

MCL-1 is a BCL-2 family member that promotes the survival of multiple cellular lineages. Wang et al. show that cardiac-specific ablation of Mcl-1 results in a rapidly fatal, dilated cardiomyopathy with abnormal mitochondria ultrastructure and defective mitochondrial respiration. Genetic ablation of proapoptotic effectors Bax and Bak rescued the lethality and impaired cardiac function but not the mitochondria abnormalities and deficient respiration. These data suggest that blocking cell death is insufficient to completely overcome the need for MCL-1 in cardiomyocytes.
Keywords: heart failure, mitochondria, MCL-1, apoptosis, BCL-2
Abstract
MCL-1 is an essential BCL-2 family member that promotes the survival of multiple cellular lineages, but its role in cardiac muscle has remained unclear. Here, we report that cardiac-specific ablation of Mcl-1 results in a rapidly fatal, dilated cardiomyopathy manifested by a loss of cardiac contractility, abnormal mitochondria ultrastructure, and defective mitochondrial respiration. Strikingly, genetic ablation of both proapoptotic effectors (Bax and Bak) could largely rescue the lethality and impaired cardiac function induced by Mcl-1 deletion. However, while the overt consequences of Mcl-1 loss were obviated by combining with the loss of Bax and Bak, mitochondria from the Mcl-1-, Bax-, and Bak-deficient hearts still revealed mitochondrial ultrastructural abnormalities and displayed deficient mitochondrial respiration. Together, these data indicate that merely blocking cell death is insufficient to completely overcome the need for MCL-1 function in cardiomyocytes and suggest that in cardiac muscle, MCL-1 also facilitates normal mitochondrial function. These findings are important, as specific MCL-1-inhibiting therapeutics are being proposed to treat cancer cells and may result in unexpected cardiac toxicity.
The death of cardiomyocytes has been linked to the initiation and progression of heart failure (Mughal et al. 2012). Since adult cardiomyocytes are terminally differentiated, they are limited in their ability to be replaced by self-renewal; therefore, the heart is particularly sensitive to the death of these cells (Kirshenbaum and Schneider 1995; Kirshenbaum et al. 1996). Indeed, even a very low rate of cardiomyocyte apoptosis (0.023% vs. 0.002% in controls) can be sufficient to cause a lethal, dilated cardiomyopathy (Wencker et al. 2003). Therefore, understanding how cell death is regulated in cardiomyocytes is critical to identifying potential therapies aimed at preventing cardiac damage and preserving cardiac function.
Members of the BCL-2 family of proteins act by either promoting or inhibiting apoptosis. Intrinsic apoptosis is triggered when stimuli activate BH3-only molecules (such as BID, BAD, BIM, PUMA, and NOXA), resulting in the activation of the multidomain proapoptotic effectors BAX and BAK and inducing mitochondrial outer membrane permeabilization. Functionally, BAX and BAK make up the obligate gateway to mitochondrial dysfunction and cell death induced by the BCL-2 family, as loss of both blocks the induction of intrinsic apoptosis (Lindsten et al. 2000; Wei et al. 2001). BAX and BAK activation is antagonized by the anti-apoptotic family members (such as BCL-2, BCL-XL, BCL-W, MCL-1, and A1) (Cheng et al. 2001; Zong et al. 2001).
Anti-apoptotic MCL-1 is particularly unique among prosurvival BCL-2 family members in that it is essential for embryonic development (Rinkenberger et al. 2000) and the survival of multiple cell lineages in the adult, including lymphocytes (Opferman et al. 2003; Dzhagalov et al. 2008; Vikstrom et al. 2010), hematopoietic stem cells (Opferman et al. 2005), neutrophils (Dzhagalov et al. 2007; Steimer et al. 2009), and neurons (Arbour et al. 2008; Malone et al. 2012). In contrast, other anti-apoptotic BCL-2 family members function in much more restricted fashions (Veis et al. 1993; Motoyama et al. 1995, 1999). MCL-1 can antagonize a subset of BH3-only family members, including BIM, PUMA, NOXA, and BID, but does not bind other BH3-only family members, including BAD and BNIP3 (Certo et al. 2006; Willis et al. 2007). The reason for MCL-1's unique nature in promoting normal physiology in so many cell lineages is still unclear, as most cell types express multiple anti-apoptotic molecules (Krajewski et al. 1995; Yang et al. 1995; Opferman et al. 2005).
BCL-2 family members play central roles in the regulation of cell death in the heart. During ischemia reperfusion, levels of anti-apoptotic BCL-2 and BCL-XL decline, and apoptotic cell death occurs (Olivetti et al. 1996; Murriel et al. 2004). Overexpression of BCL-2 or BCL-XL can block cardiac death in response to ischemia reperfusion, leading to smaller infarcts and faster recovery (Brocheriou et al. 2000; Chen et al. 2001b; Huang et al. 2003; Imahashi et al. 2004). Similarly, transgenic expression of BCL-2 was also protective in a mouse model of cardiomyopathy (Weisleder et al. 2004). Last, endogenous BCL-2 has been shown to be induced by preconditioning (exposure to short cycles of ischemia reperfusion) (Maulik et al. 1999). In contrast, a number of proapoptotic BH3-only family members (BID, PUMA, BAD, BNIP3, and NIX) have been shown to be induced by cardiac stress and have been implicated in promoting damage (Chen et al. 2001a; Kubasiak et al. 2002; Yussman et al. 2002; Murriel et al. 2004; Toth et al. 2006). These data indicate that while both pro- and anti-apoptotic regulators clearly play important roles in modulating the response of cardiomyocytes to stress, the specific genetic regulators have remained unclear.
Using Mcl-1 conditional mice, we demonstrate that ablation of Mcl-1 in cardiomyocytes results in a fatal dilated cardiomyopathy evidenced by fibrosis and a lack of contractility. Moreover, the cardiac dysfunction was accompanied by increased cell death, thus positing the question of whether MCL-1 loss in cardiomyocytes triggers an apoptotic response. Therefore, we combined the genetic ablation of the key apoptotic effector molecules (BAX and BAK) with Mcl-1 deletion and identified that codeletion of Mcl-1 and both Bax and Bak could substantially rescue the lethality and cardiomyopathy induced by MCL-1 loss. However, cardiac-specific loss of Mcl-1, Bax, and Bak still resulted in ultrastructural mitochondrial abnormalities and mitochondrial respiratory defects, indicating that MCL-1 plays additional roles beyond simply inhibiting cell death. Therefore, we propose that a critical function of MCL-1 in cardiomyocytes is to antagonize the apoptotic machinery to promote the survival of cardiac muscle cells and facilitate mitochondrial respiration and homeostasis.
Results
Cardiomyopathy induced upon constitutive Mcl-1 deletion in cardiac and skeletal muscle
To define the role of MCL-1 in the heart, we bred Mcl-1flox/flox mice with transgenic mice expressing Cre-recombinase driven by muscle creatine kinase (Ckmm) promoter that constitutively expresses in both skeletal and cardiac muscle starting at embryonic day 13 (E13) (Li et al. 2000). The resulting Mcl-1flox/flox, Ckmm-Cre mice were born at the expected Mendelian ratio, but all pups died within the first 10 d after birth (Fig. 1A; Supplemental Table 1). In contrast, no lethality was observed in wild-type mice (Fig. 1A; Supplemental Table 1). MCL-1 protein levels were significantly decreased by postnatal day 1 (P1) in the heart and skeletal muscle but not in other tissues, indicating that the deletion of Mcl-1 in this model is efficient and muscle-specific (Fig. 1B). Histological analysis of the hearts of Mcl-1flox/flox, Ckmm-Cre mice at the age of P7 exhibited thinning of the heart walls, cardiac dilation, thrombus deposition, and interstitial fibrosis (Fig. 1C). All of these features were absent in the littermate control mice, indicating that Mcl-1 deletion elicited the myocardial degeneration. In contrast to the phenotypes observed in the Mcl-1-deleted hearts, histological interrogation of skeletal muscle only revealed the rare appearance of sporadic myofiber necrosis and inflammation (Fig. 1D). Transmission electron microscopy (TEM) analysis of Mcl-1-deleted skeletal muscle did not detect any overt abnormalities (Supplemental Fig. 1). Therefore, the effects of constitutive Mcl-1 deletion were most apparent in the cardiomyocytes, with only subtle defects observed in skeletal muscle.
Figure 1.

Cardiomyopathy is induced upon constitutive Mcl-1 deletion in cardiac and skeletal muscle. (A) Survival of muscle-specific Mcl-1-deleted mice. (**) P < 0.01 by log-rank test when compared with controls. (B) Constitutive Mcl-1 deletion in heart and skeletal muscle. All tissues were harvested at P1 and immunoblotted with MCL-1 and Actin. (C) Hematoxylin and eosin (H&E)-stained longitudinal sections of the heart indicate dilated cardiomyopathy and a loss of myofibrils. Connective tissue indicative of fibrosis stained blue with the Masson's trichrome stain. Hearts were harvested at P7. Magnifications: top, 2×; middle and bottom, 40×. (D) H&E-stained longitudinal sections of the tongue skeletal muscle indicate swollen fragmented fibers with hyaline necrosis. Magnification, 40×. (E) Cardiac-specific deletion of Mcl-1 induced cell death. Caspase-3- and Caspase-7-stained sections of the heart indicate positive cells in constitutively Mcl-1-deleted hearts (harvested at P7). Magnification, 40×.
Mcl-1 deletion in adult cardiac muscle results in rapid, fatal cardiomyopathy
Since Ckmm-Cre mice express the Cre-recombinase during embryonic development, it is possible that the dramatic effects on myocardium could be due to developmental problems rather than acute damage to mature muscle. To test this hypothesis, we turned to an inducible, cardiac-specific Mcl-1-deficient mouse model in which Mcl-1 deletion can be induced during adulthood. We crossed Mcl-1flox/flox mice with Myh6-Mer-Cre-Mer (Myh-CreER) transgenic mice that express the tamoxifen-inducible CreER-recombinase under the control of a mouse, cardiac-specific α-myosin heavy chain promoter (Sohal et al. 2001). Mcl-1flox/flox, Myh-CreER mice were born at the expected Mendelian ratio and exhibited no overt phenotype in the absence of tamoxifen treatment. However, 2 wk after tamoxifen treatment of adult mice (aged 8–12 wk), we observed a significant decrease of MCL-1 protein levels specifically in the hearts of Mcl-1flox/flox, Myh-CreER mice but not littermate control mice (Mcl-1flox/wt) (Fig. 2A). Three weeks after tamoxifen initiation, echocardiographic (ECHO) analysis detected dilated cardiomyopathy with increased left ventricle (LV) interior dimension (LVID) and decreased LV posterior wall (LVPW) and interventicular septum (IVS) wall thickness of cardiac-specific Mcl-1-deficient mice but not littermate controls, including Mcl-1wt/wt, Myh-CreER mice (Fig. 2B). These data indicate that the lack of contractility is due to Mcl-1 deletion and not merely Cre activation or tamoxifen treatment. The LV contractility as measured by ejection fraction (EF) and fraction shortening (FS) was significantly decreased and worsened with time after tamoxifen treatment (Fig. 2C). Ultimately, the Mcl-1flox/flox, Myh-CreER mice exhibited overt signs of heart failure 3 wk after tamoxifen initiation, and virtually all animals succumbed shortly thereafter to heart failure (Fig. 2D). In a time-course experiment, we observed cardiac dysfunction in Mcl-1flox/flox, Myh-CreER mice as soon as 1 wk after tamoxifen administration (Supplemental Fig. 2A). Minor decreases in the cardiac function of Mcl-1flox/flox, Myh-CreER mice prior to tamoxifen administration were consistent with a low level of recombination induced by CreER leakiness (Supplemental Fig. 2B). These data indicate that the cardiac dysfunction induced by Mcl-1 deletion in adult mice is initiated quickly after deletion and rapidly leads to fatal cardiac failure.
Figure 2.

Mcl-1 deletion in adult cardiac muscle results in rapid, fatal cardiomyopathy. (A) Tamoxifen-induced Mcl-1 deletion in the heart. All tissues were harvested 2 wk after tamoxifen initiation and immunoblotted with MCL-1 and Actin. (B) Cardiac function of a Mcl-1-deleted heart. Transthoracic ECHO studies were performed 3 wk after tamoxifen initiation. M-Mode images were recorded through IVS and LVPW at the papillary muscle level to get the LV dimensions and IVS and LVPW thickness. (C) Contractility of a Mcl-1-deleted heart as measured by ECHO for LV EF and FS. The data are presented as percent of baseline (before tamoxifen treatment). (**) P < 0.01. The number of mice analyzed is indicated as n. Note: Only two Mcl-1-deleted mice were analyzed at 6 wk due to the death of most animals by this time point. Error bars represent the standard error of the mean. (D) Survival of cardiac-specific Mcl-1-deleted mice. (**) P < 0.01 by log-rank test. (E) H&E-stained cross sections of the heart indicate dilated cardiomyopathy and a loss of myofibrils. Magnifications: left, 2×; right, 40×. (F) Fibrosis in the cross-sections of the heart stained blue with the Masson's trichrome stain. Magnifications: left, 2×; right, 40×. (G) Number of TUNEL- and Caspase-3-positive cardiomyocytes upon Mcl-1 deletion. The number of mice analyzed is indicated as n. Error bars represent the standard error of the mean. The P-values are indicated comparing wild-type with Mcl-1-deleted mice by two-tailed Student's t-test.
Consistent with the ECHO data, histological sections of Mcl-1flox/flox, Myh-CreER mice harvested 3 wk after tamoxifen initiation displayed significantly dilated LVs and decreased wall thickness when compared with littermate control hearts (Fig. 2E). Higher-magnification histology revealed the loss of myofibrils in Mcl-1-deleted hearts (Fig. 2E). Furthermore, the regions with myofiber loss exhibited dramatic deposition of interstitial connective tissue, as detected by Masson's trichrome staining (Fig. 2F). These data indicate that similar to the constitutive deletion of Mcl-1 in cardiac and skeletal muscle (Ckmm-Cre), inducible deletion of Mcl-1 in adult cardiac tissue (Myh-CreER) results in a rapid and fatal cardiomyopathy.
Cardiac-specific deletion of MCL-1 induced cell death
MCL-1 is an anti-apoptotic protein, and therefore it could be predicted that loss of MCL-1 expression promotes the induction of apoptosis, as has been observed in a variety of other tissues (Opferman et al. 2003, 2005; Steimer et al. 2009). Within 1 wk after tamoxifen treatment, we observed that Mcl-1 deletion results in the loss of cardiomyocytes and a corresponding increase in fibrosis. To test whether cardiac-specific deletion of MCL-1 induces apoptosis of the cardiomyocytes, we performed terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) and Caspase-3 immunohistochemical staining in constitutive (Ckmm-Cre) Mcl-1-deleted hearts and inducible tamoxifen-treated Myh-CreER hearts. Low but significant levels of apoptosis were observed in Mcl-1-deleted hearts in both mouse models (Figs. 1E, 2G; Supplemental Fig. 2C). These data are consistent with previous reports that even low levels of cell death occurring in the heart are sufficient to result in cardiomyopathy, as adult cardiomyocytes are limited in their ability to regenerate after damage (Kirshenbaum and Schneider 1995; Kirshenbaum et al. 1996; Wencker et al. 2003).
Anti-apoptotic MCL-1 functions in part by binding proapoptotic molecules, including BAX, BAK, BIM, PUMA, NOXA, and BID, on the mitochondria to prevent the induction of cell death (Certo et al. 2006; Willis et al. 2007). Immunoblot analyses of proapoptotic molecule expression indicated that upon Mcl-1 deletion, the expression of most proapoptotic effectors at the mitochondria was unchanged, but both BAX and BIM levels were subtly elevated in Mcl-1-deleted hearts (Supplemental Fig. 3). We were unable to detect significant expression of either PUMA or NOXA, potentially as MCL-1 loss may be insufficient to induce the expression of these molecules in the heart, unlike overt stimuli like ischemia reperfusion (Toth et al. 2006; data not shown). BNIP3, a molecule that has been implicated in cardiac cell death, was also found to be induced by Mcl-1 deletion (Supplemental Fig. 3; Regula et al. 2002). Therefore, Mcl-1 deletion is accompanied by an increase in expression of a number of proapoptotic proteins, including BAX, BIM, and BNIP3.
Genetic ablation of Bax and Bak improves cardiac function and survival of cardiac-specific Mcl-1 deletion
To further confirm that apoptosis contributed to the cardiac pathology after Mcl-1 deletion, we sought to genetically disable the intrinsic apoptotic pathway in the mouse heart by deleting both proapoptotic effectors BAX and BAK (Lindsten et al. 2000; Wei et al. 2001). To do so, Mcl-1flox/flox mice were bred to Baxflox/flox, Bak-deficient mice to produce mice in which Mcl-1, Bax, and Bak could simultaneously be genetically deleted using either Ckmm-Cre or Myh-CreER (Takeuchi et al. 2005). In addition, deletion of Bax and Bak may also down-regulate cell death caused by primary necrosis (opening of mitochondrial permeability transition pore [mPTP] in the absence of cytochrome c release) by increasing the threshold for the opening of the mPTP (Whelan et al. 2012). Therefore, loss of BAX and BAK could be anticipated to block both necrotic and apoptotic cell death.
Mcl-1flox/flox, Baxflox/flox, Bak−/−, Ckmm-Cre (hereafter referred to as Ckmm triple knockout [TKO]) mice were born at the expected Mendelian ratios and, unlike Mcl-1flox/flox, Ckmm-Cre, were able to survive to adulthood (Fig. 3A). Analysis of the one mouse out of 10 Ckmm-TKO mice that revealed cardiac dysfunction at 5 wk of age and subsequently died indicated that this mouse exhibited incomplete Bax deletion (Fig. 3A). ECHO analysis was unable to detect significant cardiac dysfunction in the other Ckmm-TKO mice as compared with wild-type and Baxflox/flox, Bak−/−, Ckmm-Cre (hereafter referred to as Ckmm double knockout [DKO]) littermates up to 10 wk of age (Fig. 3B). Surprisingly, even Mcl-1flox/flox, Baxflox/wt, Bak+/−, Ckmm-Cre mice (expressing fewer Bax and Bak alleles) had an extended life span when compared with Mcl-1flox/flox, Ckmm-Cre but still exhibited impaired cardiac contractility and eventually succumbed (Fig. 3A,B). Histology of Ckmm-TKO revealed that the cardiomyopathy observed in Mcl-1flox/flox, Ckmm-Cre mice was alleviated (Fig. 3C). In contrast, Mcl-1flox/flox, Baxflox/wt, Bak+/−, Ckmm-Cre mice still exhibited loss of cardiac myofibers and interstitial fibrosis (Fig. 3C). These results indicate that the more Bax and Bak alleles remaining, the more severe the cardiac phenotype observed upon Mcl-1 deletion, suggesting that the proapoptotic effectors play an important role in the pathogenesis of cardiomyopathy induced by Mcl-1 deletion.
Figure 3.

Genetic ablation of Bax and Bak improves cardiac function and survival of cardiac-specific Mcl-1 deletion. (A) Survival of Ckmm-TKO mice and littermate control mice. (**) P < 0.01 by log-rank test when compared with Mcl-1flox/flox Ckmm-Cre mice. (B) Contractility of Mcl-1wt/wt, Mcl-1flox/flox, Baxflox/wt Bak+/− Ckmm-Cre, Ckmm-DKO, and Ckmm-TKO hearts. The data are presented as percent of baseline (before tamoxifen treatment). (*) P < 0.05 versus littermate control mice. Error bars represent the standard error of the mean. (C) H&E-stained cross-sections of the heart indicate dilated cardiomyopathy and a loss of myofibrils, and Masson's trichrome staining indicates fibrosis in the Mcl-1flox/flox Baxflox/wt Bak+/− Ckmm-Cre heart. Magnifications are indicated for each panel. All hearts were harvested at P84.
To test whether inhibition of apoptosis was capable of abrogating the cardiac damage due to inducible Mcl-1 deletion in adult hearts, we generated inducible cardiac-specific Mcl-1flox/flox, Baxflox/flox, Bak−/−, Myh-CreER (hereafter referred to as Myh-TKO) mice by crossing Mcl-1flox/flox, Myh-CreER mice with Baxflox/flox, Bak−/− mice. Immunoblot analyses indicated efficient deletion of MCL-1, BAX, and BAK in cardiac tissue of tamoxifen-treated Myh-TKO mice (Fig. 4A). In contrast to the prevalent mortality observed in Mcl-1flox/flox, Myh-CreER mice 3 wk after tamoxifen initiation, Myh-TKO mice were able to survive for >8 wk after the induction of Cre, at which point they were sacrificed for analysis (Fig. 4B). During the first week after tamoxifen treatment, Myh-TKO mice exhibited a transient but significant defect in both EF and FS that eventually resolved (Fig. 4C). Upon sacrifice, Myh-TKO hearts were comparable with littermate control Mcl-1flox/wt hearts without the appearance of overt apoptosis, interstitial fibrosis, and loss of myofibers observed in Mcl-1flox/flox, Myh-CreER hearts (Figs. 2E–G, 4D; Supplemental Fig. 2C). We also observed the induction of BIM and BNIP3 in the Myh-TKO hearts, indicating that they are upstream of BAX and BAK (Supplemental Fig. 3). Therefore, codeletion of BAX, BAK, and MCL-1 prevents heart failure and overt cardiomyopathy by blocking cell death.
Figure 4.

Genetic ablation of Bax and Bak improves cardiac function and survival of cardiac-specific Mcl-1 deletion. (A) Tamoxifen-induced Mcl-1, Bax, and Bak deletion in heart. All tissues were harvested 8 wk after tamoxifen initiation and immunoblotted with MCL-1, BAX, and BAK. Manganese superoxide dismutase (MnSOD) served as the mitochondrial marker and Actin served as the loading control. (B) Survival of Myh-TKO mice and littermate controls. (**) P < 0.01 as determined by log-rank test when compared with Mcl-1flox/flox Myh-CreER. (C) Contractility of Mcl-1wt/wt, Mcl-1flox/flox Myh-CreER, Myh-DKO, and Myh-TKO hearts. The data are presented as percent of baseline (before tamoxifen treatment). (*) P < 0.05; (**) P < 0.01. Error bars represent the standard error of the mean. (D) H&E-stained cross-sections of the heart indicate no evidence of cardiomyopathy in the Myh-TKO hearts. Masson's staining indicates no evidence of fibrosis in Myh-TKO hearts. All hearts were harvested 8 wk after tamoxifen initiation. Magnifications are as indicated.
Mitochondrial ultrastructural abnormalities and respiratory deficiency in Mcl-1-deleted hearts
Adequate cardiac function is dependent on an organized myocardium and mitochondria (Kuznetsov and Margreiter 2009); therefore, we analyzed the ultrastructure of Mcl-1-deleted hearts by TEM. In normal myocardium, mitochondria containing cristae with high electron density are elongated and tightly aligned between myofibrils (Milner et al. 2000). However, mitochondria in the Mcl-1-deleted myocardium from both mouse models appeared rounded, disorganized, and dispersed (Fig. 5A,B). The cristae structure of mitochondria also appeared frequently fragmented, disrupted, and degraded, and many mitochondria possessed large vacuoles within an intact outer mitochondrial membrane (OMM) (Fig. 5A,B). These defects were similar to those observed in the mitochondria from Mcl-1-deleted SV40-transformed murine embryonic fibroblasts (MEFs) (Perciavalle et al. 2012). Additionally, the loss of myofibrils detected by TEM was apparent from Mcl-1-deleted but not littermate control hearts (Fig. 5A,B).
Figure 5.

Mitochondrial ultrastructural abnormalities in Mcl-1-deleted hearts cannot be prevented by genetic ablation of Bax and Bak. Transmission electron micrographs indicate disrupted myocardium and disorganized mitochondria with abnormal cristae structure in A. Constitutively Mcl-1-deleted hearts (harvested at P9). Bar, 500 nm. (B) Tamoxifen-induced Mcl-1-deleted hearts (3 wk after tamoxifen initiation). Bar, 500 nm. (C) Constitutively Mcl-1-deleted Ckmm-TKO hearts (harvested at P84). (D) Tamoxifen induced Mcl-1-deleted Myh-TKO hearts (8 wk after tamoxifen initiation). For the Ckmm-TKO and Myh-TKO samples, regions with relatively normal morphology are indicated by i and iii, whereas the regions with abnormal ultrastructure are indicated by ii and iv. Bars: top panels (lower magnification), 2 μm; bottom panels (higher magnification), 500 nm.
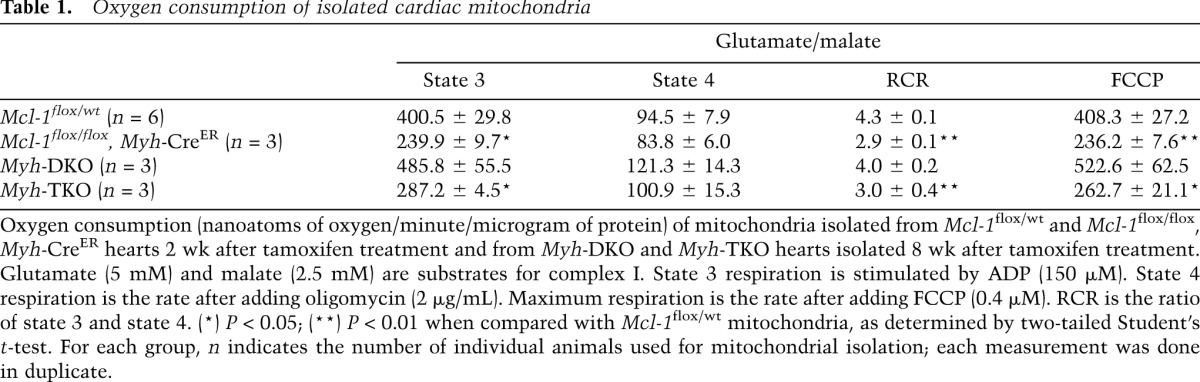
Mitochondrial cristae are enriched with proteins of the electron transport chain (ETC) that participate in mitochondrial oxidative phosphorylation and respiration (Da Cruz et al. 2003). Therefore, we sought to address whether the mitochondria isolated from Mcl-1-deleted cardiomyocytes exhibited defects in respiration by measuring the oxygen consumption rate (OCR). The mitochondria isolated from tamoxifen-treated Mcl-1flox/flox, Myh-CreER hearts revealed reduced state 3 respiration (ADP-stimulated) when compared with littermate control mitochondria (Table 1). The respiratory control ratio (RCR) of the Mcl-1-deleted mitochondria was significantly lower than that of littermate controls (Table 1). Furthermore, the maximal respiration of Mcl-1-deleted mitochondria (FCCP-uncoupled) was also reduced when compared with littermate controls (Table 1). Last, Mcl-1 deletion caused individual ETC complex enzymatic activity for NADH ubiquinone oxidoreductase (complex I) and cytochrome c oxidase (complex IV) to be reduced when assessed from isolated mitochondria but did not alter the function of succinateubiquinone oxidoreductase (complex II) (Supplemental Fig. 4A). The decreased function of the ETC components was not due to differential purification, as similar levels of mitochondrial (Prohibitin and MnSOD) and endoplasmic reticulum (BiP) contaminants were found in the preparations (Supplemental Fig. 4B). These data suggest that MCL-1 is necessary for the maintenance of normal cardiac mitochondrial respiration.
Table 1.
Oxygen consumption of isolated cardiac mitochondria
Mitochondrial dysfunction can result in elevated production of reactive oxygen species (ROS) (Wallace 1999). ROS are well known to induce mitochondrial DNA (mtDNA) mutations or deletions that have been implicated to cause cardiomyopathy (Fosslien 2003). Therefore, we investigated whether we could detect alterations in the mitochondrial genome by quantitative real-time PCR (qPCR). Consistent with previous observations made in Mcl-1-deleted MEFs and livers (Perciavalle et al. 2012), the induction of cardiac-specific Mcl-1 deletion resulted in decreased mtDNA levels when normalized to genomic DNA (Table 2). Given the drop in mtDNA, we analyzed whether reductions in the total mitochondria number in the deleted hearts could be detected. Importantly, we did not observe major differences in the total amount of mitochondrial proteins or citrate synthase enzymatic activity in Mcl-1-deleted and littermate control heart lysates, indicating that the loss of MCL-1 did not simply lead to a dramatic loss of total mitochondrial mass (Fig. 4A; Supplemental Figure 4B,C). Furthermore, there was no detectable induction of a PGC1-α-dependent mitochondrial biogenesis program in the Mcl-1-deleted hearts; instead, PGC1-α target genes were actually repressed (Supplemental Fig. 5). Such a repression of PGC1-α target genes has also been observed in several models of cardiac hypertrophy and failure (Lehman and Kelly 2002; Garnier et al. 2003; Arany et al. 2006). Thus, these data indicate that Mcl-1 deletion results in mitochondrial dysfunction, which we predict contributes to the cardiomyopathy and decreased mitochondrial respiration in our model.
Table 2.
Quantification of mtDNA from isolated cardiac muscle

Genetic ablation of Bax and Bak was insufficient to prevent mitochondrial abnormalities induced by Mcl-1 deletion
Although ECHO and histological analyses did not display significant cardiac dysfunction, analyses of the ultrastructure of TKO hearts by TEM revealed that the myocardial organization and mitochondrial ultrastructure were quite heterogeneous among different regions of the heart (Supplemental Fig. 6). Examination indicated that both interfibrillar and subsarcolemmal mitochondria appear equally affected and that the mitochondria with abnormal ultrastructure were primarily found in the endocardium and not the epicardium. Both Ckmm-TKO and Myh-TKO hearts still exhibited regions of rounded mitochondria that were disorganized and dispersed (Fig. 5C,D). The cristae structure of the damaged Ckmm-TKO and Myh-TKO mitochondria were fragmented and degraded but possessed intact OMMs (Fig. 5C,D). These defects were similar to those observed in the mitochondria from Mcl-1-deleted myocardium (Fig. 5A,B). Additionally, TEM analyses of Ckmm-TKO and Myh-TKO hearts still revealed myocardial regions that featured some loss of myofibrils, but not to the same extent as those occurring in Mcl-1-deleted hearts (Fig. 5C,D).
Since the TEM detected that Myh-TKO hearts still exhibit defective mitochondria, we tested whether Myh-TKO-isolated mitochondria displayed detectable respiratory deficiencies. To this aim, Myh-TKO, Myh-DKO, and Mcl-1-deleted cardiac mitochondria were isolated, and their ability to respire was determined by measuring OCR. As previously determined, mitochondria from Mcl-1-deleted hearts displayed decreased OCR (Table 1). In contrast, Myh-DKO mitochondria respired similarly to control mitochondria, indicating that loss of both BAX and BAK does not result in defective mitochondrial respiration (Table 1). However, despite mitigating the symptoms of Mcl-1 deletion, the mitochondria from Myh-TKO mice still exhibited defective OCR, indicating a respiratory defect (Table 1). These data suggest that MCL-1 may also play a role in promoting normal cardiac mitochondrial respiration that is distinct from its ability to inhibit apoptosis.
The mitochondrial respiratory defects induced by cardiac-specific Mcl-1 deletion alone led to the loss of mtDNA (Table 2). Therefore, we investigated whether the loss of mtDNA could be alleviated in the Myh-TKO hearts. Unlike Myh-DKO cardiac tissue, which expressed similar levels of mtDNA when compared with nondeleted littermate control hearts, the hearts from Myh-TKO mice still displayed the loss of mtDNA (Table 2). Taken together, these data indicate that even when apoptosis is genetically blocked by loss of BAX and BAK, the loss of MCL-1 expression in cardiac tissue results in mitochondrial dysfunction.
Discussion
The understanding of how cell death is regulated in cardiac tissue has emerged over recent years; however, the precise role of the individual anti-apoptotic BCL-2 family members in regulating this process has remained unclear. Genetic deletions of either Bcl-2 or Bcl-XL have not been reported to lead to overt defects in cardiac muscle development or survival (Veis et al. 1993; Motoyama et al. 1995, 1999). Conversely, overexpression of BCL-2 renders cardiac muscle resistant to death induced by a variety of stresses (Kirshenbaum and de Moissac 1997; Brocheriou et al. 2000; Chen et al. 2001b; Imahashi et al. 2004). In addition, genetic deletion of Bax and Bak significantly promoted cell survival and reduced infarct size in a mouse model of myocardial infarction (Whelan et al. 2012). Previously, it was unknown whether MCL-1 played any role in protecting cardiac muscle from death.
Here, we reveal that cardiac-specific loss of anti-apoptotic MCL-1 expression leads to a rapid and lethal cardiac dysfunction typified by cardiac dilation, lack of contractility, interstitial fibrosis, and ultrastructural abnormalities in muscle fibers and associated mitochondria. Similar outcomes were observed when Mcl-1 was deleted either during embryonic development by Ckmm-Cre or in adult mice using the inducible Myh-CreER system. Interestingly, we did not observe overt defects in the skeletal muscle upon Ckmm-Cre-mediated ablation of Mcl-1. There were low levels of muscle fiber necrosis, but no gross muscular abnormalities were observable even in the skeletal muscle of Ckmm-TKO mice in either glycolytic or aerobic muscle fiber types. One possibility is that skeletal muscle, which possesses the ability to regenerate, may be able to functionally compensate for tissue damage, unlike cardiac muscle. Additionally, it is possible that the skeletal muscle will not reveal defects until the muscle is stressed. Conversely, MCL-1 function in the skeletal muscle may not be as important as that of cardiac muscle due to compensation by other family members. Further work will be necessary to fully realize why skeletal muscle appears to be less affected by Mcl-1 deletion.
Increased cell death was detected upon Mcl-1 deletion, and the overt cardiac dysfunction and lethality of Mcl-1-deleted mice was almost completely abrogated by codeletion of Bax and Bak, the essential intrinsic apoptotic effectors (Lindsten et al. 2000). Recently, it was described that BAX can also participate in regulation of necrosis by driving mitochondrial fusion, thus lowering the threshold for the opening of the mPTP (Whelan et al. 2012). Interestingly, in a companion study in this issue of Genes & Development, Thomas et al. (2013) demonstrate that codeletion of cyclophilin D (CypD), an important regulator of the mPTP, along with Mcl-1 in mouse hearts attenuated but did not completely alleviate cardiac dysfunction. Their data indicate that inhibiting necrotic death only partially alleviates the cardiac failure observed upon Mcl-1 deletion. In contrast, our data demonstrate that codeletion of Mcl-1, Bax, and Bak in mouse hearts more completely rescues the lethality of the cardiac failure. Together, these data suggest that while loss of Mcl-1 may induce mPTP-dependent necrosis, the critical role for MCL-1 in cardiomyocytes is to prevent the induction of cell death. While primary necrosis may be occurring, it would also be alleviated by deletion of Bax and Bak (Whelan et al. 2012). In either case, the induction of Caspase-3 induced by Mcl-1 deletion indicates that cytochrome c is being released, thereby triggering apoptosis.
MCL-1 is an important survival molecule that, when genetically ablated, results in the loss of a myriad of normal cell types (Perciavalle and Opferman 2013). Our data in cardiac muscle suggest that like other cell lineages, MCL-1 is a major regulator of survival, and deletion of Mcl-1 triggers induction of BAX, BIM, and BNIP3 expression, all of which could induce cell death. However, MCL-1 plays important roles beyond normal development and maintenance of homeostasis, as it has also been shown to be highly amplified in a variety of human malignancies (Beroukhim et al. 2010). High levels of MCL-1 expression have also been associated with resistance to the new generation of BH3 mimetic cancer therapies, including ABT-263 (navitoclax), which target the prosurvival proteins BCL-2, BCL-XL, and BCL-W (Konopleva et al. 2006; van Delft et al. 2006; Nguyen et al. 2007). Therefore, a number of new modalities are in development with the goal of inhibiting MCL-1's prosurvival activity in an attempt to increase efficacy on resistant malignancies. Our findings that MCL-1 is an essential prosurvival molecule in the heart suggest that a potential unwanted toxicity associated with pharmacological MCL-1 inhibition may be cardiac toxicity. Therefore, attempts to therapeutically inhibit MCL-1 function should be careful to monitor cardiac function.
An important consequence of mitochondrial dysfunction can be the clearance of damaged mitochondria by autophagy, also known as mitophagy (Kubli and Gustafsson 2012). Simply deleting Mcl-1 may primarily trigger the induction of cardiomyocyte apoptosis, leading to loss of myofibers and the rapid cardiac dysfunction that results in the death of the mice. However, when the intrinsic cell death pathway is blocked by codeletion of Bax and Bak, it is possible that the mitochondrial dysfunction induced by Mcl-1 loss produces mitochondrial stress that drives autophagy in an attempt to alleviate the damage. Indeed, genetic deletion of MCL-1 in the nervous system has been previously reported to induce autophagy (Germain et al. 2011). However, in a companion study, Thomas et al. (2013) demonstrate that deletion of Mcl-1 in myocardium actually leads to the impairment of autophagy in the heart under both basal and stressed conditions. The mechanism for specifically how the loss of MCL-1 expression inhibits autophagy induction is still unclear from their studies and will be an important topic of future research.
Despite grossly rescuing cardiac function and prolonging mouse survival, mice lacking Mcl-1, Bax, and Bak in the heart still exhibit cardiac lesions and defects in mitochondrial function, as evidenced by deficient mitochondrial respiration and the loss of mtDNA. These data indicate that MCL-1 may be important for the long-term health and function of the heart. Indeed, the presence of lesions in the myocardium of the Myh-TKO mice suggests that MCL-1 may play an important role beyond simply inhibiting cell death. We demonstrated previously in model cell lines and liver mitochondria that different forms of MCL-1 reside in distinct mitochondrial localizations, where they exhibit separable functions (Perciavalle et al. 2012). On the OMM, MCL-1 acts to bind and sequester BH3-only family members antagonizing cell death. In contrast, N-terminally truncated MCL-1 is imported into the mitochondrial matrix, where it promotes normal mitochondrial physiology and morphology. While the separation of these roles has been observed, they have not yet been shown to be relevant in vivo. Therefore, it is possible that in cardiomyocytes, both localizations of MCL-1 may be necessary for maintaining cardiac function. Further work will be necessary to functionally reconstitute cardiomyocytes of Mcl-1-deleted mice with individual MCL-1 mutants to functionally test the contribution of MCL-1's anti-apoptotic function and its ability to promote mitochondrial function.
Importantly, these data demonstrate that MCL-1 is an essential anti-apoptotic BCL-2 family member to promote normal heart function, but our observations also have broader implications for human health. For example, we identified that a vital consideration of targeting MCL-1's protective effects in cancer therapy would be to avoid inducing cardiac toxicity. Therefore, effective therapeutic strategies will need to be carefully tailored to avoid long-term inhibition of MCL-1's anti-apoptotic functions in the heart that could induce subsequent damage. Alternatively, we would also predict that strategies aimed at enhancing or maintaining MCL-1 function in cardiomyocytes may form the basis to improve heart function after acute cardiac stress or damage. To realize these goals, it will be important to gain greater understanding of the normal mechanisms by which MCL-1 is regulated in the myocardium and identify specifically how MCL-1's functions participate in promoting heart function. However, this study represents the first step by identifying the critical anti-apoptotic player in the heart.
Materials and methods
Mice
Mcl-1 conditional and Bax conditional Bak−/− mice have been described previously (Opferman et al. 2003; Takeuchi et al. 2005). Ckmm-Cre mice (obtained from The Jackson Laboratory, strain name B6.FVB(129S4)-Tg(Ckmm-cre)5Khn/J) were described previously (Li et al. 2000). Myh-CreER mice (obtained from The Jackson Laboratory, strain name B6.FVB(129)-Tg(Myh6-cre/Esr1*)1Jmk/J) were described previously (Sohal et al. 2001). To induce deletion, mice received five doses of tamoxifen (1 μg per dose; Sigma) emulsified in sunflower oil vehicle (Sigma) by gavage. All mice were maintained on a mixed 129/C57BL6 background, and littermate controls were used in all assays. The mice were bred in accordance with the institutional animal care and use committee at St. Jude Children's Research Hospital. All mice used in the present studies were maintained in specific pathogen-free conditions, and littermate controls were used as negative controls.
ECHO
Mice were shaved and anesthetized with 0.5%–1.5% isoflurane in a 95% O2, 5% CO2 chamber prior to ECHO. ECHO was performed using VEVO-2100 high-frequency ultrasound instrumentation (Fujifilm VisualSonics, Inc.) equipped with a MS-550D transducer operating at 40 MHz. All scans were taken with the heart rate between 300 and 400 beats per minute (bpm). M-Mode data were obtained in the parasternal short axis (PSAX) orientation to determine contractility of the heart. Data were post-processed using dedicated VisualSonics software (2100-1.4.0), and direct measurements were determined for IVS, LVPW, and LV dimensions at diastole and systole. From these data, EF and FS were determined to assess contractility.
Histology
Heart and skeletal muscle tissue was fixed in 10% neutral buffered formalin, embedded in paraffin, and cut into 4-um thick sections using HM500M cryostat (Microm). The paraffin sections were stained with hematoxlyn and eosin (H&E) and Masson's trichrome stain. Paraffin sections were deparaffinized, and immunohistochemistry for detection of Capase-3 and Caspase-7 was performed using the following antibodies: anti-cleaved Caspase-3 and Caspase-7 (BioCare Medical). Anti-rabbit on rodent polymer (BioCare Medical) with deaminobenzidine detection system was used for visualization of the antigen. The number of total and positive nuclei was manually counted. Ten fields through the heart were analyzed for each animal under 20× magnification.
Western blotting and antibodies
Immunoblot analysis was performed as previously described (Stewart et al. 2010). Briefly, samples were lysed in radioimmunoprecifitation assay (RIPA) buffer (50 mM Tris-HCl at pH 7.4, 150 mM NaCl, 1% TritonX-100, 1 mM EDTA, 1% sodium deoxycholate, 0.1% SDS) containing protease inhibitors (Roche) on ice. Lysates were cleared by centrifugation, and protein concentrations were determined by BCA assay (Pierce). The following antibodies were used: anti-MCL-1 (Rockland Immunochemical), anti-BAX (Millipore), anti-BAK (Millipore), anti-Actin (Millipore), anti-MnSOD (BD Biosciences), anti-Prohibitin (Thermo Fisher Scientific), anti-BIM (Millipore), anti-BNIP3 (Cell Signaling), and anti-BID (Cell Signaling).
Isolation and purification of mouse heart mitochondria
Mice were sacrificed with CO2, and beating hearts were removed. Hearts were rinsed by ice-cold mito isolation buffer (MIB; 200 mM mannitol, 68 mM sucrose, 10 mM HEPES-KOH at pH 7.4, 10 mM KCl, 1 mM EDTA, 1 mM EGTA, 0.1% BSA) to remove the blood and then finely minced. Minced tissue was homogenized with a Polytron tissue grinder at 12,000 rpm for 2.5 sec and then hand-homogenized in a Potter-Helvehjem 10-mL homogenizer (Teflon glass) for three to five strokes. Mitochondria were isolated by differential centrifugation. Briefly, the homogenized tissue was centrifuged twice at 600g for 5 min. The resulting pellet was resuspended in MIB and centrifuged at 5500g for 10 min. The final pellet was rinsed and gently homogenized in MIB. Concentration of mitochondria was determined by Bradford assay.
Oxygen consumption
Mitochondria respiration was measured by a Clark-type oxygen electrode (Hansatech Oxygraph) in 500 μL of mito assay buffer (MAB; 70 mM sucrose, 220 mM mannitol, 10 mM Pi, 5 mM MgCl2, 2 mM Hepes, 1 mM EGTA, 0.2% BSA) at 37°C. Complex I respiration was stimulated by the addition of glutamate (5 mM) and malate (2.5 mM). State 3 respiration was stimulated by the addition of ADP (150 μM). State 4 respiration was the rate after adding oligomycin (2 μg/mL). Maximum respiration was obtained by the addition of FCCP (0.4 μM). RCR was the ratio between state 3 and state 4.
Enzymatic assays
The activity of complex I was assayed as previously described (Frost et al. 2005). Complex II activity was assayed by measuring succinate-dependent reduction in DCIP, mediated by CoQ1. The reaction was followed by absorbance at 600 nm following succinate addition (Miyadera et al. 2003). Complex IV activity was assayed by measuring oxidation of cytochrome c at 550 nm and calculated using the extinction coefficient of 19.6 mM−1cm−1 as previously described (Miro et al. 1998). Pyruvate dehydrogenase (PDH) activity was assayed by measuring the pyruvate-dependent reduction of INT, mediated by PMS, as previously described (Hinman and Blass 1981).
DNA isolation and quantification
DNA was extracted from the heart tissue of mice, and the relative copy number was quantified by analyzing the difference in threshold amplification between mtDNA and nuclear DNA. Real-time PCR was performed with SYBR Green (Bio-Rad) detection using a Bio-Rad iCycler using the ddCt method. The assay was done in triplicate for each sample.
TEM
Hearts were perfused and fixed in 2.5% gluteraldehyde in 0.1 M sodium cacodylate buffer and post-fixed in 2% osmium tetroxide in 0.1 M sodium cacodylate buffer with 0.3% potassium ferrocyanide. Tissue was stained with 4% aqueous uranyl acetate, dehydrated, infiltrated, and embedded in epoxy resin. Ultrathin sections (80 nm) were cut and imaged using a JEOL 1200 electron microscope with an AMT XR 111 camera.
Statistical considerations
Mendelian ratio significance was calculated using the χ2 test, Kaplan-Meier curve significance was calculated using the log-rank test, and ECG significance was calculated using the two-tailed Student's t-test. P-values are denoted in the figure legends.
Acknowledgments
We thank M. Johnson at the St. Jude Small Animal Imaging Facility for assistance with echocardiograms; the Cell and Tissue Imaging Facility for assistance on electron micrographs; the St. Jude Children's Research Hospital Animal Resource Center for husbandry; S. Savage for pathology assistance; and members of the St. Jude Biochemistry Department, S. Milasta, G. Quarato, R. Perciavalle, and members of the Opferman laboratory for helpful discussions. J.T.O. is supported by the National Institutes of Health (HL-102175), the American Cancer Society (RSG-10-255-01-LIB), the St. Jude Children's Research Hospital Cancer Center (Support Grant P30CA021765), and the American Lebanese Syrian Associated Charities of St. Jude Children's Research Hospital. X.W. and J.T.O. conceived the study, designed the experiments, and wrote the manuscript. X.W. performed the experiments, analyzed data, and prepared figures. M.B. and B.K. generated reagents and performed experiments. J.L. and J.D.S. performed experiments and analyzed data. C.C. supervised live animal echocardiography. S.F. assisted with electron microscopy. J.R. assessed pathology. J.T.O. supervised the project.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.215855.113.
References
- Arany Z, Novikov M, Chin S, Ma Y, Rosenzweig A, Spiegelman BM 2006. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-γ coactivator 1α. Proc Natl Acad Sci 103: 10086–10091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbour N, Vanderluit JL, Le Grand JN, Jahani-Asl A, Ruzhynsky VA, Cheung EC, Kelly MA, MacKenzie AE, Park DS, Opferman JT, et al. 2008. Mcl-1 is a key regulator of apoptosis during CNS development and after DNA damage. J Neurosci 28: 6068–6078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. 2010. The landscape of somatic copy-number alteration across human cancers. Nature 463: 899–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocheriou V, Hagege AA, Oubenaissa A, Lambert M, Mallet VO, Duriez M, Wassef M, Kahn A, Menasche P, Gilgenkrantz H 2000. Cardiac functional improvement by a human Bcl-2 transgene in a mouse model of ischemia/reperfusion injury. J Gene Med 2: 326–333 [DOI] [PubMed] [Google Scholar]
- Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, Letai A 2006. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 9: 351–365 [DOI] [PubMed] [Google Scholar]
- Chen M, He H, Zhan S, Krajewski S, Reed JC, Gottlieb RA 2001a. Bid is cleaved by calpain to an active fragment in vitro and during myocardial ischemia/reperfusion. J Biol Chem 276: 30724–30728 [DOI] [PubMed] [Google Scholar]
- Chen Z, Chua CC, Ho YS, Hamdy RC, Chua BH 2001b. Overexpression of Bcl-2 attenuates apoptosis and protects against myocardial I/R injury in transgenic mice. Am J Physiol Heart Circ Physiol 280: H2313–H2320 [DOI] [PubMed] [Google Scholar]
- Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, Korsmeyer SJ 2001. BCL-2, BCL-XL sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell 8: 705–711 [DOI] [PubMed] [Google Scholar]
- Da Cruz S, Xenarios I, Langridge J, Vilbois F, Parone PA, Martinou JC 2003. Proteomic analysis of the mouse liver mitochondrial inner membrane. J Biol Chem 278: 41566–41571 [DOI] [PubMed] [Google Scholar]
- Dzhagalov I, St John A, He YW 2007. The antiapoptotic protein Mcl-1 is essential for the survival of neutrophils but not macrophages. Blood 109: 1620–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzhagalov I, Dunkle A, He YW 2008. The anti-apoptotic Bcl-2 family member Mcl-1 promotes T lymphocyte survival at multiple stages. J Immunol 181: 521–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fosslien E 2003. Review: Mitochondrial medicine—cardiomyopathy caused by defective oxidative phosphorylation. Ann Clin Lab Sci 33: 371–395 [PubMed] [Google Scholar]
- Frost MT, Wang Q, Moncada S, Singer M 2005. Hypoxia accelerates nitric oxide-dependent inhibition of mitochondrial complex I in activated macrophages. Am J Physiol Regul Integr Comp Physiol 288: R394–R400 [DOI] [PubMed] [Google Scholar]
- Garnier A, Fortin D, Delomenie C, Momken I, Veksler V, Ventura-Clapier R 2003. Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J Physiol 551: 491–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain M, Nguyen AP, Le Grand JN, Arbour N, Vanderluit JL, Park DS, Opferman JT, Slack RS 2011. MCL-1 is a stress sensor that regulates autophagy in a developmentally regulated manner. EMBO J 30: 395–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinman LM, Blass JP 1981. An NADH-linked spectrophotometric assay for pyruvate dehydrogenase complex in crude tissue homogenates. J Biol Chem 256: 6583–6586 [PubMed] [Google Scholar]
- Huang J, Ito Y, Morikawa M, Uchida H, Kobune M, Sasaki K, Abe T, Hamada H 2003. Bcl-xL gene transfer protects the heart against ischemia/reperfusion injury. Biochem Biophys Res Commun 311: 64–70 [DOI] [PubMed] [Google Scholar]
- Imahashi K, Schneider MD, Steenbergen C, Murphy E 2004. Transgenic expression of Bcl-2 modulates energy metabolism, prevents cytosolic acidification during ischemia, and reduces ischemia/reperfusion injury. Circ Res 95: 734–741 [DOI] [PubMed] [Google Scholar]
- Kirshenbaum LA, Schneider MD 1995. Adenovirus E1A represses cardiac gene transcription and reactivates DNA synthesis in ventricular myocytes, via alternative pocket protein- and p300-binding domains. J Biol Chem 270: 7791–7794 [DOI] [PubMed] [Google Scholar]
- Kirshenbaum LA, de Moissac D 1997. The bcl-2 gene product prevents programmed cell death of ventricular myocytes. Circulation 96: 1580–1585 [DOI] [PubMed] [Google Scholar]
- Kirshenbaum LA, Abdellatif M, Chakraborty S, Schneider MD 1996. Human E2F-1 reactivates cell cycle progression in ventricular myocytes and represses cardiac gene transcription. Dev Biol 179: 402–411 [DOI] [PubMed] [Google Scholar]
- Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, Deng X, Zhai D, Shi YX, Sneed T, et al. 2006. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 10: 375–388 [DOI] [PubMed] [Google Scholar]
- Krajewski S, Bodrug S, Krajewska M, Shabaik A, Gascoyne R, Berean K, Reed JC 1995. Immunohistochemical analysis of Mcl-1 protein in human tissues. Differential regulation of Mcl-1 and Bcl-2 protein production suggests a unique role for Mcl-1 in control of programmed cell death in vivo. Am J Pathol 146: 1309–1319 [PMC free article] [PubMed] [Google Scholar]
- Kubasiak LA, Hernandez OM, Bishopric NH, Webster KA 2002. Hypoxia and acidosis activate cardiac myocyte death through the Bcl-2 family protein BNIP3. Proc Natl Acad Sci 99: 12825–12830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubli DA, Gustafsson AB 2012. Mitochondria and mitophagy: The yin and yang of cell death control. Circ Res 111: 1208–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsov AV, Margreiter R 2009. Heterogeneity of mitochondria and mitochondrial function within cells as another level of mitochondrial complexity. Int J Mol Sci 10: 1911–1929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman JJ, Kelly DP 2002. Transcriptional activation of energy metabolic switches in the developing and hypertrophied heart. Clin Exp Pharmacol Physiol 29: 339–345 [DOI] [PubMed] [Google Scholar]
- Li H, Wang J, Wilhelmsson H, Hansson A, Thoren P, Duffy J, Rustin P, Larsson NG 2000. Genetic modification of survival in tissue-specific knockout mice with mitochondrial cardiomyopathy. Proc Natl Acad Sci 97: 3467–3472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, et al. 2000. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell 6: 1389–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone CD, Hasan SM, Roome RB, Xiong J, Furlong M, Opferman JT, Vanderluit JL 2012. Mcl-1 regulates the survival of adult neural precursor cells. Mol Cell Neurosci 49: 439–447 [DOI] [PubMed] [Google Scholar]
- Maulik N, Engelman RM, Rousou JA, Flack JE 3rd, Deaton D, Das DK 1999. Ischemic preconditioning reduces apoptosis by upregulating anti-death gene Bcl-2. Circulation 100: II369–II375 [DOI] [PubMed] [Google Scholar]
- Milner DJ, Mavroidis M, Weisleder N, Capetanaki Y 2000. Desmin cytoskeleton linked to muscle mitochondrial distribution and respiratory function. J Cell Biol 150: 1283–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miro O, Cardellach F, Barrientos A, Casademont J, Rotig A, Rustin P 1998. Cytochrome c oxidase assay in minute amounts of human skeletal muscle using single wavelength spectrophotometers. J Neurosci Methods 80: 107–111 [DOI] [PubMed] [Google Scholar]
- Miyadera H, Shiomi K, Ui H, Yamaguchi Y, Masuma R, Tomoda H, Miyoshi H, Osanai A, Kita K, Omura S 2003. Atpenins, potent and specific inhibitors of mitochondrial complex II (succinate-ubiquinone oxidoreductase). Proc Natl Acad Sci 100: 473–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K, Nakayama K, Negishi I, Senju S, Zhang Q, Fujii S, et al. 1995. Massive cell death of immature hematopoietic cells and neurons in Bcl-x- deficient mice. Science 267: 1506–1510 [DOI] [PubMed] [Google Scholar]
- Motoyama N, Kimura T, Takahashi T, Watanabe T, Nakano T 1999. bcl-x prevents apoptotic cell death of both primitive and definitive erythrocytes at the end of maturation. J Exp Med 189: 1691–1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mughal W, Dhingra R, Kirshenbaum LA 2012. Striking a balance: Autophagy, apoptosis, and necrosis in a normal and failing heart. Curr Hypertens Rep 14: 540–547 [DOI] [PubMed] [Google Scholar]
- Murriel CL, Churchill E, Inagaki K, Szweda LI, Mochly-Rosen D 2004. Protein kinase Cδ activation induces apoptosis in response to cardiac ischemia and reperfusion damage: A mechanism involving BAD and the mitochondria. J Biol Chem 279: 47985–47991 [DOI] [PubMed] [Google Scholar]
- Nguyen M, Marcellus RC, Roulston A, Watson M, Serfass L, Murthy Madiraju SR, Goulet D, Viallet J, Belec L, Billot X, et al. 2007. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc Natl Acad Sci 104: 19512–19517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivetti G, Quaini F, Sala R, Lagrasta C, Corradi D, Bonacina E, Gambert SR, Cigola E, Anversa P 1996. Acute myocardial infarction in humans is associated with activation of programmed myocyte cell death in the surviving portion of the heart. J Mol Cell Cardiol 28: 2005–2016 [DOI] [PubMed] [Google Scholar]
- Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ 2003. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature 426: 671–676 [DOI] [PubMed] [Google Scholar]
- Opferman JT, Iwasaki H, Ong CC, Suh H, Mizuno S, Akashi K, Korsmeyer SJ 2005. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science 307: 1101–1104 [DOI] [PubMed] [Google Scholar]
- Perciavalle RM, Opferman JT 2013. Delving deeper: MCL-1's contributions to normal and cancer biology. Trends Cell Biol 23: 22–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perciavalle RM, Stewart DP, Koss B, Lynch J, Milasta S, Bathina M, Temirov J, Cleland MM, Pelletier S, Schuetz JD, et al. 2012. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat Cell Biol 14: 575–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regula KM, Ens K, Kirshenbaum LA 2002. Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ Res 91: 226–231 [DOI] [PubMed] [Google Scholar]
- Rinkenberger JL, Horning S, Klocke B, Roth K, Korsmeyer SJ 2000. Mcl-1 deficiency results in peri-implantation embryonic lethality. Genes & Dev 14: 23–27 [PMC free article] [PubMed] [Google Scholar]
- Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, Penninger JM, Molkentin JD 2001. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res 89: 20–25 [DOI] [PubMed] [Google Scholar]
- Steimer DA, Boyd K, Takeuchi O, Fisher JK, Zambetti GP, Opferman JT 2009. Selective roles for antiapoptotic MCL-1 during granulocyte development and macrophage effector function. Blood 113: 2805–2815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart DP, Koss B, Bathina M, Perciavalle RM, Bisanz K, Opferman JT 2010. Ubiquitin-independent degradation of antiapoptotic MCL-1. Mol Cell Biol 30: 3099–3110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O, Fisher J, Suh H, Harada H, Malynn BA, Korsmeyer SJ 2005. Essential role of BAX,BAK in B cell homeostasis and prevention of autoimmune disease. Proc Natl Acad Sci 102: 11272–11277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas RL, Roberts DJ, Kubli DA, Lee Y, Quinsay MN, Owens JB, Fischer KM, Sussman MA, Miyamoto S, Gustafsson ÅB 2013. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev (this issue). doi: 10.1101/gad.215871.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth A, Jeffers JR, Nickson P, Min JY, Morgan JP, Zambetti GP, Erhardt P 2006. Targeted deletion of Puma attenuates cardiomyocyte death and improves cardiac function during ischemia-reperfusion. Am J Physiol Heart Circ Physiol 291: H52–H60 [DOI] [PubMed] [Google Scholar]
- van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, Willis SN, Scott CL, Day CL, Cory S, et al. 2006. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 10: 389–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ 1993. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell 75: 229–240 [DOI] [PubMed] [Google Scholar]
- Vikstrom I, Carotta S, Luthje K, Peperzak V, Jost PJ, Glaser S, Busslinger M, Bouillet P, Strasser A, Nutt SL, et al. 2010. Mcl-1 is essential for germinal center formation and B cell memory. Science 330: 1095–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC 1999. Mitochondrial diseases in man and mouse. Science 283: 1482–1488 [DOI] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ 2001. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science 292: 727–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisleder N, Taffet GE, Capetanaki Y 2004. Bcl-2 overexpression corrects mitochondrial defects and ameliorates inherited desmin null cardiomyopathy. Proc Natl Acad Sci 101: 769–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S, Factor SM, Shirani J, Armstrong RC, Kitsis RN 2003. A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Invest 111: 1497–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan RS, Konstantinidis K, Wei AC, Chen Y, Reyna DE, Jha S, Yang Y, Calvert JW, Lindsten T, Thompson CB, et al. 2012. Bax regulates primary necrosis through mitochondrial dynamics. Proc Natl Acad Sci 109: 6566–6571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE, Ierino H, Lee EF, Fairlie WD, Bouillet P, et al. 2007. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 315: 856–859 [DOI] [PubMed] [Google Scholar]
- Yang T, Kozopas KM, Craig RW 1995. The intracellular distribution and pattern of expression of Mcl-1 overlap with, but are not identical to, those of Bcl-2. J Cell Biol 128: 1173–1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yussman MG, Toyokawa T, Odley A, Lynch RA, Wu G, Colbert MC, Aronow BJ, Lorenz JN, Dorn GW 2nd 2002. Mitochondrial death protein Nix is induced in cardiac hypertrophy and triggers apoptotic cardiomyopathy. Nat Med 8: 725–730 [DOI] [PubMed] [Google Scholar]
- Zong WX, Lindsten T, Ross AJ, MacGregor GR, Thompson CB 2001. BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev 15: 1481–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]