

SUMMARY
Tuberous sclerosis complex (TSC) is a multi-organ disorder caused by mutations of the TSC1 or TSC2 genes. A key function of these genes is to inhibit mTORC1 (mechanistic target of rapamycin complex 1) kinase signaling. Cells deficient for TSC1 or TSC2 have increased mTORC1 signaling and give rise to benign tumors, although, as a rule, true malignancies are rarely seen. In contrast, other disorders with increased mTOR signaling typically have overt malignancies. A better understanding of genetic mechanisms that govern the transformation of benign cells to malignant ones is crucial to understand cancer pathogenesis. We generated a zebrafish model of TSC and cancer progression by placing a heterozygous mutation of the tsc2 gene in a p53 mutant background. Unlike tsc2 heterozygous mutant zebrafish, which never exhibited cancers, compound tsc2;p53 mutants had malignant tumors in multiple organs. Tumorigenesis was enhanced compared with p53 mutant zebrafish. p53 mutants also had increased mTORC1 signaling that was further enhanced in tsc2;p53 compound mutants. We found increased expression of Hif1-α, Hif2-α and Vegf-c in tsc2;p53 compound mutant zebrafish compared with p53 mutant zebrafish. Expression of these proteins probably underlies the increased angiogenesis seen in compound mutant zebrafish compared with p53 mutants and might further drive cancer progression. Treatment of p53 and compound mutant zebrafish with the mTORC1 inhibitor rapamycin caused rapid shrinkage of tumor size and decreased caliber of tumor-associated blood vessels. This is the first report using an animal model to show interactions between tsc2, mTORC1 and p53 during tumorigenesis. These results might explain why individuals with TSC rarely have malignant tumors, but also suggest that cancer arising in individuals without TSC might be influenced by the status of TSC1 and/or TSC2 mutations and be potentially treatable with mTORC1 inhibitors.
INTRODUCTION
Tuberous sclerosis complex (TSC) is a genetic disorder caused by loss of function of the TSC1 or TSC2 genes. Individuals with this disorder have multi-organ hamartomas resulting from increased proliferation and abnormal differentiation. Although not malignant, these tumors can still cause severe clinical manifestations, particularly in the brain, kidney and lungs (Crino et al., 2006). The protein products of the TSC1 (hamartin) and TSC2 (tuberin) genes bind to each other and function together to modulate downstream signaling pathways. Rapid advances in our knowledge of TSC were catalyzed by the discovery that hamartin-tuberin normally inhibits the mechanistic target of rapamycin (mTOR; previously known as mammalian target of rapamycin) serine/threonine kinase (Tee et al., 2002). mTOR is found within two functionally and molecularly distinct complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). Further intricacies are apparent because loss of TSC1 or TSC2 gene function seems to allow broadly dysregulated mTOR activity with constitutively increased mTORC1 activity but also decreased mTORC2 signaling (Inoki et al., 2003; Jacinto et al., 2004; Manning et al., 2005; Sarbassov et al., 2005). Through a very rapid translation of basic science findings, therapies with mTORC1 inhibitors have been developed as effective treatments for some of the clinical manifestations of TSC (Davies et al., 2008; Franz et al., 2006).
It is striking that patients with TSC almost as a rule develop benign tumors instead of malignancies. In contrast, patients with PTEN mutations also have constitutive activation of mTORC1 signaling but develop frank malignancies including aggressive gliomas (Hu et al., 2005). Several models have been proposed to account for the relative lack of malignancy, including feedback inhibition of AKT by mTORC1 activation, altered mTORC2 function as well as increased rates of apoptotic cell death in TSC1- or TSC2-deficient cells. Increased cell death might then balance excessive proliferation and thus prevent the emergence of transformed cells that could ultimately form malignancies. In support of this model, either TSC1- or TSC2-deficient cells in vitro are prone to apoptosis, particularly when under metabolic stress (Choo et al., 2010).
TP53 (p53) is one of the most frequently mutated genes in human cancer (Finlay et al., 1989; Levine, 1997; Vogelstein et al., 2000). As a tumor suppressor, p53 protein regulates growth arrest and apoptosis (Vousden and Prives, 2009), and has many identified transcriptional targets (Riley et al., 2008; Veprintsev and Fersht, 2008). Approximately 50% of all human cancers have a mutation in p53 that alters its transcriptional activity (Edlund et al., 2012) (http://p53.fr/). However, many questions remain regarding downstream molecular pathways involved in tumor cell growth. Recent studies have suggested an emerging role for p53 in cellular metabolism (Maddocks and Vousden, 2011) during stress conditions, including nutrient deprivation and hypoxia. This suggests a functional interaction with the mTOR signaling pathway. In addition, ∼40–90% of human malignancies have increased mTORC1 signaling (Menon and Manning, 2008). p53 is also known to regulate transcription of the PTEN gene, an upstream inhibitor of AKT signaling, as well as TSC2 (tuberin), an upstream inhibitor of mTORC1 signaling (Feng et al., 2007; Stambolic et al., 2001). Furthermore, mTORC1 regulates the translation of p53 in vitro (Lee et al., 2007). These findings suggest mechanistic links between mTORC1 and p53 that might be important for TSC pathogenesis, and possibly explain the paucity of cancer in patients with TSC.
TRANSLATIONAL IMPACT.
Clinical issue
Tuberous sclerosis complex (TSC) is a genetic disorder caused by loss-of-function mutations of the TSC1 gene (which encodes hamartin) or the TSC2 gene (which encodes tuberin). Hamartin and tuberin bind to each other and inhibit the mechanistic target of rapamycin (mTOR) serine/threonine kinase, which is found in two functionally and molecularly distinct complexes (mTORC1 and mTORC2). Loss of TSC1 or TSC2 gene function constitutively increases mTORC1 signaling and decreases mTORC2 signaling. Strikingly, individuals with TSC develop multi-organ hamartomas that, with rare exceptions, are non-cancerous, whereas other groups of patients with constitutive activation of mTORC1 signaling develop frank malignancies. A better understanding of this apparent paradox might provide new insights into the genetic mechanisms underlying cancer pathogenesis.
Results
Previous work by this group demonstrated that tsc2 homozygous mutant zebrafish recapitulate several key aspects of TSC, including hamartoma formation in the brain and increased mTORC1 signaling. In this study, the authors generate a zebrafish model of TSC and cancer progression by placing a heterozygous mutation of the tsc2 gene in a p53 mutant background. They show that tsc2 heterozygous mutant zebrafish never develop cancers but that tsc2;p53 compound mutants develop multi-organ malignancies that are seen earlier and in more tissue types than in zebrafish that are mutant for p53 alone. Both mTORC1 and mTORC2 signaling are altered in tsc2;p53 compound mutant zebrafish compared with p53 mutant zebrafish. Moreover, there is increased angiogenesis in tumors from tsc2;p53 compound mutants than in tumors from p53 mutant zebrafish. Notably, treatment of tsc2;p53 compound mutants with rapamycin, an mTORC1 inhibitor, decreases tumor size and the caliber of tumor-associated blood vessels.
Implications and future directions
These findings suggest that the lack of progression to cancer in patients with TSC might be mediated by p53-dependent senescence and that additional genetic changes such as the loss of TP53 are likely to be required for progression to malignancy. They provide new insights into the interactions between tuberin-mTORC1 signaling and p53 during tumorigenesis and suggest that, in patients without TSC, heterozygous TSC1 or TSC2 mutations in tumors that already carry TP53 mutations might lead to the development of more severe forms of cancer. Finally, these findings suggest that inhibition of mTORC1 signaling might be a viable therapeutic option for p53-deficient tumors in patients without TCS as well as in patients with TCS.
Zebrafish have emerged as a powerful model for the study of genetic diseases as well as cancer (Feitsma and Cuppen, 2008; Goessling et al., 2007; Mione and Trede, 2010). This is due to ease of genetic manipulation in zebrafish, the aneuploid nature of cancers arising in zebrafish, and the capacity of these animals for high-throughput genetic and pharmacologic screens (Zhang et al., 2010; Zon and Peterson, 2005). We previously showed that tsc2 homozygous mutant (tsc2vu242/vu242) zebrafish recapitulate several key aspects of TSC, including control of cell size, hamartoma formation in the brain and increased mTORC1 signaling (Kim et al., 2011). p53 mutant zebrafish models showed a conserved role for p53 in tumorigenesis (Berghmans et al., 2005; Parant et al., 2010), with the p53zdf1 allele being functionally similar to DNA-binding domain mutations identified in cancers from many patients.
To define interactions of the p53 and mTOR signaling pathways, we first examined mTORC1 signaling in tumors arising from p53zdf1/zdf1 homozygous mutant zebrafish and found that it was increased compared with that found in normal tissues from wild-type fish. We then explored a potential role for mTORC1 signaling in p53-mutation-mediated tumor formation by investigating whether further increases of mTORC1 activity enhance tumorigenesis. We used tsc2vu242/+;p53zdf1/zdf1 compound mutant (tsc2;p53 compound mutant) zebrafish for this purpose. These zebrafish had altered mTORC1 and mTORC2 signaling and enhanced rates of tumorigenesis compared with that seen with p53 mutations alone. We found increased expression of Hif1-α, Hif2-α and Vegf-c that was associated with prominent angiogenesis in tumors from tsc2;p53 compound mutant compared with p53 mutant zebrafish. Transient administration of rapamycin, a potent mTORC1 inhibitor, rapidly diminished the tumor-associated blood vessels. These results provide the first in vivo evidence of a crucial role of p53 to prevent the formation of malignancies in TSC.
RESULTS
Activation of mTORC1 signaling in p53 mutant tumors
To examine the contribution of mTOR activity in tumors arising from p53 mutant zebrafish, we measured mTORC1 signaling activity in abdominal tumors spontaneously arising from homozygous p53 mutant zebrafish (Fig. 1A). Histological analyses of these p53 mutant abdominal tumors revealed spindle-like cells reminiscent of malignant sarcomas. These cells were highly invasive, destroying skeletal muscle fibers (Fig. 1B; Fig. 2A). Similar pathological features were previously described in this zebrafish model (Berghmans et al., 2005). Using immunofluorescence, we next determined expression of phosphorylated ribosomal protein S6 (phospho-S6) and phosphorylated eukaryotic initiation factor 4E-binding protein 1 (phospho-4E-BP1), well established downstream effectors of the mTOR kinase within mTORC1 (Inoki et al., 2002). These abdominal tumors had high expression of both phospho-S6 and phospho-4E-BP1 (Fig. 1C,D; supplementary material Fig. S1, upper panel) in a subset of tumor cells. Immunoblot analyses also revealed a moderate increase of mTORC1 signaling in normal appearing eyes from p53zdf1/zdf1 zebrafish compared with wild-type control eyes (data not shown). These results suggest that mTORC1 activation in p53 mutant tissues might facilitate tumor formation and/or growth.
Fig. 1.
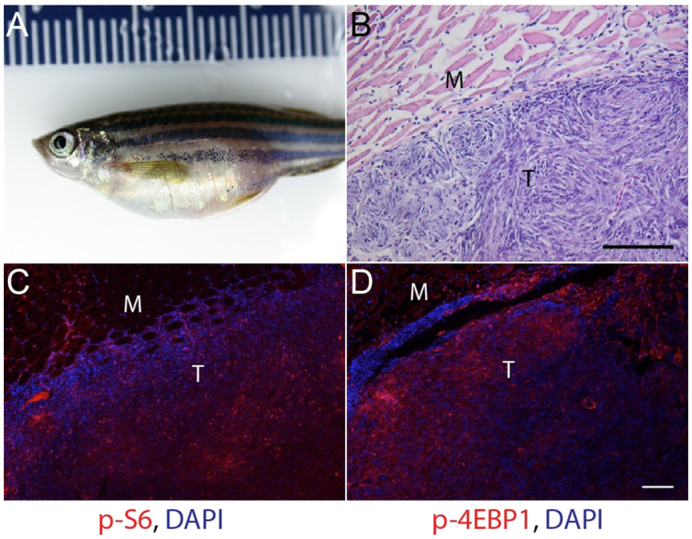
mTORC1 signaling in malignant abdominal tumors in p53zdf1/zdf1 zebrafish. (A) 1-year-old p53zdf1/zdf1 mutant fish with prominent abdominal tumor. (B) Hematoxylin and eosin (H&E) staining of a section from an abdominal tumor showing whorls of spindle-shaped cells consistent with an invasive sarcoma. (C,D) Increased expression of mTORC1 downstream effectors phospho-S6 (C) and phospho-4EBP1 (D) in abdominal tumors from p53zdf1/zdf1 zebrafish. For C and D, red fluorescence is immunosignal and blue is DAPI nuclear stain. M, muscle; T, tumor. Scale bars: 100 μm. Similar findings were seen in abdominal tumors from 10/10 p53zdf1/zdf1 zebrafish.
Fig. 2.

Increased mTORC1 signaling, enhanced tumorigenesis and altered tumor spectrum from the addition of a tsc2 heterozygous allele to a p53zdf1/zdf1 background. (A) H&E, phospho-S6 and DAPI (nuclei) staining of abdominal tumors from p53zdf1/zdf1 and tsc2;p53 compound mutant zebrafish. Left panels: invasive region of a p53zdf1/zdf1 tumor; right panels: tsc2;p53 compound mutant tumor. Circular structures are skeletal muscle bundles encircled by invasive tumor cells (pink in the H&E staining, but dark areas in the immunofluorescence images). Phospho-S6 signal is red and DAPI is white. M, muscle; T, tumor. Scale bar: 100 μm. (B) Kaplan-Meier tumor-free survival. Blue line is p53zdf1/zdf1 zebrafish, red line is tsc2;p53 compound mutant zebrafish. 50% of p53zdf1/zdf1 zebrafish had tumors at ∼550 days of life compared with 450 days for tsc2;p53 compound mutant zebrafish. n=115 and 130, respectively. This reduction in latency to tumor detection was statistically significant using the Log-rank test (P<0.0001). (C) Spectrum of tumor types from p53zdf1/zdf1 and tsc2;p53 compound mutant zebrafish. Tumor types were broadly similar with the exception of melanoma-like tumors, which were seen only in tsc2;p53 compound mutant zebrafish. (D) Normal zebrafish head and dorsal fin in the top panels and melanoma-like tumors on the head and dorsal fin in the bottom panels. Dorsal fins were sectioned at the region marked ‘1’ (normal) and ‘2’ (tumor) and stained with DAPI in D′ and D″, respectively. Scale bars: 5 mm (D); 100 μm (D″).
Heterozygous tsc2vu242 mutation enhanced mTORC1 signaling activity and tumor incidence in p53zdf1/zdf1 zebrafish
Given the increased mTORC1 signaling in p53 mutant zebrafish tumors, we hypothesized that further augmentation of mTORC1 signaling might enhance tumor formation, cell growth or tumor severity. To test this hypothesis, we introduced a heterozygous tsc2vu242 mutation into p53zdf1/zdf1 mutant zebrafish. We previously reported that heterozygous tsc2vu242 mutant zebrafish are viable and have moderately elevated mTORC1 signaling activity (Kim et al., 2011). Of note, tumors were never seen in heterozygous tsc2vu242/+ zebrafish, including in zebrafish of 2 years of age or older (n>200). In contrast, adult tsc2;p53 compound mutant zebrafish developed large sarcomatous tumors in the abdomen (Fig. 2A) as well as in other regions (Fig. 2C). Immunostaining for phospho-S6 levels confirmed prominent mTORC1 activation within tumors from compound mutant zebrafish, and these levels were further augmented when compared with tumors from p53zdf1/zdf1 zebrafish (Fig. 2A; supplementary material Fig. S1). As previously reported, these malignancies have a long latency, with 50% of p53zdf1/zdf1 zebrafish exhibiting tumors by 550 days of life (n=115 total p53zdf1/zdf1 zebrafish, Fig. 2B). In contrast, 50% of tsc2;p53 compound mutant zebrafish had tumors by 450 days of life (n=130 total tsc2;p53 compound mutant zebrafish, Fig. 2B, compared with p53zdf1/zdf1 zebrafish, Log-rank test, P<0.0001). Types of tumors were broadly similar in p53zdf1/zdf1 and tsc2;p53 compound mutant zebrafish, with the exception of melanoma-like tumors. These were seen in ∼7% of tsc2;p53 compound mutant zebrafish (Fig. 2C), but never seen in p53zdf1/zdf1 homozygous mutant zebrafish. Melanoma-like tumors were not reported in the original report of the p53zdf1/zdf1 phenotype (Berghmans et al., 2005). However, melanoma-like skin tumors were frequently observed in a different zebrafish model featuring a more deleterious p53 mutation (Parant et al., 2010).
mTORC1 and mTORC2 signaling in tumors from p53zdf1/zdf1 and tsc2;p53 compound mutant zebrafish
We next quantitated mTOR signaling levels using protein extracts from a consecutive series of abdominal tumors arising in p53zdf1/zdf1 and tsc2;p53 compound mutant zebrafish. In support of our immunostaining data presented in Figs 1 and 2, we again found increased phospho-S6 levels in a series of tumors arising from tsc2;p53 compound mutant zebrafish compared with those from p53zdf1/zdf1 zebrafish (Fig. 3A,B). When normalized for total S6, the ratio of phospho-S6 to total S6 was significantly higher in abdominal tumors arising from tsc2;p53 compound mutant zebrafish compared with p53zdf1/zdf1 zebrafish (Fig. 3D). We also looked at other tumor types and again found increased mTORC1 signaling in tsc2;p53 compound mutant zebrafish compared with tumors from p53zdf1/zdf1 zebrafish (data not shown). Finally, we quantified mTORC2 signaling in abdominal tumor extracts by immunoblotting for Akt (Ser473) levels and found no significant differences between tsc2;p53 compound mutant and p53zdf1/zdf1 zebrafish (supplementary material Fig. S2).
Fig. 3.
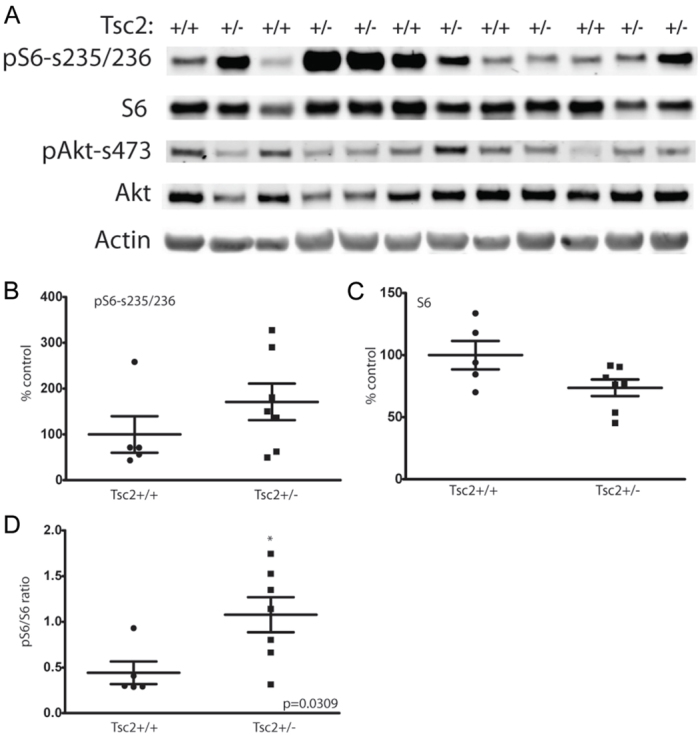
Increased mTORC1 signaling in tumors arising from tsc2;p53 compound mutant zebrafish compared with p53zdf1/zdf1 zebrafish. (A) Immunoblotting of a series of abdominal tumors comparing p53zdf1/zdf1 mutant zebrafish (lanes marked Tsc2+/+) and tsc2;p53 compound mutants (lanes marked Tsc2+/−). (B) Quantification of phospho-S6 levels were normalized to actin expression (P=0.25). (C) Quantification of total S6 levels were normalized to actin expression (P=0.06). (D) Increased ratio of normalized phospho-S6 to total S6 levels in tsc2;p53 compound mutant compared with p53zdf1/zdf1 zebrafish; asterisk denotes significance by two-tailed Student’s t-test, P=0.03. n=5 for p53 mutant and n=7 for compound mutant zebrafish, although two tumor samples from tsc2;p53 compound mutant zebrafish (lanes 4,5 and 11,12) were divided in half and processed independently for immunoblotting.
Elevated Akt kinase activity in tumors from p53zdf1/zdf1 and tsc2;p53 compound mutant zebrafish
p53 activates transcription of the PTEN gene and increased levels of PTEN protein in vitro can suppress AKT activation. This can relieve the inhibitory phosphorylation of tuberin at Ser939 and Thr1462 by AKT kinases (Manning et al., 2002; Feng et al., 2007). Although these signaling cascades have been described, genetic interactions of AKT and mTOR signaling have not been well characterized in vivo. To further address Akt activity in tumors from p53zdf1/zdf1 and tsc2;p53 compound mutant zebrafish, we examined phospho-tuberin (Ser939) using immunofluorescence. We found strongly positive cells within both tumor types, although there was a clear elevation in abdominal tumors from tsc2;p53 compound mutant zebrafish (Fig. 4A–F; supplementary material Fig. S3). We attempted to use this phospho-antibody for immunoblotting but did not obtain interpretable results. To further address Akt activity in these tumors, we also performed immunofluorescence using antibodies against total Akt, phospho-Akt (Thr308) and phospho-Akt (Ser473) (Fig. 4G–L; supplementary material Figs S4, S5). Interestingly, we found that the total Akt level was diffusely increased in tumor cells compared with adjacent muscle tissues from both p53zdf1/zdf1 and tsc2;p53 compound mutant zebrafish (Fig. 4G,J, n=8/8; supplementary material Fig. S4). Phospho-Akt (Thr308) levels were comparable, although the signal from tsc2;p53 compound mutant tumors were increased compared with p53zdf1/zdf1 tumors (Fig. 4H,K; supplementary material Fig. S5). In contrast, phospho-Akt (Ser473) levels were somewhat decreased in tumor cells (Fig. 4I,L) compared with adjacent muscle. Because Ser473 of Akt is phosphorylated by mTORC2, this reduction might be due to negative feedback by activated mTORC1 (Harrington et al., 2004; Shah et al., 2004).
Fig. 4.
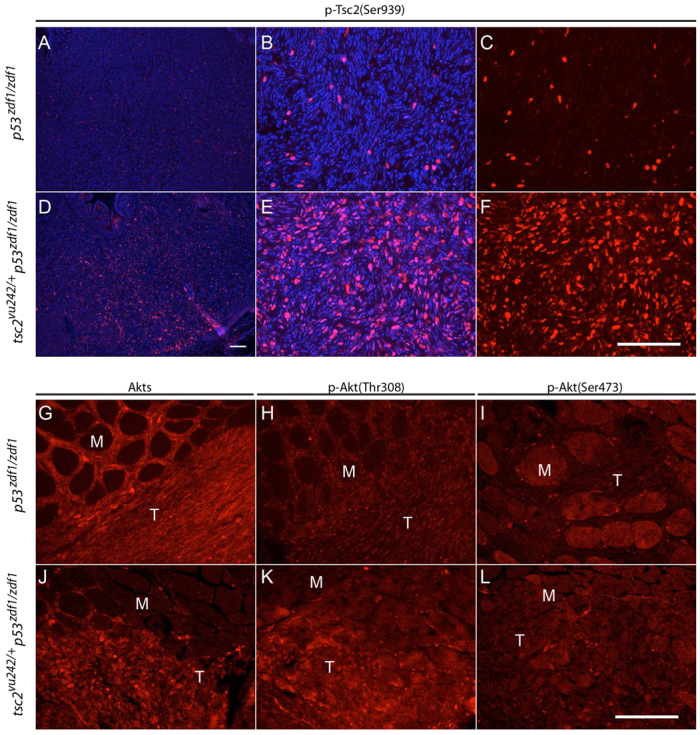
AKT activity in p53zdf1/zdf1 and tsc2;p53 compound mutant tumors. (A–F) Phospho-tuberin (p-Tsc2) staining in p53zdf1/zdf1 and tsc2;p53 compound mutant zebrafish. (B,C and E,F) Higher magnification of A and D, respectively. (C,F) Phospho-tuberin (Ser939, red); (B,E) phospho-tuberin (red) merged with DAPI (blue). (G,J) Total Akt immunofluorescence, (H,K) phospho-Akt (Thr308) immunofluorescence and (I,L) phospho-Akt (Ser473) immunofluorescence in p53zdf1/zdf1 (G–I) and tsc2;p53 compound mutant (J–L) zebrafish. M, muscle; T, tumor. Scale bars: 100 μm.
Vegf upregulation in tumors from p53zdf1/zdf1 and tsc2;p53 compound mutant zebrafish
As we assessed tumors arising in p53zdf1/zdf1 and tsc2;p53 compound mutant zebrafish, we frequently noted prominent and abnormal appearing blood vessels in the abdomen and eyes from both mutant genotypes (n=25/41 with tumors, Fig. 5A–D). This was consistently enhanced in tumors from tsc2;p53 compound mutant as compared with p53zdf1/zdf1 zebrafish. Approximately 68% of tsc2;p53 compound mutant zebrafish with abdomen and eye tumors developed thin blood vessels (n=10/25) but some abnormal vessels were also massively dilated (n=7/25, Fig. 5C,D). By comparison, about 50% of p53zdf1/zdf1 mutants with abdomen and eye tumors exhibited thin (n=5/16, Fig. 5A,B) or dilated blood vessels (n=3/16), but the degree of dilation was not as marked when compared with abnormal dilated blood vessels in tsc2;p53 mutants (comparison to tsc2;p53 mutants not statistically significant with P=0.33 by two-tailed Fisher’s exact test). These findings suggest that angiogenesis is enhanced in tumors arising from tsc2;p53 compound mutant zebrafish as compared with those arising in p53zdf1/zdf1 zebrafish. To see whether there was altered expression of angiogenic factors, we performed immunostaining for Hif1-α, Hif2-α and Vegf-c. All of these angiogenic factors were expressed in p53zdf1/zdf1 tumors with more intensive staining again seen in tsc2;p53 compound mutant zebrafish (Fig. 5E–K; supplementary material Figs S6, S7). These results suggest that further increases in mTORC1 signaling from tsc2 heterozygote zebrafish in a p53 mutant background possibly increases angiogenesis and vasodilation via upregulation of Hif1-α, Hif2-α and Vegf-c expression.
Fig. 5.
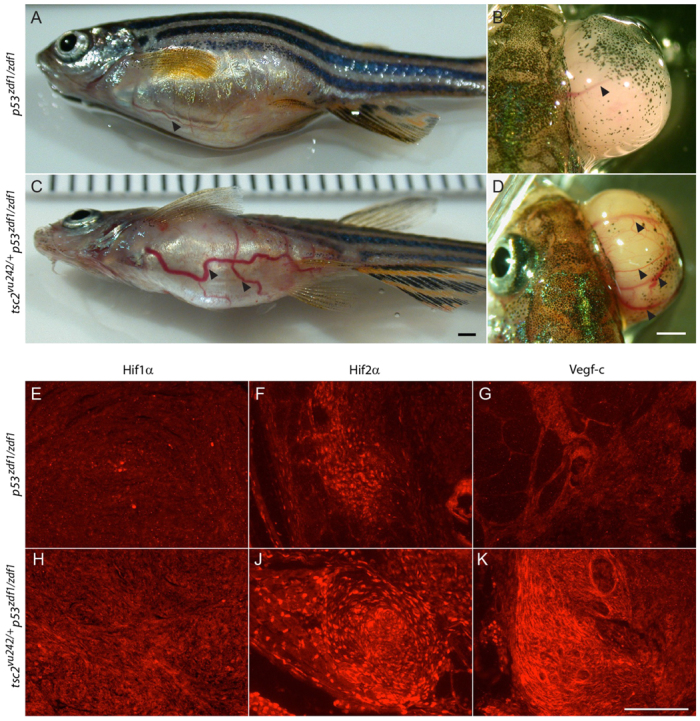
Angiogenesis and increased expression of angiogenic factors in tumors from tsc2;p53 compound mutant zebrafish. (A,B) Arrowheads indicate visible blood vessels on tumors from abdomen (A) and eye (B) from p53zdf1/zdf1 zebrafish (n=8/16). (C,D) Arrowheads indicate large and tortuous blood vessels observed in abdomen (C) and eye (D) tumors from tsc2;p53 compound mutant (n=17/25) zebrafish. Scale bars: 1 mm. (E,H) Hif1-α staining, (F,J) Hif2-α staining and (G,K) Vegf-c staining in p53zdf1/zdf1 (E–G) and tsc2;p53 compound mutant (H–K) zebrafish. Scale bar: 100 μm.
Rapamycin treatment suppresses Vegf expression in tumors from p53zdf1/zdf1 and tsc2;p53 compound mutant zebrafish
Given the increased expression of angiogenesis factors in these tumors, we next determined whether inhibition of mTORC1 signaling could suppress blood vessel formation and tumor size. Application of rapamycin daily seemed to shrink the caliber of existing blood vessels (example of an eye tumor in Fig. 6A,B). A representative vessel (Fig. 6B, black arrowheads) seemed to completely disappear at the end of both rapamycin treatment cycles (day 3 and day 14) but became visible again during the rapamycin ‘off period’ (day 6 and day 9) in the same location. This result supports a vasoconstrictive effect of rapamycin, probably through inhibition of Vegf-induced vasodilation (Kim et al., 2010). During a rapamycin 6-day ‘off period’ in tsc2;p53 compound mutant zebrafish, re-dilated vessels appeared (Fig. 6B, black arrowhead) as well as possible neoangiogenesis where vessels were not previously seen (example shown in Fig. 6B, blue arrowhead at day 6). These results suggest that activation of mTORC1 signaling is essential for neoangiogenesis and dilation of blood vessels in tumors arising in a p53zdf1/zdf1 background. To verify the effect of rapamycin treatment, we performed immunostaining with antibodies against phospho-S6, phospho-4EBP1, Akt and Vegf-c in abdominal tumors treated with vehicle (DMSO) or rapamycin daily for 2 days (Fig. 6C). Rapamycin treatment decreased mTORC1 signaling as evidenced by lower levels of phospho-S6 and phospho-4E-BP1 in rapamycin-treated tumors (Fig. 6C). Furthermore, we found that total Akt expression levels were also downregulated by rapamycin, although this suppression was mainly observed in the inner region of tumors.
Fig. 6.

Rapamycin treatment reduced blood vessel caliber in tumors from p53zdf1/zdf1 and tsc2;p53 compound mutant zebrafish. (A,B) Representative tumors from rapamycin-treated fish. (A) Rapamycin treatment of p53zdf1/zdf1 zebrafish harboring a tumor in its trunk. Arrowheads indicate visible vascular structure on the tumor surface. (B) Rapamycin treatment cycle (on day 0 to 3, 9 to 14, off day 4 to 8) lasting 2 weeks in a compound mutant zebrafish with an expending eye tumor. Pictures were taken at indicated days. Black arrowheads indicate vascular structures that disappeared during rapamycin treatment. Blue arrowhead indicates apparent new vessels formed during the rapamycin-off period. (C) H&E staining, and phospho-S6, phospho-4EBP1, Akt and Vegf-c expression (antibody signal shown as white) in p53zdf1/zdf1 and tsc2;p53 compound mutant abdomen tumors treated with DMSO or rapamycin for 2 days (n=3). Scale bar: 100 μm. (D) Rapamycin shrinks tumors in p53zdf1/zdf1 zebrafish. A representative angioma-like tumor was seen near the pectoral fin and treated daily with rapamycin. After 3 days of treatment, a clear decrease in size was noted. The tumor continued to shrink over the 1 week total period of rapamycin treatment. Scale bar: 1 mm.
mTORC1 inhibitors decrease tumor size
Given the rapid effect of rapamycin on blood vessel caliber and regression, we investigated whether continued treatment could reduce the size of tumors. We treated several types of rapidly growing tumors and found either stabilization of tumor size or a reduction in size during daily rapamycin treatments (Fig. 6; supplementary material Fig. S7). This effect was seen in tumors from p53zdf1/zdf1 zebrafish as well as those from tsc2;p53 compound mutants. In all cases, there was rapid resumption of tumor growth after rapamycin was stopped.
DISCUSSION
This study was designed to investigate why patients with TSC almost never develop cancer despite high levels of mTORC1 signaling in multiple organs. This is in contrast to the malignancies typically seen in patients with increased mTORC1 signaling due to PTEN mutations (Hu et al., 2005; Inoki et al., 2005). Various models have been proposed to explain this striking discrepancy, including feedback inhibition of AKT through mTORC1 and direct or indirect reductions of mTORC2 activity (Manning et al., 2005). Alternatively, decreased cell proliferation and enhanced cell death in TSC1- or TSC2-deficient cells might compensate for any excessive proliferation induced by increased mTORC1 activity. This model is also supported by complications reported during the generation of mouse embryonic fibroblasts (MEFs) from Tsc2 knockout mice (Zhang et al., 2003). Although derived Tsc2 mutant MEFs initially grew as expected, they rapidly underwent senescence and died. However, the authors circumvented this problem by crossing the Tsc2 knockout allele into p53-deficient mice. The resulting Tsc2/p53 mutant embryos were then used to generate MEFs that were capable of continuous growth. Our in vivo results using a zebrafish animal model of TSC further support that senescence in Tsc2 mutant cells might be mediated by p53. The loss of both tsc2 and p53 gene function might then permit uncontrolled proliferation leading to malignancies.
There are multiple types of benign tumors in TSC, including brain subependymal giant cell astrocytomas (SEGAs), angiomyolipomas in the kidney and rhabdomyomas in the heart. One notable exception is renal cell carcinoma (RCC). Although RCC is rarely seen in patients with TSC, it has been reported in relatively young patients (Lendvay et al., 2002). This is potentially quite relevant to our results because RCCs usually harbor p53 mutations (Gad et al., 2007; Habib et al., 2011; Noon et al., 2010). This further suggests that, in TSC, an additional mutation of p53 might allow progression to malignancy. Direct support for this model will require analyses of p53 status in RCC obtained from patients with TSC. Additional deep sequencing of TSC-associated hamartomas for any p53 mutations would also support or refute this model.
Our finding of enhanced tumor formation in tsc2;p53 compound mutant zebrafish is supported by previous reports of substantial crosstalk between p53, AKT and mTOR signaling during tumorigenesis. For example, p53 activates transcription of PTEN, a phosphoinositide phosphatase that normally inhibits AKT signaling (Feng et al., 2007). In addition, AKT and mTORC1 signaling is frequently activated in ovarian (Altomare et al., 2004) and lung (Memmott and Dennis, 2010) cancers, which usually have p53 mutations. Furthermore, p53 in vitro can inhibit mTOR signaling via the hamartin-tuberin complex (Feng et al., 2005). A recent study also indicated that signaling components downstream of mTOR can modulate p53 through the signaling of ribosomal S6 kinase to MDM2, a ubiquitin ligase that controls p53 stability (Lai et al., 2010). Thus, the p53 signaling pathway incorporates AKT and mTORC1 (Levine et al., 2006). However, interactions between p53 and mTOR pathway components during tumor formation have not been previously well studied in vivo.
Differential activation of Hif1-α and Hif2-α by mTORC1 and mTORC2 has been reported using in vitro assays (Toschi et al., 2008). The expression of Hif1-α and Hif2-α was dependent on Akt3 and Akt2, respectively. The effect of rapamycin treatment to blood vessel maintenance is also indicative of an mTORC1-dependent regulation of proangiogenic factors. However, the rapid return of blood vessels when rapamycin treatment was stopped supports a vasoconstrictive mechanism that has been previously suggested (Kim et al., 2010). Vegf-c expression, one of the central regulators of blood vessel formation, was also suppressed by rapamycin treatment, this affect was also most apparent in the inner part of the tumor (Fig. 6C). Altogether, these results suggest that mTORC1 activation plays an important role in tumor angiogenesis in p53 mutant tissues. In support of this model, we also found increased blood vessel size in the livers of tsc2 homozygous mutants compared with control Tg(Fli1:EGFP) zebrafish larvae (supplementary material Fig. S8). An additional feedback pathway that results from tuberin deficiency is increased PTEN activity via a rapamycin-dependent mechanism (Bonneau and Longy, 2000; Mahimainathan et al., 2009; Mehenni et al., 2005). We are currently testing this potential mechanism by placing a tsc2 heterozygous allele in a pten mutant background and again analyzing resultant zebrafish for the development of malignant tumors. AMPK might also be involved as this kinase normally interacts with p53 and tuberin to modulate cell proliferation. We found that activated phospho-AMPK (Thr172) was increased in tumor cells from both p53zdf1/zdf1 and tsc2;p53 compound mutant zebrafish (supplementary material Fig. S9). This might be due to a feedback loop uncovered in mutant zebrafish, because activated AMPK normally phosphorylates tuberin to activate it and inhibit mTORC1 signaling.
Of note, we have never seen sarcomas arising in tsc2 heterozygous zebrafish. This is broadly consistent to what is observed in human patients with TSC as well as rodent models of this disorder. tsc2;p53 compound mutant zebrafish have an significantly decreased time to tumor detection compared with p53zdf1/zdf1 zebrafish. However, it is possible that tumors arising in tsc2;p53 compound mutant zebrafish are simply faster growing, leading to their earlier detection. This is possible, although even within the oldest groups of zebrafish (>2 years old), tsc2;p53 compound mutant zebrafish always exhibited more tumors than p53zdf1/zdf1 zebrafish.
We conclude that additional genetic changes such as loss of TP53 are probably required for the progression to malignancy in TSC. Our results might also have implications for non-TSC patients if additional tumor-specific heterozygous TSC1 or TSC2 mutations in the setting of existing or subsequent TP53 mutations can potentially lead to more severe forms of cancer. The expected increased mTORC1 signaling in this scenario suggests that mTORC1 inhibitors could be effective therapies for such cancers. Further application of full exome and genome sequencing will likely uncover additional evidence of genetic interactions between TP53 and both TSC1 and TSC2 in a variety of human cancers.
MATERIALS AND METHODS
Zebrafish strains
Zebrafish strains used in this study included tsc2vu242/+ (Kim et al., 2011) and p53zdf1/zdf1 (Berghmans et al., 2005). Five pairs of tsc2;p53 compound mutant zebrafish (tsc2vu242/+;p53zdf1/zdf1) and p53zdf1/zdf1 adult fish were mated and embryos raised at 28.5°C. 5-month-old zebrafish siblings were genotyped and p53zdf1/zdf1 and tsc2;p53 compound mutant zebrafish identified and raised for up to 20 months in separate tanks. For genotyping of tsc2vu242/+, we amplified a 151 bp fragment by PCR using forward primer 5′-CCAGCACCACCTGCAGTCTGG-3′ and reverse primer 5′-CTCTTGGGCAGAGCAGAGAAGTTGG-3′ flanking the mutation site. The point mutation was confirmed by absence of a HpyCh4IV restriction site. Tg(Fli1:EGFP) zebrafish (Lawson and Weinstein, 2002) were obtained from the Zebrafish International Resource Center (ZIRC). All experiments were approved by the Vanderbilt Institutional Animal Care and Use Committee (IACUC).
Tumor analyses
For 550 days, adult fish (total of 110 p53zdf1/zdf1 and 130 tsc2;p53 compound mutant zebrafish) were monitored twice per week for tumor development or death. Fish noted to have a prominent tumor were euthanized in 20% MESAB and dissected into four pieces (head, anterior abdomen, posterior abdomen and tail). Pieces containing tumor were fixed in 4% paraformaldehyde for 2 days at 4°C. Fixed bodies were embedded in 1.2% agarose/5% sucrose and saturated in 30% sucrose. Tissue blocks were frozen in 2-methyl butane. 10-μm sections were collected on microscope slides using a Leica cryostat. Sections were then kept at −80°C prior to use.
Immunofluorescence
To minimize staining variation, ten tumor sections of each p53zdf1/zdf1 and tsc2;p53 compound mutant zebrafish were concurrently processed in a Sequenza™ Slide Rack (Thermo Scientific) (results are shown in supplementary material Figs S1–S3). Sections were rehydrated in 1× PBS for 20 minutes at room temperature and blocked in 5% sheep serum/1× PBS for 2 hours. Antibodies used were against: phospho-S6 ribosomal protein (Ser235/236) (Cell Signaling #2215), phospho-4E-BP1 (Thr37/46) (Cell Signaling #2855), phospho-tuberin (Ser939) (Cell Signaling #3615), Akt (Cell Signaling #9272), phospho-Akt (Thr308) (Cell Signaling #2965), phospho-Akt (Ser473) (Cell Signaling #9271), phospho-AMPK (Thr172) (Cell Signaling #2535), Hif1-α (Abcam ab8366), Hif2-α (Thermo Scientific FA1-16510) and Vegf-c (Ana Spec 55877). Antibodies were diluted 1:300 and added to sections overnight at 4°C. Cy3-labeled goat anti-rabbit secondary antibody (Jackson ImmunoResearch) was incubated overnight at 4°C. Sections were then washed with 1× PBS for 1 hour and mounted in VECTASHIELD with DAPI (Vector Laboratories). Images were acquired using a Zeiss Axio Imager Z1 and Zeiss AxioCam MRm digital camera. Digital images were then processed using Adobe Photoshop CS5. Images received only minor modifications of contrast with both control and mutant sections always processed in parallel.
Rapamycin treatment
After anesthesia of zebrafish with MESAB, 10 μl of rapamycin in DMSO (10 mg/ml, LC Laboratories) was added to a gill for 10 seconds. Fish were then placed back in water where they rapidly recovered. To monitor angiogenesis in zebrafish with tumors, we treated once a day for 1–2 weeks. For zebrafish with abdominal tumors (Fig. 6), rapamycin treatment was performed for 2 days total.
Immunoblots
Tumors and control tissues (mixture of organs derived from brain, kidney, intestine and liver of wild-type zebrafish) were lysed in RIPA cell lysis buffer (10 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% Triton X-100, 1% sodium deoxycholate with protease and phosphatase inhibitors added), using a motorized pestle grinder to aid in cell disruption. Protein concentrations were determined using a BCA protein assay, loading buffer was added to each sample and heated at 80°C for 10 minutes prior to loading on a gel and electrophoresed using the Invitrogen X-Cell II™ system. Gels were transferred to PVDF membranes, blocked in Odyssey buffer (Li-COR #927-40000) for 1 hour and incubated with primary antibodies overnight. Primary antibodies were against: phospho-S6 ribosomal protein (Ser240/244; Cell Signaling #2215; 1:1000), S6 ribosomal protein (Cell Signaling #2217; 1:1000), phospho-GSK-3α/β (Ser21/9; Cell Signaling #9331; 1:1000) and β-actin as a loading control (Sigma-Aldrich #A5441; 1:2000). Secondary antibodies include IRDye® 800CW goat anti-mouse IgG (Li-COR #926-32210; 1:10,000) and IRDye® 680RD goat anti-rabbit IgG (Li-COR #926-68071; 1:10,000). Fluorescent-tagged secondary antibodies were visualized with an Odyssey fluorescence scanner and digitized band densities quantitated using ImageJ (version 1.46).
Supplementary Material
Acknowledgments
We thank Drs Lilliana Solnica-Krezel and Steve Hann for encouragement and helpful discussions.
Footnotes
COMPETING INTERESTS
The authors declare that they do not have any competing or financial interests.
AUTHOR CONTRIBUTIONS
S.-H.K. developed the concept, performed experiments, and wrote and edited the manuscript; M.L.K. and L.R.B. performed experiments; R.P.C. performed experiments and edited the manuscript; and K.C.E. developed the concept, and wrote and edited the manuscript.
FUNDING
This work was supported by the Department of Defense, USA (DOD) grant W81XWH-10-1-0854 to K.C.E.
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.011494/-/DC1
REFERENCES
- Altomare D. A., Wang H. Q., Skele K. L., De Rienzo A., Klein-Szanto A. J., Godwin A. K., Testa J. R. (2004). AKT and mTOR phosphorylation is frequently detected in ovarian cancer and can be targeted to disrupt ovarian tumor cell growth. Oncogene 23, 5853–5857 [DOI] [PubMed] [Google Scholar]
- Berghmans S., Murphey R. D., Wienholds E., Neuberg D., Kutok J. L., Fletcher C. D., Morris J. P., Liu T. X., Schulte-Merker S., Kanki J. P., et al. (2005). tp53 mutant zebrafish develop malignant peripheral nerve sheath tumors. Proc. Natl. Acad. Sci. USA 102, 407–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonneau D., Longy M. (2000). Mutations of the human PTEN gene. Hum. Mutat. 16, 109–122 [DOI] [PubMed] [Google Scholar]
- Choo A. Y., Kim S. G., Vander Heiden M. G., Mahoney S. J., Vu H., Yoon S. O., Cantley L. C., Blenis J. (2010). Glucose addiction of TSC null cells is caused by failed mTORC1-dependent balancing of metabolic demand with supply. Mol. Cell 38, 487–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crino P. B., Nathanson K. L., Henske E. P. (2006). The tuberous sclerosis complex. N. Engl. J. Med. 355, 1345–1356 [DOI] [PubMed] [Google Scholar]
- Davies D. M., Johnson S. R., Tattersfield A. E., Kingswood J. C., Cox J. A., McCartney D. L., Doyle T., Elmslie F., Saggar A., de Vries P. J., et al. (2008). Sirolimus therapy in tuberous sclerosis or sporadic lymphangioleiomyomatosis. N. Engl. J. Med. 358, 200–203 [DOI] [PubMed] [Google Scholar]
- Edlund K., Larsson O., Ameur A., Bunikis I., Gyllensten U., Leroy B., Sundström M., Micke P., Botling J., Soussi T. (2012). Data-driven unbiased curation of the TP53 tumor suppressor gene mutation database and validation by ultradeep sequencing of human tumors. Proc. Natl. Acad. Sci. USA 109, 9551–9556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feitsma H., Cuppen E. (2008). Zebrafish as a cancer model. Mol. Cancer Res. 6, 685–694 [DOI] [PubMed] [Google Scholar]
- Feng Z., Zhang H., Levine A. J., Jin S. (2005). The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 102, 8204–8209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z., Hu W., de Stanchina E., Teresky A. K., Jin S., Lowe S., Levine A. J. (2007). The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 67, 3043–3053 [DOI] [PubMed] [Google Scholar]
- Finlay C. A., Hinds P. W., Levine A. J. (1989). The p53 proto-oncogene can act as a suppressor of transformation. Cell 57, 1083–1093 [DOI] [PubMed] [Google Scholar]
- Franz D. N., Leonard J., Tudor C., Chuck G., Care M., Sethuraman G., Dinopoulos A., Thomas G., Crone K. R. (2006). Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann. Neurol. 59, 490–498 [DOI] [PubMed] [Google Scholar]
- Gad S., Lefèvre S. H., Khoo S. K., Giraud S., Vieillefond A., Vasiliu V., Ferlicot S., Molinié V., Denoux Y., Thiounn N., et al. (2007). Mutations in BHD and TP53 genes, but not in HNF1beta gene, in a large series of sporadic chromophobe renal cell carcinoma. Br. J. Cancer 96, 336–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goessling W., North T. E., Zon L. I. (2007). New waves of discovery: modeling cancer in zebrafish. J. Clin. Oncol. 25, 2473–2479 [DOI] [PubMed] [Google Scholar]
- Habib S. L., Yadav A., Mahimainathan L., Valente A. J. (2011). Regulation of PI 3-K, PTEN, p53, and mTOR in malignant and benign tumors deficient in tuberin. Genes Cancer 2, 1051–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington L. S., Findlay G. M., Gray A., Tolkacheva T., Wigfield S., Rebholz H., Barnett J., Leslie N. R., Cheng S., Shepherd P. R., et al. (2004). The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J. Cell Biol. 166, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X., Pandolfi P. P., Li Y., Koutcher J. A., Rosenblum M., Holland E. C. (2005). mTOR promotes survival and astrocytic characteristics induced by Pten/AKT signaling in glioblastoma. Neoplasia 7, 356–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K., Li Y., Zhu T., Wu J., Guan K. L. (2002). TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 4, 648–657 [DOI] [PubMed] [Google Scholar]
- Inoki K., Li Y., Xu T., Guan K. L. (2003). Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 17, 1829–1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K., Corradetti M. N., Guan K. L. (2005). Dysregulation of the TSC-mTOR pathway in human disease. Nat. Genet. 37, 19–24 [DOI] [PubMed] [Google Scholar]
- Jacinto E., Loewith R., Schmidt A., Lin S., Rüegg M. A., Hall A., Hall M. N. (2004). Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 6, 1122–1128 [DOI] [PubMed] [Google Scholar]
- Kim D. D., Kleinman D. M., Kanetaka T., Gerritsen M. E., Nivaggioli T., Weber D., Durán W. N. (2010). Rapamycin inhibits VEGF-induced microvascular hyperpermeability in vivo. Microcirculation 17, 128–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. H., Speirs C. K., Solnica-Krezel L., Ess K. C. (2011). Zebrafish model of tuberous sclerosis complex reveals cell-autonomous and non-cell-autonomous functions of mutant tuberin. Dis. Model. Mech. 4, 255–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai K. P., Leong W. F., Chau J. F., Jia D., Zeng L., Liu H., He L., Hao A., Zhang H., Meek D., et al. (2010). S6K1 is a multifaceted regulator of Mdm2 that connects nutrient status and DNA damage response. EMBO J. 29, 2994–3006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson N. D., Weinstein B. M. (2002). In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev. Biol. 248, 307–318 [DOI] [PubMed] [Google Scholar]
- Lee C. H., Inoki K., Karbowniczek M., Petroulakis E., Sonenberg N., Henske E. P., Guan K. L. (2007). Constitutive mTOR activation in TSC mutants sensitizes cells to energy starvation and genomic damage via p53. EMBO J. 26, 4812–4823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lendvay T. S., Broecker B., Smith E. A. (2002). Renal cell carcinoma in a 2-year-old child with tuberous sclerosis. J. Urol. 168, 1131–1132 [DOI] [PubMed] [Google Scholar]
- Levine A. J. (1997). p53, the cellular gatekeeper for growth and division. Cell 88, 323–331 [DOI] [PubMed] [Google Scholar]
- Levine A. J., Feng Z., Mak T. W., You H., Jin S. (2006). Coordination and communication between the p53 and IGF-1-AKT-TOR signal transduction pathways. Genes Dev. 20, 267–275 [DOI] [PubMed] [Google Scholar]
- Maddocks O. D., Vousden K. H. (2011). Metabolic regulation by p53. J. Mol. Med. (Berl.) 89, 237–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahimainathan L., Ghosh-Choudhury N., Venkatesan B., Das F., Mandal C. C., Dey N., Habib S. L., Kasinath B. S., Abboud H. E., Ghosh Choudhury G. (2009). TSC2 deficiency increases PTEN via HIF1alpha. J. Biol. Chem. 284, 27790–27798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning B. D., Tee A. R., Logsdon M. N., Blenis J., Cantley L. C. (2002). Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol. Cell 10, 151–162 [DOI] [PubMed] [Google Scholar]
- Manning B. D., Logsdon M. N., Lipovsky A. I., Abbott D., Kwiatkowski D. J., Cantley L. C. (2005). Feedback inhibition of Akt signaling limits the growth of tumors lacking Tsc2. Genes Dev. 19, 1773–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehenni H., Lin-Marq N., Buchet-Poyau K., Reymond A., Collart M. A., Picard D., Antonarakis S. E. (2005). LKB1 interacts with and phosphorylates PTEN: a functional link between two proteins involved in cancer predisposing syndromes. Hum. Mol. Genet. 14, 2209–2219 [DOI] [PubMed] [Google Scholar]
- Memmott R. M., Dennis P. A. (2010). The role of the Akt/mTOR pathway in tobacco carcinogen-induced lung tumorigenesis. Clin. Cancer Res. 16, 4–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon S., Manning B. D. (2008). Common corruption of the mTOR signaling network in human tumors. Oncogene 27 Suppl. 2, S43–S51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mione M. C., Trede N. S. (2010). The zebrafish as a model for cancer. Dis. Model. Mech. 3, 517–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noon A. P., Vlatković N., Polański R., Maguire M., Shawki H., Parsons K., Boyd M. T. (2010). p53 and MDM2 in renal cell carcinoma: biomarkers for disease progression and future therapeutic targets? Cancer 116, 780–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parant J. M., George S. A., Holden J. A., Yost H. J. (2010). Genetic modeling of Li-Fraumeni syndrome in zebrafish. Dis. Model. Mech. 3, 45–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley T., Sontag E., Chen P., Levine A. (2008). Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 9, 402–412 [DOI] [PubMed] [Google Scholar]
- Sarbassov D. D., Guertin D. A., Ali S. M., Sabatini D. M. (2005). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 [DOI] [PubMed] [Google Scholar]
- Shah O. J., Wang Z., Hunter T. (2004). Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr. Biol. 14, 1650–1656 [DOI] [PubMed] [Google Scholar]
- Stambolic V., MacPherson D., Sas D., Lin Y., Snow B., Jang Y., Benchimol S., Mak T. W. (2001). Regulation of PTEN transcription by p53. Mol. Cell 8, 317–325 [DOI] [PubMed] [Google Scholar]
- Tee A. R., Fingar D. C., Manning B. D., Kwiatkowski D. J., Cantley L. C., Blenis J. (2002). Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc. Natl. Acad. Sci. USA 99, 13571–13576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toschi A., Lee E., Gadir N., Ohh M., Foster D. A. (2008). Differential dependence of hypoxia-inducible factors 1 alpha and 2 alpha on mTORC1 and mTORC2. J. Biol. Chem. 283, 34495–34499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veprintsev D. B., Fersht A. R. (2008). Algorithm for prediction of tumour suppressor p53 affinity for binding sites in DNA. Nucleic Acids Res. 36, 1589–1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B., Lane D., Levine A. J. (2000). Surfing the p53 network. Nature 408, 307–310 [DOI] [PubMed] [Google Scholar]
- Vousden K. H., Prives C. (2009). Blinded by the light: the growing complexity of p53. Cell 137, 413–431 [DOI] [PubMed] [Google Scholar]
- Zhang H., Cicchetti G., Onda H., Koon H. B., Asrican K., Bajraszewski N., Vazquez F., Carpenter C. L., Kwiatkowski D. J. (2003). Loss of Tsc1/Tsc2 activates mTOR and disrupts PI3K-Akt signaling through downregulation of PDGFR. J. Clin. Invest. 112, 1223–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G., Hoersch S., Amsterdam A., Whittaker C. A., Lees J. A., Hopkins N. (2010). Highly aneuploid zebrafish malignant peripheral nerve sheath tumors have genetic alterations similar to human cancers. Proc. Natl. Acad. Sci. USA 107, 16940–16945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zon L. I., Peterson R. T. (2005). In vivo drug discovery in the zebrafish. Nat. Rev. Drug Discov. 4, 35–44 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.