative stress. J Cell Sci. 2008;121:1046–1053. doi: 10.1242/jcs.019372. [DOI] [PubMed] [Google Scholar]
- Allsopp RC, Chang E, Kashefi-Aazam M, Rogaev EI, Piatyszek MA, Shay JW, Harley CB. Telomere shortening is associated with cell division in vitro and in vivo. Exp Cell Res. 1995;220:194–200. doi: 10.1006/excr.1995.1306. [DOI] [PubMed] [Google Scholar]
- Ben-Porath I, Weinberg RA. The signals and pathways activating cellular senescence. Int J Biochem Cell Biol. 2005;37:961–976. doi: 10.1016/j.biocel.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- Britt-Compton B, Wyllie F, Rowson J, Capper R, Jones RE, Baird DM. Telomere dynamics during replicative senescence are not directly modulated by conditions of oxidative stress in IMR90 fibroblast cells. Biogerontology. 2009 doi: 10.1007/s10522-009-9216-4. [DOI] [PubMed] [Google Scholar]
- Brouilette S, Singh RK, Thompson JR, Goodall AH, Samani NJ. White cell telomere length and risk of premature myocardial infarction. Arterioscler Thromb Vasc Biol. 2003;23:842–846. doi: 10.1161/01.ATV.0000067426.96344.32. [DOI] [PubMed] [Google Scholar]
- Brown JP, Wei W, Sedivy JM. Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science. 1997;277:831–834. doi: 10.1126/science.277.5327.831. [DOI] [PubMed] [Google Scholar]
- Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- Chen J, Goligorsky MS. Premature senescence of endothelial cells: Methusaleh’s dilemma. Am J Physiol Heart Circ Physiol. 2006;290:H1729–H1739. doi: 10.1152/ajpheart.01103.2005. [DOI] [PubMed] [Google Scholar]
- Chen JH, Ozanne SE, Hales CN. Methods of cellular senescence induction using oxidative stress. Methods Mol Biol. 2007;371:179–189. doi: 10.1007/978-1-59745-361-5_14. [DOI] [PubMed] [Google Scholar]
- Cho KA, Park SC. Caveolin-1 as a prime modulator of aging: a new modality for phenotypic restoration? Mech Ageing Dev. 2005;126:105–110. doi: 10.1016/j.mad.2004.09.029. [DOI] [PubMed] [Google Scholar]
- Chretien A, Piront N, Delaive E, Demazy C, Ninane N, Toussaint O. Increased abundance of cytoplasmic and nuclear caveolin 1 in human diploid fibroblasts in H(2)O(2)-induced premature senescence and interplay with p38alpha(MAPK) FEBS Lett. 2008;582:1685–1692. doi: 10.1016/j.febslet.2008.04.026. [DOI] [PubMed] [Google Scholar]
- Cichowski K, Hahn WC. Unexpected pieces to the senescence puzzle. Cell. 2008;133:958–961. doi: 10.1016/j.cell.2008.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasari A, Bartholomew JN, Volonte D, Galbiati F. Oxidative stress induces premature senescence by stimulating caveolin-1 gene transcription through p38 mitogen-activated protein kinase/Sp1-mediated activation of two GC-rich promoter elements. Cancer Res. 2006;66:10805–10814. doi: 10.1158/0008-5472.CAN-06-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Flora S, Izzotti A, D’Agostini F, Balansky RM. Mechanisms of N-acetylcysteine in the prevention of DNA damage and cancer, with special reference to smoking-related end-points. Carcinogenesis. 2001;22:999–1013. doi: 10.1093/carcin/22.7.999. [DOI] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erusalimsky JD, Kurz DJ. Cellular senescence in vivo: its relevance in ageing and cardiovascular disease. Exp Gerontol. 2005;40:634–642. doi: 10.1016/j.exger.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Farhat N, Thorin-Trescases N, Voghel G, Villeneuve L, Mamarbachi M, Perrault LP, Carrier M, Thorin E. Stress-induced senescence predominates in endothelial cells isolated from atherosclerotic chronic smokers. Can J Physiol Pharmacol. 2008;86:761–769. doi: 10.1139/Y08-082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick AL, Kronmal RA, Gardner JP, Psaty BM, Jenny NS, Tracy RP, Walston J, Kimura M, Aviv A. Leukocyte telomere length and cardiovascular disease in the cardiovascular health study. Am J Epidemiol. 2007;165:14–21. doi: 10.1093/aje/kwj346. [DOI] [PubMed] [Google Scholar]
- Forsyth NR, Evans AP, Shay JW, Wright WE. Developmental differences in the immortalization of lung fibroblasts by telomerase. Aging Cell. 2003;2:235–243. doi: 10.1046/j.1474-9728.2003.00057.x. [DOI] [PubMed] [Google Scholar]
- Frank PG, Lee H, Park DS, Tandon NN, Scherer PE, Lisanti MP. Genetic ablation of caveolin-1 confers protection against atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:98–105. doi: 10.1161/01.ATV.0000101182.89118.E5. [DOI] [PubMed] [Google Scholar]
- Furukawa A, Tada-Oikawa S, Kawanishi S, Oikawa S. H2O2 accelerates cellular senescence by accumulation of acetylated p53 via decrease in the function of SIRT1 by NAD + depletion. Cell Physiol Biochem. 2007;20:45–54. doi: 10.1159/000104152. [DOI] [PubMed] [Google Scholar]
- Gorbunova V, Seluanov A, Pereira-Smith OM. Expression of human telomerase (hTERT) does not prevent stress-induced senescence in normal human fibroblasts but protects the cells from stress-induced apoptosis and necrosis. J Biol Chem. 2002;277:38540–38549. doi: 10.1074/jbc.M202671200. [DOI] [PubMed] [Google Scholar]
- Grasselli A, Nanni S, Colussi C, Aiello A, Benvenuti V, Ragone G, Moretti F, Sacchi A, Bacchetti S, Gaetano C, Capogrossi MC, Pontecorvi A, Farsetti A. Estrogen receptor-alpha and endothelial nitric oxide synthase nuclear complex regulates transcription of human telomerase. Circ Res. 2008;103:34–42. doi: 10.1161/CIRCRESAHA.107.169037. [DOI] [PubMed] [Google Scholar]
- Haendeler J, Hoffmann J, Diehl JF, Vasa M, Spyridopoulos I, Zeiher AM, Dimmeler S. Antioxidants inhibit nuclear export of telomerase reverse transcriptase and delay replicative senescence of endothelial cells. Circ Res. 2004;94:768–775. doi: 10.1161/01.RES.0000121104.05977.F3. [DOI] [PubMed] [Google Scholar]
- Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345:458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- Houben JM, Moonen HJ, van Schooten FJ, Hageman GJ. Telomere length assessment: biomarker of chronic oxidative stress? Free Radic Biol Med. 2008;44:235–246. doi: 10.1016/j.freeradbiomed.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Jeyapalan JC, Sedivy JM. Cellular senescence and organismal aging. Mech Ageing Dev. 2008;129:467–474. doi: 10.1016/j.mad.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawanishi S, Oikawa S. Mechanism of telomere shortening by oxidative stress. Ann N Y Acad Sci. 2004;1019:278–284. doi: 10.1196/annals.1297.047. [DOI] [PubMed] [Google Scholar]
- Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- Klimova TA, Bell EL, Shroff EH, Weinberg FD, Snyder CM, Dimri GP, Schumacker PT, Budinger GR, Chandel NS. Hyperoxia-induced premature senescence requires p53 and pRb, but not mitochondrial matrix ROS. FASEB J. 2009;23:783–794. doi: 10.1096/fj.08-114256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruszewski M, Szumiel I. Sirtuins (histone deacetylases III) in the cellular response to DNA damage–facts and hypotheses. DNA Repair (Amst) 2005;4:1306–1313. doi: 10.1016/j.dnarep.2005.06.013. [DOI] [PubMed] [Google Scholar]
- Ksiazek K, Mikula-Pietrasik J, Olijslagers S, Jorres A, von Zglinicki T, Witowski J. Vulnerability to oxidative stress and different patterns of senescence in human peritoneal mesothelial cell strains. Am J Physiol Regul Integr Comp Physiol. 2009;296:R374–R382. doi: 10.1152/ajpregu.90451.2008. [DOI] [PubMed] [Google Scholar]
- Ksiazek K, Passos JF, Olijslagers S, Saretzki G, Martin-Ruiz C, von Zglinicki T. Premature senescence of mesothelial cells is associated with non-telomeric DNA damage. Biochem Biophys Res Commun. 2007;362:707–711. doi: 10.1016/j.bbrc.2007.08.047. [DOI] [PubMed] [Google Scholar]
- Kurz DJ, Decary S, Hong Y, Trivier E, Akhmedov A, Erusalimsky JD. Chronic oxidative stress compromises telomere integrity and accelerates the onset of senescence in human endothelial cells. J Cell Sci. 2004;117:2417–2426. doi: 10.1242/jcs.01097. [DOI] [PubMed] [Google Scholar]
- Langley E, Pearson M, Faretta M, Bauer UM, Frye RA, Minucci S, Pelicci PG, Kouzarides T. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J. 2002;21:2383–2396. doi: 10.1093/emboj/21.10.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy MZ, Allsopp RC, Futcher AB, Greider CW, Harley CB. Telomere end-replication problem and cell aging. J Mol Biol. 1992;225:951–960. doi: 10.1016/0022-2836(92)90096-3. [DOI] [PubMed] [Google Scholar]
- Lin WW, Lin YC, Chang TY, Tsai SH, Ho HC, Chen YT, Yang VC. Caveolin-1 expression is associated with plaque formation in hypercholesterolemic rabbits. J Histochem Cytochem. 2006;54:897–904. doi: 10.1369/jhc.5A6869.2006. [DOI] [PubMed] [Google Scholar]
- Lou Z, Chen J. Cellular senescence and DNA repair. Exp Cell Res. 2006;312:2641–2646. doi: 10.1016/j.yexcr.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Matsushita H, Chang E, Glassford AJ, Cooke JP, Chiu CP, Tsao PS. eNOS activity is reduced in senescent human endothelial cells: Preservation by hTERT immortalization. Circ Res. 2001;89:793–798. doi: 10.1161/hh2101.098443. [DOI] [PubMed] [Google Scholar]
- Medrano EE, Im S, Yang F, Abdel-Malek ZA. Ultraviolet B light induces G1 arrest in human melanocytes by prolonged inhibition of retinoblastoma protein phosphorylation associated with long-term expression of the p21Waf-1/SDI-1/Cip-1 protein. Cancer Res. 1995;55:4047–4052. [PubMed] [Google Scholar]
- Michan S, Sinclair D. Sirtuins in mammals: insights into their biological function. Biochem J. 2007;404:1–13. doi: 10.1042/BJ20070140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I. Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation. 2002;105:1541–1544. doi: 10.1161/01.cir.0000013836.85741.17. [DOI] [PubMed] [Google Scholar]
- Naka K, Tachibana A, Ikeda K, Motoyama N. Stress-induced premature senescence in hTERT-expressing ataxia telangiectasia fibroblasts. J Biol Chem. 2004;279:2030–2037. doi: 10.1074/jbc.M309457200. [DOI] [PubMed] [Google Scholar]
- Ogami M, Ikura Y, Ohsawa M, Matsuo T, Kayo S, Yoshimi N, Hai E, Shirai N, Ehara S, Komatsu R, Naruko T, Ueda M. Telomere shortening in human coronary artery diseases. Arterioscler Thromb Vasc Biol. 2004;24:546–550. doi: 10.1161/01.ATV.0000117200.46938.e7. [DOI] [PubMed] [Google Scholar]
- Ota H, Akishita M, Eto M, Iijima K, Kaneki M, Ouchi Y. Sirt1 modulates premature senescence-like phenotype in human endothelial cells. J Mol Cell Cardiol. 2007;43:571–579. doi: 10.1016/j.yjmcc.2007.08.008. [DOI] [PubMed] [Google Scholar]
- Passos JF, Von Zglinicki T. Oxygen free radicals in cell senescence: are they signal transducers? Free Radic Res. 2006;40:1277–1283. doi: 10.1080/10715760600917151. [DOI] [PubMed] [Google Scholar]
- Rajendrasozhan S, Yang SR, Kinnula VL, Rahman I. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;177:861–870. doi: 10.1164/rccm.200708-1269OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samani NJ, Boultby R, Butler R, Thompson JR, Goodall AH. Telomere shortening in atherosclerosis. Lancet. 2001;358:472–473. doi: 10.1016/S0140-6736(01)05633-1. [DOI] [PubMed] [Google Scholar]
- Shi Q, Hubbard GB, Kushwaha RS, Rainwater D, Thomas CA, 3rd, Leland MM, Vandeberg JL, Wang XL. Endothelial senescence after high-cholesterol, high-fat diet challenge in baboons. Am J Physiol Heart Circ Physiol. 2007;292:H2913–H2920. doi: 10.1152/ajpheart.01405.2006. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Halicka HD, Traganos F, Seiter K, Darzynkiewicz Z. Induction of ATM activation, histone H2AX phosphorylation and apoptosis by etoposide: relation to cell cycle phase. Cell Cycle. 2007;6:371–376. doi: 10.4161/cc.6.3.3835. [DOI] [PubMed] [Google Scholar]
- van der Harst P, van der Steege G, de Boer RA, Voors AA, Hall AS, Mulder MJ, van Gilst WH, van Veldhuisen DJ. Telomere length of circulating leukocytes is decreased in patients with chronic heart failure. J Am Coll Cardiol. 2007;49:1459–1464. doi: 10.1016/j.jacc.2007.01.027. [DOI] [PubMed] [Google Scholar]
- Vasa M, Breitschopf K, Zeiher AM, Dimmeler S. Nitric oxide activates telomerase and delays endothelial cell senescence. Circ Res. 2000;87:540–542. doi: 10.1161/01.res.87.7.540. [DOI] [PubMed] [Google Scholar]
- Vaziri H, Benchimol S. Reconstitution of telomerase activity in normal human cells leads to elongation of telomeres and extended replicative life span. Curr Biol. 1998;8:279–282. doi: 10.1016/s0960-9822(98)70109-5. [DOI] [PubMed] [Google Scholar]
- Voghel G, Thorin-Trescases N, Farhat N, Mamarbachi AM, Villeneuve L, Fortier A, Perrault LP, Carrier M, Thorin E. Chronic treatment with N-acetyl-cystein delays cellular senescence in endothelial cells isolated from a subgroup of atherosclerotic patients. Mech Ageing Dev. 2008;129:261–270. doi: 10.1016/j.mad.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voghel G, Thorin-Trescases N, Farhat N, Nguyen A, Villeneuve L, Mamarbachi AM, Fortier A, Perrault LP, Carrier M, Thorin E. Cellular senescence in endothelial cells from atherosclerotic patients is accelerated by oxidative stress associated with cardiovascular risk factors. Mech Ageing Dev. 2007;128:662–671. doi: 10.1016/j.mad.2007.09.006. [DOI] [PubMed] [Google Scholar]
- von Zglinicki T. Oxidative stress shortens telomeres. Trends Biochem Sci. 2002;27:339–344. doi: 10.1016/s0968-0004(02)02110-2. [DOI] [PubMed] [Google Scholar]
- von Zglinicki T, Saretzki G, Ladhoff J, d’Adda di Fagagna F, Jackson SP. Human cell senescence as a DNA damage response. Mech Ageing Dev. 2005;126:111–117. doi: 10.1016/j.mad.2004.09.034. [DOI] [PubMed] [Google Scholar]
- Wang C, Jurk D, Maddick M, Nelson G, Martin-Ruiz C, von Zglinicki T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell. 2009;8:311–323. doi: 10.1111/j.1474-9726.2009.00481.x. [DOI] [PubMed] [Google Scholar]
- Westphal CH, Dipp MA, Guarente L. A therapeutic role for sirtuins in diseases of aging? Trends Biochem Sci. 2007;32:555–560. doi: 10.1016/j.tibs.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Yang J, Chang E, Cherry AM, Bangs CD, Oei Y, Bodnar A, Bronstein A, Chiu CP, Herron GS. Human endothelial cell life extension by telomerase expression. J Biol Chem. 1999;274:26141–26148. doi: 10.1074/jbc.274.37.26141. [DOI] [PubMed] [Google Scholar]
- Yang L, Suwa T, Wright WE, Shay JW, Hornsby PJ. Telomere shortening and decline in replicative potential as a function of donor age in human adrenocortical cells. Mech Ageing Dev. 2001;122:1685–1694. doi: 10.1016/s0047-6374(01)00280-9. [DOI] [PubMed] [Google Scholar]
- Zafarullah M, Li WQ, Sylvester J, Ahmad M. Molecular mechanisms of N-acetylcysteine actions. Cell Mol Life Sci. 2003;60:6–20. doi: 10.1007/s000180300001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Aviv H, Gardner JP, Okuda K, Patel S, Kimura M, Bardeguez A, Aviv A. Loss of chromosome 13 in cultured human vascular endothelial cells. Exp Cell Res. 2000;260:357–364. doi: 10.1006/excr.2000.4997. [DOI] [PubMed] [Google Scholar]