

Abstract
Mucopolysaccharidosis type IIIB (MPS IIIB) or Sanfilippo Syndrome type B is a lysosomal storage disease resulting from the deficiency of N-acetyl glucosaminidase (NAGLU) activity. We previously showed that intracranial adeno-associated virus (AAV) -based gene therapy results in partial improvements of several aspects of the disease. In an attempt to further correct the disease, MPS IIIB mice were treated at 2–4 days of age with intracranial AAV2/5-NAGLU (IC-AAV), intravenous lentiviral-NAGLU (IV-LENTI) or the combination of both (BOTH). The BOTH group had the most complete biochemical and histological improvements of any treatment group. Compared to untreated MPS IIIB animals, all treatments resulted in significant improvements in motor function (rotarod) and hearing (auditory-evoked brainstem response). In addition, each treatment group had a significantly increased median life span compared to the untreated group (322 days). The combination arm had the greatest increase (612 days), followed by IC-AAV (463 days) and IV-LENTI (358 days). Finally, the BOTH group had nearly normal circadian rhythm measures with improvement in time to activity onset. In summary, targeting both the systemic and central nervous system disease of MPS IIIB early in life appears to be the most efficacious approach for this inherited metabolic disorder.
Keywords: Sanfilippo, gene therapy, behavioral
Introduction
N-acetyl-glucosaminidase deficiency (Sanfilippo Syndrome type B, Mucopolysaccharidosis IIIB) typically causes a pediatric onset disease characterized phenotypically, by progressive motor and cognitive deterioration, and histologically by accumulation of lysosomal inclusions in most tissues1. No current treatment is approved in humans. After the gene was identified 2,3, a murine model of MPS IIIB was created 4. The MPS IIIB mouse shares many of the biochemical, histological and clinical features with the human disease 4,5.
Several groups have demonstrated the ability of either intracranial or systemic gene therapy approaches to reduce lysosomal distention in the brains of MPS IIIB mice 6–11. Our group also demonstrated improvements in histology with corresponding improvements in neurologic function and lifespan using intracranial gene therapy 12. Intracranial delivery has thus far demonstrated the most consistent improvement in disease progression. Increases of approximately 30% in lifespan have been observed with CNS-directed therapies. An intracranial gene therapy approach is now being pursued in a larger animal model of MPS IIIB 13. Systemic-targeted gene therapy was shown to reduce lysosomal storage in peripheral organs. However, none of these single approaches completely eradicates intra-cytoplasmic inclusions or normalizes the disease phenotype. We previously attempted a combination of CNS-directed gene therapy and bone marrow transplantation (BMT) with little or no benefit seen for the BMT arms. However, the level of chimerism was relatively low and toxicities from the radiation conditioning were evident in the transplant arm. In other lysosomal storage disease models, therapies to the CNS and the periphery, with enzyme replacement, BMT or gene therapy have shown greatly improved disease correction especially when initiated in the neo-natal period 14–17.
Therefore, we hypothesized that neonatal combination therapy directed to both the CNS and the periphery would provide better correction of the disease, especially if higher systemic levels of NAGLU activity could be attained. We describe here the benefits obtained from each mode of gene therapy and the synergistic effect of combining intracranial AAV-NAGLU and systemic lentiviral-NAGLU gene therapy.
Results
Treatments
All gene therapy injections were performed in mice pups at 2–4 days of age. Intracranial AAV-NAGLU treatment was performed as described previously with six direct injections of 2 μl each into frontal, temporal and cerebellar regions of the brain of vector at a concentration of 1.5 × 1012 viral particles/ml 5,12. Intravenous lentiviral-NAGLU injections were also performed as described previously by injection of 100 μl of 1.6 × 108 infectious units/ml viral aliquot into the superficial temporal vein 18
NAGLU activity
Biochemical analysis of N-acetyl-glucosaminidase activity for various organs was determined in mice from each group at ~8 months of age and compared to untreated MPS IIIB animals (MPS IIIB NO TX) (Fig. 1). The MPS IIIB animals receiving only intravenous lentiviral vector treatment (MPS IIIB IV-LENTI) had detectable NAGLU activity in all organs assayed (<2% of normal in the brains (p<0.05) and kidneys (NS), 11% in heart (p<0.01), 12% in lung (NS), 34% in liver (p<0.001), 34% in spleen (p<0.05) and 28% in the serum (NS)[data not shown]). Conversely, animals treated only with intracranial AAV (MPS IIIB IC-AAV) had approximately 200% (p<0.05), 3% (NS), and 5% (NS) of normal activity in the brain, liver, and serum (data not shown), respectively, and little or no activity in the spleen, heart, lung, or kidney. Combination therapy (MPS IIIB BOTH) yielded NAGLU activity levels of 424% for brain(p<0.01), 13.6% for liver (p<0.001), 6.7% for heart (p<0.01), 19.8% for spleen (p<0.01), 2.9% for lung (p<0.05), 42% for serum (NS) and less than 1% in the kidney (NS). Normal mice treated with both therapies (NORMAL BOTH) had no significant change in NAGLU activity from normal mice for the visceral organs (p >0.05 for every comparison).
Figure 1.

NAGLU activity was measured in Brain, Liver, Heart, Spleen, Kidney, and Lung from untreated and treated MPS IIIB mice. Mean activity levels +SEM for each group relative to the Normal NO TX group are depicted in the graphs. NO TX, no treatment; IV-LENTI, intravenous lentiviral NAGLU vector; IC-AAV, intracranial adeno-associated viral NAGLU vector; BOTH, both IV-LENTI and IC-AAV treatments. * indicates p<0.05 **indicates p<0.01, *** indicates p<0.001 compared to MPS IIIB NO TX group
Secondary enzyme activity
In most lysosomal storage disorders other lysosomal enzymes, such as β-glucuronidase (GUSB), are elevated. The resolution of this secondary elevation has been used as a surrogate for therapeutic response. Secondary elevations of GUSB activity in the various groups are depicted in Figure 2 and statistically compared to the MPS IIIB NO TX group. Untreated MPS IIIB (MPS IIIB NO TX) animals had significant elevations in GUSB compared to normal mice for all organs assayed: brain (358%, p<0.001), liver (175%, p<0.01), spleen (343%, p<0.05), heart (619%, p<0.05), lung (645%, p<0.001), and kidney (439%, p<0.001). IC-AAV mice had significant reductions (nearly normal) in secondary elevations compared to untreated MPS IIIB mice in the brain (p<0.0001), 387% normal in the lung (p<0.01), but no significant reductions in the other visceral organs. IV-LENTI-treated mice had significant reductions in GUSB activity in the brain (228%, p<0.01), kidney (217%, p<0.05), and lung (227%, p<0.0001). Combination treatment resulted in normalization of levels in the brain (95%, p<0.0001), and significant reductions in the kidney (188%, p<0.01) and lung (183%, p<0.0001). β-Glucuronidase activity in the spleen and heart were reduced in the combination arm but did not reach significance (p=0.052 and 0.099, respectively). The combination-treated wild type animals remained within the normal range except for the brain with a significant reduction to 82% of normal.
Figure 2.

Secondary elevation of β-glucuronidase activity in Brain, Liver, Heart, Spleen, Kidney, and Lung from untreated and treated MPS IIIB mice. Mean activity levels +SEM for each group relative to the Normal NO TX group are depicted in the graphs. Group designations are identical to those in Figure 1. (* p<0.05 ** p<0.01, *** p<0.001, **** p<0.0001compared to MPS IIIB NO TX group)
Histology
MPS IIIB BOTH mice had the best response in the CNS, with marked reduction in lysosomal storage in glial and meningeal cells as well as in cortical neurons as depicted in Figure 3. These same cell types in MPS IIIB IC-AAV mice had a less striking but definite response to the intracranial therapy. MPS IIIB IV-LENTI mice had no response in neurons and only mild reduction in storage in glial and meningeal cells (Table 1). The spleen and liver in treated mice showed minimal or no response with IC-AAV therapy only (Table 1). However, there was a clear and similar reduction in storage in these sites in both the IV-LENTI and BOTH groups. As a group, the mice treated with combined IC-AAV and IV-LENTI therapy had the best response with the greatest reduction in storage.
Figure 3.

Lysosomal inclusions in Parietal Cortex. Representative sections of parietal cortex of A) MPS IIIB, B) MPS IIIB IV-LENTI, C) MPS IIIB IC-AAV, D) MPS IIIB BOTH mice are shown. Black arrows indicate neuronal lysosomal inclusions and white arrows indicate lysosomal distension in glial cells.
Table 1.
Lysosomal storage in brain and viscera by light microscopy in MPS IIIB mice after treatment with intracranial AAV, intravenous lentiviral or combined gene therapy
| Tissue | IC-AAV (n=4) | IV-Lenti (n=4) | BOTH (n=3) |
|---|---|---|---|
| Liver | |||
| Hepatocytes | NC | NC-↓ | NC-↓↓ |
| Kupffer Cells | NC-↓ | ↓-↓↓ | ↓-↓↓ |
| Spleen | |||
| Sinus lining cells | NC-↓ | ↓↓-↓↓↓ | ↓↓↓ |
| Brain | |||
| Cortical Neurons | NC-↓↓ | NC | ↓↓-↓↓↓ |
| Meninges | ↓-↓↓ | ↓ | ↓↓↓ |
| Glia | ↓↓-↓↓↓ | NC-↓ | ↓↓↓ |
NC: no change in lysosomal storage from age-matched untreated MPS IIIB mouse
Slight reduction in storage,
Moderate reduction in storage,
Marked reduction in storage, all compared to age-matched untreated MPS IIIB mouse
Glycosaminoglycan analysis
In order to assess the lysosomal storage more quantitatively, the Sensi-Pro NRE assay was performed on brain homogenates of adult mice from each group in order to measure the pathologic glycosaminoglycan (pGAG) accumulation of heparan sulfate 19–21. Figure 4 depicts the reduction in brain heparan sulfate by each treatment. Compared to the MPS IIIB NO TX group, MPS IIIB IV-LENTI had no significant reduction in pGAG (85% of NO TX, p=NS). MPS IIIB IC-AAV (24% of NO TX, p<0.01) and MPS IIIB BOTH (18% of NO TX, p<0.01) had significantly reduced pGAG. NORMAL mice had no detectable pGAG.
Figure 4.

Heparan sulfate levels in the brain of untreated and treated mice. Pathologic glycosaminoglycan (pGAG) levels in brain homogenates +SEM for each group are depicted in pg/ug of protein. (N=4–8 for each group, NS–not significant, ** p<0.01, **** p<0.0001 compared to the MPS IIIB NO TX group)
Circadian activity
Long term running wheel recordings from 14 to 24 weeks of age were performed with mice from each experimental group under a 12-hour light: 12-hour dark cycle. We previously described a difference between untreated MPS IIIB and normal animals in two circadian measurements; percentage of activity occurring during the light phase (greater in MPS IIIB) and the time from lights off to activity onset (shorter in MPS IIIB) 5. Similar differences were observed in the current study, however percent activity during the light phase did not reach statistical significance (figure not shown) after correction for multiple comparisons between the MPS IIIB NO TX (mean = 15.14%) and the Normal NO TX group (mean = 9.60%)(p = 0.14). While the average activity of the MPS IIIB IC-AAV (mean = 14.23%) and MPS IIIB BOTH (mean = 12.03%) groups were increasingly numerically improved from the MPS IIIB NO TX group and closer to the Normal NO TX group, they were not statistically significantly different after correction for multiple groups.
The time of daily activity onset (phase angle of entrainment) was significantly earlier in the MPS IIIB NO TX animals compared to Normal NO TX mice (p = 0.007; Fig. 5). There was no difference in the phase angle of entrainment between the MPS IIIB NO TX and the MPS IIIB IV-LENTI groups. However, the MPS IIIB IC-AAV trended toward improvement (p=0.078) and MPS IIIB BOTH was significantly improved relative to the untreated MPS IIIB mice (p=0.003). The MPS IIIB BOTH group phase angle is not different than the Normal NO TX (p=0.710)
Figure 5.

Time of activity onset. Mean times of day of activity onset are plotted + SEM. Light offset at 19:00 is indicated with grey shading. Note that untreated MPS IIIB mice and MPS IIIB mice treated with lenti started their daily running about 15 minutes before lights off whereas MPS IIIB mice treated with AAV or both Lenti and AAV started running about 30 minutes after lights off, similar to normal, untreated mice. (n=6 for each group, ** p<0.01 compared to MPS IIIB NO TX group)
Untreated MPS IIIB and normal animals had no difference in total daily activity level similar to published results (Heldermon et al 2010). No other treatment group was different than normal or MPS IIIB untreated mice for total activity.
The combination treated normal animals were no different than the normal untreated animals for any circadian parameter tested.
Auditory function
Auditory-evoked brainstem response (ABR) thresholds were performed on each group at ~8–8.5 months of age. Similar to our previous findings MPS IIIB NO TX mice had significantly diminished hearing compared to normal animals, reaching the maximum sound output level of the equipment at several frequencies without generating a response (Fig. 6). Each of the MPS IIIB treatment groups had a consistently lower response threshold at all frequencies tested compared to MPS IIIB NO TX mice. Based on a group wise comparison of the treatment groups, there appeared to be a trend towards lower thresholds in the IC-AAV group compared to the IV-LENTI group. MPS IIIB BOTH mice were significantly improved compared to either IC-AAV or IV-LENTI mice but remains significantly different than the Normal NO TX group (p<0.001). Surprisingly, the Normal BOTH animals also had diminished hearing compared to Normal NO TX animals. This presumably reflects some adverse effect of virus administration on hearing, but the Normal BOTH animals still had demonstrably better hearing thresholds than MPS IIIB mice.
Figure 6.

Hearing sensitivity in decibels of treated and untreated MPS IIIB and Normal mice. Auditory-evoked brainstem responses (ABR) recording was performed at 5, 10, 20, 28.3, and 40 kHz on 9–10 animals per group at 8–9 months of age. Each group’s mean decibel threshold to detect the audible stimuli +SD is shown. All treatment groups are improved when compared to MPS IIIB NO TX group over all frequencies. (p<0.001 by repeated measure ANOVA)
Motor function
We previously demonstrated IC-AAV treatment could delay the progression of motor deficits in MPS IIIB mice as measured using a rocking rotarod. In the current study, we demonstrate that each of the treatment interventions significantly delayed the progression of motor dysfunction compared to MPS IIIB NO TX animals (Fig. 7). The median time to a latency of ≤60 seconds on the rod increased from ~40 weeks in MPS IIIB NO TX animals, to ~78 weeks in MPS IIIB BOTH mice. MPS IIIB IC-AAV mice reached the median latency at ~62 weeks and MPS IIIB IV-LENTI mice reached that latency at ~52 weeks. The time for the MPS IIIB BOTH mice to reach the median latency was significantly greater than any other MPS IIIB group. The median had not been reached for either Normal group at 90 weeks of age and is statistically longer than the MPS IIIB BOTH group (p=0.05).
Figure 7.

Rocking rotarod performance Log Rank analysis. A latency of 60 seconds or less was used as a threshold to represent motor dysfunction in the Log Rank analysis that is graphed. All groups (n=6–10) are significantly longer than MPS IIIB NO TX. MPS IIIB BOTH is significantly longer than IC-AAV (p<0.05) and IV-LENTI (p<0.001). There is no difference between Normal NO TX and Normal BOTH groups so the latter is not shown for figure clarity. (** p<0.01, *** p<0.001 compared to MPS IIIB NO TX group)
Life span
We showed previously that IC-AAV treatment prolongs the life of MPS IIIB animals by >100 days. We find a similar increase in median lifespan of IC-AAV in this study (Fig. 8). All treatment groups demonstrate a significant increase in median survival relative to MPS IIIB NO TX (322 days). MPS IIIB IV-LENTI median survival increased to 358 days, MPS IIIB IC-AAV to 452 days, and MPS IIIB BOTH to 612 days. There are no apparent differences in survival between the Normal and Normal BOTH groups up to 720 days of age (Data not shown), and median survival has not been reached in these groups, and is significantly longer than any MPS IIIB treatment group.
Figure 8.
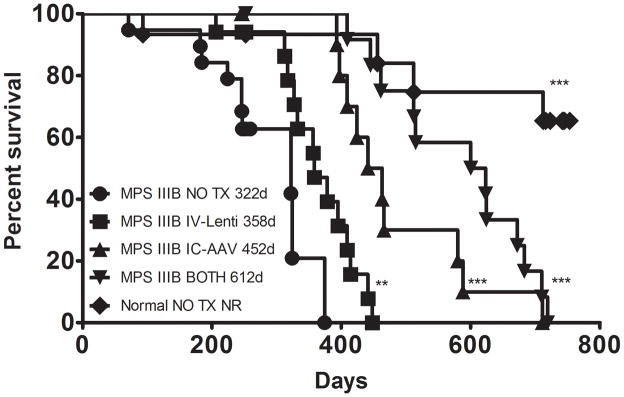
Survival. All treated mice were analyzed for median survival by Kaplan-Meier analysis and plots are shown. All treated groups had significantly improved survival compared to MPS IIIB NO TX. MPS IIIB BOTH is significantly longer than IC-AAV (p<0.05) and IV-LENTI (p<0.001). There is no difference between Normal NO TX and Normal BOTH groups so the latter is not shown for figure clarity. (** p<0.01, *** p<0.001 compared to MPS IIIB NO TX group)
Adverse events
We had the opportunity to examine at least 4 mice from all groups at ~280 days of age and 2 MPS IIIB IC-AAV, 2 MPS IIIB BOTH, and 9 Normal BOTH mice from 588–726 days of age for tumor formation. None of the mice examined had demonstrable tumors on gross examination.
Discussion
The most important measures of clinical benefit for any therapy are quality of life and quantity of life. We have chosen the neonatal timeframe for treatment because lysosomal storage disease treatment by bone marrow transplant in humans has demonstrated that the earliest possible treatment must be pursued to prevent irreversible neurologic damage. This is likely to be the most efficacious intervention timeframe in humans once newborn screening for this disorder becomes commonplace. We have demonstrated that neonatal treatment with either an intravenous injection of a lentiviral vector, intracranial injection of an AAV vector or the combination of both can significantly improve surrogates for quality of life (motor function, hearing, and circadian rhythm) and increase life span. In all cases the systemic approach was less efficacious than the intracranial approach. This is not surprising given the predominantly neurologic manifestations of MPS IIIB. However, the human disease has manifestations that are not in the CNS, such as hepatomegaly, hernias, diarrhea and ear infections with effects on hearing that may be a combination of central and peripheral neurologic effects and ear structure abnormalities22–26.
Additionally some of the benefit in the IV-LENTI group may be related to the very small amount of activity observed in the brains of these animals, presumable from a small amount of lentiviral transduction across the blood brain barrier as has been seen by other investigators27. This may be supported by the relatively small decrease in brain heparan sulfate in this group, which was not statistically significant. We also cannot rule out the possibility that greater benefit might be achievable from the systemic approach if we obtained higher expression of NAGLU by either using a higher or repeated dose of vector. Although we were able to attain supra-normal levels in the brain with the combination of systemic and intracranial treatments, the levels of NAGLU activity were less than half normal in all other organs assayed. Interestingly in our study the combination approach consistently yielded lower organ activity than the systemic monotherapy and higher brain activity than the CNS directed monotherapy. This may be due to disruption of the blood brain barrier during the CNS injections allowing systemic vector to shunt to the brain reducing the other organ viral exposure. We attempted to determine the relative contribution of lenti-viral and AAV transduction in the brain. However, due to the presence of repetitive elements, identical inserts, and poor DNA quality after homogenization neither the qPCR nor ISH approaches to quantify AAV versus lenti virus were succesful. We cannot rule out the possibility that the combination approach animals received a greater vector copy number than the IC-AAV group, despite the same injection volume from the same lot of virus, that could explain the differences between the groups.
We observed that some organs, such as lung, heart and kidney seem more resistant to transduction as has been observed by other gene therapy investigations 27 but still have substantial reduction of secondary enzyme elevation. This abrogation of secondary elevation may be through low level NAGLU enzyme cross correction, from more highly transduced organs such as the liver or spleen. However, this low level cross correction is not enough to markedly reduce lysosomal inclusions histologically. Perhaps the level of cross correction needed to reduce secondary enzyme elevations is lower than what is needed to markedly reduce lysosomal distention. This has been observed with liver directed gene therapy for MPS VII as well 17
The adverse effects on ABR thresholds following virus administration in the Normal group most likely did not result from AAV since this group was not adversely affected in our prior study which evaluated IC AAV given in the same manner. Whether the adverse effects are attributable to a direct effect of lentivirus or rather toxic effects of excess NAGLU to normal hearing is not yet known. However, this could be addressed by determination of long term effects on hearing structure and function after systemic lentiviral treatment of Normal mice with either NAGLU or a reporter enzyme.
As we originally hypothesized, the combination approach appears to have the greatest benefit. Although MPS IIIB is a simple monogenic disease, it has a complex biochemical, histological and clinical phenotype. Since NAGLU is expressed in virtually every cell of the body, most tissues have some lysosomal storage. Consequently, multiple tissues must be treated to provide the maximum benefit. Interestingly, the combination therapy appears to be additive for some measures that affect quality of life such as hearing and motor function, and synergistic for measures of circadian function (difference in time to activity onset of 2 minutes with IV-LENTI, 22 minutes with IC-AAV and 38 minutes with BOTH) and overall survival (difference in median survival of 36 days with IV-LENTI, 130 days with IC-AAV and 290 days with BOTH). Not surprisingly, the combination treatment was not bested by either single therapy in any functional assessment. Unfortunately, despite long term functional improvements and substantial benefits in survival of the combination approach the progression of disease during the last several weeks of life for each of the groups remained similar with poor coat care, urinary retention and loss of balance.
The combination therapy approach using two different vectors in the current study is not the only dual approach that can be envisioned. We selected a lentiviral vector for the systemic approach for its efficacy at producing sustained expression of a protein product and the lack of known immune inactivation in human trials 28–30. A higher sustained level of NAGLU production may have been obtained with lentiviral transduced bone marrow selected for high enzyme level as has been demonstrated by others31. Alternatively, a systemic AAV9 vector approach may yield similar systemic benefits with a single peripheral injection site 32, however this serotype has not yet been used in humans. In contrast to lentivirus trials, human systemic AAV gene therapy approaches have been limited by immune responses to the virus-infected cells. The systemic AAV approach needs further study to avoid immune inactivation that has been seen in human trials 33,34.
In addition to determining the efficacy of a dual gene therapy approach, the safety of AAV and lentiviral vectors requires additional study. Several recent studies have demonstrated the long-term safety of systemic delivery of AAV vectors in both rodents and non-human primates 35,36. However, a number of studies have also shown an increase in tumorigenesis in AAV-treated animals 37–42. Given recent findings regarding recombinant lentiviral integration events near active genes 36, it seems prudent to monitor for tumor formation in future pre-clinical experiments and human trials We have not observed any hepatocellular carcinoma or other malignancies in the current study despite long follow-up. Finally, other combination approaches could also prove to be efficacious 43. Several studies have shown that combining hematopoietic stem cell-mediated therapy with gene therapy or substrate reduction therapy greatly increases the efficacy in the murine model of Krabbe disease 16,44 and metachromatic leukodystrophy 45. It was also shown that the addition of a small molecule anti-inflammatory or substrate reduction agents enhanced the efficacy of both neuronal stem cell- and hematopoietic stem cell-mediated transplantation in the murine model of Sandhoff disease 46,47. The caveat to this approach is the relatively high morbidity and mortality of stem cell transplant methods in humans from the conditioning regimens, infections, and for allogeneic transplants from graft versus host disease.
In conclusion, combination neonatal intracranial and systemic NAGLU gene therapy provides significant and clinically meaningful therapeutic benefit in a mouse model of MPS IIIB. However, study of additional interventions is warranted since the current strategy is not completely corrective.
Materials and Methods
Viral constructs
AAV-NAGLU was constructed as previously described with a huNAGLU cDNA (gift of Elizabeth Neufeld), AAV2 genome, CMV enhancer, chicken β-actin promoter, SV40 poly-A signal and 3′ untranslated region from the rabbit β-globin gene. Vector was produced at the University of Florida Vector Core with a pseudotype AAV5 capsid, and diluted to 1.5 × 1012 viral particles/ml with lactated Ringer’s prior to freezing at −85 C.
Lentiviral-NAGLU was constructed with the same huNAGLU cDNA into the MND vector plasmid (kind gift from D. Kohn) with a delta-gag, central polypurine tract and the Myeloproliferative sarcoma virus enhancer, negative control region deleted, Dl587rev primer-binding site substituted (MND) promoter, SV40 poly-A tail, and delta u3 3′LTR. Vector was produced in the Sands’ lab with a four plasmid system in 293T cells and concentrated to 1.6 × 108 infectious units/ml prior to aliquoting and freezing at −85 C.
Mice
C57BL/6 NAGLU-deficient mice (kind gift from E. Neufeld) were maintained by strict sibling mating by M.S.S. at Washington University School of Medicine. Genotyping was done on tissue of newborn mice by enzyme assay or NAGLU exon 6 and neomycin insertion cassette PCR. All procedures on animals were in accordance with the guidelines of Institutional Animal Care and Use Committee at Washington University in St. Louis.
Treatments
At 2–4 days of age all mice were allocated to treatment groups: untreated MPS IIIB (MPS IIIB NO TX, n=19, 15 males), MPS IIIB treated with intracranial AAV-NAGLU (MPS IIIB IC-AAV, n=19, 10 males), MPS IIIB treated with intravenous lentiviral NAGLU (MPS IIIB IV-LENTI, n=19, 7 males), MPS IIIB treated with both intracranial AAV-NAGLU and intravenous lentiviral NAGLU (MPS IIIB BOTH, n=16, 9 males), normal untreated (Normal NO TX, n=15, 10 males) and normal with both intracranial AAV-NAGLU and intravenous lentiviral NAGLU (Normal BOTH, n=17, 8 males).
All gene therapy injections were performed in mice pups at 2–4 days of age. Intracranial AAV-NAGLU treatment was performed as described previously with six direct injections of 2 μl each into frontal, temporal and cerebellar regions of the brain using a 32-gauge needle 5,12. Intravenous lentiviral-NAGLU injections were also performed as described previously by injection of 100 μl of viral aliquot into the superficial temporal vein 18. When combined, the AAV injections were performed first and the systemic injections were performed within 5–60 minutes.
Histology and Biochemistry
Mice from each group (n=3–8 for MPS IIIB NO TX and MPS IIIB BOTH, and n=4–8 all other groups) between 242 and 259 days of age were sacrificed by CO2 asphyxiation. Liver, spleen, kidneys, heart, lung, and brain were harvested. Part of each organ was immersion fixed in 2% gluteraldehyde/4% paraformaldehyde in phosphate buffered saline and part was flash frozen in liquid nitrogen and stored at − 85C until mechanical homogenization in 10mM Tris (pH 7.5), 150mM NaCl, 1mM DTT, and 0.2% Triton X-100. Fixed tissue was embedded in Spurr’s resin and one-micron-thick sections were stained with toluidine blue prior to blinded evaluation of lysosomal storage and vacuolization.
Cell debris was pelleted and supernatants were collected for enzyme assays of NAGLU and GUSB activity. Duplicate NAGLU assays were performed using 20 μl of supernatant added to 40 μl of 0.2mM 4-methylumbelliferone-N-acetyl-α-D-glucopyranoside (Sigma), 0.1M NaC2H3O2, 0.5 mg/ml bovine serum albumin and incubated at 37 C 48. Reactions were stopped with 1 ml of 0.2M Na2CO3, 0.32M glycine. Substrate cleavage was determined at excitation 365nm and emission 448nm using a Hitachi F-2000 Flourescence Spectrophotometer using a standard curve of 0.5 to 5 nm/ml. Specific activity was corrected for protein concentration. Duplicate GUSB assays were performed similarly using the 4-methylumbelliferone enzyme assay method previously described 49. Activity levels were analyzed by one way ANOVA for comparison of treatments to NO TX groups after confirmation of a difference between NORMAL and MPS IIIB NO TX groups by Student’s t-test. Barnes-Forsythe Test of equal variances was satisfied as not significant for organ comparisons of brain, liver, heart, lung, spleen and kidney of GUSB.
Glycosaminoglycan analysis
Homogenized brain samples (N=4–8 for each group, all from mice older than 200 days) were coded with a numerical identifier and sent blinded to Zacharon Pharmaceuticals Inc. (San Diego, CA) for pathologic GAG (pGAG) analysis using Sensi-Pro Non-Reducing End (NRE) assay. pGAG are the GAG fragments present due to the deficiency of the specific lysosomal enzyme. The Sensi-Pro NRE assay is a highly specific and sensitive assay that uses High Performance Liquid Chromatography (HPLC) to quantitate the reduction in lysosomal GAG accumulation, by labeling and quantifying the non-reducing ends (NRE) of these GAG fragments. In MPSIIIB, the pGAG markers are unique NRE-derived trisaccharides that terminate in N-acetylglucosamine due to the lysosomal deficiency in N-acetylglucosaminidase as described in detail by Lawerence et al. 20,21. The GAGs were extracted and purified by DEAE chromatography, digested with heparin lyases, fluorescently labeled and analyzed as previously described 19. One way ANOVA was used to compare picograms of pGAG per microgram of protein for each treatment group to the MPS IIIB NO TX group.
Circadian assessments
Six male mice from each group were studied from 14 to 24 weeks of age as previously described 5. All mice were housed individually in cages with a running wheel within light-tight ventilated chambers illuminated by fluorescent bulbs (F30T12-SP41-RS, General Electric, USA, 3.9 × 1017 to 6.9 × 1018 photons/s/m2) at the bottom of the cages. Wheel running activity was recorded in 1 min bins (Clocklab, Actimetrics, Evanston, IL) while mice were exposed to a light-dark (LD) schedule (lights on at 7:00a.m. and off at 7:00 p.m.).
We analyzed phase angle of entrainment (delay between daily light offset and onset of activity), total daily activity, and proportion of daily activity in the light phase of the photocycle using Clocklab 50. Statistical analysis was by repeated measures ANOVA.
Auditory evaluation
ABRs were performed at 8–8.5 months of age using procedures similar to those described previously (Heldermon et al., 2007, 2010). Mice were anesthetized with ketamine/xylazine (85/15 mg/kg, i.p.) while core temperature was maintained at 37.0 ± 1.0 °C by a thermostatically regulated heating pad monitored via a rectal probe (Yellow Springs). Platinum needle electrodes (Grass, West Warwick, RI) were placed subcutaneously in the back, vertex and behind the right ear while connecting to a Grass P15 differential amplifier (100–10,000 Hz, 100x). A Cambridge Electronic Design Micro 1401 (Cambridge Electronic Design, Cambridge, England) running SIGNAL™ and custom averaging software digitized the signal at 30 kHz. Toneburst stimuli at 5, 10, 20, 28.3 and 40 kHz were delivered 1000 times at 20/second using an Alpine SPS-OEOA coaxial speaker (Crutchfield, Charlottesville, VA) located 10 cm lateral to the right ear. At each test frequency, the stimulus level was reduced in 5 dB minimum steps to determine the minimum sound pressure level required for visual detection of Wave I. Repeated measure ANOVA with Dunnets multiple comparison correction was performed to compare each treatment group to the MPS IIIB NO TX control. Separate one way ANOVA was performed for data from all groups at each test frequency, followed by Bonferroni multiple comparisons tests. Sample sizes by group were as follows: MPS IIIB NO TX, n= 10; MPS IIIB IC-AAV, n= 9; MPS IIIB IV-LENTI, n= 9; MPS IIIB BOTH, n= 10; Normal NO TX, n= 10; Normal BOTH, n= 10.
Motor function assessment
As we described previously in this model (Heldermon et al., 2007, Heldermon 2010), mice were trained on a rotarod (UGO Basile, Varese, Italy) moving at a speed of 10 rpm that switched direction after each full rotation for up to 180 seconds per attempt, with three attempts per day. Tests were done every 28 days from 196 up to 672 days of age (MPS IIIB NO TX, n= 6; MPS IIIB IC-AAV, n= 9; MPS IIIB IV-LENTI, n= 10; MPS IIIB BOTH, n= 10; Normal NO TX, n= 9; Normal BOTH, n= 8). Longest latency to fall from the rotarod of the three attempts was used for comparisons. Data were interpreted using a Log Rank test for time to latency of <60 seconds on rotarod.
Life span
All treated animals were analyzed by intention to treat with a Gehan–Wilcoxon test. Kaplan-Meier curves were generated to assess the effect of treatments on survival.
Acknowledgments
This work was funded in part by NIH grants NS043205 (M.S.S), HD055461 (M.S.S), K08 DK085141-01 (C.D.H) and by the Sanfilippo Children’s Research Foundation (C.D.H.).
Footnotes
Conflict of interest.
Jillian R. Brown and Brett E. Crawford are full time employees and shareholders of Zacharon Pharmaceuticals. All other authors have no conflict of interest to report.
References
- 1.Yogalingam G, Hopwood JJ. Molecular genetics of mucopolysaccharidosis type IIIA and IIIB: Diagnostic, clinical, and biological implications. Hum Mutat. 2001;18:264–281. doi: 10.1002/humu.1189. [DOI] [PubMed] [Google Scholar]
- 2.Weber B, Blanch L, Clements PR, Scott HS, Hopwood JJ. Cloning and expression of the gene involved in Sanfilippo B syndrome (mucopolysaccharidosis III B) Hum Mol Genet. 1996;5:771–777. doi: 10.1093/hmg/5.6.771. [DOI] [PubMed] [Google Scholar]
- 3.Zhao HG, Li HH, Bach G, Schmidtchen A, Neufeld EF. The molecular basis of Sanfilippo syndrome type B. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:6101–6105. doi: 10.1073/pnas.93.12.6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li HH, Yu WH, Rozengurt N, Zhao HZ, Lyons KM, Anagnostaras S, et al. Mouse model of Sanfilippo syndrome type B produced by targeted disruption of the gene encoding alpha-N-acetylglucosaminidase. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:14505–14510. doi: 10.1073/pnas.96.25.14505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heldermon CD, Hennig AK, Ohlemiller KK, Ogilvie JM, Herzog ED, Breidenbach A, et al. Development of sensory, motor and behavioral deficits in the murine model of Sanfilippo syndrome type B. PLoS ONE. 2007;2:e772. doi: 10.1371/journal.pone.0000772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fu H, Samulski RJ, McCown TJ, Picornell YJ, Fletcher D, Muenzer J. Neurological correction of lysosomal storage in a mucopolysaccharidosis IIIB mouse model by adeno-associated virus-mediated gene delivery. Mol Ther. 2002;5:42–49. doi: 10.1006/mthe.2001.0514. [DOI] [PubMed] [Google Scholar]
- 7.Cressant A, Desmaris N, Verot L, Brejot T, Froissart R, Vanier MT, et al. Improved behavior and neuropathology in the mouse model of Sanfilippo type IIIB disease after adeno-associated virus-mediated gene transfer in the striatum. J Neurosci. 2004;24:10229–10239. doi: 10.1523/JNEUROSCI.3558-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fu H, Kang L, Jennings JS, Moy SS, Perez A, Dirosario J, et al. Significantly increased lifespan and improved behavioral performances by rAAV gene delivery in adult mucopolysaccharidosis IIIB mice. Gene Ther. 2007;14:1065–1077. doi: 10.1038/sj.gt.3302961. [DOI] [PubMed] [Google Scholar]
- 9.Di Natale P, Di Domenico C, Gargiulo N, Castaldo S, Gonzalez YRE, Mithbaokar P, et al. Treatment of the mouse model of mucopolysaccharidosis type IIIB with lentiviral-NAGLU vector. Biochem J. 2005;388:639–646. doi: 10.1042/BJ20041702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fu H, DiRosario J, Kang L, Muenzer J, McCarty DM. Restoration of central nervous system alpha-N-acetylglucosaminidase activity and therapeutic benefits in mucopolysaccharidosis IIIB mice by a single intracisternal recombinant adeno-associated viral type 2 vector delivery. J Gene Med. 2010;12:624–633. doi: 10.1002/jgm.1480. [DOI] [PubMed] [Google Scholar]
- 11.McCarty DM, DiRosario J, Gulaid K, Muenzer J, Fu H. Mannitol-facilitated CNS entry of rAAV2 vector significantly delayed the neurological disease progression in MPS IIIB mice. Gene Ther. 2009;16:1340–1352. doi: 10.1038/gt.2009.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heldermon CD, Ohlemiller KK, Herzog ED, Vogler C, Qin E, Wozniak DF, et al. Therapeutic efficacy of bone marrow transplant, intracranial AAV-mediated gene therapy, or both in the mouse model of MPS IIIB. Mol Ther. 2010;18:873–880. doi: 10.1038/mt.2010.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ellinwood NM, Ausseil J, Desmaris N, Bigou S, Liu S, Jens JK, et al. Safe, Efficient, and Reproducible Gene Therapy of the Brain in the Dog Models of Sanfilippo and Hurler Syndromes. Mol Ther. 2010;19:251–259. doi: 10.1038/mt.2010.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vogler C, Levy B, Grubb JH, Galvin N, Tan Y, Kakkis E, et al. Overcoming the blood-brain barrier with high-dose enzyme replacement therapy in murine mucopolysaccharidosis VII. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:14777–14782. doi: 10.1073/pnas.0506892102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sferra TJ, Backstrom K, Wang C, Rennard R, Miller M, Hu Y. Widespread correction of lysosomal storage following intrahepatic injection of a recombinant adeno-associated virus in the adult MPS VII mouse. Mol Ther. 2004;10:478–491. doi: 10.1016/j.ymthe.2004.05.029. [DOI] [PubMed] [Google Scholar]
- 16.Lin D, Donsante A, Macauley S, Levy B, Vogler C, Sands MS. Central Nervous System-directed AAV2/5-Mediated Gene Therapy Synergizes with Bone Marrow Transplantation in the Murine Model of Globoid-cell Leukodystrophy. Mol Ther. 2007;15:44–52. doi: 10.1038/sj.mt.6300026. [DOI] [PubMed] [Google Scholar]
- 17.Donsante A, Levy B, Vogler C, Sands MS. Clinical response to persistent, low-level beta-glucuronidase expression in the murine model of mucopolysaccharidosis type VII. J Inherit Metab Dis. 2007;30:227–238. doi: 10.1007/s10545-007-0483-4. [DOI] [PubMed] [Google Scholar]
- 18.Sands MS, Barker JE. Percutaneous intravenous injection in neonatal mice. Lab Anim Sci. 1999;49:328–330. [PubMed] [Google Scholar]
- 19.Deakin JA, Lyon M. A simplified and sensitive fluorescent method for disaccharide analysis of both heparan sulfate and chondroitin/dermatan sulfates from biological samples. Glycobiology. 2008;18:483–491. doi: 10.1093/glycob/cwn028. [DOI] [PubMed] [Google Scholar]
- 20.Lawrence R, Brown JR, Al-Mafraji K, Lamanna WC, Beitel JR, Boons GJ, et al. Disease-specific non-reducing end carbohydrate biomarkers for mucopolysaccharidoses. Nature chemical biology. 2012;8:197–204. doi: 10.1038/nchembio.766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lawrence R, Olson SK, Steele RE, Wang L, Warrior R, Cummings RD, et al. Evolutionary differences in glycosaminoglycan fine structure detected by quantitative glycan reductive isotope labeling. The Journal of biological chemistry. 2008;283:33674–33684. doi: 10.1074/jbc.M804288200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yogalingam G, Weber B, Meehan J, Rogers J, Hopwood JJ. Mucopolysaccharidosis type IIIB: characterisation and expression of wild-type and mutant recombinant alpha-N-acetylglucosaminidase and relationship with sanfilippo phenotype in an attenuated patient. Biochim Biophys Acta. 2000;1502:415–425. doi: 10.1016/s0925-4439(00)00066-1. [DOI] [PubMed] [Google Scholar]
- 23.Colville GA, Watters JP, Yule W, Bax M. Sleep problems in children with Sanfilippo syndrome. Dev Med Child Neurol. 1996;38:538–544. doi: 10.1111/j.1469-8749.1996.tb12114.x. [DOI] [PubMed] [Google Scholar]
- 24.Gorlin R. Genetic hearing loss associated with endocrine and metabolic disorders. In: Gorlin RJ, Toriello HV, Cohen MM, editors. Hereditary Hearing Loss and its Syndromes. Oxford Monographs on Medical Genetics. Vol. 28. Oxford University Press; New York: 1995. pp. 318–354. [Google Scholar]
- 25.Bredenkamp JK, Smith ME, Dudley JP, Williams JC, Crumley RL, Crockett DM. Otolaryngologic manifestations of the mucopolysaccharidoses. Ann Otol Rhinol Laryngol. 1992;101:472–478. doi: 10.1177/000348949210100605. [DOI] [PubMed] [Google Scholar]
- 26.Leung LS, Weinstein GW, Hobson RR. Further electroretinographic studies of patients with mucopolysaccharidoses. Birth Defects Orig Artic Ser. 1971;7:32–40. [PubMed] [Google Scholar]
- 27.Yoshimitsu M, Sato T, Tao K, Walia JS, Rasaiah VI, Sleep GT, et al. Bioluminescent imaging of a marking transgene and correction of Fabry mice by neonatal injection of recombinant lentiviral vectors. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:16909–16914. doi: 10.1073/pnas.0407572101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Biffi A, Bartolomae CC, Cesana D, Cartier N, Aubourg P, Ranzani M, et al. Lentiviral vector common integration sites in preclinical models and a clinical trial reflect a benign integration bias and not oncogenic selection. Blood. 2011;117:5332–5339. doi: 10.1182/blood-2010-09-306761. [DOI] [PubMed] [Google Scholar]
- 29.Wang GP, Levine BL, Binder GK, Berry CC, Malani N, McGarrity G, et al. Analysis of lentiviral vector integration in HIV+ study subjects receiving autologous infusions of gene modified CD4+ T cells. Mol Ther. 2009;17:844–850. doi: 10.1038/mt.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cartier N, Hacein-Bey-Abina S, Bartholomae CC, Veres G, Schmidt M, Kutschera I, et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818–823. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- 31.Zheng Y, Ryazantsev S, Ohmi K, Zhao HZ, Rozengurt N, Kohn DB, et al. Retrovirally transduced bone marrow has a therapeutic effect on brain in the mouse model of mucopolysaccharidosis IIIB. Mol Genet Metab. 2004;82:286–295. doi: 10.1016/j.ymgme.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 32.Fu H, Dirosario J, Killedar S, Zaraspe K, McCarty DM. Correction of Neurological Disease of Mucopolysaccharidosis IIIB in Adult Mice by rAAV9 Trans-Blood-Brain Barrier Gene Delivery. Mol Ther. 2011 doi: 10.1038/mt.2011.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moss RB, Milla C, Colombo J, Accurso F, Zeitlin PL, Clancy JP, et al. Repeated aerosolized AAV-CFTR for treatment of cystic fibrosis: a randomized placebo-controlled phase 2B trial. Hum Gene Ther. 2007;18:726–732. doi: 10.1089/hum.2007.022. [DOI] [PubMed] [Google Scholar]
- 34.Hasbrouck NC, High KA. AAV-mediated gene transfer for the treatment of hemophilia B: problems and prospects. Gene Ther. 2008;15:870–875. doi: 10.1038/gt.2008.71. [DOI] [PubMed] [Google Scholar]
- 35.Nathwani AC, Rosales C, McIntosh J, Rastegarlari G, Nathwani D, Raj D, et al. Long-term Safety and Efficacy Following Systemic Administration of a Self-complementary AAV Vector Encoding Human FIX Pseudotyped With Serotype 5 and 8 Capsid Proteins. Mol Ther. 2011;19:876–885. doi: 10.1038/mt.2010.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H, Malani N, Hamilton SR, Schlachterman A, Bussadori G, Edmonson SE, et al. Assessing the potential for AAV vector genotoxicity in a murine model. Blood. 2010;117:3311–3319. doi: 10.1182/blood-2010-08-302729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reiss J, Hahnewald R. Molybdenum cofactor deficiency: Mutations in GPHN, MOCS1, and MOCS2. Hum Mutat. 2010;32:10–18. doi: 10.1002/humu.21390. [DOI] [PubMed] [Google Scholar]
- 38.Bell P, Moscioni AD, McCarter RJ, Wu D, Gao G, Hoang A, et al. Analysis of tumors arising in male B6C3F1 mice with and without AAV vector delivery to liver. Mol Ther. 2006;14:34–44. doi: 10.1016/j.ymthe.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 39.Embury JE, Frost S, Charron CE, Madrigal E, Perera O, Poirier AE, et al. Hepatitis Virus Protein X-Phenylalanine Hydroxylase fusion proteins identified in PKU mice treated with AAV-WPRE vectors. Gene Therapy and Molecular Biology. 2008;12:69–76. [Google Scholar]
- 40.Donsante A, Miller DG, Li Y, Vogler C, Brunt EM, Russell DW, et al. AAV vector integration sites in mouse hepatocellular carcinoma. Science. 2007;317:477. doi: 10.1126/science.1142658. [DOI] [PubMed] [Google Scholar]
- 41.Donsante A, Vogler C, Muzyczka N, Crawford JM, Barker J, Flotte T, et al. Observed incidence of tumorigenesis in long-term rodent studies of rAAV vectors. Gene Ther. 2001;8:1343–1346. doi: 10.1038/sj.gt.3301541. [DOI] [PubMed] [Google Scholar]
- 42.Rosas LE, Grieves JL, Zaraspe K, La Perle KM, Fu H, McCarty DM. Patterns of scAAV vector insertion associated with oncogenic events in a mouse model for genotoxicity. Mol Ther. 2012;20:2098–2110. doi: 10.1038/mt.2012.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hawkins-Salsbury JA, Reddy AS, Sands MS. Combination therapies for lysosomal storage disease: is the whole greater than the sum of its parts? Hum Mol Genet. 2011;20:R54–60. doi: 10.1093/hmg/ddr112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Biswas S, LeVine SM. Substrate-reduction therapy enhances the benefits of bone marrow transplantation in young mice with globoid cell leukodystrophy. Pediatr Res. 2002;51:40–47. doi: 10.1203/00006450-200201000-00009. [DOI] [PubMed] [Google Scholar]
- 45.Biffi A, Capotondo A, Fasano S, del Carro U, Marchesini S, Azuma H, et al. Gene therapy of metachromatic leukodystrophy reverses neurological damage and deficits in mice. The Journal of clinical investigation. 2006;116:3070–3082. doi: 10.1172/JCI28873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jeyakumar M, Norflus F, Tifft CJ, Cortina-Borja M, Butters TD, Proia RL, et al. Enhanced survival in Sandhoff disease mice receiving a combination of substrate deprivation therapy and bone marrow transplantation. Blood. 2001;97:327–329. doi: 10.1182/blood.v97.1.327. [DOI] [PubMed] [Google Scholar]
- 47.Lee JP, Jeyakumar M, Gonzalez R, Takahashi H, Lee PJ, Baek RC, et al. Stem cells act through multiple mechanisms to benefit mice with neurodegenerative metabolic disease. Nat Med. 2007;13:439–447. doi: 10.1038/nm1548. [DOI] [PubMed] [Google Scholar]
- 48.Marsh J, Fensom AH. 4-Methylumbelliferyl alpha-N-acetylglucosaminidase activity for diagnosis of Sanfilippo B disease. Clin Genet. 1985;27:258–262. doi: 10.1111/j.1399-0004.1985.tb00217.x. [DOI] [PubMed] [Google Scholar]
- 49.Sands MS, Barker JE, Vogler C, Levy B, Gwynn B, Galvin N, et al. Treatment of murine mucopolysaccharidosis type VII by syngeneic bone marrow transplantation in neonates. Lab Invest. 1993;68:676–686. [PubMed] [Google Scholar]
- 50.Herzog ED, Aton SJ, Numano R, Sakaki Y, Tei H. Temporal precision in the mammalian circadian system: a reliable clock from less reliable neurons. J Biol Rhythms. 2004;19:35–46. doi: 10.1177/0748730403260776. [DOI] [PubMed] [Google Scholar]