

Abstract
The most common form of congenital adrenal hyperplasia is steroid 21-hydroxylase deficiency (21OHD). When the nonclassical (mild) form is included, 21OHD is the most common genetic disease in human beings. With the advent of pharmaceutical preparation of glucocorticoids starting in the 1960s and newborn screening starting in the 1990s, the majority of children with 21OHD are reaching adulthood, which has yielded a cohort of patients with, in essence, a new disease. Only recently have some data emerged from cohorts of adults with 21OHD, and in some centers, experience with the management of these patients is growing. These patients suffer from poor health, infertility, characteristic tumors in the adrenal glands and gonads, and consequences of chronic glucocorticoid therapy. Their care is fragmented and inconsistent, and many stop taking their medications out of frustration. Internal medicine residents and endocrinology fellows receive little training in their care, which further discourages their seeking medical attention. Adults with 21OHD have a different physiology from patients with Addison's disease or other androgen excess states, and their needs are different than those of young children with 21OHD. Consequently, their care requires unorthodox treatment and monitoring strategies foreign to most endocrine practitioners. Our goal for this article is to review their physiology, complications, and needs in order to develop rational and effective treatment and monitoring strategies.
Accreditation and Credit Designation Statements.
The Endocrine Society is accredited by the Accreditation Council for Continuing Medical Education to provide continuing medical education for physicians. The Endocrine Society has achieved Accreditation with Commendation.
The Endocrine Society designates this JCEM Journal-based CME activity for a maximum of 1 AMA PRA Category 1 Credits™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
Learning Objectives
Upon completion of this educational activity, participants should be able to:
interpret laboratory data for men and women with CAH and determine if their control is at goal.
explain the molecular genetics of classical and nonclassical CAH and provide genetic counseling to these patients and family members.
adjust therapy regimens for men and women with CAH appropriate for their treatment goals.
recognize when treatments other than glucocorticoids are appropriate.
Disclosure Policy
Authors, editors, and Endocrine Society staff involved in planning this JCEM Journal-based CME activity are required to disclose to The Endocrine Society and to learners any relevant financial relationship(s) of the individual or spouse/partner that have occurred within the last 12 months with any commercial interest(s) whose products or services are discussed in the CME content. The Endocrine Society has reviewed all disclosures and resolved all identified conflicts of interest.
The following author reported no relevant financial relationships:
Wiebke Arlt, M.D., has no relevant financial relationships.
The following author reported relevant financial relationships:
Richard J. Auchus, M.D., Ph.D., provided contracted research for Janssen Pharmaceuticals.
The following JCEM Editors reported relevant financial relationships:
The Editor-in-Chief, Leonard Wartofsky, M.D., is a Consultant for Asurogen, Genzyme, and IBSA, and is on the Speaker's Bureau for Genzyme. Kenneth Burman, M.D., is a Consultant for Medscape and UpToDate; a Reviewer for the Endocrine Fellows Foundation; and has received Institutional Grants for Research from Amgen, Eisei, and Pfizer. Samuel Dagogo-Jack, M.D., is a Consultant for Merck and Novo Nordisk; a Grantee for the American Diabetes Association, AstraZeneca, Boehringer Ingelheim, National Institutes of Health, and Novo Nordisk; and a Grant Reviewer for the American Diabetes Association and National Institutes of Health. Silvio Inzucchi, M.D., is a Consultant/Advisor for Boehringer Ingelheim, Genentech, Janssen, Merck, and Takeda; has DSMB Activity with Amgen, Esai, and Gilead; and receives CME support from Abbott, Amylin, Boeringher-Ingelheim, Merck, and Takeda. Kieren Mather, M.D., received an Investigator-initiated Grant from Novo Nordisk. Lynnette Nieman, M.D., is an Author/Editor for UpToDate, and receives Research Support from HRA-Pharmaceutical.
The following JCEM Editors reported no relevant financial relationships: Paolo Beck-Peccoz, M.D.; David Ehrmann, M.D.; David Handelsman, Ph.D.; Michael Kleerekoper, M.D.; Merrily Poth, M.D.; Constantine Stratakis, M.D.
Endocrine Society staff associated with the development of content for this activity reported no relevant financial relationships.
Acknowledgement of Commercial Support
JCEM Journal-based CME activities are not supported by grants, other funds, or in-kind contributions from commercial supporters.
Instructions
The estimated time to complete each JCEM Journal-based CME activity, including review of material, is 1 hour. Instructions for completing this activity can be found at https://www.endocrine.org/education-and-practice-management/continuing-medical-education/journal-cme.
If you have questions about this JCEM Journal-based CME activity, please direct them to education@endocrine.org.
Activity release date: July 2013
Activity expiration date: July 2015
Case Presentation
J.W. is a 29-year-old woman born with genital ambiguity due to classic 21-hydroxylase deficiency (21OHD). She collapsed while watching a sporting event on a hot, humid day and was rushed by ambulance to the emergency department; she was poorly responsive, with a systolic blood pressure of 85 mm Hg, heart rate of 135 beats/min, and temperature of 37°C. She was a short, overweight woman with excessive facial and body hair, frontal balding, and small hands and feet. After receiving 2 L of iv normal saline, she awoke and told a nurse that she had “Addison's disease.” She was given an iv bolus of 100 mg hydrocortisone sodium succinate; 1 hour later, her blood pressure normalized, and she felt markedly better. On further questioning, she gave the staff the name of a pediatric endocrinologist she had seen until age 25, who prescribed hydrocortisone 3 times daily and fludrocortisone acetate. Her care had then been transferred to an internal medicine endocrinologist who 4 years earlier had prescribed prednisone, 10 mg twice daily, and 9α-fludrocortisone acetate (9FCA), 0.1 mg/d. She had neither taken these medications nor returned for follow-up because, she said, “he wanted me to take too much medicine.” The staff called the office of her former pediatric endocrinologist, who explained that the patient has classic 21OHD. Laboratory values obtained the following morning are shown in Table 1. The patient agreed to resume adrenal replacement therapy and was scheduled to see a new endocrinologist 3 weeks later.
Table 1.
Laboratory Data for Patient J.W.
| Analyte | Value |
|---|---|
| Serum potassium | 5.5 mEq/L |
| Plasma renin activity | 48 ng/mL/h = 13.3 ng/L/s |
| P4 | 26 ng/mL = 83 nmol/L |
| 17OHP | 32 800 ng/dL = 991 nmol/L |
| AD | 1660 ng/dL = 58 nmol/L |
| T | 265 ng/dL = 9.2 nmol/L |
| DHEAS | 65 μg/dL = 1.8 μmol/L |
Background
Nomenclature and altered steroidogenesis
The variants of congenital adrenal hyperplasia (CAH) are genetic diseases in which cortisol production is impaired by mutations in the genes encoding 1 of the enzymes or cofactor proteins required for cortisol biosynthesis (Table 2). Adrenocortical hyperplasia occurs due to loss of cortisol negative feedback, thus increasing ACTH secretion, which is both tropic and trophic for the adrenal cortex. Each form of CAH has its unique clinical and biochemical features, which derive from the lack of cortisol but sometimes also from excess and/or deficiency of mineralocorticoids and androgens. In addition, the cortisol precursor steroids accumulating above the enzymatic block might have biological activities of their own or follow other “overflow” pathways to alternative steroid products (Figure 1).
Table 2.
Forms of CAH
| Gene Defect | High Steroids | Low Steroids |
|---|---|---|
| CYP21A2 | P4, 17OHP, 21dF, 19C | Aldo, DOC, B, S, F |
| CYP17A1 | P4, DOC, B | Aldoa, S, F, 19C |
| CYP11B1 | P4, DOC, S, 19C | Aldoa, F |
| HSD3B2 | Delta-5>>P4, 17OHP, AD | S, F, other Delta-4b |
| POR | P4>>DOC, B, 17OHP | F, 19C |
| StAR, CYP11A1 | None | All |
Abbreviations: 21dF, 21-deoxycortisol; 19C, 19-carbon steroids; Aldo, aldosterone; DOC, 11-deoxycorticosterone; B, corticosterone; S, 11-deoxycortisol; F, cortisol; Delta-5, 3-hydroxy-5-ene steroids; Delta-4, 3-keto-4-ene steroids.
Aldo low due to suppressed plasma renin activity, secondary to high DOC.
Complex patterns; for example, T is high in females and low in males.
Figure 1.
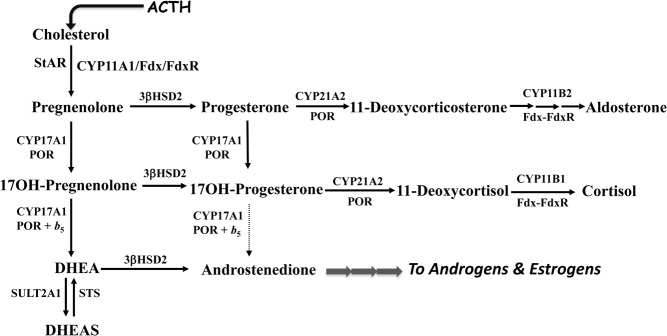
Adrenal steroid biosynthesis pathways and enzymatic defects causing CAH. The enzymes and proteins required for normal cortisol synthesis are shown: StAR, the steroid acute regulatory protein; CYP11A1, cholesterol side chain cleavage enzyme or P450scc; CYP17A1, steroid 17-hydroxylase/17,20-lyase or P450c17; 3βHSD2, 3β-hydroxysteroid dehydrogenase/isomerase type 2; POR, P450-oxidoreductase; and CYP21A2, steroid 21-hydroxylase or P450c21. Also shown are Fdx and FdxR, ferredoxin and ferredoxin reductase, respectively, which are the electron transfer proteins for the mitochondrial cytochrome P450 enzymes in the CYP11 family; SULT2A1, steroid sulfotransferase type 2A1 or DHEA sulfotransferase; and STS, steroid sulfatase. The 19-carbon androgen precursors DHEA and DHEAS are metabolized to active androgens and estrogens via other enzymes in the adrenal and peripheral tissues. Dotted line from 17OHP to AD indicates poor conversion by human CYP17A1, even in the presence of cytochrome b5 (b5), compared to conversion of 17OH-pregnenolone (17-hydroxypregnenolone) to DHEA.
The most common form of CAH, found in 1:16 000 newborns, is 21OHD (1). Traditionally, 21OHD is defined as “classic” if the defect is severe enough to cause cortisol insufficiency or “nonclassic” if cortisol production is normal but with excessive precursor accumulation to overcome the partial enzymatic block (Figure 2), yielding the characteristic elevation of 17-hydroxyprogesterone (17OHP) and more specifically 21-deoxycortisol, which is only generated in large amounts in the absence of substantial CYP21A2 function. Classic 21OHD has been dichotomously grouped as “salt wasting” and “simple virilizing,” depending on whether or not the newborn child spontaneously develops hypotensive crises without provocation, which reflects severe aldosterone deficiency. For adults with 21OHD, this subtyping of classic 21OHD is misleading and should be avoided (Table 3). Whereas virtually all patients with classic 21OHD are ascertained as children, primarily in the first year of life and now often by newborn screening (2), nonclassic 21OHD is diagnosed occasionally in childhood or at newborn screening, most commonly in adolescence and young adulthood, and sometimes in adulthood. Furthermore, many patients with nonclassic 21OHD are never diagnosed, and those with cryptic disease rarely require treatment. The focus of this article is adults with classic 21OHD, and we will include a limited discussion of nonclassic 21OHD. It is important to recognize that the distinction between classic and nonclassic 21OHD is somewhat arbitrary and that in reality a continuous spectrum exists between severe and mild disease, based on the severity of the underlying CYP21A2 mutations (3).
Figure 2.
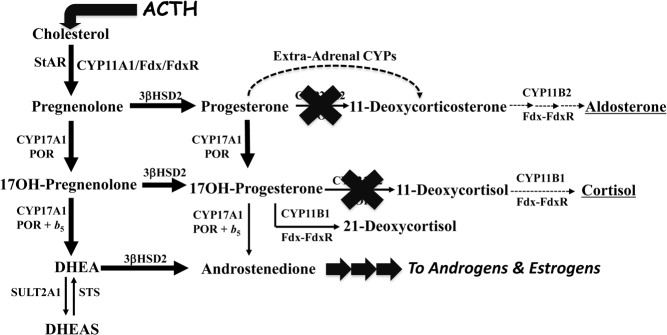
Altered steroidogenesis in 21OHD. The block at CYP21A2 (bold X) and raised ACTH cause 21-deoxysteroids to accumulate and to undergo conversion to androgens and estrogens (darkened lines), as well as significant amounts of 21-deoxycortisol due to conversion of 17OHP by CYP11B1, a reaction that is insignificant in the presence of a functional CYP21A2 enzyme. Some 11-deoxycorticosterone is formed by peripheral conversion of P4 by hepatic cytochrome P450 (CYP) enzymes.
Table 3.
Suggested Working Classification of 21OHD in Adults
| Classic 21OHD |
| Nonclassic 21OHD |
| With normal cortisol production |
| With partial cortisol deficiency |
The manifestations of 21OHD derive not only from cortisol and aldosterone deficiency, but also from androgen excess due to shunting of accumulating precursors to 19-carbon steroids. Glucocorticoid and mineralocorticoid replacement is therefore not only administered to restore fluid and electrolyte balance but also to reduce ACTH production, thereby attenuating the adrenal 19-carbon steroid synthesis. In children, adherence to a carefully adjusted treatment regimen optimizes growth and sexual maturation, although sexual precocity and short stature are still common (4).
The changing needs and concerns in adults with 21OHD
Upon completion of pubertal development and attainment of adult height, the auxological and clinical parameters used to guide therapy are no longer germane, but the need for lifelong treatment persists. With adolescence, the patient must gradually assume primary responsibility for his/her care and eventually stop seeing the pediatric endocrinologist. The needs and concerns shift as well to include family planning and long-term health quality. Patients with 21OHD might be at increased risk for other health problems, due both to their underlying disease and to their treatments. Lifelong glucocorticoid therapy might predispose patients with 21OHD to glucose intolerance, bone loss, and cardiovascular disease, and this risk is likely to be higher than for patients with autoimmune adrenal insufficiency, who do not require glucocorticoid dosing to control of androgen excess.
Some data have emerged over the last decade from cohorts of adults with 21OHD (5–7), although no long-term studies including significant numbers of patients with other forms of CAH have appeared. In contrast to the vulnerability to hypotension as children, the prevalence of hypertension appears to be increased in adolescents (8) and adults (6, 7) with 21OHD. Several studies have shown reduced bone mineral density in adults with 21OHD (9, 10). Although osteopenia is common, osteoporosis is uncommon (5, 7), possibly because the detrimental influence of glucocorticoids is in part mitigated by increased exposure to androgens and estrogens. Adults with 21OHD tend to be short and are often obese, which might predispose to the metabolic syndrome and adverse cardiovascular risk (11). Some studies confirm an increased prevalence of glucose intolerance, obesity, and dyslipidemia in adults with 21OHD (6), as well as gestational diabetes in over 20% of pregnant women with 21OHD. In 1 study, bone mass correlated inversely with the cumulative dose of glucocorticoid (12). Consequently, a major priority when managing adults with CAH is to minimize long-term consequences of both the disease itself and its treatment.
Tumors in adults with 21OHD
The chronic enlargement of the adrenal glands in 21OHD is associated with increased prevalence of adrenal tumors, including massive myelolipomas (13–15).
About 30–50% of men with 21OHD develop testicular adrenal rest tumors (TARTs) (7, 16), and the prevalence increases when control is poor, particularly for men with the most severe deficiency. TARTs are thought to arise from cells of adrenal origin, which travel with the primordial gonad after the adrenal and gonadal cells separate within the urogenital ridge during fetal development. Women with 21OHD also develop adrenal rests in or near the ovaries, particularly after bilateral adrenalectomy (17, 18).
The term “adrenal rest tumor” should not be construed as a neoplasm because these cells are hyperplastic and do not show increased proliferation. TARTs originate in the rete testis (19) and are thought to expand after chronic ACTH stimulation, just like the adrenal cortex itself. As a result of this expansion, the testis suffers from compression, functional impairment, and eventually fibrosis. It is therefore important to avoid confusing TARTs with primary testicular tumors when discussing a case with the patient and colleagues from other disciplines. In the United Kingdom Congenital Adrenal Hyperplasia Adult Study Executive cohort, 4 of 65 adult men with CAH had previously undergone unilateral orchidectomy for presumed testicular cancer or Leydig cell tumor, with histology findings consistent with TARTs (5).
Subfertility in adults with 21OHD
Men and women with 21OHD suffer from subfertility, and although attention has focused on women and androgen excess, the men are probably more commonly and severely affected (16, 20, 21). Although the adrenal androgen excess does not cause contrasexual changes in men, gonadotropin suppression occurs and causes testicular atrophy and infertility (22). A young adult male with classic 21OHD in poor control might appear healthy and normally virilized, but if testicular atrophy and suppressed gonadotropins are found, this man might still be paradoxically prepubertal due to adrenal-derived androgen excess. Male subfertility can also occur as the consequence of TART expansion (16, 20, 21). Women with both classic and nonclassic 21OHD often suffer from infertility, even when having regular monthly menses, for reasons discussed below.
Transition of care
Transition of care and coordination of services among providers for adults with 21OHD remains a major problem, particularly for those who require psychological attention due to impaired body image and issues of sexuality consequent to the manifestations of disordered sex development and associated surgeries. Many patients reach adulthood not understanding why they require chronic adrenal replacement therapy, and often these patients stop all treatment for years, similar to J.W. in the case presentation above (Table 4). In one clinic, less than half of the referred young adults with 21OHD attended 2 or more appointments with the adult endocrinologist (23). Miraculously, most of these patients appear relatively well and nicely tanned, with the exception of salt craving and heat intolerance, until they become seriously ill and suffer an adrenal crisis (24). Unskilled providers might reinforce their nonadherence to medication by erroneously concluding that patients who stop therapy without spontaneous crisis have been misdiagnosed and are not vulnerable to adrenal crisis. Herein lies one of the paradoxes of classic 21OHD: unlike children with the same disease or adults with autoimmune Addison's disease, many adults with 21OHD maintain relatively normal blood pressure and functional capacity without treatment. This observation suggests that either other adrenal-derived steroids substitute for aldosterone and/or cortisol, or that a physiological compensation for low corticosteroid activity at the target organs has occurred over time. It is important to reassess mineralocorticoid requirements at the point of transition because this need might differ significantly between the adult and childhood phases. Efforts to improve transitions and coordination of care have begun (25, 26) but are not widely implemented.
Table 4.
Transition of Care Issues
| Factors associated with discontinuing adrenal replacement therapy |
| Poor understanding of disease |
| Less vulnerable to crises than patients with Addison's disease |
| Limited training of practitioners in adult care for 21OHD |
| Impediments to good health |
| Need for multidisciplinary, coordinated care |
| Chronic consequences of disease and treatments |
| Limited training of practitioners in adult care for 21OHD |
In addition, adults with 21OHD require cancer screening, immunizations, and general health maintenance like any other adult. Common symptoms and conditions, such as fatigue and obesity, might be incorrectly attributed to their adrenal disease or their treatment, either by the physician or the patient. Long-term consequences of vaginal reconstruction surgery in women with 21OHD include vaginal strictures and fistulas as well as frequent urinary tract infections (27). Consequently, the approach to the adult with 21OHD requires knowledge of the patient's prior treatment, the natural history of the disease, and the influence of various treatments on adrenal and gonadal steroid flux. Treatment decisions should be based on the needs and desires of the patient, with internally consistent data from careful physical examination and appropriate laboratory evaluation.
Assessment
Diagnosis, genetics, and nonclassic 21OHD
Although most adults with classic 21OHD will already have received a diagnosis, many women with nonclassic 21OHD are not diagnosed until they are evaluated for androgen excess and irregular menses and/or subfertility. The diagnosis of nonclassic 21OHD is based on a follicular-phase 17OHP >30 nmol/L (>1000 ng/dL) after cosyntropin stimulation (28). Unlike the patient with classic 21OHD, adrenal replacement is not required for normal health, so treatment is entirely goal-oriented for the untoward manifestations of androgen excess. Glucocorticoid therapy should be minimized to accomplish the goal for the requisite period of time, such as restoring fertility, and 1 retrospective series found better outcomes when hydrocortisone is continued until delivery (29). Alternative means of controlling androgen excess and menstrual irregularity, including antiandrogens and oral contraceptives, should be considered for long-term management during times when fertility is not desired. The diagnosis and treatment of nonclassic CAH has been reviewed in detail elsewhere (30, 31).
A few women, typically compound heterozygotes for P30L and null alleles, have marked androgen excess but not severe enough to virilize, and these patients straddle the classic/nonclassic distinction. Genotype-phenotype analysis has revealed a strong correlation between genotype and the presence and severity of glucocorticoid and mineralocorticoid deficiency; however, the correlation with the severity of virilization at birth in girls with 46,XX disordered sex development is not as strong (for review, see Ref. 32). A recent cohort study in 153 patients found that impaired health status in adults with CAH did not correlate with genotype but was mostly acquired as a consequence of glucocorticoid treatment (33). No prospective studies are available either to support or to refute the utility of genotype as a guide to treatment. Genetic counseling, however, should be offered to all affected individuals and their family members, and genotyping is useful in many cases, particularly when the history and laboratory data do not distinguish classic from nonclassic 21OHD or when the patient presents with limited past medical record and chronic overtreatment, as well as in all those seeking fertility, including patients with nonclassic 21OHD. In 1 study, 70% of patients with nonclassic 21OHD were compound heterozygotes and carriers for classic 21OHD (34). Because the prevalence of nonclassic 21OHD is at least 1:1000 (35), high prevalence of carrier status for classic 21OHD suggests that the less severely affected homozygotes infrequently come to medical attention. Among parents of children with classic 21OHD, 4% are affected with cryptic or undiagnosed nonclassic 21OHD (36).
The initial visit
Before the initial visit, it is important to review available records for the diagnosis and treatment. Questions such as, “At what age were you diagnosed?,” “When did you stop growing?,” and “Did you have any genital surgeries?” are helpful if records are incomplete. It is likewise critical to gauge how much the patient understands about the disease and to establish the goals, concerns, and limitations to compliance with therapy. The medical history should include lapses in medication (intentional or otherwise), recent episodes of adrenal crisis and/or increased glucocorticoids, reasons for switching from hydrocortisone to longer-acting glucocorticoids if relevant, and training in emergency hydrocortisone injection. The evaluation should include a review of any adrenal or gonadal imaging studies and a brief sexual history with open-ended questions. Men should be asked if they perform testicular exams, and women should be asked about menses and use of depilation methods.
Physical examination
Physical examination should carefully assess signs of iatrogenic Cushing syndrome, including disproportionate supraclavicular and dorsocervical fat pads, facial and upper neck plethora, facial rounding, muscle weakness, violaceous striae, bruising, and skin thinning, which is perhaps the most sensitive finding as in patients treated chronically with dexamethasone. In addition to height and weight, blood pressure and heart rate should be obtained in both the seated and standing positions. Men should have a testicular examination to assess atrophy and presence of TARTs, although most TARTs will only be identified with ultrasound imaging. Women require an evaluation of androgen-dependent body hair growth and acne in the context of depilation treatments, whereas only a brief external genital examination is necessary initially. Gynecological or urological consultation regarding the current genital anatomy should be considered.
Laboratory testing
If recent comprehensive laboratory test results are not available, a good baseline assessment is appropriate for the first visit. Perhaps the greatest misconception among providers for adults with 21OHD concerns the importance of serum 17OHP as a guide to therapy. Although 17OHP is used for diagnosis of all types of 21OHD, 17OHP should not be the sole or dominant parameter used for guiding the management of 21OHD in the adult. Although 17OHP and androgens tend to correlate, 17OHP is typically 100 to 1000 times higher and much more variable than androgens. As a result, androgens can be normal or slightly high despite markedly elevated 17OHP. Consequently, suppressing 17OHP into the normal range invariably results in overtreatment and should not be a treatment goal. Medication need not be interrupted for laboratory testing if results are interpreted in the context of medication schedule. Withholding the morning dose of glucocorticoid before phlebotomy elevates precursors and androgens to their maximal “worst case” values but might underestimate average control on current treatment. When control is poor, androgens will be high before and after each dose; when control is good, the differences will be slight. Most importantly, laboratory data are always secondary to clinical judgment in making treatment decisions.
A complete laboratory panel is shown in Table 5, paired with the information gained from each analyte. For unknown reasons, dehydroepiandrosterone (DHEA) sulfate (DHEAS), the dominant ACTH-dependent 19-carbon steroid product of the adrenal, is easily suppressed by almost any glucocorticoid therapy (37) and cannot be used to gauge control. DHEAS is often paradoxically low or low-normal even in 21OHD patients under poor control; consequently, poor compliance or low drug exposure should be considered if the DHEAS is not suppressed. Electrolytes and plasma renin activity or concentration, in conjunction with orthostatic blood pressures, assess volume status, and thus the need for and adequacy of fludrocortisone therapy.
Table 5.
Laboratory monitoring for the adult with CAH due to classic 21OHD
| Analyte | Physiology | Goals & Comments |
|---|---|---|
| Men and Women | ||
| Plasma renin | Volume status | Low to normal unless hypertension; spironolactone raises renin, hydrocortisone lowers renin |
| Potassium | Mineralocorticoid sufficiency | Should be normal |
| Sodium | Mineralocorticoid sufficiency | Should be normal |
| Testosterone | Total androgen production | Combined adrenal & gonadal |
| Androstenedione | Reflects adrenal androgen excess | Assess with testosterone |
| SHBG | Testosterone binding protein | Raised by estrogen therapy |
| DHEAS | Major adrenal 19-carbon steroid | Should be low or suppressed |
| 17OHP | Highly variable | Should not be low or normal |
| Men | ||
| Gonadotropins | Gonadal axis integrity | Low if adrenal androgen excess |
| Semen analysis | Fertility | Normal result means appropriate control |
| Women | ||
| Progesterone | Adrenal and corpus luteum | Normalize for fertility (<2 nmol/L) during follicular phase |
In women, total T and SHBG are used to determine free and bioavailable T by calculation (38, 39), and these values are the most important parameters to gauge androgen excess. Because women with 21OHD often develop a secondary polycystic ovary syndrome (40), a high T does not necessarily mean poor control. Androstenedione (AD) and the AD/T ratio help to ascertain how much T derives from the adrenal vs the ovary. In normal females, AD originates from both the adrenals and the ovaries, but in patients with 21OHD, most AD is usually of adrenal origin. As a rule of thumb, the AD/T ratio in regularly cycling women is <2, but in hyperthecosis or with T-producing tumors, the ratio is <1. If the AD/T ratio is >4 in a woman with 21OHD, most of the T is of adrenal origin. In women with 21OHD who are infertile or who have chronic amenorrhea, progesterone (P4), which accumulates upstream of 17OHP in the adrenal, should be assessed as well. Excessive adrenal-derived P4 creates a functional chronic luteal phase, even despite ovulation, which impairs both fertility and menses. For women with 21OHD attempting to conceive, the single most important parameter is maintaining the P4 <2 nmol/L (<0.6 ng/mL) (41). The SHBG remains relatively stable for an individual, unless estrogen therapy is started or stopped, significant weight change occurs, or glucocorticoid therapy is markedly changed. In essence, it is of predominant importance to assess the androgen equilibrium, whereas 17OHP measurements are of lesser value—primarily to identify overtreatment, when 17OHP is normal or low.
In men with 21OHD, the same principles for evaluating volume status and androgen production apply, with some caveats to the latter. The total T and SHBG cannot be used to assess control without simultaneous AD and gonadotropins (LH, FSH). In men, the AD/T ratio is normally <0.2 because most of the T derives from the testis, in which AD is almost completely reduced to T by 17β-hydroxysteroid dehydrogenase type 3 (17βHSD3) (42, 43). The adrenals, ovary, and peripheral tissues lack 17βHSD3, and secondary enzymes such as 17βHSD5 (AKR1C3) and 17βHSD1 convert AD to T or DHEA to androst-5-ene-3β,17β-diol poorly in comparison. Consequently, an AD/T ratio >0.5 in a man with 21OHD indicates that a significant fraction of T is of adrenal origin, and if LH and FSH are suppressed with AD/T >1, most of the T is of adrenal origin. A normal semen analysis is the “gold standard” for normal androgen physiology in the male; however, the semen analysis will remain abnormal for months to years in the man with 21OHD and TARTs or testicular atrophy after an extended period of poor control (44) and might never return to normal despite good control (21). Testis-sparing surgery affords long-term control of TART growth but usually does not restore fertility (45), probably because the mass effect impairs blood flow to the spermatic tubules enough to cause severe damage (16). Consequently, an AD level greater than the T level in the presence of testicular masses in a man with 21OHD is a sign that current therapy is inadequate to maintain fertility.
Imaging
Most sources recommend annual testicular sonography in males with 21OHD to screen for TARTs, which is more sensitive than physical examination (46). Recommendations for adrenal imaging and for bone densitometry have not been established. As a rule of thumb, patients who have been consistently well-controlled since childhood do not need routine adrenal imaging, whereas imaging should be considered for patients with a long history of poor control, inconsistent therapy, or difficult-to-control disease. Analogously, dual-photon x-ray absorptiometry should be considered for patients taking chronic dexamethasone 0.5 mg/d or higher, having Cushingoid stigmata, or following a long period of suppressed 17OHP and androgens.
Therapy
Glucocorticoids
Adults with 21OHD receive a wide range of glucocorticoid treatment regimens, which does not correlate with CYP21A2 genotype or disease severity as children (7, 33, 47). This divergence derives from a lack of best practice models, the spectrum of practice patterns and biases of individual physicians, and limited training in the management of adults with 21OHD, even among otherwise very skilled endocrinologists. Another contribution derives from antecedent control and the amount of adrenal hyperplasia upon reaching adulthood; patients who have remained poorly controlled throughout life require higher glucocorticoid doses to manage subsequently throughout adulthood. Lastly, patient preference, related to frequency of dosing and tolerance of androgen or glucocorticoid excess, is likewise an important factor. Besides these practical matters, the variability in response to specific therapies among adults with 21OHD is astounding. Some adults with 21OHD are overtreated with 15 mg hydrocortisone daily, whereas others are grossly undertreated with 5–10 mg prednisolone twice daily. This “monogenic disease” with a clearly defined pathophysiology and biochemistry demonstrates the inherently polygenic nature of all diseases and the importance of individualizing therapy based on each patient's goals and responses.
As a general principle, there is no reason to indiscriminately abandon hydrocortisone therapy upon reaching adulthood. In fact, 3 or sometimes 2 divided doses of hydrocortisone daily provide good control in most adults with 21OHD who have been compliant with treatment throughout adolescence. The main obstacle to hydrocortisone is adherence to multiple daily doses, but most patients with Addison's disease take 2–3 daily hydrocortisone doses without difficulty. Cost is sometimes cited as a reason for using prednisone or dexamethasone in the United States, where a month's supply costs only a few dollars, whereas even generic hydrocortisone is 10 times more costly for 5- and 10-mg tablets. The generic hydrocortisone 20-mg scored pills, however, are only slightly more costly than dexamethasone.
If a patient is in good control during adolescence taking hydrocortisone and demonstrates the insight and commitment to remain strictly compliant, we encourage them to continue this regimen. Three doses of 5–10 mg with each meal is generally necessary yet rarely causes iatrogenic Cushing syndrome, weight gain, or sleep disturbance in most patients. Most patients starting in good control will remain in good control with this treatment, but others will not, mainly because hydrocortisone—even a large dose at bedtime—does not last long enough to blunt the rise of ACTH early in the morning (48–50). For these reasons, large doses at night and “inverse diurnal rhythm” dosing with hydrocortisone should be avoided in adults with 21OHD. In this respect, the challenge of managing the adult with 21OHD is similar to managing the adult with type 1 diabetes mellitus: the first-morning value (adrenal steroids or glucose) is the most important and the hardest to control; regular insulin given at bedtime will not accomplish this goal and causes problems!
If control does not meet the goal for that patient with hydrocortisone alone, our preference is to add the minimum amount of long-acting glucocorticoid necessary to restore control to goal. This next step is analogous to adding bedtime NPH insulin to an adult with type 2 diabetes mellitus when oral agents alone do not reach treatment goals. Prednisolone, 0.5–2 mg, is usually sufficient to achieve the desired control, and a good starting dose is 1 mg. The smallest tablet available in the United States is a 5-mg size; consequently, doses below 2.5 mg might require the use of the liquid form, whereas in many European countries 1- and 2-mg prednisolone tablets are available. If the prednisolone is contributing more to the 21OHD control than the hydrocortisone, it is then reasonable to transition to twice daily prednisolone; a single morning dose of prednisolone is occasionally sufficient (51). We discourage the use of the prodrug prednisone because at the low doses used for adrenal replacement, its conversion to the active drug prednisolone by 11β-hydroxysteroid dehydrogenase activity is inconsistent.
Without question, dexamethasone is the most effective oral glucocorticoid for suppressing adrenal-derived androgen excess in 21OHD, but it is also likely to cause iatrogenic Cushing syndrome. Once again, the interindividual variation in response, both to the androgen suppression and side effects, is broad. A few patients tolerate 0.5 mg/d or more with minimal untoward consequences, but many become floridly Cushingoid with marked weight gain and disfiguring violaceous striae, which never resolve completely. This experience prompts some to refuse all treatments, because in their minds, the side effects are not worth the slight improvement in daily function. A typical sequence is a switch from hydrocortisone to prednisone for convenience, but often the prednisone is ineffective—due to poor conversion to prednisolone—and then dexamethasone is started, with good androgen suppression but horrific physical consequences. Thus, initial doses of dexamethasone should be 0.25–0.375 mg/d.
Thus, we reserve dexamethasone for only very few specific situations, most prominently the man with TARTs, where continuous ACTH suppression for several months might shrink these tumors and reinitiate fertility if caught early (44). In order to administer enough dexamethasone at bedtime to provide complete adrenal suppression and replacement throughout the following day, supraphysiological doses (>0.75 mg) must be used, but high doses are not necessary to restore sperm production if given in divided doses such as 0.25 mg twice daily. Alternatively, 0.1 mg dexamethasone at night plus daytime hydrocortisone 10 mg three times a day successfully shrank TARTs and improved semen quality without side effects in 1 man with 21OHD and infertility (52). Patients rarely tolerate higher dexamethasone doses chronically due to sleep disturbance, weight gain, and skin changes, and these doses should only be used when lower doses are tolerated but ineffective. Similarly, women with 21OHD might require some bedtime dexamethasone to sufficiently suppress follicular phase P4 when preferable treatments such as prednisolone fail. In the United States, the smallest dexamethasone tablet is 0.5 mg, so patients must quarter tiny tablets or use a liquid form to take such low doses. When dexamethasone is used to enhance fertility, a discussion of the potential risks to the fetus should precede its use. Dexamethasone use for prenatal treatment of affected fetuses with 21OHD has been employed but is considered experimental for these reasons (28).
A second use of dexamethasone is in women with nonclassic 21OHD who suffer from prominent androgen excess, irregular menses, and infertility. These patients require normalization—but not complete suppression—of adrenal androgens in the early morning yet do not require adrenal replacement by day. A very simple, effective, and safe regimen for this purpose utilizes the fact that dexamethasone suppresses adrenal androgen production longer than cortisol synthesis. Thus, 0.25 mg dexamethasone, administered at bedtime every other day or 3 days a week, is usually sufficient to achieve satisfactory androgen reduction (31), but the dexamethasone-free days avoid the side effects associated with daily therapy. The same caveats about dexamethasone use during gestation apply to women with nonclassic 21OHD as for classic 21OHD.
Mineralocorticoid replacement
In contrast to the salt-wasting tendency of all children with classic 21OHD, adolescents (8) and adults (6) with classic 21OHD are prone to developing hypertension. Although obesity is also common among adults with 21OHD, the blood pressure does not correlate with body mass index (7). Once again, the range in dose requirements varies considerably among genotypically similar adults with classic 21OHD, based on diet, concurrent medications, and other factors; mineralocorticoid requirements also change in an individual between childhood and adulthood. Patients who take dexamethasone or prednisolone generally require more 9FCA than those who take hydrocortisone (cortisol), which also acts as a mineralocorticoid, with 40 mg hydrocortisone having equivalent mineralocorticoid activity as 0.1 mg 9FCA (53). The goal of mineralocorticoid replacement is to maintain plasma renin activity as low as possible without causing hypertension or hypokalemia and without orthostasis or salt craving. A typical dose of 9FCA is 0.05–0.2 mg once daily. Adults with nonclassic 21OHD do not require mineralocorticoid replacement.
Additional medical and mechanical therapies for women with 21OHD
Women with 21OHD often develop a secondary polycystic ovary syndrome with ovarian androgen excess (40); consequently, menstrual irregularity and elevated androgens do not always indicate poor control. For women who are not attempting to conceive, oral contraceptive pills provide several benefits. Oral contraceptive pills reduce androgen production from the ovaries, regularize menses if P4 is not chronically elevated, and most importantly raise SHBG, which significantly lowers the fraction of bioavailable and free T. Spironolactone is widely used to treat hirsutism and acne because it is an inexpensive, safe, and effective androgen antagonist. In the woman with classic 21OHD, however, its mineralocorticoid antagonist activity of spironolactone usually requires increasing the 9FCA dose to balance volume status. Flutamide has been employed off-label as part of a multidrug regimen in children with classic 21OHD (54), but this drug is expensive and has significant gastrointestinal toxicities. Only anecdotal information exists for the use of 5α-reductase inhibitors or other androgen antagonists in women with 21OHD. For small problem areas, topical 13.9% eflornithine cream reliably slows hair growth, but it is expensive and difficult to use over large areas. Other mechanical depilation methods, including shaving, plucking, waxing, electrolysis, and laser therapies are effective as well. Terminal hair follicles, which are formed during periods of elevated androgens, never revert back to vellus hairs and only become thinner with a longer resting phase when control is improved.
For women with all forms of 21OHD attempting to conceive, these alternative therapies are contraindicated, and glucocorticoid therapy must be intensified. Hyperandrogenemic oligoanovulation is common in women with 21OHD (40), and even when ovulating regularly, persistently high adrenal-derived P4 prevents withdrawal bleeding (48) and renders the endometrial tissue and cervical mucus unfavorable for fertilization (55). For women with 21OHD attempting to conceive, the P4 should be suppressed to below 2 nmol/L (0.6 ng/mL), which almost always requires at least a small evening dose of long-acting prednisolone and in rare cases dexamethasone. Using this approach, with 2 or 3 divided doses of prednisolone or hydrocortisone, 1 study found the fecundity rate for women with classic 21OHD to be 91%, not different from the general population (41).
Adrenalectomy, TART removal, and vaginal reconstruction surgeries
Although bilateral adrenalectomy has been suggested as a definitive therapy for children with classic 21OHD in poor control, this approach has fallen out of favor and should be used only in exceptional circumstances. Unilateral or bilateral adrenalectomy has been employed to restore fertility in otherwise compliant men (56) and women (57) with classic 21OHD, respectively. Bilateral adrenalectomy trades 1 form of adrenal insufficiency for a more severe form, and the patient should not be promised that androgen excess is permanently cured. For men with classic 21OHD, TART tissue is not cured by adrenalectomy, and they still require corticosteroid replacement to prevent TART growth and to maintain normal physiology. Even women with classic 21OHD can develop adrenal rest tumors in the pelvic area after bilateral adrenalectomy, with recurrent androgen excess (17, 18). We have observed marked and symptomatic reductions in blood pressure after bilateral adrenalectomy in adults with classic 21OHD, suggesting that adrenal-derived mineralocorticoids defend against volume depletion common in Addison's disease. Adrenalectomy becomes the only option for the management of massive myelolipomas, which will continue to grow and cause symptoms from mass effect if not removed (58). Surgical TART removal effectively treats the mass effect in the testes but has not been shown to improve fertility (45).
Most women with classic 21OHD never attempt pregnancy, for many reasons, including vaginal stenosis (59) and dyspareunia (60). Although vaginal reconstruction surgery is often performed during early childhood, a growing trend is to delay surgery until the patient is old enough to participate in the decision. In severely virilized girls, traditional skin or bowel grafts do not always give satisfactory long-term results (61), whereas buccal mucosa grafts appear promising but lack long-term outcomes (62, 63). Some women with 21OHD who have undergone vaginal reconstruction suffer frequent bladder infections or require surgical resection of abscesses, cysts, or scar tissue.
Controversies and Areas of Uncertainty
See Table 6 and Supplemental Data (published on The Endocrine Society's Journals Online web site at http://jcem.endojournals.org).
Table 6.
Controversies and Areas of Uncertainty
| Optimal glucocorticoid replacement regimens |
| Hydrocortisone dosing, controlled-release preparations, sc pump |
| Other glucocorticoids |
| Use of antiandrogens and androgen biosynthesis inhibitors |
| Goals for optimal laboratory parameters |
| Incorporation of steroid profiling by mass spectrometry |
| Initial assessment of young men with 21OHD, including sperm banking |
| Management recommendations during menopausal transition |
| Indications for additional surgeries |
Back to Our Patient
Our patient did return for follow-up, and she explained that she feared the side effects of prednisone, which she read on the patient information from the pharmacy. Due to her job, she did not think that she could take several doses of hydrocortisone each day. She noted that she had more energy on treatment again but that the androgen excess and daily shaving did not bother her. After reviewing the options, she agreed to take 2.5 mg prednisolone upon arising and again at suppertime, plus 9FCA 0.1 mg daily. At her next visit 3 months later, she had been exercising on her days off and had lost 1 kg. She was having regular monthly menses, was not sexually active, and was pleased with her quality of life. She had no Cushingoid stigmata, and she was wearing medical alert identification. Her laboratory data showed a plasma renin activity of 3.5 ng/mL/h (1 ng/L/s), normal electrolytes, and total T of 62 ng/dL (2.2 nmol/L). Her treatment goals were met, and no changes were made.
Conclusion
We have a great deal to learn about the management of adults with 21OHD. The dangers of chronic overtreatment are probably the major threat to the long-term health of these adults, but adrenal crises occur with undertreatment, which makes patients also more prone to suffering from TARTs and adrenal tumors. Treatment regimens and goals should be individualized, and these targets often change throughout a patient's life. The concept of employing 2 different glucocorticoids—one for adrenal replacement and the other for androgen suppression—is foreign to an internist's parsimonious way of thinking, but in many cases, this regimen is the best approach currently to optimize treatment while minimizing side effects. Laboratory data for adults with 21OHD are only a guide, which is less important than the clinical evaluation. Adults with 21OHD have many unmet medical needs, and additional research to the natural history and optimal interventions is urgently needed to improve outcomes. The challenges we face to achieve these goals include improving the training of providers, developing new medical or surgical treatment strategies, and delivering coordinated care throughout the patients' lifetime.
Acknowledgments
We thank our pediatric, internal medicine, and reproductive endocrinology colleagues, surgeons, urologists, gynecologists, psychologists, and others with whom we have shared discussions and management challenges for adults with 21OHD. Finally, we are indebted to the patients with 21OHD who have allowed us to participate in their care and who have shared their experiences with us. We learn a tremendous amount from both of these groups at every encounter, and these interactions have shown us that we have only begun this journey.
R.J.A. is supported by grants R01-GM086596 from the National Institutes of Health and a Clinician Scientist Award in Translational Research (no. 1005954) from the Burroughs-Wellcome Fund.
Disclosure Summary: R.J.A. was contracted to conduct research by Janssen Pharmaceuticals. W.A. has nothing to disclose.
Footnotes
- AD
- androstenedione
- CAH
- congenital adrenal hyperplasia
- DHEA
- dehydroepiandrosterone
- DHEAS
- DHEA sulfate
- 9FCA
- 9α-fludrocortisone acetate
- 17β-HSD3
- 17β-hydroxysteroid dehydrogenase type 3
- 21OHD
- 21-hydroxylase deficiency
- 17OHP
- 17-hydroxyprogesterone
- P4
- progesterone
- TART
- testicular adrenal rest tumor.
Reference
- 1. Speiser PW, White PC. Congenital adrenal hyperplasia. N Engl J Med. 2003;349:776–788 [DOI] [PubMed] [Google Scholar]
- 2. Therrell BL, Jr, Berenbaum SA, Manter-Kapanke V, et al. Results of screening 1.9 million Texas newborns for 21-hydroxylase-deficient congenital adrenal hyperplasia. Pediatrics. 1998;101:583–590 [DOI] [PubMed] [Google Scholar]
- 3. Krone N, Arlt W. Genetics of congenital adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab. 2009;23:181–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Joint LWPES/ESPE CAH Working Group Consensus statement on 21-hydroxylase deficiency from the Lawson Wilkins Pediatric Endocrine Society and the European Society for Paediatric Endocrinology. J Clin Endocrinol Metab. 2002;87:4048–4053 [DOI] [PubMed] [Google Scholar]
- 5. Arlt W, Willis DS, Wild SH, et al. Health status of adults with congenital adrenal hyperplasia: a cohort study of 203 patients. J Clin Endocrinol Metab. 2010;95:5110–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Falhammar H, Filipsson H, Holmdahl G, et al. Metabolic profile and body composition in adult women with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2007;92:110–116 [DOI] [PubMed] [Google Scholar]
- 7. Finkielstain GP, Kim MS, Sinaii N, et al. Clinical characteristics of a cohort of 244 patients with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2012;97:4429–4438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nebesio TD, Eugster EA. Observation of hypertension in children with 21-hydroxylase deficiency: a preliminary report. Endocrine. 2006;30:279–282 [DOI] [PubMed] [Google Scholar]
- 9. Falhammar H, Filipsson H, Holmdahl G, et al. Fractures and bone mineral density in adult women with 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2007;92:4643–4649 [DOI] [PubMed] [Google Scholar]
- 10. Sciannamblo M, Russo G, Cuccato D, Chiumello G, Mora S. Reduced bone mineral density and increased bone metabolism rate in young adult patients with 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2006;91:4453–4458 [DOI] [PubMed] [Google Scholar]
- 11. Kim MS, Merke DP. Cardiovascular disease risk in adult women with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Semin Reprod Med. 2009;27:316–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chakhtoura Z, Bachelot A, Samara-Boustani D, et al. Impact of total cumulative glucocorticoid dose on bone mineral density in patients with 21-hydroxylase deficiency. Eur J Endocrinol. 2008;158:879–887 [DOI] [PubMed] [Google Scholar]
- 13. Ravichandran R, Lafferty F, McGinniss MJ, Taylor HC. Congenital adrenal hyperplasia presenting as massive adrenal incidentalomas in the sixth decade of life: report of two patients with 21-hydroxylase deficiency. J Clin Endocrinol Metab. 1996;81:1776–1779 [DOI] [PubMed] [Google Scholar]
- 14. Reisch N, Scherr M, Flade L, et al. Total adrenal volume but not testicular adrenal rest tumor volume is associated with hormonal control in patients with 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2010;95:2065–2072 [DOI] [PubMed] [Google Scholar]
- 15. Nermoen I, Rorvik J, Holmedal SH, et al. High frequency of adrenal myelolipomas and testicular adrenal rest tumours in adult Norwegian patients with classical congenital adrenal hyperplasia because of 21-hydroxylase deficiency. Clin Endocrinol (Oxf). 2011;75:753–759 [DOI] [PubMed] [Google Scholar]
- 16. Claahsen-van der Grinten HL, Otten BJ, Hermus AR, Sweep FC, Hulsbergen-van de Kaa CA. Testicular adrenal rest tumors in patients with congenital adrenal hyperplasia can cause severe testicular damage. Fertil Steril. 2008;89:597–601 [DOI] [PubMed] [Google Scholar]
- 17. Tiosano D, Vlodavsky E, Filmar S, Weiner Z, Goldsher D, Bar-Shalom R. Ovarian adrenal rest tumor in a congenital adrenal hyperplasia patient with adrenocorticotropin hypersecretion following adrenalectomy. Horm Res Paediatr. 2010;74:223–228 [DOI] [PubMed] [Google Scholar]
- 18. Crocker MK, Barak S, Millo CM, et al. Use of PET/CT with cosyntropin stimulation to identify and localize adrenal rest tissue following adrenalectomy in a woman with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2012;97:E2084–E2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bouman A, Hulsbergen-van de Kaa C, Claahsen-van der Grinten HL. Prevalence of testicular adrenal rest tissue in neonates. Horm Res Paediatr. 2011;75:90–93 [DOI] [PubMed] [Google Scholar]
- 20. Cabrera MS, Vogiatzi MG, New MI. Long term outcome in adult males with classic congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2001;86:3070–3078 [DOI] [PubMed] [Google Scholar]
- 21. Reisch N, Flade L, Scherr M, et al. High prevalence of reduced fecundity in men with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2009;94:1665–1670 [DOI] [PubMed] [Google Scholar]
- 22. Tiitinen A, Välimäki M. Primary infertility in 45-year-old man with untreated 21-hydroxylase deficiency: successful outcome with glucocorticoid therapy. J Clin Endocrinol Metab. 2002;87:2442–2445 [DOI] [PubMed] [Google Scholar]
- 23. Gleeson H, Davis J, Jones J, O'Shea E, Clayton PE. The challenge of delivering endocrine care and successful transition to adult services in adolescents with congenital adrenal hyperplasia: experience in a single centre over 18 years. Clin Endocrinol (Oxf). 2013;78:23–28 [DOI] [PubMed] [Google Scholar]
- 24. Reisch N, Willige M, Kohn D, et al. Frequency and causes of adrenal crises over lifetime in patients with 21-hydroxylase deficiency. Eur J Endocrinol. 2012;167:35–42 [DOI] [PubMed] [Google Scholar]
- 25. Auchus RJ, Witchel SF, Leight KR, et al. Guidelines for the development of comprehensive care centers for congenital adrenal hyperplasia: guidance from the CARES Foundation Initiative. Int J Pediatr Endocrinol. 2010;2010:275213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Godbout A, Tejedor I, Malivoir S, Polak M, Touraine P. Transition from pediatric to adult healthcare: assessment of specific needs of patients with chronic endocrine conditions. Horm Res Paediatr. 2012;78:247–255 [DOI] [PubMed] [Google Scholar]
- 27. Sircili MH, de Mendonca BB, Denes FT, Madureira G, Bachega TA, e Silva FA. Anatomical and functional outcomes of feminizing genitoplasty for ambiguous genitalia in patients with virilizing congenital adrenal hyperplasia. Clinics (Sao Paulo). 2006;61:209–214 [DOI] [PubMed] [Google Scholar]
- 28. Speiser PW, Azziz R, Baskin LS, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2010;95:4133–4160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bidet M, Bellanne-Chantelot C, Galand-Portier MB, et al. Fertility in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2010;95:1182–1190 [DOI] [PubMed] [Google Scholar]
- 30. Moran C, Azziz R, Weintrob N, et al. Reproductive outcome of women with 21-hydroxylase-deficient nonclassic adrenal hyperplasia. J Clin Endocrinol Metab. 2006;91:3451–3456 [DOI] [PubMed] [Google Scholar]
- 31. Witchel SF, Azziz R. Nonclassic congenital adrenal hyperplasia. Int J Pediatr Endocrinol. 2010;2010:625105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Krone N, Braun A, Roscher AA, Knorr D, Schwarz HP. Predicting phenotype in steroid 21-hydroxylase deficiency? Comprehensive genotyping in 155 unrelated, well defined patients from southern Germany. J Clin Endocrinol Metab. 2000;85:1059–1065 [DOI] [PubMed] [Google Scholar]
- 33. Krone N, Rose IT, Willis DS, et al. Genotype-phenotype correlation in 153 adult patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency: analysis of the United Kingdom Congenital Adrenal Hyperplasia Adult Study Executive (CaHASE) cohort. J Clin Endocrinol Metab. 2013;98:E346–E354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Finkielstain GP, Chen W, Mehta SP, et al. Comprehensive genetic analysis of 182 unrelated families with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2011;96:E161–E172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Speiser PW, Dupont B, Rubinstein P, Piazza A, Kastelan A, New MI. High frequency of nonclassical steroid 21-hydroxylase deficiency. Am J Hum Genet. 1985;37:650–667 [PMC free article] [PubMed] [Google Scholar]
- 36. Nandagopal R, Sinaii N, Avila NA, et al. Phenotypic profiling of parents with cryptic nonclassic congenital adrenal hyperplasia: findings in 145 unrelated families. Eur J Endocrinol. 2011;164:977–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rezvani I, Garibaldi LR, Digeorge AM, Artman HG. Disproportionate suppression of dehydroepiandrosterone sulfate (DHEAS) in treated patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Pediatr Res. 1983;17:131–134 [DOI] [PubMed] [Google Scholar]
- 38. Rosner W, Auchus RJ, Azziz R, Sluss PM, Raff H. Position statement: utility, limitations, and pitfalls in measuring testosterone: An Endocrine Society Position Statement. J Clin Endocrinol Metab. 2007;92:405–413 [DOI] [PubMed] [Google Scholar]
- 39. Vermeulen A, Verdonck L, Kaufman JM. A critical evaluation of simple methods for the estimation of free testosterone in serum. J Clin Endocrinol Metab. 1999;84:3666–3672 [DOI] [PubMed] [Google Scholar]
- 40. Barnes RB, Rosenfield RL, Ehrmann DA, et al. Ovarian hyperandrogynism as a result of congenital adrenal virilizing disorders: evidence for perinatal masculinization of neuroendocrine function in women. J Clin Endocrinol Metab. 1994;79:1328–1333 [DOI] [PubMed] [Google Scholar]
- 41. Casteràs A, De Silva P, Rumsby G, Conway GS. Reassessing fecundity in women with classical congenital adrenal hyperplasia (CAH): normal pregnancy rate but reduced fertility rate. Clin Endocrinol (Oxf). 2009;70:833–837 [DOI] [PubMed] [Google Scholar]
- 42. Andersson S, Moghrabi N. Physiology and molecular genetics of 17β-hydroxysteroid dehydrogenases. Steroids. 1997;62:143–147 [DOI] [PubMed] [Google Scholar]
- 43. Khan N, Sharma KK, Andersson S, Auchus RJ. Human 17β-hydroxysteroid dehydrogenases types 1, 2, and 3 catalyze bi-directional equilibrium reactions, rather than unidirectional metabolism, in HEK-293 cells. Arch Biochem Biophys. 2004;429:50–59 [DOI] [PubMed] [Google Scholar]
- 44. Claahsen-van der Grinten HL, Otten BJ, Sweep FC, Hermus AR. Repeated successful induction of fertility after replacing hydrocortisone with dexamethasone in a patient with congenital adrenal hyperplasia and testicular adrenal rest tumors. Fertil Steril. 2007;88:705.e5–e8 [DOI] [PubMed] [Google Scholar]
- 45. Claahsen-van der Grinten HL, Otten BJ, Takahashi S, et al. Testicular adrenal rest tumors in adult males with congenital adrenal hyperplasia: evaluation of pituitary-gonadal function before and after successful testis-sparing surgery in eight patients. J Clin Endocrinol Metab. 2007;92:612–615 [DOI] [PubMed] [Google Scholar]
- 46. Avila NA, Premkumar A, Merke DP. Testicular adrenal rest tissue in congenital adrenal hyperplasia: comparison of MR imaging and sonographic findings. AJR Am J Roentgenol. 1999;172:1003–1006 [DOI] [PubMed] [Google Scholar]
- 47. Han TS, Stimson RH, Rees DA, et al. Glucocorticoid treatment regimen and health outcomes in adults with congenital adrenal hyperplasia. Clin Endocrinol (Oxf). 2013;78(2):197–203 [DOI] [PubMed] [Google Scholar]
- 48. Rosenfield RL, Bickel S, Razdan AK. Amenorrhea related to progestin excess in congenital adrenal hyperplasia. Obstet Gynecol. 1980;56:208–215 [PubMed] [Google Scholar]
- 49. Winterer J, Chrousos GP, Loriaux DL, Cutler GB., Jr Effect of hydrocortisone dose schedule on adrenal steroid secretion in congenital adrenal hyperplasia. J Pediatr. 1985;106:137–142 [DOI] [PubMed] [Google Scholar]
- 50. Charmandari E, Matthews DR, Johnston A, Brook CG, Hindmarsh PC. Serum cortisol and 17-hydroxyprogesterone interrelation in classic 21-hydroxylase deficiency: is current replacement therapy satisfactory? J Clin Endocrinol Metab. 2001;86:4679–4685 [DOI] [PubMed] [Google Scholar]
- 51. Caldato MC, Fernandes VT, Kater CE. One-year clinical evaluation of single morning dose prednisolone therapy for 21-hydroxylase deficiency. Arq Bras Endocrinol Metabol. 2004;48:705–712 [DOI] [PubMed] [Google Scholar]
- 52. Mouritsen A, Juul A, Jorgensen N. Improvement of semen quality in an infertile man with 21-hydroxylase deficiency, suppressed serum gonadotropins and testicular adrenal rest tumours. Int J Androl. 2010;33:518–520 [DOI] [PubMed] [Google Scholar]
- 53. Oelkers W, Diederich S, Bahr V. Diagnosis and therapy surveillance in Addison's disease: rapid adrenocorticotropin (ACTH) test and measurement of plasma ACTH, renin activity, and aldosterone. J Clin Endocrinol Metab. 1992;75:259–264 [DOI] [PubMed] [Google Scholar]
- 54. Merke DP, Keil MF, Jones JV, Fields J, Hill S, Cutler GB., Jr Flutamide, testolactone, and reduced hydrocortisone dose maintain normal growth velocity and bone maturation despite elevated androgen levels in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2000;85:1114–1120 [DOI] [PubMed] [Google Scholar]
- 55. Lo JC, Schwitzgebel VM, Tyrrell JB, et al. Normal female infants born of mothers with classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 1999;84:930–936 [DOI] [PubMed] [Google Scholar]
- 56. Scaroni C, Favia G, Lumachi F, et al. Unilateral adrenal tumor, erectile dysfunction and infertility in a patient with 21-hydroxylase deficiency: effects of glucocorticoid treatment and surgery. Exp Clin Endocrinol Diabetes. 2003;111:41–43 [DOI] [PubMed] [Google Scholar]
- 57. Ogilvie CM, Rumsby G, Kurzawinski T, Conway GS. Outcome of bilateral adrenalectomy in congenital adrenal hyperplasia: one unit's experience. Eur J Endocrinol. 2006;154:405–408 [DOI] [PubMed] [Google Scholar]
- 58. McGeoch SC, Olson S, Krukowski ZH, Bevan JS. Giant bilateral myelolipomas in a man with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2012;97:343–344 [DOI] [PubMed] [Google Scholar]
- 59. Nordenskjöld A, Holmdahl G, Frisen L, et al. Type of mutation and surgical procedure affect long-term quality of life for women with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2008;93:380–386 [DOI] [PubMed] [Google Scholar]
- 60. Gastaud F, Bouvattier C, Duranteau L, et al. Impaired sexual and reproductive outcomes in women with classical forms of congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2007;92:1391–1396 [DOI] [PubMed] [Google Scholar]
- 61. Stikkelbroeck NM, Beerendonk CC, Willemsen WN, et al. The long term outcome of feminizing genital surgery for congenital adrenal hyperplasia: anatomical, functional and cosmetic outcomes, psychosexual development, and satisfaction in adult female patients. J Pediatr Adolesc Gynecol. 2003;16:289–296 [DOI] [PubMed] [Google Scholar]
- 62. Lin WC, Chang CY, Shen YY, Tsai HD. Use of autologous buccal mucosa for vaginoplasty: a study of eight cases. Hum Reprod. 2003;18:604–607 [DOI] [PubMed] [Google Scholar]
- 63. Samuelson ML, Baker LA. Autologous buccal mucosa vulvovaginoplasty for high urogenital sinus. J Pediatr Urol. 2006;2:486–488 [DOI] [PubMed] [Google Scholar]