

Myeloid cell leukemia-1 (MCL-1) is an anti-apoptotic BCL-2 protein highly expressed in myocardium. Thomas et al. show that ablation of MCL-1 in the adult heart leads to rapid cardiomyopathy and death. Mitochondria in myocytes were swollen and exhibited reduced respiration. Mice lacking MCL-1 and cyclophilin D, an essential regulator of the mitochondrial permeability transition pore, exhibited delayed progression to heart failure and extended survival. Induction of autophagy was impaired in MCL-1-deficient hearts. These results demonstrate that MCL-1 is essential for mitochondrial homeostasis and autophagy induction in the heart.
Keywords: autophagy, BCL-2, heart failure, MCL-1, mitochondria, necrosis
Abstract
Myeloid cell leukemia-1 (MCL-1) is an anti-apoptotic BCL-2 protein that is up-regulated in several human cancers. MCL-1 is also highly expressed in myocardium, but its function in myocytes has not been investigated. We generated inducible, cardiomyocyte-specific Mcl-1 knockout mice and found that ablation of Mcl-1 in the adult heart led to rapid cardiomyopathy and death. Although MCL-1 is known to inhibit apoptosis, this process was not activated in MCL-1-deficient hearts. Ultrastructural analysis revealed disorganized sarcomeres and swollen mitochondria in myocytes. Mitochondria isolated from MCL-1-deficient hearts exhibited reduced respiration and limited Ca2+-mediated swelling, consistent with opening of the mitochondrial permeability transition pore (mPTP). Double-knockout mice lacking MCL-1 and cyclophilin D, an essential regulator of the mPTP, exhibited delayed progression to heart failure and extended survival. Autophagy is normally induced by myocardial stress, but induction of autophagy was impaired in MCL-1-deficient hearts. These data demonstrate that MCL-1 is essential for mitochondrial homeostasis and induction of autophagy in the heart. This study also raises concerns about potential cardiotoxicity for chemotherapeutics that target MCL-1.
The BCL-2 family proteins are important regulators of mitochondrial integrity and the intrinsic pathway of apoptosis. Upon activation, the proapoptotic members promote permeabilization of the outer mitochondrial membrane, which results in the release of proapoptotic proteins such as cytochrome c and subsequent activation of caspases (Gustafsson and Gottlieb 2007). In contrast, anti-apoptotic members such as BCL-2, BCL-XL, and myeloid cell leukemia-1 (MCL-1) promote cell survival by inhibiting the proapoptotic BCL-2 proteins. Aberrant expression of the anti-apoptotic BCL-2 family members is one of the defining features of cancer cells and is also strongly associated with resistance to current therapies (Kang and Reynolds 2009). Thus, these proteins are currently major targets in the development of new therapeutics to improve treatment outcomes for cancer patients. Studies have also established BCL-2 and BCL-XL as important prosurvival molecules in the heart, and elevated levels of these proteins protect against ischemia/reperfusion (I/R) injury (Huang et al. 2003; Imahashi et al. 2004). BCL-2 also protects against p53-mediated apoptosis in cardiac myocytes (Kirshenbaum and de Moissac 1997) and increases the calcium threshold for permeability transition pore opening in heart mitochondria (Zhu et al. 2001). Although MCL-1 is highly expressed in the heart (Wu et al. 1997), its role in this tissue has not been previously characterized.
MCL-1, BCL-2, and BCL-XL are often coexpressed in the same tissue, but knockout studies have revealed that they have distinct physiological roles. For instance, BCL-2 knockout mice are viable but display growth retardation, renal failure, and apoptosis of lymphocytes (Veis et al. 1993). BCL-XL is required for brain development, and BCL-XL-deficient mice do not survive past embryonic day 13.5 (Motoyama et al. 1995). MCL-1 is also essential for embryonic development, and global deletion results in lethality before embryonic day 4 (Rinkenberger et al. 2000). Despite functional overlap with BCL-2 and BCL-XL, MCL-1 is distinguished by its short half-life and lack of a BH4 domain (Krajewski et al. 1995; Zhou et al. 1997). PEST sequences in the nonhomologous N-terminal region facilitate rapid proteosomal degradation in response to cellular stress (Thomas et al. 2010).
Recent studies have demonstrated that the BCL-2 proteins can also regulate autophagy (Pattingre et al. 2005; Maiuri et al. 2007). Autophagy is a cellular recycling process that facilitates protein turnover and organelle maintenance in cells (Levine and Kroemer 2008). During this process, cytosolic contents are sequestered in autophagosomes and delivered to lysosomes for degradation. Basal autophagy is essential for tissue homeostasis. In the heart, disruption of this process leads to accumulation of aberrant mitochondria and cardiac dysfunction (Nakai et al. 2007). In addition, Danon disease, caused by LAMP-2 deficiency, disrupts fusion between autophagosomes and lysosomes and leads to a lethal cardiomyopathy (Nishino et al. 2000). Autophagy is also rapidly induced by physiologic or mechanical stress in the heart (Matsui et al. 2007; Zhu et al. 2007). By clearing damaged organelles that can harm the cell and providing amino and fatty acids to support energy production, autophagy helps sustain cardiac myocyte viability during stress.
In this study, we generated inducible, myocyte-specific Mcl-1 knockout mice to investigate the functional role of MCL-1 in the adult heart. We discovered that ablation of Mcl-1 in adult myocytes led to mitochondrial dysfunction, impaired autophagy, and rapid development of severe heart failure. These findings suggest that MCL-1 is critical for cardiac homeostasis and have broad clinical implications for the design of potential chemotherapeutic antagonists of MCL-1.
Results
Loss of MCL-1 leads to rapid contractile dysfunction, cardiac hypertrophy, and early mortality
Initial characterization demonstrated that MCL-1 is highly expressed in the heart and regulates cell death in cardiac myocytes. MCL-1 exists as two different isoforms that are localized in the outer mitochondrial membrane and the matrix, respectively (Perciavalle et al. 2012). Both isoforms of MCL-1 are highly expressed in the heart (Fig. 1A). After an infarct, the outer membrane form is rapidly degraded in the border zone, whereas the matrix form is unchanged (Fig. 1B). The levels of MCL-1 are restored 24 h after the infarct. Overexpression of MCL-1 also protects against doxorubicin-mediated cell death in neonatal myocytes (Supplemental Fig. S1), confirming that MCL-1 is an important prosurvival protein in myocytes. To further investigate the functional role of MCL-1 in the myocardium, we generated inducible, myocyte-specific Mcl-1 knockout mice by crossing Mcl-1flox/flox mice with transgenic mice expressing MerCreMer under the control of the α-myosin heavy chain (α-MHC) promoter to permit tamoxifen-inducible deletion of the Mcl-1 allele in the adult heart. The Mcl-1flox/flox;MerCreMer+ (Mcl-1f/fCre+) mice were born at the expected Mendelian frequency and were indistinguishable in appearance from control Mcl-1flox/flox;MerCreMer- (Mcl-1f/f) littermates. To delete Mcl-1 in cardiac myocytes, 8- to 10-wk-old mice were administered tamoxifen for 5 d. Western blotting of heart lysates (Fig. 1C) confirmed a reduction in MCL-1 protein levels in hearts of Mcl-1f/fCre+ mice without affecting MCL-1 levels in skeletal muscle. Surprisingly, echocardiographic analysis of tamoxifen-treated Mcl-1f/fCre+ mice revealed severe contractile dysfunction and left ventricular dilation as early as 1 wk after initiating tamoxifen injection (Fig. 1D,E). Strikingly, all Mcl-1f/fCre+ mice died within 29 d after the initiation of tamoxifen treatment (median survival 16 d) (Fig. 1F). Cardiac function was not impaired in tamoxifen-treated Mcl-1WT/WTMerCreMer+ mice, confirming that the cardiac dysfunction was not due to increased Cre recombinase activity (Supplemental Table 1).
Figure 1.

Cardiomyocyte-specific ablation of Mcl-1 in the adult heart leads to contractile dysfunction and death. (A) Western blot analysis of MCL-1 in control hearts and in the border zone of the infarct. (B) Quantitation of outer mitochondrial membrane (OMM) and matrix MCL-1 isoforms (n = 5–6). (C) Western blot analysis for MCL-1 in heart and skeletal muscle after 2 wk. (D) Representative M-Mode echocardiograms at 2 wk. (E) Echocardiographic analysis revealed reduced ejection fraction (EF) and fractional shortening (FS) as well as enlarged left ventricular diastolic dimension (LVDd) in Mcl-1f/fCre+ mice. n = 5–14; (*) P < 0.05. (F) Kaplan-Meyer survival curve (P < 0.05). Data are mean ± standard error of the mean (SEM).
Mcl-1f/fCre+ hearts were enlarged by gross morphology (Fig. 2A), and ventricle to body and lung to body weight ratios were significantly increased compared with Mcl-1f/f hearts 2 wk after tamoxifen administration (Fig. 2B). Increased ventricle weight indicates cardiac hypertrophy, while atrial enlargement and increased lung weight are signs of cardiac insufficiency and pulmonary edema. Additional evidence of cardiac hypertrophy included significantly increased myocyte fiber size (Fig. 2C,D) and increased expression of ventricular hypertrophy markers atrial natriuretic factor (ANF) and brain natriuretic peptide (BNP) (Fig. 2E) in Mcl-1f/fCre+ hearts. In addition, expression of α-MHC decreased while expression of β-MHC increased upon loss of MCL-1 (Fig. 2F). This switch from adult to fetal MHC isoforms indicates reactivation of fetal gene programs that are associated with pathological cardiac hypertrophy.
Figure 2.

Ablation of Mcl-1 in the adult heart leads to cardiac hypertrophy. (A) Mcl-1f/fCre+ hearts are enlarged on gross morphology by 1 wk. (B) Ventricle weight and lung weight to body weight ratios from Mcl-1f/f and Mcl-1f/fCre+ mice 2 wk after initiating tamoxifen. n = 15–17. (C) Representative images of wheat germ agglutinin (WGA) staining at 2 wk. Bar, 35 μm. (D) Quantitation of myocyte cross-sectional area (n = 4) at 2 wk. (E) Analysis of hypertrophy markers by qPCR at 2 wk. n = 9–10. (F) MHC isotype switching in Mcl-1flox/floxCre+ hearts at 2 wk shown by qPCR. n = 9–10. Data are mean ± SEM.
MCL-1 deficiency leads to cardiac fibrosis and inflammation
Histological analysis revealed extensively increased left ventricular chamber dimensions and severe ventricular wall thinning in Mcl-1f/fCre+ hearts 2 wk after tamoxifen treatment. Left atrial thrombosis was also evident in the Mcl-1f/fCre+ heart, demonstrating impaired hemodynamics in this heart (Fig. 3A,B). At the tissue level, H&E-stained cross-sections revealed myofibrillar disarray and myocyte degeneration in Mcl-1f/fCre+ hearts (Fig. 3C). Masson's trichrome staining of heart sections demonstrated extensive interstitial fibrosis (Fig. 3D,E), and quantitative PCR (qPCR) showed increased transcription of collagen mRNA in Mcl-1f/fCre+ hearts (Fig. 3F). Finally, MCL-1 deficiency led to increased myocardial inflammation. Mcl-1f/fCre+ hearts exhibited increased CD45+ staining (consistent with leukocyte infiltration) as well as elevated inflammation markers (Fig. 3G,H). Thus, loss of MCL-1 results in rapid, extensive myocardial tissue damage and inflammation.
Figure 3.

Ablation of Mcl-1 leads to cardiomyocyte disorganization, fibrosis, and inflammation. (A) H&E staining shows thinning of the left ventricular wall in Mcl-1f/fCre+ hearts 2 wk after tamoxifen injection. Bar, 5 mm. (B) Left atrial thrombus in the Mcl-1f/fCre+ heart. Bars: left, 500 μm; right, 100 μm. (C) Representative H&E staining shows marked myocyte disorganization and degeneration in Mcl-1f/fCre+ hearts at 2 wk. Bars: left, 500 μm; right, 100 μm. (D) Masson's trichrome staining reveals extensive fibrosis in the left ventricle (LV) of Mcl-1f/fCre+ hearts at 2 wk. Bars: left, 500 μm; right, 100 μm. Quantitation of fibrosis (n = 3) (E) and analysis of Col1A1 mRNA levels at 2 wk (n = 9–10) (F). (G) Representative CD45 staining at 2 wk. Bar, 50 μm. (H) Analysis of inflammatory markers by qPCR at 2 wk (n = 5–8). Data are mean ± SEM.
MCL-1 deficiency does not activate apoptosis
Since MCL-1 is an anti-apoptotic protein, we investigated whether apoptosis of myocytes contributed to damage in MCL-1-deficient hearts. Activation of apoptosis was assessed in hearts by terminal deoxynucleotidyl transferase UTP nick end labeling (TUNEL) and immunostaining for cleaved (active) caspase-3 in heart sections. Surprisingly, the number of TUNEL-positive nuclei (Fig. 4A; Supplemental Fig. S2) and myocytes positive for cleaved caspase-3 (Fig. 4B; Supplemental Fig. S3) was not significantly increased after deletion of Mcl-1. Moreover, we did not detect an increase in caspase-3 activity (Fig. 4C), cleaved caspase-3 (Fig. 4D), or cleavage of PARP (Fig. 4E) in heart lysates from Mcl-1f/fCre+ mice after tamoxifen treatment. In response to proapoptotic stimuli, apoptosis-inducing factor (AIF) is released from mitochondria and translocates to the nucleus, where it causes caspase-independent cell death (Susin et al. 1999). MCL-1-deficient hearts did not have increased levels of AIF in the nucleus (Fig. 4F). Next, we assessed whether loss of MCL-1 caused changes in other BCL-2 proteins. We found that BCL-2 and BCL-XL were significantly up-regulated upon deletion of Mcl-1 (Fig. 4G,H). Although proapoptotic BAX was also up-regulated in Mcl-1f/fCre+ hearts after tamoxifen, the BCL-2/BAX ratio was not altered (Supplemental Fig. S4). MCL-1 is known to interact with the BH3-only proteins BIM and NOXA, preventing them from activating apoptosis. However, the levels of these proteins at the mitochondria were unchanged upon loss of MCL-1 (Fig. 4I). Thus, it is possible that both BIM and NOXA are neutralized by BCL-2 and/or BCL-XL in the absence of MCL-1. Similarly, the level of BH3-only protein BNIP3 was unchanged. Thus, these data suggest that enhanced apoptosis is not a significant cause of the cardiac damage observed in MCL-1-deficient hearts.
Figure 4.

Ablation of Mcl-1 does not result in activation of apoptosis. (A) Quantitation of TUNEL positive nuclei in heart sections. n = 3–4. (B) Quantitation of myocytes positive for cleaved caspase-3 in heart sections. n = 3–4. (C) Caspase-3 activity assay at 2 wk. Positive control consists of HL-60 cells treated with 0.5 μg/mL actinomycin D for 24 h. n = 3–6; (*) P < 0.001 vs. Mcl-1f/f. (D) Western blot for cleaved caspase-3 at 2 wk. Positive control consists of HL-1 cells treated with 1 μM staurosporine for 8 h. (E) Western blot analysis for PARP cleavage. (F) Western blot analysis for AIF in cytosolic, mitochondrial, and nuclear fractions at 2 wk. (G) Western blot analysis for BCL-2 family proteins at 2 wk in Mcl-1f/f and Mcl-1f/fCre+ hearts. (H) Quantitation of BCL-2 protein levels. n = 7; (*) P < 0.05 vs. Mcl-1f/f. (I) Western blot analysis for BH3-only proteins in mitochondrial fractions at 2 wk. Data are mean ± SEM.
Loss of MCL-1 in myocytes leads to mitochondrial permeability transition and necrosis
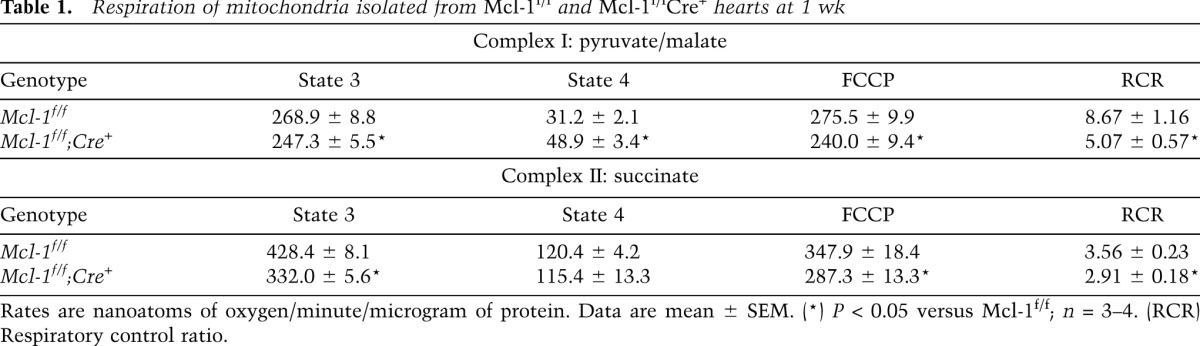
Next, we used transmission electron microscopy (TEM) to examine MCL-1-deficient hearts at the ultrastructural level. Mcl-1f/fCre+ myocytes showed extensive mitochondrial damage 1 wk after tamoxifen administration (Fig. 5A). Most mitochondria appeared fragmented and exhibited extensive swelling and cristae remodeling. Analysis of mitochondrial fission and fusion proteins revealed no changes in Mfn1/2, Opa1, Fis1, and Drp1 levels (Supplemental Fig. S5). Drp1 was undetectable in the mitochondrial fraction from Mcl-1f/fCre+ hearts, indicating that mitochondrial fission was not activated. The TEM also revealed mitochondria with ruptured outer mitochondrial membranes, an indication that these mitochondria had undergone opening of the mitochondrial permeability transition pore (mPTP), a prominent feature in necrotic cell death. Other signs of necrosis included the presence of cellular debris in the extracellular space from ruptured myocytes and the presence of infiltrating inflammatory cells. Lactate dehydrogenase activity, a marker of acute cardiac injury, was also significantly elevated in the blood of MCL-1-deficient mice at 1 wk (Fig. 5B). Further analysis confirmed that isolated MCL-1-deficient cardiac mitochondria were swollen at baseline and exhibited modest calcium-induced swelling compared with wild-type cardiac mitochondria (Fig. 5C). Interestingly, the mPTP inhibitor cyclosporine A initially blocked Ca2+-mediated swelling in MCL-1-deficient mitochondria, but after 15 min, it was no longer effective in preventing swelling. This suggests that these mitochondria can swell independently of mPTP opening. Moreover, addition of recombinant MCL-1 to mitochondria isolated from Mcl-1f/fCre+ hearts did not reduce baseline swelling and had no effect on Ca2+-mediated swelling, suggesting that MCL-1 is not a direct regulator of the mPTP (Supplemental Fig. S6). In addition, we assessed mitochondrial respiration in mitochondria isolated from Mcl-1f/f and Mcl-1f/fCre+ hearts 1 wk after tamoxifen treatment. Mitochondria isolated from Mcl-1f/fCre+ hearts had reduced respiration rates compared with Mcl-1f/f (Table 1). There was also a significant reduction in the respiratory control ratio between Mcl-1f/f and Mcl-1f/fCre+ mitochondria, confirming that mitochondria from MCL-1-deficient hearts are dysfunctional. Although mitochondrial respiration was significantly reduced in the knockouts, we expected to see a much more severe decrease in function. However, this modest decrease is most likely due to selection of healthy mitochondria during the isolation process. Most of the damaged mitochondria are lost during the isolation process, and as a result, the defects in respiration are often underestimated. However, analysis of the specific enzyme activities confirmed an ∼40% reduction in complex I and an ∼50% reduction in complex II activities in MCL-1-deficient mitochondria (Fig. 5D). These data indicate that loss of MCL-1 resulted in impairment of mitochondria, opening of the mPTP, and necrotic cell death.
Figure 5.

Ablation of Mcl-1 leads to degeneration of mitochondria. (A) Ultrastructural images reveal the presence of swollen mitochondria (white arrows) and lipid accumulation (black arrows) in MCL-1-deficient hearts 1 wk after tamoxifen injection. Bars: top panels, 2 μm; bottom left, 0.5 μm; bottom center, 1 μm; bottom right, 0.5 μm. (B) LDH activity in blood at 1 wk. n = 5–6. (C) Swelling of isolated mitochondria in the presence of 250 μM calcium with or without 1 μM CsA 1 wk after tamoxifen injection. (D) Complex I and complex II activities in isolated mitochondria at 1 wk. n = 4.
Table 1.
Respiration of mitochondria isolated from Mcl-1f/f and Mcl-1f/fCre+ hearts at 1 wk
To further investigate the role of mPTP opening and necrosis in Mcl-1f/fCre+ hearts, we generated Ppif−/−Mcl-1flox/floxCre+ double-knockout mice. Ppif encodes for cyclophilin D (CypD), which is an essential component of the mPTP (Baines et al. 2005). Although ejection fraction (EF) and fractional shortening (FS) in Ppif−/−Mcl-1f/fCre+ mice were still significantly reduced compared with Ppif−/−Mcl-1f/f mice 1 wk after tamoxifen treatment (Fig. 6A,B), LDH activity in blood was not significantly increased at this time point (Fig. 6C). Cardiac function in Ppif−/−Mcl-1f/fCre+ mice was significantly better compared with Mcl-1f/fCre+ mice at 1 wk (EF = 69.24 ± 4.98% *, FS = 39.57 ± 3.78%** for Ppif−/−Mcl-1f/fCre+ vs. EF = 47.54 ± 6.27%, FS = 24.31 ± 3.68% for Mcl-1f/fCre+; [*,**] P < 0.05), but the functional differences between these groups were no longer significant at 2 wk (EF = 61.81 ± 4.82%#, FS = 34.53 ± 3.68%## for Ppif−/−Mcl-1f/fCre+ vs. EF = 47.31 ± 5.57%, FS = 24.78 ± 3.33% for Mcl-1f/fCre+; [#,##] P > 0.05). These mice also developed cardiac hypertrophy and pulmonary edema (Fig. 6D) but had extended survival (up to 40 d, with median survival 37 d) compared with Mcl-1f/fCre+ mice (median survival 16 d) (Fig. 6E). Histological analysis of H&E-stained sections revealed myofibrillar disarray similar to what we observed in Mcl-1f/fCre+ hearts (Fig. 6F). Consistent with previous reports (Baines et al. 2005), mitochondria from both Ppif−/−Mcl-1f/f and Ppif−/−Mcl-1f/fCre+ hearts were resistant to Ca2+-mediated swelling (Fig. 6G). Similar to Mcl-1f/fCre+ mitochondria, Ppif−/−Mcl-1f/fCre+ mitochondria were already swollen at baseline, indicating that mitochondrial swelling occurs even in the absence of a functional mPTP. Next, we investigated whether lack of a functional mPTP led to activation of apoptosis upon loss of MCL-1. However, we found no increase in TUNEL-positive myocytes (Fig. 6H; Supplemental Fig. S7) or detectable levels of cleaved caspase-3 in heart sections (Fig. 6I; Supplemental Fig. S8). Thus, lack of a functional mPTP delays but does not rescue the development of heart failure upon loss of MCL-1.
Figure 6.

Ablation of Mcl-1 in Ppif−/− mice delays development of heart failure and death. (A) Representative M-Mode echocardiograms at 2 wk. (B) Echocardiographic analysis of Ppif−/−Mcl-1f/f and Ppif−/−Mcl-1f/fCre+ mice. n = 5–19; (*) P < 0.05. (C) LDH activity in blood at 1 wk. n = 5–6. (D) Ventricle weight and lung weight to body weight ratios at 2 wk after initiating tamoxifen. n = 17–21. (E) Kaplan-Meyer survival curve. P < 0.05. (F) Representative H&E staining shows myocyte disorganization and degeneration in Ppif−/−Mcl-1f/fCre+ hearts at 2 wk. Bars: left, 500 μm; right, 100 μm. (G) Swelling of isolated mitochondria in the presence of 0, 0.5, or 1 mM calcium at 1 wk. (H) Quantitation of TUNEL-positive nuclei in heart sections. n = 3. (I) Quantitation of myocytes positive for cleaved caspase-3 in heart sections. n = 3. Data are mean ± SEM.
Loss of MCL-1 leads to impaired autophagy
Although autophagy is normally induced to remove damaged organelles and protein aggregates, no autophagosomes were identified in tamoxifen-treated Mcl-1f/fCre+ hearts by electron microscopy. The lack of autophagy was striking given the extent of myocardial damage and mitochondrial dysfunction in MCL-1-deficient myocytes. To investigate whether autophagy was impaired in MCL-1-deficient hearts, we assessed the levels of LC3II/LC3I and p62 in Mcl-1f/f and Mcl-1f/fCre+ hearts 2 wk after tamoxifen administration. The conversion of LC3I to LC3II is indicative of autophagosome formation (Kabeya et al. 2000), whereas the p62 protein is degraded by autophagy and accumulates in cells when autophagic flux is reduced or impaired (Komatsu et al. 2007). We found significantly reduced LC3II/LC3I ratios and accumulation of p62 in Mcl-1f/fCre+ hearts 2 wk after tamoxifen injection (Fig. 7A,B). Total LC3 levels were unaffected, indicating that the loss of MCL-1 impaired LC3 processing rather than LC3 expression (Fig. 7B). Mcl-1f/fCre+ hearts also exhibited reduced levels of Beclin-1, an essential protein for formation of autophagic isolation membranes (Supplemental Fig. S9). Ubiquitin is used to mark proteins and organelles that need to be removed by autophagosomes, and disruption of autophagy leads to accumulation of ubiquitin-containing aggregates (Nakai et al. 2007). We found a significant increase in protein ubiquitination, indicating accumulation of autophagic substrates in Mcl-1f/fCre+ hearts (Supplemental Fig. S9). The PTEN-induced putative kinase 1 (PINK1)/Parkin pathway is important in regulating autophagy of dysfunctional mitochondria in myocytes (Kubli and Gustafsson 2012). Although Parkin levels increased in Mcl-1f/fCre+ cytosolic fractions, Parkin was not found on the damaged mitochondria in Mcl-1f/fCre+ hearts (Fig. 7C). We also found that PINK1, which is required for recruitment of Parkin to mitochondria, was significantly reduced in MCL-1-deficient mitochondria (Fig. 7D,E). These data indicate that loss of MCL-1 also disrupts mitochondrial autophagy. LAMP-2 is important for lysosomal function and plays a role in chaperone-mediated autophagy (Bandyopadhyay et al. 2010). Interestingly, LAMP2 was increased in MCL-1-deficient hearts (Fig. 7F), suggesting that there might be a compensatory increase in alternative degradation pathways.
Figure 7.

Ablation of Mcl-1 in the adult heart leads to impaired autophagy. (A) Representative Western blots of heart lysates prepared at 2 wk after initiation of tamoxifen administration. (B) Quantitation analyses of LC3II/I, p62, and total LC3 levels. n = 6–13. (C) Western blots for Parkin in cytosolic and mitochondrial fractions at 1 wk. (D) Western blot for PINK1 in the mitochondrial fraction at 1 wk. (E) Quantitation of PINK1 levels in mitochondria. n = 3. (F) Analysis of LAMP2 levels at 2 wk in heart lysates. (G) Assessment of autophagic flux in vivo using chloroquine 1 wk after tamoxifen treatment. (H) Quantitation of LC3II/I levels in a chloroquine study. n = 4–6. (I) Representative Western blot of heart lysates after resting or acute swimming. (J) Quantitation of LC3II/I levels in swimming study. n = 4–8. (K) Quantitation of p62 levels in a swimming study. n = 3–4. Data are mean ± SEM.
Since reduced LC3II levels can be caused by decreased autophagosome formation or excessive degradation, we investigated the effect of MCL-1 deficiency on autophagic flux. One week after tamoxifen injection, Mcl-1f/f and Mcl-1f/fCre+ mice were injected with chloroquine, which inhibits fusion between autophagosomes and lysosomes and causes LC3II to accumulate if autophagosome formation is intact (Iwai-Kanai et al. 2008). Accumulation of LC3II occurred in Mcl-1f/f hearts after chloroquine treatment as expected, but no such increase was observed in Mcl-1f/fCre+ hearts (Fig. 7G,H). Recent studies have shown that acute exercise causes induction of autophagy in the myocardium (He et al. 2012). To investigate whether autophagy could still be induced in MCL-1-deficient hearts, we subjected Mcl-1f/fCre+ mice to swimming 1 wk after tamoxifen injections. We observed enhanced LC3 processing and p62 degradation in Mcl-1f/f hearts but not in Mcl-1f/fCre+ hearts (Fig. 7I–K). Taken together, these results demonstrate that the loss of MCL-1 impairs autophagosome formation in cardiac myocytes. Notably, in contrast to the 2-wk time point, we did not see a significant reduction in the LC3II/I ratio at 1 wk (Fig. 7H,J). However, there was still a significant accumulation of p62 at 1 wk (Fig. 7K), suggesting that baseline autophagy is still impaired and progressively worsens after loss of MCL-1.
Although our data suggest that MCL-1 is required for induction of autophagy in cardiac myocytes, overexpression of MCL-1 has been reported to inhibit autophagy in neurons (Germain et al. 2011). Similarly, anti-apoptotic BCL-2 and BCL-XL have been shown to inhibit autophagy in mammalian cells (Pattingre et al. 2005; Maiuri et al. 2007). Therefore, we investigated the effect of MCL-1 on induction of autophagy in neonatal rat cardiac myocytes. Interestingly, overexpression of MCL-1 had no effect on the number of GFP-LC3-positive autophagosomes or LC3II/I levels in myocytes after glucose deprivation (Supplemental Fig. S10). These findings suggest that MCL-1 does not function as a negative regulator of autophagy. These characteristics distinguish MCL-1 from other anti-apoptotic BCL-2 proteins and suggest a potential unique role for MCL-1 in myocytes.
Discussion
Loss of terminally differentiated cardiac myocytes results in a reduced ability to sustain contractile function and can lead to the development of heart failure. Thus, increased understanding of pathways that regulate survival of myocytes is critical for the identification of new therapeutic targets to treat or prevent heart failure. Our studies demonstrate that MCL-1 is essential for cardiac homeostasis in the adult heart and that loss of MCL-1 in cardiac myocytes leads to rapid development of heart failure. Surprisingly, loss of MCL-1 does not substantially activate apoptosis. Instead, our data suggest that it leads to mitochondrial dysfunction, opening of the mPTP, and subsequent necrotic cell death. Another striking finding was that loss of MCL-1 leads to impaired autophagy both under basal conditions and in response to physiological stress. Thus, these findings suggest that in addition to its anti-apoptotic role, MCL-1 is critical in maintaining mitochondrial function and initiating autophagy in cardiac myocytes under normal conditions.
Although the anti-apoptotic BCL-2 proteins are well known to regulate the intrinsic pathway of apoptosis, recent studies suggest that this family also plays important roles in other cellular processes, including autophagy (Pattingre et al. 2005; Maiuri et al. 2007) and mitochondrial morphology (Berman et al. 2009; Perciavalle et al. 2012). Studies have provided some evidence that MCL-1 has functions beyond regulating apoptosis. For instance, global deletion of Mcl-1 results in peri-implantation embryonic lethality, but Mcl-1−/− embryos display normal nuclear architecture and no increase in apoptosis (Rinkenberger et al. 2000). MCL-1 is also important in facilitating mitochondrial fusion and for proper assembly of the F1F0-ATP synthase (Perciavalle et al. 2012). Our data show clearly that loss of MCL-1 does not result in massive loss of myocytes due to apoptosis. Instead, loss of MCL-1 leads to mitochondrial swelling and rupturing of the outer mitochondrial membrane. Mcl-1 deletion also resulted in cardiac inflammation, another consequence of necrosis (Frangogiannis 2012). CypD is an important regulator of the mPTP, and mice deficient in CypD are resistant to stresses that promote necrotic cell death (Baines et al. 2005). Interestingly, we found that CypD deletion delayed development of cardiac dysfunction and significantly extended survival in MCL-1-deficient mice. These findings suggest that opening of the mPTP and necrotic cell death contribute to the loss of myocytes and development of heart failure but are not the underlying cause of the heart failure observed in the MCL-1-deficient mice. Our data also suggest that MCL-1 is not a direct regulator of the mPTP. Rather, opening of the mPTP most likely occurs as a consequence of mitochondrial impairment due to MCL-1 deficiency.
Wencker et al. (2003) demonstrated that low levels of apoptosis can lead to a lethal cardiomyopathy. We are not excluding the possibility that very low levels of apoptosis may contribute to lethality in MCL-1-deficient mice. However, the lethal cardiac phenotype in MCL-1 knockout mice is exceptionally rapid, indicating that low levels of apoptosis alone are insufficient to produce this phenotype. The absence of significant apoptosis is not surprising, since this process is energy-dependent. Our data show that loss of MCL-1 leads to defects in mitochondrial respiration, which will lead to reduced cellular ATP levels and potentially an inability undergo apoptosis. Interestingly, a companion study by Wang et al. (2013) in this issue of Genes & Development also reports that loss of MCL-1 leads to rapid development of heart failure, and they discovered that mice lacking BAX/BAK were more resistant to loss of MCL-1 than the CypD knockout mice in our study. Although the triple BAX/BAK/MCL-1 knockout mice had improved cardiac function and survival, this study also found little evidence of apoptosis upon deletion of MCL-1 in heart tissue (Wang et al. 2013). Thus, the results from our study and the study by Wang et al. (2013) suggest the intriguing possibility that BAX/BAK are master regulators of mitochondria-mediated cell death. In fact, a recent study by Whelan et al. (2012) found that BAX is an essential regulator of mPTP opening and necrotic cell death in the heart. They reported that the BAX/BAK double-knockout mice are resistant to both apoptotic and necrotic cell death. These studies confirm that the regulation of cell survival and death by BCL-2 family proteins is much more complex than initially anticipated. Similarly, our studies suggest that BNIP3 induces both apoptosis and necrosis via BAX/BAK (Kubli et al. 2007; Quinsay et al. 2010b). We also discovered that BNIP3 induces permeabilization of the inner mitochondrial membrane, as evident by calcein AM release from the matrix in both wild-type and CypD-deficient cells (Quinsay et al. 2010b). BNIP3 is also an important regulator of mitochondrial autophagy, where it acts as an autophagy receptor to tether mitochondria to the forming autophagosomes (Hanna et al. 2012). This complex dual role is also evident in studies with BNIP3-deficient mice. These mice are more resistant to myocardial I/R (Diwan et al. 2007) but accumulate dysfunctional mitochondria in myocytes with aging (Dorn 2010).
Our study also demonstrates that loss of MCL-1 leads to impaired induction of autophagy. Studies have found that autophagy is essential for homeostasis in the heart both under baseline conditions and in response to stress (Matsui et al. 2007; Nakai et al. 2007). Autophagy is also very important in clearing damaged mitochondria in myocytes before they cause harm to the cell (Kubli et al. 2013). Thus, mitochondrial dysfunction combined with the inability to clear damaged mitochondria via autophagy as a result of MCL-1 deficiency will quickly be detrimental to the myocyte and accelerate development of heart failure. It is currently unclear how loss of MCL-1 impairs autophagy. Our data indicate that the PINK1/Parkin pathway might be affected in MCL-1-deficient hearts, but it is still unknown whether this pathway can still be activated in response to additional stress. We are currently investigating whether MCL-1 regulates the PINK1/Parkin pathway in myocytes. The finding that MCL-1 deficiency results in impaired autophagy is unexpected, since the anti-apoptotic BCL-2 proteins have been reported to inhibit induction of autophagy by sequestering Beclin-1 (Pattingre et al. 2005; Maiuri et al. 2007). A previous study found that deletion of MCL-1 in cortical neurons led to activation of autophagy (Germain et al. 2011). However, this study found that the levels of other BCL-2 proteins did not change upon loss of MCL-1 in neurons. In contrast, we found a compensatory up-regulation of BCL-2 and BCL-XL after deletion of Mcl-1 in the heart, which could repress autophagy via increased interaction with Beclin-1. Further studies are needed to determine exactly how loss of MCL-1 impairs induction of autophagy in myocytes.
A number of BCL-2 family protein inhibitors have entered clinical trials for the treatment of leukemia, lymphoma, and solid tumors (Azmi et al. 2011). Some of these specifically target MCL-1 in an effort to counter its anti-apoptotic effects (Dash et al. 2011). Unfortunately, our study suggests that antagonizing MCL-1 may lead to significant cardiotoxicity. By compromising both autophagy and mitochondrial function, MCL-1 inhibitors are likely to affect the cells' energy supply. Although these effects on mitochondria may potentiate chemotherapeutic cytotoxicity, the Warburg effect reduces dependence on mitochondrial respiration in many malignancies by up-regulating glycolytic machinery (Bensinger and Christofk 2012). In contrast, cardiac myocytes rely on mitochondria for ATP to maintain contraction and exhibit the greatest mitochondrial density of any tissue in the body, making the heart especially vulnerable to antagonists that target MCL-1. The story of double-edged chemotherapeutics with cardiotoxicity is familiar from our experience with anthracyclines such as doxorubicin. BCL-2 family inhibitors may become useful chemotherapeutics, but their use in patients will require a keen understanding of the mechanisms of MCL-1 in the heart.
In summary, our results demonstrate that the loss of MCL-1 leads to rapid mitochondrial dysfunction, impaired autophagy, and heart failure in the absence of stress. To our knowledge, this is the first study that selectively deletes an anti-apoptotic BCL-2 family protein in the heart and explores the function of MCL-1 in that organ. Our study also suggests that therapies that preserve endogenous MCL-1 have therapeutic potential for cardiovascular disease. In addition, chemotherapeutics that target MCL-1 must be carefully designed to avoid cardiotoxicity.
Materials and methods
Mouse surgeries and cardiac-restricted ablation of the Mcl-1 gene
All animal experiments were carried out in accordance with institutional guidelines and approved by the Institutional Animal Care and Use Committee of the University of California at San Diego. Adult mice were subjected to myocardial infarction by permanently ligating the left anterior descending coronary artery (LAD) as described previously (Kubli et al. 2013). Briefly, 12- to 16-wk-old C57Bl/6J mice were anesthetized with isoflurane, intubated, and ventilated (Harvard Apparatus). The LAD was ligated with an 8-0 silk suture, and the chest was immediately closed up.
Mice harboring a homozygous conditional null mutation in the Mcl-1 gene (Mcl-1flox/flox) (Opferman et al. 2003) were crossed to transgenic αMHC-MerCreMer mice (Sohal et al. 2001) that express Cre recombinase under the transcriptional control of the cardiomyocyte-specific α-MHC promoter to generate conditional cardiomyocyte-specific Mcl-1 knockout mice. The Mcl-1flox/flox;MerCreMer+ mice were crossed with CypD-null mice (Ppif−/−) (Baines et al. 2005) to create Ppif−/−Mcl-1flox/floxMerCreMer+ double knockout mice. All mice were on a C57BL/6J background, and Cre littermates were always used as controls. To selectively delete Mcl-1 in cardiomyocytes, 8- to 10-wk-old mice were treated with 80 mg/kg tamoxifen for 5 d (Koitabashi et al. 2009). We found that this dose resulted in the most rapid and effective knockdown of the Mcl-1 gene without causing cardiotoxicity.
Echocardiography
Echocardiography was carried out as described previously using a VisualSonics Vevo 770 with an RMV707B 15- to 45-MHz imaging transducer (Kubli et al. 2013). Mice were anesthetized with 0.5%–1.5% isoflurane in 95% O2 and 5% CO2 and placed on a recirculating water warming pad. All scans were taken with heart rates >500 beats per minute (bpm).
qPCR for hypertrophy markers
RNA was extracted using Trizol (Ambion) according to the manufacturer's method. cDNA synthesis was carried out with the Verso cDNA synthesis kit (Thermo Scientific) using 1 μg of RNA and oligo dT primers according to manufacturer's protocol. qPCR was then carried out using standard TaqMan primers and TaqMan Universal Mastermix II (Applied Biosystems) on a 7500 Fast Real-Time PCR system (Applied Biosystems). Fold difference was calculated according to the comparative CT (2−ΔΔCT) method using GAPDH as a control (Schmittgen and Livak 2008).
Histological analysis and immunofluorescence
Hearts were arrested in diastole with 200 mM cold KCl, fixed in 4% paraformaldehyde, dehydrated in 70% ethanol, and embedded in paraffin. Sections were stained with H&E or Masson's trichrome. To determine the size of cardiomyocytes, transverse sections were labeled with fluorescein isothiocyanate (FITC)-labeled wheat germ agglutinin (WGA) (Sigma-Aldrich), nuclei were counterstained with DAPI (Vector Laboratories), and the cross-sectional area of myocytes in the left ventricular wall was determined using Image Pro Plus software as described previously (Xiang et al. 2011). TUNEL staining was performed using the In Situ Cell Death Detection kit (Roche Applied Science) according to the manufacturer's instructions. To detect activated caspase-3, heart sections were incubated with an antibody specific for cleaved caspase-3 (Cell Signaling Technology) and tropomyosin (Sigma) overnight. Nuclei were counter-stained with Hoechst 33342. Sections from three to four hearts in each group were used in the staining, and at least five different fields/section were analyzed in each experiment.
LDH and caspase-3 activity assay
LDH activity was determined in blood samples cleared by centrifugation at 3000g for 10 min at 4°C. LDH activity was quantified using a colorimetric assay according to the manufacturer's instructions (Biovision, no. K726-500). Caspase-3 activity was determined in whole-heart homogenates using a fluorescent caspase-3 activity assay (EMD Biosciences). HL-60 cells treated with 0.5 μg/mL actinomycin D for 24 h were used as a positive control. Fluorescence was measured using a Molecular Devices SpectraMax M3 spectrophotometer.
Western blot analysis
Hearts were homogenized in lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM EGTA, 1 mM EDTA, 1% Triton X-100, plus Complete protease inhibitor cocktail [Roche]) and cleared by centrifugation at 20,000g for 20 min at 4°C. Samples were run on Invitrogen NuPAGE Bis-Tris gels and transferred to nitrocellulose membranes. Membranes were incubated with the indicated antibodies and imaged with a Bio-Rad ChemiDoc XRS+ imager. Antibodies used were directed against MCL-1 (1:1000; Rockland, no. 600-401-394), BCL-2 (1:1000; BD Biosciences), BCL-XL (1:100; Santa Cruz Biotechnology, no. sc-8392), BAK (1:500; Millipore Upstate Biotechnology, no. 06-536), BAX (1:1000; Cell Signaling, no. 2772), LC3 (1:1000; Cell Signaling, no. 4108), p62 (1:1000; ARP, no. 03-GP62-C), cleaved caspase-3 (1:1000; Cell Signaling, no. 9664), GAPDH (1:1000; Cell Signaling, no. 2118), actin (1:1000; Sigma, no. A4700), tubulin (1:1000; Sigma, no. T6074), ubiquitin (1:100; Santa Cruz Biotechnology, no. sc-8017), AIF (1:1000; Cell Signaling, no. 5318), BNIP3 (1:1000; Sigma, no. B7931), p84 (1:1000; Genetex, no. GTX70220), complex III subunit core 2 (1:1000; Invitrogen, no. 459220), Tim23 (1:1000; BD Biosciences, no. 611222), PINK1 (1:500; Cayman, no. 10006283), Bim (1:500; Cell Signaling, no. 2819), and NOXA (1:200; Novus, no. NB600-1159).
Electron microscopy
TEM was performed on heart sections as previously described (Quinsay et al. 2010b). The hearts were fixed in 2.5% glutaraldehyde in 0.1 M cacodylate buffer, post-fixed in 1% osmium tetroxide, treated with 0.5% tannic acid and 1% sodium sulfate, cleared in 2-hydroxypropyl methacrylate, and embedded in LX112 (Ladd Research). Sections were mounted on copper slot grids coated with parlodion and stained with uranyl acetate and lead citrate for examination on a Philips CM100 electron microscope (FEI).
Mitochondrial swelling assay, oxygen consumption, and complex I/II activity
Cardiac mitochondria were isolated from tamoxifen-treated Mcl-1f/f,Mcl-1f/fCre+, Ppif−/−Mcl-1f/f, and Ppif−/−Mcl-1f/fCre+ mice at 1 wk. The mitochondrial swelling was carried out as described previously (Quinsay et al. 2010a). Briefly, 60 μg of mitochondria was suspended in a volume of 100 μL of homogenization buffer (250 mM sucrose, 5 mM KH2PO4, 2 mM MgCl2, 10 mM MOPS at pH 7.4, 1 mM EGTA, 0.1% fatty acid-free bovine serum albumin [BSA]) and added to a 96-well plate. Mitochondrial swelling was monitored by measuring absorbance on a plate reader (Molecular Devices SpectraMax M3) at 520 nm. The procedures used for mitochondrial respiration have been described previously (Kubli et al. 2013). Mitochondrial oxygen consumption was recorded at 30°C using a Clark-type oxygen electrode (Hansatech Oxygraph) in 600 μL of respiration buffer (140 mM KCl, 1 mM EGTA, 10 mM MOPS at pH 7.4, 10 mM MgCl2, 5 mM KH2PO4, 0.2% fatty acid-free BSA). Complex I activity was measured using 200 μg of mitochondria with 5 mM pyruvate and 5 mM malate as a substrate. Complex II activity was measured with 5 mM succinate as a substrate. ADP-stimulated respiration rate (state 3) was measured after the addition of 2 mM ADP, oligomycin-insensitive (state 4) was measured after the addition of 2 μM oligomycin, and the maximal respiration rate was measured after uncoupling the mitochondria with 2 μM FCCP. Rates were calculated as nanoatoms of oxygen/minute/microgram of protein. As a measure of mitochondrial integrity, the respiratory control ratio (RCR) (state 3 divided by state 4) was calculated. The data reported represent three to four independent mitochondrial preparations comprising two hearts each. Individual complex I and II activities were measured as described (Frazier and Thorburn 2012). To assess Complex I activity, mitochondria were resuspended in buffer containing 50 mM KH2PO4, 5 mM MgCl2, 50 μM NADH, 1 mM KCN, 10 μM Antimycin A, and 0.1% docecyl maltoside. Oxidation of NADH by complex I was recorded at 340 nm in a plate reader upon addition of 50 μM CoQ1. To assess complex II activity, mitochondria were resuspended in 50 mM KH2PO4, 5 mM MgCl2, 50 μM CoQ1, 80 μM DCIP, 1 mM KCN, 10 μM Antimycin A, and 0.1% docecyl maltoside. Reduction in DCIP was recorded at 600 nm upon addition of 10 mM succinate. The enzymatic activities were calculated using absorption coefficients of 6.22 mM/cm for NADH and 21 mM/cm for DCIP.
In vivo autophagy experiments
Mcl-1flox/flox;MerCreMer+ and Mcl-1flox/flox;MerCreMer- littermates were administered tamoxifen for 5 d. One week after the first injection (day 7), mice were injected with 60 mg/kg chloroquine (Sigma) or vehicle to block autophagic flux. Six hours later, hearts were harvested for Western blotting experiments as described above. One week after initiating tamoxifen administration, mice were subjected to two 10-min swimming sessions in a 37°C water bath separated by a 2-h resting period in between. On day 8, the two swimming periods were extended to 20 min. On day 9, the mice were subjected to swimming for 120 min followed by immediate euthanasia and tissue collection.
Statistical analyses
All values are expressed as mean ± standard error of mean (SEM). Statistical analyses were performed using Student's t-test. Survival was assessed using the Kaplan-Meier method and log-rank test. P < 0.05 was considered significant.
Acknowledgments
The Ppif−/− mice were generously provided by Dr. Jeffrey D. Molkentin (Cincinnati Children's Hospital Medical Center). This work was supported by NIH grants R01HL087023 and R01HL101217 (to A.B.G.) and a predoctoral fellowship from the American Heart Association's Western States Affiliate (R.L.T.).
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.215871.113.
References
- Azmi AS, Wang Z, Philip PA, Mohammad RM, Sarkar FH 2011. Emerging Bcl-2 inhibitors for the treatment of cancer. Expert Opin Emerg Drugs 16: 59–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, et al. 2005. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434: 658–662 [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay U, Sridhar S, Kaushik S, Kiffin R, Cuervo AM 2010. Identification of regulators of chaperone-mediated autophagy. Mol Cell 39: 535–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensinger SJ, Christofk HR 2012. New aspects of the Warburg effect in cancer cell biology. Semin Cell Dev Biol 23: 352–361 [DOI] [PubMed] [Google Scholar]
- Berman SB, Chen YB, Qi B, McCaffery JM, Rucker EB 3rd, Goebbels S, Nave KA, Arnold BA, Jonas EA, Pineda FJ, et al. 2009. Bcl-xL increases mitochondrial fission, fusion, and biomass in neurons. J Cell Biol 184: 707–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash R, Azab B, Quinn BA, Shen X, Wang XY, Das SK, Rahmani M, Wei J, Hedvat M, Dent P, et al. 2011. Apogossypol derivative BI-97C1 (Sabutoclax) targeting Mcl-1 sensitizes prostate cancer cells to mda-7/IL-24-mediated toxicity. Proc Natl Acad Sci 108: 8785–8790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diwan A, Krenz M, Syed FM, Wansapura J, Ren X, Koesters AG, Li H, Kirshenbaum LA, Hahn HS, Robbins J, et al. 2007. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J Clin Invest 117: 2825–2833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn GW 2nd 2010. Mitochondrial pruning by Nix and BNip3: An essential function for cardiac-expressed death factors. J Cardiovasc Transl Res 3: 374–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangogiannis NG 2012. Regulation of the inflammatory response in cardiac repair. Circ Res 110: 159–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier AE, Thorburn DR 2012. Biochemical analyses of the electron transport chain by spectrophotometry. Methods Mol Biol 837: 49–62 [DOI] [PubMed] [Google Scholar]
- Germain M, Nguyen AP, Le Grand JN, Arbour N, Vanderluit JL, Park DS, Opferman JT, Slack RS 2011. MCL-1 is a stress sensor that regulates autophagy in a developmentally regulated manner. EMBO J 30: 395–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson AB, Gottlieb RA 2007. Bcl-2 family members and apoptosis, taken to heart. Am J Physiol Cell Physiol 292: C45–C51 [DOI] [PubMed] [Google Scholar]
- Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson ÅB 2012. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem 287: 19094–19104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, An Z, Loh J, Fisher J, Sun Q, et al. 2012. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 481: 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Ito Y, Morikawa M, Uchida H, Kobune M, Sasaki K, Abe T, Hamada H 2003. Bcl-xL gene transfer protects the heart against ischemia/reperfusion injury. Biochem Biophys Res Commun 311: 64–70 [DOI] [PubMed] [Google Scholar]
- Imahashi K, Schneider MD, Steenbergen C, Murphy E 2004. Transgenic expression of Bcl-2 modulates energy metabolism, prevents cytosolic acidification during ischemia, and reduces ischemia/reperfusion injury. Circ Res 95: 734–741 [DOI] [PubMed] [Google Scholar]
- Iwai-Kanai E, Yuan H, Huang C, Sayen MR, Perry-Garza CN, Kim L, Gottlieb RA 2008. A method to measure cardiac autophagic flux in vivo. Autophagy 4: 322–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T 2000. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19: 5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang MH, Reynolds CP 2009. Bcl-2 inhibitors: Targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res 15: 1126–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirshenbaum LA, de Moissac D 1997. The bcl-2 gene product prevents programmed cell death of ventricular myocytes. Circulation 96: 1580–1585 [DOI] [PubMed] [Google Scholar]
- Koitabashi N, Bedja D, Zaiman AL, Pinto YM, Zhang M, Gabrielson KL, Takimoto E, Kass DA 2009. Avoidance of transient cardiomyopathy in cardiomyocyte-targeted tamoxifen-induced MerCreMer gene deletion models. Circ Res 105: 12–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al. 2007. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131: 1149–1163 [DOI] [PubMed] [Google Scholar]
- Krajewski S, Bodrug S, Krajewska M, Shabaik A, Gascoyne R, Berean K, Reed JC 1995. Immunohistochemical analysis of Mcl-1 protein in human tissues. Differential regulation of Mcl-1 and Bcl-2 protein production suggests a unique role for Mcl-1 in control of programmed cell death in vivo. Am J Pathol 146: 1309–1319 [PMC free article] [PubMed] [Google Scholar]
- Kubli DA, Gustafsson ÅB 2012. Mitochondria and mitophagy: The yin and yang of cell death control. Circ Res 111: 1208–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubli DA, Ycaza JE, Gustafsson AB 2007. Bnip3 mediates mitochondrial dysfunction and cell death through Bax and Bak. Biochem J 405: 407–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK, Jimenez R, Petrosyan S, Murphy AN, Gustafsson AB 2013. Parkin deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem 288: 915–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Kroemer G 2008. Autophagy in the pathogenesis of disease. Cell 132: 27–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N, et al. 2007. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J 26: 2527–2539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J 2007. Distinct roles of autophagy in the heart during ischemia and reperfusion: Roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res 100: 914–922 [DOI] [PubMed] [Google Scholar]
- Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K, Nakayama K, Negishi I, Senju S, Zhang Q, Fujii S, et al. 1995. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science 267: 1506–1510 [DOI] [PubMed] [Google Scholar]
- Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, et al. 2007. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 13: 619–624 [DOI] [PubMed] [Google Scholar]
- Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T, Mora M, Riggs JE, Oh SJ, Koga Y, et al. 2000. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 406: 906–910 [DOI] [PubMed] [Google Scholar]
- Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ 2003. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature 426: 671–676 [DOI] [PubMed] [Google Scholar]
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B 2005. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122: 927–939 [DOI] [PubMed] [Google Scholar]
- Perciavalle RM, Stewart DP, Koss B, Lynch J, Milasta S, Bathina M, Temirov J, Cleland MM, Pelletier S, Schuetz JD, et al. 2012. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat Cell Biol 14: 575–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinsay MN, Lee Y, Rikka S, Sayen MR, Molkentin JD, Gottlieb RA, Gustafsson AB 2010a. Bnip3 mediates permeabilization of mitochondria and release of cytochrome c via a novel mechanism. J Mol Cell Cardiol 48: 1146–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinsay MN, Thomas RL, Lee Y, Gustafsson AB 2010b. Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy 6: 855–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinkenberger JL, Horning S, Klocke B, Roth K, Korsmeyer SJ 2000. Mcl-1 deficiency results in peri-implantation embryonic lethality. Genes Dev 14: 23–27 [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3: 1101–1108 [DOI] [PubMed] [Google Scholar]
- Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, Penninger JM, Molkentin JD 2001. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res 89: 20–25 [DOI] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zmzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Constantini P, Loeffler M, et al. 1999. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397: 441–446 [DOI] [PubMed] [Google Scholar]
- Thomas LW, Lam C, Edwards SW 2010. Mcl-1; the molecular regulation of protein function. FEBS Lett 584: 2981–2989 [DOI] [PubMed] [Google Scholar]
- Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ 1993. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell 75: 229–240 [DOI] [PubMed] [Google Scholar]
- Wang X, Bathina M, Lynch J, Koss B, Calabrese C, Frase S, Schuetz JD, Rehg JE, Opferman JT 2013. Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev (this issue) doi: 10.1101/gad.215855.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S, Factor SM, Shirani J, Armstrong RC, Kitsis RN 2003. A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Invest 111: 1497–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan RS, Konstantinidis K, Wei AC, Chen Y, Reyna DE, Jha S, Yang Y, Calvert JW, Lindsten T, Thompson CB, et al. 2012. Bax regulates primary necrosis through mitochondrial dynamics. Proc Natl Acad Sci 109: 6566–6571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CF, Bishopric NH, Pratt RE 1997. Atrial natriuretic peptide induces apoptosis in neonatal rat cardiac myocytes. J Biol Chem 272: 14860–14866 [DOI] [PubMed] [Google Scholar]
- Xiang SY, Vanhoutte D, Del Re DP, Purcell NH, Ling H, Banerjee I, Bossuyt J, Lang RA, Zheng Y, Matkovich SJ, et al. 2011. RhoA protects the mouse heart against ischemia/reperfusion injury. J Clin Invest 121: 3269–3276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Qian L, Kozopas KM, Craig RW 1997. Mcl-1, a Bcl-2 family member, delays the death of hematopoietic cells under a variety of apoptosis-inducing conditions. Blood 89: 630–643 [PubMed] [Google Scholar]
- Zhu L, Yu Y, Chua BH, Ho YS, Kuo TH 2001. Regulation of sodium-calcium exchange and mitochondrial energetics by Bcl-2 in the heart of transgenic mice. J Mol Cell Cardiol 33: 2135–2144 [DOI] [PubMed] [Google Scholar]
- Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA, Hill JA 2007. Cardiac autophagy is a maldaptive response to hemodynamic stress. J Clin Invest 177: 1782–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]