

SUMMARY
The expansion of repressive epigenetic marks has been implicated in heterochromatin formation during embryonic development, but the general applicability of this mechanism is unclear. Here we show that nuclear rearrangement of repressive histone marks H3K9me3 and H3K27me3 into nonoverlapping structural layers characterizes senescence-associated heterochromatic foci (SAHF) formation in human fibroblasts. However, the global landscape of these repressive marks remains unchanged upon SAHF formation, suggesting that in somatic cells, heterochromatin can be formed through the spatial repositioning of pre-existing repressively marked histones. This model is reinforced by the correlation of presenescent replication timing with both the subsequent layered structure of SAHFs and the global landscape of the repressive marks, allowing us to integrate microscopic and genomic information. Furthermore, modulation of SAHF structure does not affect the occupancy of these repressive marks, nor vice versa. These experiments reveal that high-order heterochromatin formation and epigenetic remodeling of the genome can be discrete events.
INTRODUCTION
The high-order structure of chromatin plays a critical role in genome function, such as in dynamic gene regulation and phenotypic plasticity (Misteli, 2007; Fraser and Bickmore, 2007). In the interphase nucleus, two forms of chromatin can be distinguished based on their compaction: condensed heterochromatin (HC) and diffuse euchromatin (EC). EC contains transcriptionally active or competent genes, while in HC transcription is repressed. HC has been divided into constitutive HC (cHC) and facultative HC (fHC). cHC includes centromeres and telomeres and is a permanent state, whereas fHC switches between HC and EC states depending on the biological context (Trojer and Reinberg, 2007).
In addition to the cytological definition of chromatin states, biochemical marks have been described, including specific histone modifications that often are mutually exclusive between HC and EC (Grewal and Rice, 2004). Histone H3K9me3 is a hallmark of cHC, while H3K27me3 is associated with fHC. H3K9me3 and H3K27me3 are recognized by the chromodomain of heterochromatin protein 1 (HP1) and polycomb (Pc) proteins, respectively, components of distinct gene silencing complexes (Campos and Reinberg, 2009).
Dose-dependent correlations of these marks to both HC structure and gene silencing have been observed (Trojer and Reinberg, 2007). H3K9me3 is recognized by HP1 proteins, which recruit SUV39H histone methyltransferases, leading to the “spreading” of H3K9me3 during cHC formation (Bannister et al., 2001; Lachner et al., 2001; Nakayama et al., 2001). Similarly, formation of the inactive X chromosome (Xi), the best-studied fHC model in mammals, is accompanied by a number of chromatin modulations, including H3K27me3 and the accumulation of Pc group proteins (Plath et al., 2003; Silva et al., 2003). Despite these correlations, the function of repressive histone marks with regard to changes in high-order chromatin structure is not fully understood.
We have previously described a unique HC structure in human fibroblasts, namely senescence-associated heterochromatic foci (SAHFs) (Narita et al., 2003). SAHFs are readily visible as distinct DAPI-dense foci, which are enriched for cHC markers and exclude EC markers. These initial studies suggested a correlation between SAHF formation and gene expression, in part through the p16/RB pathway (Narita et al., 2003; Zhang et al., 2007). SAHFs have been widely used as a marker of senescence, which has led to the identification of a number of effectors involved in the senescence program (Adams, 2007). However, despite the functional and practical relevance of SAHFs in senescence, neither a detailed characterization of SAHF structure nor a comprehensive analysis of the chromatin states within SAHFs has been elucidated. Here we demonstrate that SAHF formation results in a concentric chromatin architecture, not only segregating the chromatin of individual chromosomes into HC and EC regions, but also concentrating H3K9me3 and H3K27me3 in nonoverlapping layers within the condensed HC. Regardless of the SAHF phenotype, the chromosome-wide profiles of these repressive histone marks are largely unchanged. This is in contrast to the dynamic changes in repressive marks during embryonic differentiation (Hawkins et al., 2010). We also find that the replication timing (RT) of presenescent proliferating cells is spatially associated with the layered structure of SAHFs. In addition, we find a substantial correlation between the linear chromosome-wide profiles of RT and the repressive histone marks, allowing us to integrate microscopic and genomic information for these repressive histone marks. These data indicate that SAHFs are formed through coassociation of at least three types of chromatin and suggest that high-order HC formation and redistribution of classical HC markers can be discrete events.
RESULTS
Multilayer Structure of Chromatin in SAHFs
To characterize SAHFs in detail, we immunolabeled different combinations of histone marks in oncogenic H-RasV12-induced senescent (RIS) IMR90 human fibroblasts. RIS was initiated with a 4-hydroxytamoxifen (4OHT)-inducible system, in which H-rasV12 is fused to the estrogen receptor (ER) ligand-binding domain (ER:Ras) (Young et al., 2009). RIS is established about 6 days after 4OHT addition, when the majority of the cells form SAHFs (Figure 1A). Consistent with previous reports, methylation on H3K9, probed by antibodies against H3K9me2 (a fHC marker) and H3K9me3, localized to SAHFs (Narita et al., 2003, 2006). However, the enrichment of H3K9me3 was limited to the core of the DAPI-dense SAHFs, while H3K9me2 covered the whole area of SAHFs, suggesting that SAHF chromatin is composed of at least two layers (Figure 1B).
Figure 1. SAHF Chromatin Segregates into H3K9me3 Core and H3K27me3 Ring.

(A) ER:Ras-expressing IMR90 cells were assessed for SAHFs and expression of the proteins indicated. Gr, growing (no 4OHT); RIS, d6 Ras-induced senescent cells. p16 and HMGA2, senescence markers; Cyclin A, a cell-cycle marker. Data are shown as mean ± SEM (n > 3).
(B–D) Confocal images for indicated histone marks in cells indicated. The region indicated by the rectangle is magnified. The arrow indicates the path over which the fluorescent intensity was profiled (B and D). Arrowheads depict the Xi (C). The number represents the proportion of Xi, in SAHF-positive cells, displaying the typical H3K9me3 core with associated H3K27me3 ring (percent ± SEM, 176 cells from three independent experiments were assessed) (C).
(E and F) Electron spectroscopic imaging (ESI) for phosphorous (P), nitrogen (N), and the N minus P (N – P), to delineate chromatin (P, yellow) and nonchromosomal (ribonucleo)protein (N – P, blue). The region indicated by the rectangle is magnified. The arrow indicates the path over which the intensity was profiled (E). Small, bright objects around the core represent gold particles labeling H3K27me3. The N/P ratio was calculated for the H3K9me3 core (core; green dashed line), the H3K27me3 ring (ring; red dashed line), and for perinuclear heterochromatin (peri; see Figure S1F) (F). Data are shown as mean ± SEM (16 SAHFs from three cells from two preparations). *p < 0.001.
See also Figure S1.
In marked contrast to H3K9me3, H3K27me3 exhibited a “ring” structure, surrounding the “H3K9me3 core” with no obvious overlap in the majority of SAHF-positive cells (Figure 1C and Figures S1A and S1B available online). In growing cells both marks showed an irregular pattern with prominent perinuclear enrichment for H3K9me3 (Figure 1C). As expected H3K4me3 and H3K9/14 acetylation, EC marks, were excluded from the ring structure (Figure S1C). In addition, the repressive layers of SAHFs excluded an additional layer for H3K36me3, which has been reported to correlate with transcriptional elongation (Figure 1D) (Kolasinska-Zwierz et al., 2009). Furthermore, localization of endogenous HP1γ and GFP-fused M33 (mouse Pc1) was restricted to the H3K9me3 and H3K27me3 layers, respectively, confirming the segregation of these two repressive structures within SAHFs (Figure S1D).
It has been shown that pericentromeric cHC localizes to the peripheral regions of DAPI-dense SAHFs (Narita et al., 2003; Zhang et al., 2007; Funayama et al., 2006). We probed for centromere protein A (CENP-A) together with H3K27me3. CENP-A mostly localized to the periphery of the SAHF-core (patterns 1 and 2 in Figure S1E) with ~16% of CENP-A being outside the H3K27me3 layer (pattern 3 in Figure S1E), confirming the polar localization of pericentromeric cHC within SAHFs.
We next examined chromatin “compactness” in these repressive layers, in both SAHFs and typical perinuclear HC regions, using electron spectroscopic imaging (ESI), which allows us to directly and quantitatively measure the density of chromatin fibers (Figures 1E and S1F) (Ahmed et al., 2010). Integrative phosphorus and nitrogen density analyses revealed that the chromatin fiber in the SAHF core was highly packed, even more so than perinuclear HC (Figures 1E, 1F, and S1F). Chromatin in the H3K27me3 ring was less densely packed than H3K9me3-rich regions, exhibiting protein-rich structures interspersed within this domain (Figures 1E and 1F). These data suggest that the SAHF core HC is distinct from peri-nuclear HC, despite of being enriched in similar marks, such as H3K9me3.
Thus, SAHF formation involves the segregation of chromatin featuring distinct epigenetic components and chromatin compactness.
The Xi in Senescent Cells Exhibits a Subchromosomal SAHF-like Structure
The human Xi harbors both H3K9me3- and H3K27me3-enriched domains, and a nonoverlapping distribution of H3K9me3 and H3K27me3 has been shown in mitotic Xi (Chadwick and Willard, 2004; Chadwick, 2007). To gain further insight into the structural relationship between H3K9me3 and H3K27me3 within HC, we compared the distribution of these marks between SAHFs and Xi.
SAHFs represent single chromosomes (Zhang et al., 2007; Funayama et al., 2006). Consistent with this idea, the Xi corresponded to a single condensed focus in RIS cells (Xi/S, where S indicates senescence) (Figure 1C). In addition, chr X painting by DNA FISH revealed that, while the active X chromosome (Xa) and the Xi in growing cells (Xa/G and Xi/G, respectively, where G indicates growing) can be readily identified by their condensation state (Figure 2A, left panels), the Xa in RIS cells (Xa/S) also forms a single SAHF, making it difficult to distinguish the Xa/S from the Xi/S by DAPI staining (Figure 2A, right panels). We next performed X-specific alpha satellite DNA FISH coupled with immunolabeling of H3K27me3. The Xa/S exhibited a similar H3K27me3 staining pattern to other SAHFs in the same nuclei, indicating that each allele of the X chromosome shows a distinct distribution of repressive marks upon SAHF formation, as in mitotic chromosomes (Figure 2B).
Figure 2. Subchromosomal SAHF Formation in Xi.

(A) Confocal images of DNA FISH with a chr X paint probe in the indicated conditions, as in Figure 1.
(B) Confocal images of DNA FISH with a chr X alpha satellite probe (arrows) and immunofluorescence for H3K27me3. Xa/S and Xi/S represent the active and inactive chr X, respectively.
(C) Merged confocal images for H3K27me3 and H3K9me3. The right hand panels are magnified images of the regions indicated by the rectangles and their corresponding schemes. Proportion of Xi/S displaying a clear H3K9me3 core with H3K27me3 ring (percent ± SEM); 35 Xi/S from four independent experiments were counted.
Interestingly, we identified a SAHF-like H3K9me3 region that was surrounded by an H3K27me3 ring within the majority of the Xi/S, but not Xi/G. This suggests that a part of the Xi is still susceptible to SAHF formation and that, therefore, SAHF remodeling does not necessarily involve whole chromosomes as previously proposed (Figure 2C).
Gene-Specific and Global Analyses for Histone Marks in Growing and RIS Cells
We reasoned that the segregation of histone marks in SAHFs might allow us to effectively link our microscopic data to the primary structure of the genome. We performed chromatin immunoprecipitation with genome-wide deep sequencing (ChIP-seq) using recently developed monoclonal antibodies against repressive (H3K9me3, H3K9me2, and H3K27me3), and active (H3K4me3 and H3K36me3) marks in growing and RIS IMR90 cells (Figure S2). In agreement with previous studies, we found distinct distributions of reads for these histone marks throughout the genome (Table S1) (Bernstein et al., 2005; Kim et al., 2005; Barski et al., 2007; Mikkelsen et al., 2007; Cui et al., 2009; Pauler et al., 2009; Hawkins et al., 2010). Regardless of the condition, both active marks were highly gene associated: more than 70% of H3K36me3 was mapped in genic regions with poor association with promoters, while H3K4me3 showed the highest promoter association. H3K27me3 showed higher gene- and promoter-association than H3K9me3. A similar trend was also observed with the RSEG method (Table S1) (Song and Smith, 2011).
Consistent with previous reports, occupancy of active and repressive histone marks altered between growing and RIS in some genic regions, such as p16INK4A and CCNA2 (Figures 1A, 3A, and 3B) (Narita et al., 2003; Barradas et al., 2009; Agger et al., 2009). At a genome-wide level, genes that were differentially associated with each mark between growing and RIS were determined with DiffBind, and those genes were examined for their expression pattern (Figure 3C). While the alteration in H3K9me3 occupancy in genic regions did not translate into differential gene expression between growing and RIS cells overall, alterations of all the other marks were substantially correlated with gene expression changes (Figure 3C).
Figure 3. Genome-wide Mapping of Histone Marks in Growing and RIS IMR90 Cells.

(A and B) Genome browser representations (A) and read counts (B) of ChIP-seq data for the indicated histone marks at the CCNA2 and p16INK4A loci. Gr, growing; RIS, d6 Ras-induced senescent. Read counts are shown for 500 bp upstream and 5,000 bp downstream of each transcription start site (TSS).
(C) Distribution of gene expression values for genes with greatest changes in read counts for each histone mark indicated. Read counts were determined as in (B). The 1,000 genes with the greatest increase (black) or decrease (gray) in number of reads in RIS over Gr cells were plotted for the distribution of differential expression fold changes (FC, RIS/Gr) from the corresponding expression microarray data sets. Vertical red lines indicate the mean fold changes.
(D and E) Read density cluster analysis of 48 ChIP-seq samples (Table S2) for the indicated histone marks, with a window size of 500 Kb (D). The correlation between the indicated histone marks is plotted against different window sizes (mean ± SEM) (E).
We next examined the correlation between the global profiles of the histone marks used. We performed cluster analyses on the ChIP-seq data based on the read density for each histone mark, using different enrichment intervals throughout the genome for 48 ChIP-seq data sets (Figures 3D and 3E and Table S2). While all data were generated in IMR90 cells using the same antibody for each mark (Figure S2), cellular conditions vary: in addition to growing and RIS, we also included cells lacking SAHFs using stable RNA interference against high mobility group AT-hook 1 (HMGA1) or the retinoblastoma (RB) protein. Both proteins are essential for SAHF formation (described in detail later) (Narita et al., 2003, 2006). In all chosen window sizes, the same marks clustered together regardless of chromatin phenotype, while H3K9me2 was substantially correlated with both distinct clusters of H3K9me3 and H3K27me3 (Figures 3D and 3E, and data not shown). This is reminiscent of our imaging data showing that H3K9me2 covers both nonoverlapping H3K9me3 and H3K27me3 layers in SAHFs (Figure 1B). With a 500 Kb interval, the negative correlation between H3K9me3 and H3K27me3 was most pronounced (Figure 3E). Considering the well-established mutually exclusive pattern of these repressive marks, this resolution of ChIP-seq data may well reflect their microscopic pattern.
Spatial Rearrangement of Repressive Marks during SAHF Formation
To visualize the chromosome-wide landscape of repressive marks H3K9me3 and H3K27me3, we normalized the ChIP-seq data for the number of total reads and plotted the reads with a 1 Kb window size with smoothing over 1,000 units. In RIS cells both repressive marks were represented as domains of a few hundred Kb to a few Mb (Figure 4A), which are comparable to the window size that recapitulated the negative correlation between these repressive marks in the read density cluster analysis (Figure 3E). Indeed, an overlay of H3K9me3 and H3K27me3 profiles over chr 20 in RIS cells captured their mutually exclusive and alternate patterns (Figure 4A). These landscapes also showed a substantial correlation with domains determined by the RSEG method, which emphasizes domain boundaries (top bars in Figure 4A). To quantitatively support the visual inspection, we evaluated the extent of the correlation between profiles of the repressive marks in each chromosome (Figure 4A). Surprisingly, the landscapes of both H3K9me3 and H3K27me3 were remarkably unchanged despite the dramatic foci formation during RIS (Figures 4B). This is in marked contrast to the spreading of these repressive marks during embryonic stem cell (ESC) differentiation (Hawkins et al., 2010). We reanalyzed the published ChIP-seq data and confirmed that our method can recapitulate the alteration of the repressive marks between human ESCs and fibroblasts (IMR90 cells), although less pronounced in H3K27me3 (Figure 4C) (Hawkins et al., 2010). Our data imply that spreading of the repressive marks is not a necessity for HC formation, at least in somatic cells.
Figure 4. Spatial Repositioning of Pre-existing Repressive Histone Marks to Form SAHFs.
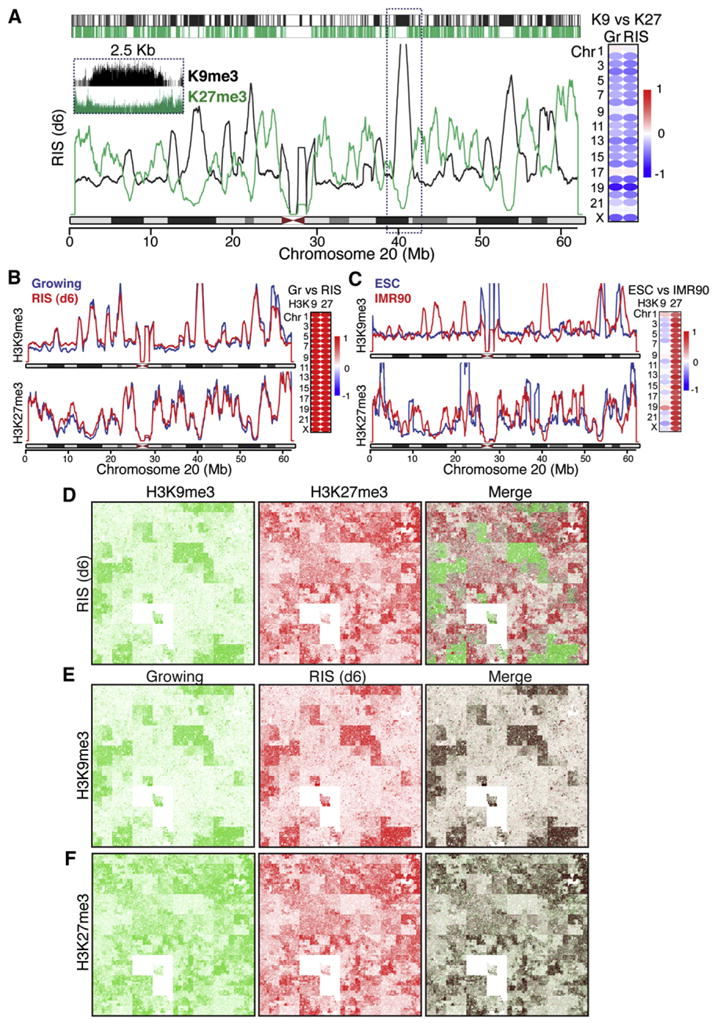
(A) Chromosome-wide landscape of repressive histone marks. The window size is 1 Kb, and the smoothing unit is 1,000. Top: Significantly enriched regions for each mark, determined by RSEG, are sketched with a 1 Kb window size. The browser shot of the region indicated by the rectangle is shown in the inset. Spearman correlations between H3K9me3 versus H3K27me3 in Growing (Gr) or Ras-induced senescent (RIS) cells are shown for all chromosomes.
(B) Unaltered landscape of H3K9me3 and H3K27me3 marks between Gr and RIS cells on chromosome 20. Chromosome-wide spearman correlations between Gr and RIS cells for each mark are shown.
(C) Marked changes in the landscape of H3K9me3 and H3K27me3 between ESCs and IMR90 cells on chromosome 20. Publicly available ChIP-seq data (Hawkins et al., 2010) were reanalyzed as in (B).
(D–F) Chromosome-wide profiles of the ChIP-seq data visualized by Hilbert curves for the indicated marks and cells. The merged images were generated with Photoshop, using blending mode “darken.”
See also Figure S3.
To compare the spatial relationship of the repressive marks in more detail, we applied space-filling graphs (Hilbert curves), which convert one-dimensional (1D) data into a 2D spatial pattern through mathematical “folding” without loosing resolution (Figure S3A) (Anders, 2009). The Hilbert curves for these marks showed distinct patterns: H3K4me3 exhibited “puncta,” while H3K36me3, H3K27me3, and H3K9me3 formed wider “areas” (Figures S3B and S3C). Consistent with the data shown in Table S1, substantial fractions of the H3K4me3 puncta and H3K36me3 areas of RIS cells were associated with overlaid RefSeq transcripts, while H3K27me3 and H3K9me3 showed moderate and poor association with the transcribed regions, respectively (Figures S3B and S3C). The mutually exclusive pattern between H3K9me3 and H3K27me3 was also recapitulated (Figure 4D), and each repressive mark showed a remarkable similarity between growing and RIS (Figure 4E), with only active marks showing substantial differences (e.g., the H3K4me3 pattern in the first exon of the p16INK4A in Figure S3B and the H3K36me3 pattern in gene-rich regions in Figure S3C).
Thus, while both active and repressive marks are redistributed during RIS in some genic regions, the global pattern of these repressive marks is highly static, strongly suggesting that a 3D repositioning of pre-existing H3K9me3 and H3K27me3 is involved in SAHF formation.
Replication Timing Is Correlated with the Layered Structure of SAHFs
To visualize the dynamic spatial rearrangement of the genome, we monitored genomic regions with different RT during SAHF formation. ER:Ras expressing IMR90 cells were first arrested at the G1/S border by a double thymidine block. After releasing the synchronization, newly synthesized DNA was labeled with EdU (5-ethyny-2-deoxyuridine) and BrdU (5-bromo-2-deoxyuridine). A 3 hr EdU pulse and a 2 hr chase with normal media was followed by another 3 hr BrdU pulse, and then 4OHT was added to induce ER:Ras expression. The cells were harvested 2 days later when ~10% of the cells formed SAHFs. As expected, in growing cells the late replicating DNA showed enrichment in the DAPI-dense regions, whereas early replicating DNA showed a more diffused pattern (Figure 5A). Strikingly, the majority of SAHF-positive cells that retained both EdU/BrdU showed a pattern similar to that of H3K9/27me3 staining (Figures 5A–5C).
Figure 5. Spatiotemporal Correlation between SAHF Architecture and Replication Timing.
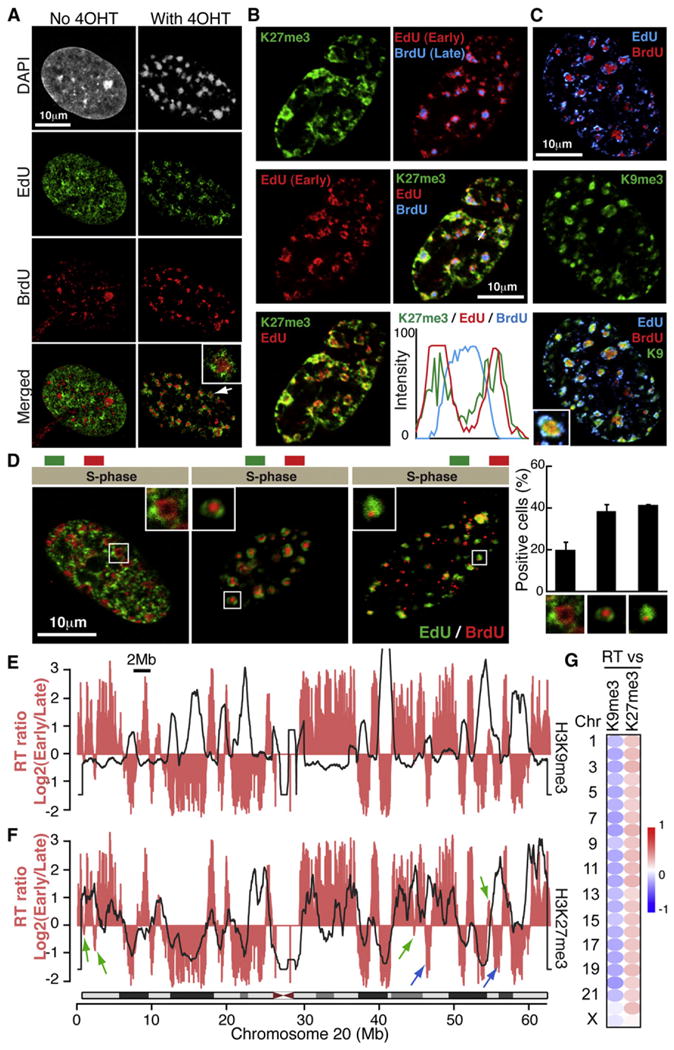
(A) Radial distribution of DNA during SAHF formation according to replication timing (RT). After release of synchronization at the G1-S border, early- and late-replicating DNA was labeled by EdU and BrdU, respectively, followed by Ras induction. Confocal images for EdU/BrdU are shown. The focus indicated by the arrow is magnified.
(B and C) Confocal images for the costaining of EdU/BrdU and repressive histone marks. Intensity profiles of fluorescence along the white line indicated in the merged panel is shown (B).
(D) Three spatiotemporal patterns of RT in RIS cells. Asynchronous cells were labeled with EdU/BrdU, followed by Ras induction as in (A). Data are shown as mean ± SEM (131 cells from three independent experiments were assessed).
(E and F) Global association between RT and indicated marks. The landscape for the RT ratio (pink) and the indicated histone marks (black lines) in growing IMR90 cells were overlaid for chromosome 20. Regions that show a negative correlation between RT and H3K27me3 profiles are indicated by blue arrows. Green arrows indicate mid-replicating regions.
(G) Spearman correlations between RT and repressive marks in all chromosomes.
See also Figure S4.
To gain more comprehensive insight into the relationship between RT and SAHF structure, we labeled asynchronously growing cells using the same “pulse-chase-pulse” method followed by ER:Ras induction. As shown in Figure 5D, we observed three patterns in cells that formed SAHFs. Nearly 40% of these nuclei exhibited the H3K9/27me3 pattern (Figure 5D, middle panel). In about 20% of SAHF-positive cells, BrdU regions formed a ring-like structure, which was surrounded by EdU regions, reminiscent of the H3K36/27me3 pattern (Figure 5D, left panel). These data suggest an intimate relationship between RT and SAHF structure with a unidirectional distribution of RT between the SAHF cores and euchromatic regions. We also observed very late replicating small dots that often accumulated at the periphery of SAHFs (Figure 5D, right panel). Although the nature of these very late replicating regions is unclear, our data indicate that SAHFs are formed largely through a concentric reorganization of the genome.
To directly visualize the relocalization of the genome during SAHF formation, we performed time-lapse experiments after consecutive pulses of Alexa488-dUTP and Cy3-dUTP by glass beads loading (Hayashi-Takanaka et al., 2009). We introduced these fluorescent thymidine analogs 2 days after 4OHT addition to avoid the mitotic phase that precedes senescence. Figure S4 shows a representative series, in which we captured the last S phase before SAHF formation. At earlier time points, late-S foci (Cy3) were located at the nuclear periphery and around nucleoli, and early-to-mid S foci (Alexa488) were scattered in the interior of the nucleus. Late-S foci became more intense and interiorly located, forming SAHF-core-like structures surrounded by early-to-mid S foci at the late time points without cell division (Figure S4, left panel). Together, these data indicate that there is a spatiotemporal correlation between the concentric multilayer structure of SAHFs and presenescent RT.
SAHF Layers and the Genome-wide Chromatin Landscape
We next analyzed genome-wide RT profiles. As previously described (Hiratani et al., 2008), IMR90 cells were pulse labeled with BrdU and sorted into early and late S phase populations by flow cytometry. BrdU-substituted nascent DNA from these populations was immunoprecipitated, differentially labeled, and cohybridized to a whole-genome oligonucleotide microarray. To visualize the landscape of the RT profile, we plotted “RT ratio,” Log2 (early/late), for each probe (Figures 5E and 5F). In agreement with previous studies (Hiratani et al., 2008; Ryba et al., 2010), the size of the “replication domains” in IMR90 cells ranged from a few hundred Kb to several Mb, a range similar to the domains found for the repressive marks in the chromosome-wide landscapes (Figure 4A).
To evaluate the relationship between the repressive marks and RT, we overlaid their chromosome-wide landscapes (Figures 4B, 5E, and 5F). Visual inspection suggests that, in IMR90 cells, early replicating regions correlate negatively with the H3K9me3-enriched regions within chromosome bodies (Figure 5E). Considering the static nature of H3K9me3 during senescence (Figure 4B) and the spatial association between RT and the repressive mark layers within SAHFs (Figures 5B and 5C), our data suggest that late replicating regions with high H3K9me3 are repositioned to the SAHF core during senescence. Furthermore, we also observed substantial correlation between H3K27me3 and early replication, with some clear exceptions (Figure 5F, blue arrows). In addition, the “mid-replicating” regions, which are consistent with fHC (Hiratani et al., 2009), were often correlated with H3K27me3 enrichment (Figure 5F, green arrows). This implies that substantial stretches of DNA with early-to-mid RT and high H3K27me3 relocate to the H3K27me3 ring upon SAHF formation. The landscape pattern of H3K9me3 and H3K27me3 showed negative and positive correlations with RT, respectively, in nearly all autosomes, although these correlations were modest (Figure 5G). Altogether, both 3D and 1D correlations between RT and the repressive histone marks reinforce the dynamic coassociation of these repressive marks during SAHF formation.
Disruption of SAHF Structure Does Not Affect H3K9me3 and H3K27me3 Landscapes
Our data not only provide mechanistic insight into SAHF formation, but also imply that H3K9me3 and H3K27me3 deposition and high-order HC formation can be separable events. To test this idea, we examined whether or not the landscape of these repressive marks changes when SAHFs are disrupted (Figure 6A). We previously showed that the architectural protein HMGA1 is an essential structural component of SAHFs, which is required for both the establishment and maintenance of SAHFs (Narita et al., 2006). Depletion of HMGA1 before (by retroviral sh-HMGA1) or after (by lentiviral sh-HMGA1) SAHF formation efficiently disrupted SAHFs (Figures 6B, and 6C). However, the chromosome-wide repressive mark landscape was unaltered (Figures 6D and 6E).
Figure 6. Independence of High-Order Heterochromatin Compaction and Repressive Histone Marks in SAHFs.
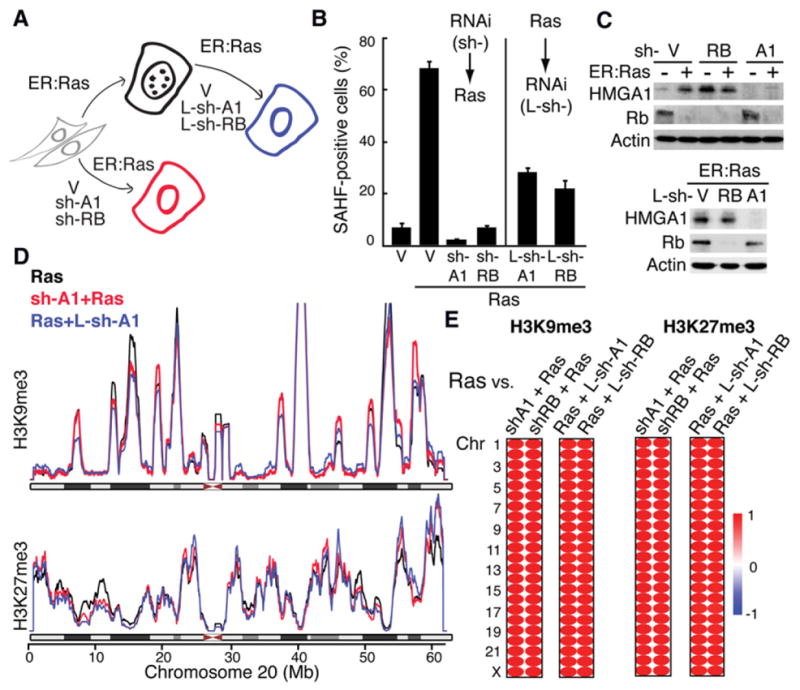
(A) Overview of experiments: SAHF formation can be prevented by either sh-HMGA1 (sh-A1) or sh-RB during RIS. SAHFs can also be disrupted by lentivirus-mediated shRNAs (L-sh-A1 or L-sh-RB) after RIS establishment.
(B) SAHF counts for the experiments described in (A). Data are shown as mean ± SEM.
(C) Western blots for the proteins indicated, in the conditions indicated in (A).
(D) Chromosomal landscapes plotted as in Figure 4 for H3K9me3 and H3K27me3 in the conditions indicated in (A).
(E) Spearman correlations between Ras cells and the cells indicated.
We also examined the effect of the depletion of RB, another essential factor for SAHF establishment (Narita et al., 2003). We confirmed that RB is also required for SAHF maintenance (Figures 5B and 5C). Although the role of RB in SAHF integrity is unknown, it has been shown that RB associates with SUV39H1 and HP1 and that RB mediates H3K9me3 deposition, at least in some gene promoters (Nielsen et al., 2001; Ait-Si-Ali et al., 2004). However, RB depletion failed to induce an alteration in the global landscape of H3K9me3 or H3K27me3, suggesting that RB-mediated SAHF formation is not dependent on a global change of H3K9me3. Depletion of HMGA1 or RB alone is not sufficient for complete RIS bypass, thus outgrowth of the nonsenescent cells is unlikely. These data support the “dissociation” between high-order HC structure and these repressive histone marks.
H3K9/27me3 Marks Are Not Necessary for SAHF Formation
We next examined the effect of the depletion of H3K9me3 and H3K27me3 on SAHF formation. To reduce the global level of H3K9me3 or H3K27me3, we used overexpression of JMJD2D (a demethylase that preferentially demethylates H3K9me3) or knockdown of SUZ12 (a component of Pc repressive complex 2), respectively (Shin and Janknecht, 2007) (Figures S5A and S5B). Upon Ras induction, H3K9me3- or H3K27me3-depleted cells still exhibited DAPI-dense SAHFs to a similar extent to the control RIS cells (Figure 7). To avoid potential bias resulting from heterogeneity in the levels of the repressive marks, we quantitated repressive marks at a single cell level by laser scanning cytometry. Cells selected for the lowest intensity of either repressive mark exhibited SAHFs upon ER:Ras induction (Figure S5C), suggesting that, although SAHF formation is mediated through spatial rearrangement of pre-existing H3K9me3 and H3K27me3 regions of the genome, these marks are not a prerequisite for the process of SAHF formation. Therefore, expansion of these repressive histone marks can be a discrete and separable event from HC compaction, at least in somatic cells.
Figure 7. Perturbation of the Repressive Histone Marks Does Not Prevent SAHF Formation.
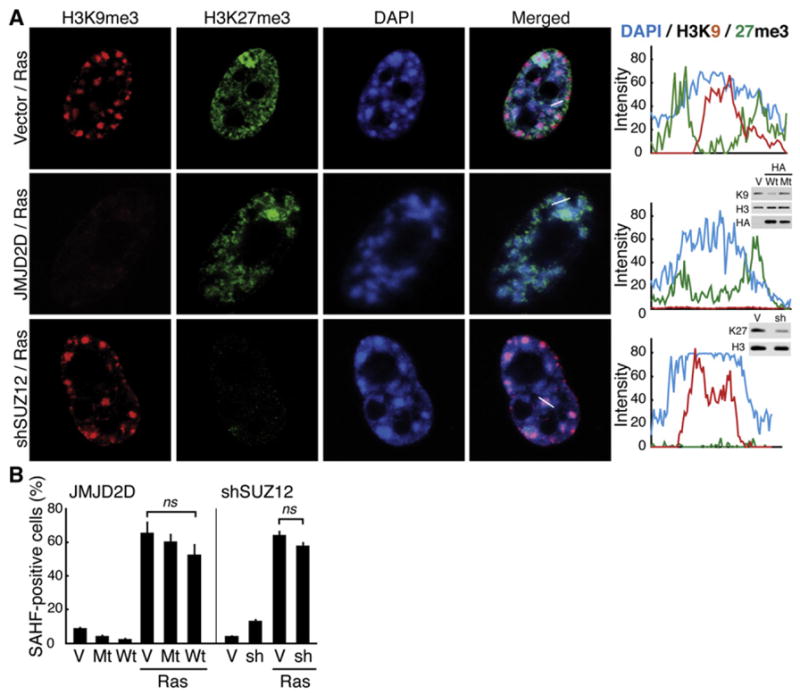
(A) Confocal images for the marks indicated. ER:Ras-expressing cells were infected with retroviruses expressing HA-JMJD2D (a demethylase of H3K9me3) or sh-SUZ12, and then ER:Ras was induced by 4OHT for 6 days. Intensity profiles of fluorescence along the white lines indicated in the merged panels are shown. Western blots show the extent of the reduction for each mark. Wt, wild-type HA-JMJD2D; Mt, a catalytically inactive mutant, H192A.
(B) Quantitative analysis of the effect of the depletion of each repressive mark on SAHF formation. Data are shown as mean ± SEM.
See also Figure S5.
DISCUSSION
Association between chromatin domains sharing similar epigenetic states has been suggested by cell biological methods (Clemson et al., 2006; Shopland et al., 2006; Simonis et al., 2006; Sinclair et al., 2010). This model has recently been reinforced by genome-wide chromosome conformation capture analyses (Lieberman-Aiden et al., 2009). In addition, genome-wide combinatorial analyses of chromatin proteins and histone marks have classified chromatin into its principle types (Filion et al., 2010; Kharchenko et al., 2011). However, a method to visualize dynamic links between microscopic and epigenomic data has not been established, largely due to the lack of a suitable model system. In this study, the nonoverlapping layer structure of distinct epigenetic marks within SAHF provides us with a tool to fill the gap between microscopic and epigenomic data. The readily manipulative nature of SAHFs revealed that landscapes of the repressive marks are highly static regardless of the chromatin phenotype, thus SAHFs are formed through 3D repositioning of repressively marked chromatin. This is further supported by the spatiotemporal association between presenescence RT and the segregation of the repressive marks during SAHF formation, as well as the substantial correlation between chromosome-wide landscapes of RT and the repressive marks. These data confirm dynamic co-associations within at least two repressive chromatin types during SAHF formation. Although more detailed analysis of the components of each layer remains to be performed, our data extend the chromatin type concept and the model that common properties of the chromatin domains may allow for their coassociation during chromatin folding.
Our data also imply that spreading of H3K9me3 and H3K27me3, hallmarks of cHC and fHC, respectively, is not involved in SAHF formation. Spreading of repressive marks has been implicated in HC formation, but this notion is largely based on data using unicellular organisms or, in higher eukaryotes, developmental systems. Indeed, the spreading of H3K9me3 and H3K27me3 has been confirmed upon ESC differentiation (Hawkins et al., 2010). Although we do not exclude that other repressive marks might spread during SAHFs formation, the mechanisms of HC formation during ESC differentiation and RIS are distinct, representing the spreading and 3D repositioning, respectively, of at least these two hallmarks of HC. It is tempting to speculate that, once the static profiles of these repressive marks are established during development or differentiation, the machinery for HC formation might be shifted to the repositioning model. However, our data do not exclude the possibility that different genomic regions might adopt different models depending on the cellular context, thus they are not necessarily mutually exclusive models at a genome-wide level. It is also possible that the landscapes of the repressive marks differ between cell types. Interestingly, a comparison between normal human hepatocytes and fibroblasts revealed a better correlation in the H3K9me3 landscape than in the H3K27me3 landscape; hinting at cross-lineage similarity and specificity in the H3K9me3 and H3K27me3 patterns, respectively (Figures S3E–S3G).
Correlation between H3K9me3 or H3K27me3 and RT has not been clear at a genome-wide level. RT has been negatively correlated with H3K9me2, but to a much lesser extent with H3K9me3 in human lymphoblastoid cells, and variable results have been reported on the association between RT and H3K27me3 depending on the cell system (Thurman et al., 2007; Ryba et al., 2010). In addition to the differences in the experimental systems used, this might be due to the intermediate nature of H3K27me3 in terms of its association with gene density and promoter occupancy compared to H3K4me3 and H3K9me3 (Table S1). The substantial exceptions in the H3K27me3/RT correlation, as well as the association of H3K27me3 with mid-replicating regions in our data (Figure 5F), might also contribute to the mixed observations reported. Although how presenescence RT is remotely associated with SAHF formation remains to be elucidated, the correlation between the repressive marks and RT found in this study may provide a platform to study how RT reflects chromatin structure and gene expression.
We have previously shown that HMGA1 is a structural component of SAHFs, and is essential for their structural integrity (Narita et al., 2006). In addition to this architectural aspect, the present study provides another layer to the mechanism, namely the 3D repositioning of repressively marked chromatin. Interestingly our data suggest that H3K9me3 and H3K27me3 are not required, as such, for SAHF formation (Figure 7). In addition, a previous report has shown that SAHFs are still formed when 50%–80% of the endogenous HP1 protein is removed from chromatin (Zhang et al., 2007). This suggests that H3K9me3/HP1, hallmarks of cHC, are dispensable for formation of the highly compacted SAHF core, although other repressive histone marks may possibly be involved in this process. Alternatively, chromatin proteins that are correlated with, but do not require, repressive histone marks to contribute to HC formation might be involved. Consistent with this possibility, chromatin types, which are defined independently of the histone marks, are still closely correlated with histone marks (Filion et al., 2010). Our ESI data indicate that the H3K27me3 ring is relatively protein rich (Figure 1E), and exploring layer specific proteins might provide insight into the mechanism(s) behind high-order chromatin type coassociation.
In interphase nuclei, chromosome territories have been shown to be transcriptionally permeable (Cremer and Cremer, 2010), and a sequence-based transient and probabilistic folding model has been proposed (Shopland et al., 2006), possibly reflecting the diverse and dynamic regulation of gene activity within chromosome territories. In contrast, SAHFs create transcriptionally impermeable and more uniformly structured HC. RIS and SAHF formation are progressive processes, where the most dramatic changes in gene expression are likely to occur before senescence/SAHF establishment. Thus, such a clear separation of active and repressive regions might facilitate not only the silencing of specific genes, but also gene expression pattern stabilization at the cost of dynamic gene regulation.
EXPERIMENTAL PROCEDURES
ESI Analysis
Correlative light microscopy/ESI microscopy analysis was performed as described previously (Ahmed et al., 2010). ESI procedure has been fully described before. Immunogold labeling was performed with Aurion Ultra Small ImmunoGold Reagents (Electron Microscopy Sciences).
Replication Timing Assays
For the pulse-chase-pulse experiments, cells were synchronized at the G1-S border by a double thymidine block. Upon release, early (0–3 hr) and late (5–8 hr) replicating DNA was labeled using EdU (Invitrogen) and BrdU (Sigma), respectively. The same labeling procedure was also applied to asynchronous cells. ER:Ras was induced by adding 4OHT immediately after BrdU removal. Cells were harvested 2 days later. EdU was stained with the Click iT EdU Alexa Fluor 488 (or 647) Cell Proliferation Assay kit (Invitrogen) after BrdU IF.
ChIP-Seq
ChIP-seq was performed as described (Schmidt et al., 2009). In short, the immunoprecipitated DNA was end repaired, A tailed, ligated to the sequencing adapters, amplified by 18 cycles of PCR, and size selected, followed by single-end sequencing on an Illumina Genome Analyzer. Methods for data analysis and visualization can be found in the Supplemental Experimental Procedures. Normal human liver was obtained from a patient who had undergone partial liver resection for colorectal cancer metastasis following local research ethics committee approval.
Genome-wide BrdU-IP/Chip
RT was profiled and analyzed as described (Hiratani et al., 2008) using a human whole-genome triplex microarray with one probe every 2.5 kb (Roche Nimble-Gen, 090210_HG18_WG_CGH_v3.1_HX3; 719,690 oligonucleotide probes). A complete replication-timing data set for all probes is downloadable and is graphically displayed at http://www.replicationdomain.org (Weddington et al., 2008).
Laser Scanning Cytometry
Laser Scanning Cytometry was performed on an iCys Research Imaging Cytometer (CompuCyte, Cambridge, MA). The intensity of immunolabeled histone marks was measured over the nuclei stained with DAPI.
Supplementary Material
Acknowledgments
We thank R. Didier for assistance with flow cytometry; A. Straight (CENPA Ab), D. Reinberg (H3K27me2/3 Ab), M. Ferguson-Smith (sorted chromosomes), and I. Roberts (a-satellite vector) for reagents; A. Young, M. Hoare, J. Nakayama, and K. Hamada for critical reading of the manuscript; L. Blackburn for editing; and the CRI core facilities (Genomics, Bioinformatics, and Microscopy) for technical support. We thank the Cambridge NIHR Biomedical Research Centre, who fund and support the Addenbrooke’s Hospital Tissue Bank. This work was supported by the University of Cambridge, Cancer Research UK, Hutchison Whampoa, the UK Medical Research Council, the Human Frontier Science Program (M.N. and R.S.), JST CREST and grants-in-aid from the MEXT of Japan (H.K. and Y.H.-T.), the Canadian Institutes of Health Research (D.P.B.-J.), and National Institute of General Medicine grants GM083337 and GM085354 (D.M.G.). D.P.B.-J. holds the Canada Research Chair in Molecular and Cellular Imaging.
Footnotes
ACCESSION NUMBERS
Data described here have been deposited in the GEO database with accession numbers GSE38448 (ChIP-seq and microarray) and GSE38460 (RT).
Supplemental Information includes Supplemental Experimental Procedures, five figures, and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.molcel.2012.06.010.
References
- Adams PD. Remodeling of chromatin structure in senescent cells and its potential impact on tumor suppression and aging. Gene. 2007;397:84–93. doi: 10.1016/j.gene.2007.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agger K, Cloos PAC, Rudkjaer L, Williams K, Andersen G, Christensen J, Helin K. The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A-ARF locus in response to oncogene- and stress-induced senescence. Genes Dev. 2009;23:1171–1176. doi: 10.1101/gad.510809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed K, Dehghani H, Rugg-Gunn P, Fussner E, Rossant J, Bazett-Jones DP. Global chromatin architecture reflects pluripotency and lineage commitment in the early mouse embryo. PLoS ONE. 2010;5:e10531. doi: 10.1371/journal.pone.0010531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ait-Si-Ali S, Guasconi V, Fritsch L, Yahi H, Sekhri R, Naguibneva I, Robin P, Cabon F, Polesskaya A, Harel-Bellan A. A Suv39h-dependent mechanism for silencing S-phase genes in differentiating but not in cycling cells. EMBO J. 2004;23:605–615. doi: 10.1038/sj.emboj.7600074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S. Visualization of genomic data with the Hilbert curve. Bioinformatics. 2009;25:1231–1235. doi: 10.1093/bioinformatics/btp152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–124. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- Barradas M, Anderton E, Acosta JC, Li S, Banito A, Rodriguez-Niedenführ M, Maertens G, Banck M, Zhou M-M, Walsh MJ, et al. Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes Dev. 2009;23:1177–1182. doi: 10.1101/gad.511109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, McMahon S, Karlsson EK, Kulbokas EJ, 3rd, Gingeras TR, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120:169–181. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Campos EI, Reinberg D. Histones: annotating chromatin. Annu Rev Genet. 2009;43:559–599. doi: 10.1146/annurev.genet.032608.103928. [DOI] [PubMed] [Google Scholar]
- Chadwick BP. Variation in Xi chromatin organization and correlation of the H3K27me3 chromatin territories to transcribed sequences by microarray analysis. Chromosoma. 2007;116:147–157. doi: 10.1007/s00412-006-0085-1. [DOI] [PubMed] [Google Scholar]
- Chadwick BP, Willard HF. Multiple spatially distinct types of facultative heterochromatin on the human inactive X chromosome. Proc Natl Acad Sci USA. 2004;101:17450–17455. doi: 10.1073/pnas.0408021101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemson CM, Hall LL, Byron M, McNeil J, Lawrence JB. The X chromosome is organized into a gene-rich outer rim and an internal core containing silenced nongenic sequences. Proc Natl Acad Sci USA. 2006;103:7688–7693. doi: 10.1073/pnas.0601069103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremer T, Cremer M. Chromosome territories. Cold Spring Harb Perspect Biol. 2010;2:a003889. doi: 10.1101/cshperspect.a003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui K, Zang C, Roh TY, Schones DE, Childs RW, Peng W, Zhao K. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell. 2009;4:80–93. doi: 10.1016/j.stem.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filion GJ, van Bemmel JG, Braunschweig U, Talhout W, Kind J, Ward LD, Brugman W, de Castro IJ, Kerkhoven RM, Bussemaker HJ, van Steensel B. Systematic protein location mapping reveals five principal chromatin types in Drosophila cells. Cell. 2010;143:212–224. doi: 10.1016/j.cell.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser P, Bickmore W. Nuclear organization of the genome and the potential for gene regulation. Nature. 2007;447:413–417. doi: 10.1038/nature05916. [DOI] [PubMed] [Google Scholar]
- Funayama R, Saito M, Tanobe H, Ishikawa F. Loss of linker histone H1 in cellular senescence. J Cell Biol. 2006;175:869–880. doi: 10.1083/jcb.200604005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal SIS, Rice JC. Regulation of heterochromatin by histone methylation and small RNAs. Curr Opin Cell Biol. 2004;16:230–238. doi: 10.1016/j.ceb.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Hawkins RD, Hon GC, Lee LK, Ngo Q, Lister R, Pelizzola M, Edsall LE, Kuan S, Luu Y, Klugman S, et al. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell. 2010;6:479–491. doi: 10.1016/j.stem.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi-Takanaka Y, Yamagata K, Nozaki N, Kimura H. Visualizing histone modifications in living cells: spatiotemporal dynamics of H3 phosphorylation during interphase. J Cell Biol. 2009;187:781–790. doi: 10.1083/jcb.200904137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiratani I, Ryba T, Itoh M, Yokochi T, Schwaiger M, Chang C-W, Lyou Y, Townes TM, Schübeler D, Gilbert DM. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 2008;6:e245. doi: 10.1371/journal.pbio.0060245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiratani I, Takebayashi S-I, Lu J, Gilbert DM. Replication timing and transcriptional control: beyond cause and effect—part II. Curr Opin Genet Dev. 2009;19:142–149. doi: 10.1016/j.gde.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, Alekseyenko AA, Schwartz YB, Minoda A, Riddle NC, Ernst J, Sabo PJ, Larschan E, Gorchakov AA, Gu T, et al. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature. 2011;471:480–485. doi: 10.1038/nature09725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, Barrera LO, Zheng M, Qu C, Singer MA, Richmond TA, Wu Y, Green RD, Ren B. A high-resolution map of active promoters in the human genome. Nature. 2005;436:876–880. doi: 10.1038/nature03877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolasinska-Zwierz P, Down T, Latorre I, Liu T, Liu XS, Ahringer J. Differential chromatin marking of introns and expressed exons by H3K36me3. Nat Genet. 2009;41:376–381. doi: 10.1038/ng.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachner M, O’Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410:116–120. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T. Beyond the sequence: cellular organization of genome function. Cell. 2007;128:787–800. doi: 10.1016/j.cell.2007.01.028. [DOI] [PubMed] [Google Scholar]
- Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science. 2001;292:110–113. doi: 10.1126/science.1060118. [DOI] [PubMed] [Google Scholar]
- Narita M, Nũnez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–716. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- Narita M, Narita M, Krizhanovsky V, Nũnez S, Chicas A, Hearn SA, Myers MP, Lowe SW. A novel role for high-mobility group a proteins in cellular senescence and heterochromatin formation. Cell. 2006;126:503–514. doi: 10.1016/j.cell.2006.05.052. [DOI] [PubMed] [Google Scholar]
- Nielsen SJ, Schneider R, Bauer UM, Bannister AJ, Morrison A, O’Carroll D, Firestein R, Cleary M, Jenuwein T, Herrera RE, Kouzarides T. Rb targets histone H3 methylation and HP1 to promoters. Nature. 2001;412:561–565. doi: 10.1038/35087620. [DOI] [PubMed] [Google Scholar]
- Pauler FM, Sloane MA, Huang R, Regha K, Koerner MV, Tamir I, Sommer A, Aszodi A, Jenuwein T, Barlow DP. H3K27me3 forms BLOCs over silent genes and intergenic regions and specifies a histone banding pattern on a mouse autosomal chromosome. Genome Res. 2009;19:221–233. doi: 10.1101/gr.080861.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plath K, Fang J, Mlynarczyk-Evans SK, Cao R, Worringer KA, Wang H, de la Cruz CC, Otte AP, Panning B, Zhang Y. Role of histone H3 lysine 27 methylation in X inactivation. Science. 2003;300:131–135. doi: 10.1126/science.1084274. [DOI] [PubMed] [Google Scholar]
- Ryba T, Hiratani I, Lu J, Itoh M, Kulik M, Zhang J, Schulz TC, Robins AJ, Dalton S, Gilbert DM. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res. 2010;20:761–770. doi: 10.1101/gr.099655.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D, Wilson MD, Spyrou C, Brown GD, Hadfield J, Odom DT. ChIP-seq: using high-throughput sequencing to discover protein-DNA interactions. Methods. 2009;48:240–248. doi: 10.1016/j.ymeth.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S, Janknecht R. Activation of androgen receptor by histone demethylases JMJD2A and JMJD2D. Biochem Biophys Res Commun. 2007;359:742–746. doi: 10.1016/j.bbrc.2007.05.179. [DOI] [PubMed] [Google Scholar]
- Shopland LS, Lynch CR, Peterson KA, Thornton K, Kepper N, Hase JV, Stein S, Vincent S, Molloy KR, Kreth G, et al. Folding and organization of a contiguous chromosome region according to the gene distribution pattern in primary genomic sequence. J Cell Biol. 2006;174:27–38. doi: 10.1083/jcb.200603083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva J, Mak W, Zvetkova I, Appanah R, Nesterova TB, Webster Z, Peters AHFM, Jenuwein T, Otte AP, Brockdorff N. Establishment of histone h3 methylation on the inactive X chromosome requires transient recruitment of Eed-Enx1 polycomb group complexes. Dev Cell. 2003;4:481–495. doi: 10.1016/s1534-5807(03)00068-6. [DOI] [PubMed] [Google Scholar]
- Simonis M, Klous P, Splinter E, Moshkin Y, Willemsen R, de Wit E, van Steensel B, de Laat W. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C) Nat Genet. 2006;38:1348–1354. doi: 10.1038/ng1896. [DOI] [PubMed] [Google Scholar]
- Sinclair P, Bian Q, Plutz M, Heard E, Belmont AS. Dynamic plasticity of large-scale chromatin structure revealed by self-assembly of engineered chromosome regions. J Cell Biol. 2010;190:761–776. doi: 10.1083/jcb.200912167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Q, Smith AD. Identifying dispersed epigenomic domains from ChIP-Seq data. Bioinformatics. 2011;27:870–871. doi: 10.1093/bioinformatics/btr030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman RE, Day N, Noble WS, Stamatoyannopoulos JA. Identification of higher-order functional domains in the human ENCODE regions. Genome Res. 2007;17:917–927. doi: 10.1101/gr.6081407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojer P, Reinberg D. Facultative heterochromatin: is there a distinctive molecular signature? Mol. Cell. 2007;28:1–13. doi: 10.1016/j.molcel.2007.09.011. [DOI] [PubMed] [Google Scholar]
- Weddington N, Stuy A, Hiratani I, Ryba T, Yokochi T, Gilbert DM. Replication Domain: a visualization tool and comparative database for genome-wide replication timing data. BMC Bioinformatics. 2008;9:530. doi: 10.1186/1471-2105-9-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young ARJ, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JFJ, Tavaré S, Arakawa S, Shimizu S, Watt FM, Narita M. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23:798–803. doi: 10.1101/gad.519709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Chen W, Adams PD. Molecular dissection of formation of senescence-associated heterochromatin foci. Mol Cell Biol. 2007;27:2343–2358. doi: 10.1128/MCB.02019-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.