

Abstract
We have developed a coculture system that establishes DLK+ fetal hepatic progenitors as the authentic supportive cells for expansion of hematopoietic stem (HSCs) and progenitor cells. In 1-week cultures supplemented with serum and supportive cytokines, both cocultured DLK+ fetal hepatic progenitors and their conditioned medium supported rapid expansion of hematopoietic progenitors and a small increase in HSC numbers. In 2- and 3-week cultures DLK+ cells, but not their conditioned medium, continuously and significantly (>20-fold) expanded both hematopoietic stem and progenitor cells. Physical contact between HSCs and DLK+ cells was crucial to maintaining this long-term expansion. Similar HSC expansion (approximately sevenfold) was achieved in cocultures using a serum-free, low cytokine-containing medium. In contrast, DLK− cells are incapable of expanding hematopoietic cells, demonstrating that hepatic progenitors are the principle supportive cells for HSC expansion in the fetal liver.
During early development, hematopoietic stem cells (HSCs) are found successively in multiple embryonic sites [1,2]. In vertebrates, the aorta-gonad-mesonephros (AGM) region was identified as a major initial site for de novo generation of adult type HSCs [3]. Additional sites such as the placenta, vitelline and umbilical vessels, and the yolk sac also harbor adult HSCs during early stages of development [4–6]. Following the generation of definitive HSCs, fetal liver quickly becomes the unique center for hematopoietic stem and progenitor cell expansion. In the mouse, HSCs start to migrate into the fetal liver around embryonic day 11.5. Between embryonic day 12.5 (E12.5) and E16.5, they not only self-renew to expand in numbers, but also undergo rapid differentiation to generate vast numbers of hematopoietic progenitors [1]. The number of competitive repopulating units in each fetal liver increases by 38-fold during these 5 days [7]. After birth, HSCs migrate into bone marrow and soon became quiescent. They self-renew only to replenish the ones that are lost owing to differentiation, and a portion of adult bone marrow HSCs are extremely quiescent throughout adulthood [8,9].
A central theme of HSC biology is that the fate of HSCs is controlled by their surrounding microenvironmentsdthe HSC niches [10,11]—and much effort has been devoted to understanding the HSC niches in adult bone marrow. Many types of cells, including osteoblasts [12,13], endothelial cells [14], leptin receptor-expressing perivascular cells [15], reticular CAR cells [16], Nestin+ mesenchymal stem cells [17], and nonmyelinated Schwann cells [18], are located adjacent to HSCs and might regulate HSC functions. In stark contrast, little is known of the cells that support HSC expansion in the fetal liver.
Stem cell factor (SCF) is a key membrane-bound growth factor that meditates the interaction between stromal cells and its receptor, c-Kit, on the surfaces of HSCs [19–21]. Using flow cytometry, we purified fetal liver SCF+DLK+ cells, which consist of 1%–2% of total E15.5 liver cells [22]. These are the major cell type in the fetal liver that expresses several known stem cell supportive cytokines, including Thrombopoietin (TPO), SCF, and CXCL12[23,24]. SCF+DLK+ cells are a subset of fetal hepatic progenitors that express high levels of α-fetoprotein (AFP) and albumin (ALB), two specific markers of fetal hepatic progenitor cells [22]. We therefore hypothesized that fetal liver hepatic progenitors are the major supportive stromal cells for HSC expansion.
In this study, we report the establishment of a coculture system using DLK+ fetal liver hepatic progenitors that closely mimics hematopoietic stem and progenitor cell expansion in the fetal liver. These hepatic progenitors support the rapid expansion of hematopoietic progenitors in 1-week cocultures and significantly expand HSCs during 2- and 3-week cocultures. Our results provide direct proof that hepatic progenitors are the principle supportive cells for the expansion of hematopoietic stem and progenitors in the fetal liver and establish an ex vivo system for investigating the details of HSC function in the developing embryo.
Methods
Mice
CD45.2 and CD45.1 mice of C57BL/6 background were purchased from the Jackson Laboratory or the National Cancer Institute, respectively, and were maintained at the animal facility of the Whitehead Institute for Biomedical Research. CD45.2 Tg(AFP-GFP) mice were gifts from Dr. Margaret Baron (Mt. Sinai School of Medicine). All animal experiments were performed with the approval of the Massachusetts Institute of Technology Committee on Animal Care.
Magnetic bead purification of fetal liver DLK+ cells
Embryonic day 15.5 fetal liver cells were dispersed into single cells by pipetting and treated with collagenase and DNAase I as described previously [25]. Ammonium chloride (StemCell Technologies, Vancouver, BC, Canada) was used to lyse erythrocytes and the remaining cells were suspended in Hank’s balanced solution (StemCell Technologies) with 2% fetal bovine serum and incubated with CD16/32 antibody (eBioscience, San Diego, CA, USA) to block nonspecific binding. The cells were next incubated with FITC-conjugated DLK1 antibody (MBL International, Woburn, MA, USA) and anti-FITC magnetic beads (Miltenyi Biotec, Auburn, CA, USA) for 15 minutes each. DLK+ cells were separated using an autoMACS Magnetic Separator (Miltenyi) using a double-column setting.
FACS sorting of bone marrow HSCs
We purified SLAM+ (CD150+CD48−CD41−) HSCs according to a previous publication, with some modifications [14]. Bone marrow cells were flushed from the femur and tibia from 8– 10-week-old mice and filtered through a 70-μm nylon strainer (BD Biosciences, Franklin Lakes, NJ, USA). Cells were treated with ammonium chloride, and lineage positive cells were depleted using a mouse hematopoietic progenitor (stem) cell enrichment kit (BD Biosciences). The remaining lineage-negative cells were incubated with APC-conjugated CD150 (BioLegend, San Diego, CA, USA), FITC conjugated CD48 (BioLegend) and FITC conjugated CD41 (eBioscience) antibodies for 15 min. Single cells with the surface phenotype of CD150+CD48−CD41− were isolated using a BD Biosciences FACSAria1 cell sorter.
Coculture with DLK+ fetal hepatic progenitors
For 1-week coculture experiments with DLK+ cells in serum-containing medium, 5000 purified DLK+ cells were cultured in one well of a 96-well gelatin-coated plate (BD Biosciences) containing 170 mL Iscove’s modified Dulbecco’s medium (IMDM) with 10% fetal bovine serum, 50 μmol/L β-mercaptoethanol, and penicillin-streptomycin (Life Technologies, Carlsbad, CA, USA) added. The plates were incubated at 37°C for 2 days to allow hepatic cells to attach to the bottom of the wells and then carefully washed to remove all the cells that did not attach to the plates. In initial experiments, 2-day conditioned medium was filtered using 0.22-μm syringe-driven filter units (Millipore, Billerica, MA, USA) and added back to the wells. In later experiments, 170 μL fresh medium was added into each well directly, because we had shown that conditioned medium from DLK+ cells was dispensable for ex vivo HSC expansion. In either case, a cocktail of cytokines including 50 ng/mL SCF, 20 ng/mL TPO, and 50 ng/mL FLT3L (all from Peprotech, Rocky Hill, NJ, USA) supplemented the cultures. One hundred SLAM+ cells were sorted directly into each well and incubated at 37°C for 7 days before transplantation.
For 2-week coculture experiments, cells expanded from 50 SLAM+ cells after a 1-week coculture were transferred to one well of a six-well gelatin-coated plate (BD Biosciences) containing 125,000 purified DLK+ cells in 2.5 mL IMDM plus 10% FBS supplemented with the cytokine cocktail. These DLK+ cells have previously been cultured for 2 days in IMDM plus 10% serum medium and carefully washed as described earlier. For week 3 of coculture, the cells from 2-week cocultures were diluted 40-fold and transferred to freshly prepared six-well gelatin-coated plates containing 125,000 DLK+ cells per well.
Two-week coculture with DLK+ cells in serum-free, low-cytokine medium was performed in a similar way, except that greater numbers of DLK+ cells were plated into each well. For the first week of the coculture, 170 μL StemSpan medium (StemCell Technologies) supplemented with 10 ng/mL SCF and 2 ng/mL TPO and penicillin-streptomycin was added onto a washed DLK+ cell layer (20,000 cells/well) and cocultured with sorted SLAM+ cells (200 cells/well) in 96-well gelatin-coated plates. For week 2, the progeny of 200 SLAM+ cells were transferred to one well of a six-well gelatin-coated plate coated with 300,000 washed DLK+ cells. For each transplantation experiment, cells from at least three individual wells were pooled together.
Competitive repopulating analysis of HSC activity
For competitive repopulation analysis, 10 SLAM+CD45.1 HSCs without culture or the progeny 10 SLAM+ cells after culture were mixed with 100,000 freshly isolated total bone marrow CD45.2+ cells (unless otherwise notified) and injected intravenously into mice irradiated with a lethal dose of 1000 rad. Peripheral blood samples were collected at indicated times after transplantation and analyzed with antibodies against CD45.1 (donor), CD45.2 (recipient), B220 (B cells), Thy1.2 (T cells), Gr-1 (granulocytes), and CD11b (granulocytes and monocytes).
For competitive secondary transplantations, the bone marrow cells from recipient mice were harvested 4 months after transplantation. Five million total bone marrow cells from each recipient mouse were injected directly into one lethally irradiated secondary recipient mouse.
Immunocytochemistry and microscopy
The method for the immunocytochemistry study of purified DLK+ cells is the same as that described previously [22]. DLK+ cells purified by magnetic beads method were stained with antibodies to DLK1 (MBL International) together with antibodies to ALB (Abcam, Cambridge, UK), AFP (Santa Cruz Biotechnology, Santa Cruz, CA, USA), or biotin-conjugated antibody to SCF (AbD Serotec, Kidlington, UK). Secondary antibodies were DyLight 488 conjugated donkey anti-rat antibody, Rhodamine Red X (RRX)-conjugated donkey anti-goat or anti-rabbit antibodies, or RRX-conjugated streptavidin (all from Jackson Immunoresearch). DAPI was added into the mounting solution. A Perkin-Elmer UltraView spinning disk confocal microscope was used to view the fluorescent signals.
Images of cultured DLK+ cells purified from the fetal livers of Tg(AFP-GFP) mice were obtained using a Nikon Eclipse TS100 fluorescence microscope (original magnification ×100) and taken using SPOT software.
Results
Establishment of a coculture system that can expand HSCs
The most direct way to prove that fetal hepatic progenitors are bona fide supportive cells for HSC expansion is to establish a coculture assay that expands HSCs ex vivo. Initially, we cocultured FACS-sorted SCF+DLK+ cells with purified SLAM+ (CD150+CD48−CD41−) [14] fetal liver HSCs for 5 days. Although SCF+DLK+ cells were able to maintain fetal liver HSCs numbers in short-term ex vivo culture, as judged by transplantation experiments, there was no net expansion of HSCs [22]. Several factors likely contributed to this lack of HSC expansion. First, more than 90% of sorted SCF+DLK+ cells died within 24 hours of culture, presumably because of the stress caused by FACS sorting. To increase the numbers and survival of hepatic progenitors, we used magnetic beads to purify DLK+ cells from the fetal liver (Supplementary Figure 1A, online only, available at www.exphem.org). Using collagenase to treat fetal liver cells before magnetic bead selection, we were able to isolate DLK+ cells to greater than 70% purity. (Supplementary Figure 1A, online only, available at www.exphem.org). Most of the contaminating cells appeared to be hematopoietic, because they comprised of vast majority of the cells in the fetal liver. This fraction comprised approximately 5% of total E15.5 fetal liver cells, and only a fraction (~56% and 24%) express ALB and SCF, respectively (Fig. 1A). However, almost every DLK+ cell is also AFP+ (Fig. 1A) and is thus a hepatic cell, consistent with previous studies on fetal rat liver [26]. Furthermore, quantitative polymerase chain reaction (qPCR) analysis shows that DLK+ cells are highly enriched for expression of AFP and ALB, two specific markers for hepatic cells. Markers for other cell types in the fetal liver such as endothelial cells, mesenchymal cells, Kupffer cells, and bile duct epithelial cells are not enriched (Fig. 1B). Therefore, purified fetal liver DLK+ cells are specifically enriched for hepatic progenitor cells.
Figure 1.

Purified DLK+ fetal hepatic progenitors support the growth of hematopoietic cells in ex vivo culture. (A) Double immunocytochemistry for DLK (green) and ALB, AFP, or SCF (red) expression in fetal liver DLK+ cells purified by magnetic beads. Only a fraction of DLK+ cells express SCF and ALB, but almost every DLK+ cell is also AFP+. (B) Relative enrichment of mRNAs encoding markers for hepatic progenitors (AFP, ALB), endothelial cells (VECAD, TIE2), mesenchymal cells (α-SMA, VIM, FSP1), Kupffer cells (F4/80) and bile duct epithelial cells (CK-19, CDH1) in purified DLK+ versus DLK− cells. The mRNA level of each gene was determined by qPCR and normalized against ribosomal RNA. The ratio of mRNA levels in DLK+ cells versus DLK− cells is plotted (n 5 3 with SEM). (C) Hematopoietic cells contaminating the DLK+ cells expanded adjacent to fetal hepatic cells in serum containing medium; 25,000 DLK+ cells purified from Tg (AFP-GFP) mice were cultured in one well of a 96-well plate in IMDM medium plus 10% serum. The survival of fetal hepatic progenitors in ex vivo culture was tracked by their expression of GFP (bottom panels). Clusters of small, round hematopoieitc cells start to grow adjacent to GFP+ hepatic cells at day 10 and continue to expand through day 14 (Differential Interference Contrast microscopy; top panels).
Hepatocytes are notoriously difficult to culture; therefore, it is important to find a condition that can both support the expansion of HSCs and sustain hepatic progenitors for an extended period of time. We first determined the survival of purified DLK+ fetal hepatic progenitors in several culture media; we used fetal liver DLK+ cells purified from Tg(AFP-GFP) mice so that live fetal liver hepatic progenitors can be identified by their expression of GFP protein. We found that hepatic progenitors survived best in medium with serum and reasonably well in serum-free StemSpan SFEM medium (StemCell Technologies;Supplementary Figure 1B, online only, available at www. exphem.org), both of which are also capable of supporting hematopoietic stem or progenitor cells with the addition of supportive cytokines [27–29]. Growing in regular cell culture plates, GFP+ hepatic cells form cell clusters of various sizes (Supplementary Figure 1B, top row, online only, available at www.exphem.org). Growing on gelatin-coated plates, the cells spread and form monolayers (Supplementary Figure 1B, bottom row, online only, available at www.exphem.org).
At E15.5, more than 90% of fetal liver cells are hematopoietic; therefore, purified DLK+ cells inevitably contain some hematopoietic cells. Without supportive cytokines, these cells cannot survive in either serum or StemSpan medium. However, when we cultured purified DLK+ cells in serum-containing medium for 10 days, clusters of small and round hematopoietic cells started to appear adjacent to GFP-positive hepatic cells and continued expanding through day 14 (Fig. 1C). In contrast, we found little accumulation of hematopoietic cells around GFP+ cells in serum-free Stem-Span medium (Supplementary Figure 1C, online only, available at www.exphem.org). This result indicates that fetal hepatic progenitors have the ability to support some hematopoietic stem or progenitor cells for an extended period of time in serum-containing medium during ex vivo culture.
As a result, we chose to culture DLK+ cells in serum-containing medium supplemented with 50 ng/mL SCF, 20 ng/mL TPO, and 50 ng/mL FLT3L (STF medium [IMDM + 10% bovine serum albumin supplemented with previously mentioned cytokines]) to support expansion of HSCs. DLK+ cells were plated on gelatin-coated plates for 2 days to allow the hepatic cells to attach and spread (Supplementary Figure 1B, online only, available at www.exphem.org), after which the culture plates were washed carefully to remove nonadherent cells. Another concern was that ex vivo–cultured DLK+ cells lose expression of many cytokines, including IGF2, DLK1, and Angptl3. (Supplementary Figure 2, online only, available at www.exphem.org). To maximize the chance of HSC-supportive factors remaining in the culture, we also added back-filtered, 2-day conditioned medium (CM) from DLK+ cells to some of the cocultures (Fig. 2A).
Figure 2.

DLK+ fetal hepatic progenitors promote a rapid expansion of hematopoietic progenitors and HSCs during short-term culture. (A–D) One-week coculture with fetal liver DLK+ cells resulted in a modest expansion of HSCs. (A) Scheme of the coculture experiment: 25,000 purified DLK+ cells (purified without collagenase treatment) were cultured in serum-containing medium on gelatin coated plates for 2 days. The plates were washed, and the conditioned medium (CM) was added back together with 50 ng/mL SCF, 20 ng/mL TPO, and 50 ng/mL FLT3L. One hundred CD45.1+SLAM+ HSCs were placed into each well and cocultured for 1 week. The progeny of 50 CD45.1+SLAM+ cells were transplanted together with 200,000 freshly isolated CD45.2+ bone marrow cells into each recipient mouse. Fifty sorted HSCs were also transplanted directly (no culture control) or cultured for 1 week in serum-containing medium with cytokines (cytokines-only control). (B) HSCs cocultured with DLK+ cells showed an increase of repopulating activity over the no-culture control (p < 0.002) and cytokines-only control (p < 0.05). Each data point in the figure represents the percentage repopulation of one recipient mouse, which is calculated as the percentage of CD45.1+ cells in the peripheral blood divided by total number of CD45.1+ plus CD45.2+ cells. (n = 5–10). (C) Scatter plot showing the percentage of CD45.1+ donor -derived B cells (B220+), T cells (Thy1.2+), and myeloid cells (Gr-1+/CD11b+) in the peripheral blood 4 months after transplantion. (D) An exemplary FACS analysis of donor-derived (CD45.1+) multilineage reconstitution at 6 months after transplantation in mice injected with HSCs cocultured with DLK+ cells. (E–G) Two-day conditioned medium (CM) from DLK+ cells rapidly expanded hematopoietic progenitors and slightly expanded HSCs. CD45.1+SLAM+ HSCs were cocultured for 1 week with 5000 DLK+ cells (collagenase treated) with or without adding CM, or cultured in CM alone. (E) The numbers of hematopoietic cells expanded from a single SLAM+ cell. (F) The numbers of hematopoietic progenitors generated from one SLAM+ cell determined by colony-forming assays as described previously [37].
Thus, sorted SLAM+ bone marrow HSCs from CD45.1 mice were cocultured with DLK+ cells from CD45.2 mice with CM for 1 week, and the nonadherent hematopoietic cells derived from 50 SLAM+ cells were transplanted into CD45.2 recipient mice. HSCs cocultured with DLK+ cells showed a clear increase of donor-derived peripheral nucleated blood cells relative to uncultured SLAM cells (p < 0.002) at 1 and 4 months after transplantation (Fig. 2B). Importantly, the percentages of donor-derived B, T, and myeloid cells were all increased in the cocultured cells (Fig. 2C and 2D). These results suggest that there is an expansion of both short-term and long-term reconstituting HSCs after coculture.
We also tested the ability of DLK+ cells or their conditioned medium to expand hematopoietic progenitor cells. After 1 week, SLAM+ cells cultured in STF medium (cytokines only control) increased in number by 90-fold (Fig. 2E). When SLAM+ cells were cultured in CM or with DLK+ cells, an additional fourfold to ninefold expansion was seen. Colony-forming assays showed that both DLK+ cells and their conditioned medium promoted a 10- to 30-fold increase of all types of hematopoietic progenitors, relative to culture only with cytokines (Fig. 2F). These results suggest that DLK+ fetal hepatic progenitors can both expand HSCs and promote their differentiation into hematopoietic progenitors in ex vivo culture.
DLK+ cells support long-term expansion of HSCs
To examine whether HSCs can be expanded by DLK+ cells beyond a 1-week culture, we extended the coculture experiment to 3 weeks. Cells were transferred onto a new layer of DLK+ cells at the beginning of each week. SLAM+ cells cultured in CM alone expanded rapidly at the beginning, but proliferation slowed at the end of week 2 and then halted (Fig. 3A and 3B). In contrast, HSCs cocultured with DLK+ cells with or without CM continued to expand in numbers for 3 weeks (Fig. 3A and 3B). At the end of week3, one SLAM+ cell cocultured with DLK+ stromal cells produced nearly 3 million progeny—a more than 200-fold increase over HSCs cultured with cytokines alone and approximately 100-fold higher than those cultured in CM.
Figure 3.

Fetal DLK+ hepatic progenitors support long-term expansion of HSCs. The coculture experiment shown in Figures 2E–2G was extended to 3 weeks. (A) Numbers of cells generated from one SLAM+ HSC after 1, 2, and 3 weeks of culture with DLK+ cells plus CM, with DLK+ cells alone, with CM alone, or with cytokines only. (B) Representative phase contrast micrographs of the progeny of 50–100 SLAM+ HSCs cultured with DLK+ cells, CM, or cytokines alone for 2 weeks in one well of a six-well plate. Also shown are DLK+ cells cultured alone with no HSCs added (bottom right). (C) Scatter plots showing the percentage of donor-derived CD45.1+ peripheral blood cells in mice transplanted with the progeny of 10 SLAM+ HSCs after a 2-week culture (n = 7–8). (D) The percentage of donor-derived blood cells in mice at successive times after transplantation with the progeny of 10 SLAM+ HSCs cultured with DLK+ cells, CM, or cytokines only. The data for months 2 and 4 after transplantation are the same as in (C). The average percentage repopulation of each group of recipient mice at the indicated times after transplantation was calculated and shown as trend lines. DLK+ cells, but not CM, expanded long-term HSCs that maintain their repopulating ability after 6 months. (E) Three-week coculture further expanded HSCs. Comparison of the percentage reconstitution from the progeny of one SLAM+ cell after a 2-week culture (n = 8) with those after 3 weeks of coculture (n = 5). One mouse transplanted with cells expanded from 3 weeks of coculture died because of anemia between 1 and 2 months after transplantation (open circle).
To test whether long-term coculture with DLK+ cells further expanded HSCs, we transplanted the progeny of 10 SLAM+ cells after a 2-week culture. Only half of the mice transplanted with 10 uncultured SLAM+ cells were able to reconstitute irradiated mice (Fig. 3C). In contrast, every mouse transplanted with HSCs cocultured with DLK+ cells had a significant amount (>2%) of donor-derived nucleated blood cells at 1 and 4 months after transplantation, whether or not CM was included, indicating a clear expansion of HSCs (Fig. 3C). Donor-derived reconstitution in these mice was similar at 6 months after transplant (Fig. 3D). The recipient mice were kept for more than 10 months with no incidence of leukemia. In contrast, HSCs cultured in CM for 2 weeks only slightly increased their repopulating activity at 1 month after transplant. The percentage of donor-derived cells in peripheral blood continued to decline over time, and there was no longer a noticeable difference between HSCs cultured in CM and cytokines only at 6 month after transplantation (Fig. 3D). Therefore, HSCs cultured in CM lost their long-term repopulating ability after 2 weeks and were not able to continue expansion during week 3. In contrast, HSCs continued to expand at week 3 when cocultured with DLK+ cells (Fig. 3E). Recipient mice transplanted with HSCs cocultured for 3 weeks have high levels (between 28% and 85%) of donor-derived blood cells at one month after transplantation, indicating a large expansion of short-term HSCs. The percentage of donor-derived peripheral blood cells decreased over time, but were still present in significant levels (between 4% and 23%) in every recipient mouse at both 4 and 6 months after transplantation (Fig. 3E). Because all mice transplanted with the progeny of only one SLAM+ cell after a 3-week coculture were reconstituted, we can calculate using Poisson statistics that, compared with uncultured SLAM cell, coculture with DLK+ cells for 3 weeks resulted in a minimum of a 20-fold increase in HSC numbers. These results suggest that although factors secreted by DLK+ cells are capable of promoting HSC expansion in a short-term (1 week) coculture, direct cell-cell contact is required for HSCs to continue their expansion in long-term culture. It is likely that membrane-bound signaling molecules on the surface of DLK+ cells are important to maintain HSCs in an undifferentiated state.
Coculture with DLK+ cells in serum-free, low-cytokine medium expanded HSCs that can long-term self-renew and efficiently reconstitute all blood lineages
The vast majority of mice transplanted with HSCs expanded by long-term coculture with DLK+ cells remained healthy at 10 months after transplantation. However, occasional transplanted mice died less than 2 months after transplantation. These dead mice had a high percentage of donor-derived cells in the peripheral blood at 1 month after transplantation and appeared to be anemic. One example is shown in Figure 3E (open cycle); 85% of blood cells from this mouse were donor derived at 1 month after transplantation. A closer inspection found that all recipient mice exhibited a temporary defect in donor-derived myeloid reconstitution at 2 months after transplantation (Supplementary Figures 3A–3C, online only, available at www.exphem.org). This subtle myeloid, and perhaps also erythroid, reconstituting problem is not caused by the DLK+ cells because SLAM+ cells cultured in medium containing cytokines only also exhibited a similar problem (Supplementary Figures 3D and 3E, online only, available at www.exphem.org). Although the exact reason for this myeloid reconstituting defect is unclear, the prolonged exposure to serum or high levels of mitotic cytokines such as TPO are the major suspects.
Because DLK+ cells are able to survive in serum-free StemSpan medium for an extended period of time (Supplementary Figure 1B, online only, available at www.exphem.org), we tested whether coculture in serum-free, low-cytokine media could allow the expansion of HSCs without compromising their myeloid differentiation potential. We cocultured 100 CD 45.1 SLAM+ HSCs together with CD 45.2+DLK+ cells in StemSpan medium supplemented with either 10 ng/mL SCF or 10 ng/mL SCF plus 2 ng/mL TPO for 2 weeks, followed by bone marrow transplantation. In both conditions, donor-derived blood cells were present in all recipient mice 4 months after transplantation (Fig. 4A). We found that HSCs cultured in serum-free, low-cytokine medium no longer have myeloid reconstitution defects at 2 months after transplantation (Supplementary Figure 3F, online only, available at www.exphem.org) and these expanded HSCs could efficiently reconstitute B cells, T cells, and myeloid lineages (Fig. 4B). We next extracted the bone marrow cells from each recipient mouse and transplanted them into one secondary recipient mouse. We found that most of the secondary recipients contained significant levels of donor-derived cells in their peripheral blood 3 months after secondary transplantation (Fig. 4A). This result indicates that most of the expanded HSCs still maintain truly long-term self-renewing ability. These experiments suggest that in proper culture conditions, DLK+ cells are capable of expanding HSCs that can both long-term self-renew and efficiently reconstitute all blood lineages.
Figure 4.

A serum-free, low cytokine coculture system expands HSCs with normal lymphoid and myeloid reconstituting ability. (A) One hundred SLAM+ CD45.1 HSCs were cocultured with 10,000 CD45.2 DLK+ cells for the first week and with 300,000 DLK+ cells for the second week in a serum-free medium supplemented with 10 ng/mL SCF or 10 ng/mL SCF plus 2 ng/mL TPO. The progeny of 10 HSCs were transplanted into each recipient mouse; nucleated cells in peripheral blood were analyzed after 1 and 4 months. After 4 months, bone marrow cells from each recipient mouse were transplanted into secondary recipients. Significant levels of donor-derived blood cells can be found in most (6 of 7) of the secondary recipient mice at 3 months after transplantation. (B) Representative FACS analysis of donor-derived multilineage reconstitution 4 months after first transplantation. (C–E) SLAM+ cells were cocultured with 20,000 DLK+ cells for the first week and with 300,000 DLK+ cells for the second week (A) in serum-free medium supplemented with 10 ng/mL SCF and 2 ng/mL TPO. (C) Numbers of hematopoietic cells generated from one SLAM+ HSC after a 2-week culture with DLK+ cells or cytokines only in serum-free medium. (D) Scatter plots comparing the percentage of donor-derived peripheral blood cells from mice transplanted with the progeny of 10 uncultured SLAM+ HSCs or 10 cultured for 2 weeks with cytokines alone or cocultured with DLK+ cells in SFLC medium. (E) Multilineage reconstitution of peripheral blood cells at 4 months after transplantation by HSCs cocultured with DLK+ cells for 2 weeks. (F) Limiting dilution assay of the repopulating activity of SLAM+ HSCs before and after coculture. Poisson statistical analysis was used to estimate competitive repopulating units within each condition (n = 9–10 mice transplanted at each dose per condition; 56 mice total). The plot shows the percentage of recipient mice containing less than 1% donor-derived nucleated peripheral blood cells 4 months after transplantation versus the number of SLAM+ cells initially added to the culture.
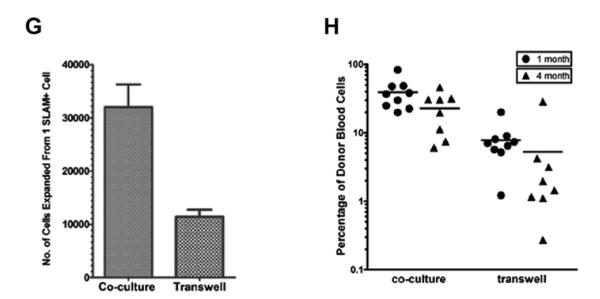
(G–H) Cell-cell contact between HSCs and DLK+ cells is required for the continual expansion of HSCs in SFLC medium. SLAM+ cells were cocultured with DLK+ cells for 1 week. Their progeny were either cocultured with DLK+ cells, or transferred to 0.4-mm porous transwell cell culture inserts placed on top of DLK+ cells, and cultured for 1 week. (G) Direct coculture with DLK+ cells generated approximately threefold the number of hematopoietic cells compared with transwell plates. (H) Transplant assay showing that HSCs cultured in transwell plates had dramatically reduced reconstitution ability at 1 month after transplantation.
We next quantified the level of HSC expansion in serum-free cocultures. Because DLK+ cells do not proliferate in serum-free medium, we could add twice the numbers of DLK+ cells without overcrowding the culture. We compared uncultured HSCs with those cultured in cytokines only (10 ng/mL SCF, 2 ng TPO) and those cocultured with DLK+ cells. After a 2-week coculture, HSCs cocultured with DLK+ cells generated nearly approximately 50-fold more hematopoietic cells than did cultures with cytokines only (Fig. 4C). Transplant assays showed that HSCs cultured with cytokines alone completely lost their repopulating activity after 2 weeks, and those cocultured with DLK+ cells had a clear increase of HSC numbers over uncultured HSCs (Fig. 4D). The expanded HSCs reconstituted B, T, and myeloid lineages equally well (Fig. 4E). We next performed a limiting dilution assay to assess the magnitude of HSC expansion after a 2-week coculture. The frequency of long-term repopulating HSCs in uncultured SLAM+ cells was 1 per 11.4 at 4 months after transplantation (95% confidence interval: 1 in 9 to 1 in 15) compared with 1 per 1.7 (95% confidence interval: 1 in 1.3 to 1 in 2.1) after the 2-week culture (Fig. 4F), determined using L-calc software (Stem Cell Technologies, Vancouver, BC, Canada). Therefore, coculture with DLK+ cells for 2 weeks in serum-free medium results in a sevenfold increase of HSC number. To evaluate the importance of the cell-cell contact between DLK+ cells and HSCs in supporting HSC expansion in serum-free medium, we separated HSCs from DLK+ cells by 0.4 mm transwell cell culture inserts (Millipore, Billerica, MA, USA). These porous cell culture inserts allow secreted protein factors to reach cells cultured inside the transwells, but prevent the direct contact between cells grown inside and outside of the transwells. SLAM+ cells were cocultured with DLK+ cells for 1 week, and their progeny were transferred onto cell culture inserts and placed on top of gelatin-coated plates cultured with DLK+ cells. After 1 week, the number of cells expanded in transwell plates was threefold less than the number of cells expanded by coculture (Fig. 4G). Transplantation assays showed a dramatic decrease in donor-derived reconstitution of peripheral blood cells when HSCs were placed in transwell plates compared to cultures in which the two cell types were in direct contact. (Fig. 4H). Considering these results and our earlier findings (Fig. 3C), it is clear that the contact between HSCs and their hepatic stromal cells is critical for HSC expansion in long-term culture.
DLK− cells failed to expand hematopoietic cells
To eliminate the possibility that the HSC expansion we saw was actually mediated by contaminating DLK− cells, we examined whether DLK− cells could also support HSC and hematopoietic progenitor expansion in ex vivo culture. A 2-week coculture with DLK− cells in serum-free, low-cytokine medium completely failed to significantly expand hematopoietic cell numbers (Fig. 5A and 5B). Similar results were also obtained in serum containing medium (Supplementary Figure 4, online only, available at www.exphem.org). Transplant assays showed that there was no expansion of HSCs (Fig. 5C) when they were cocultured with DLK− cells (Fig. 5C). Therefore, DLK+ fetal hepatic progenitors are the major cell population in the fetal liver that supports expansion of HSCs.
Figure 5.

DLK+ cells are the primary supporting cells for HSC expansion in the fetal liver. Comparison of the ability of DLK+ and DLK− cells in supporting the expansion of hematopoietic cells in serum-free medium. (A) Representative phase contrast images of 200 HSCs cocultured with DLK+ or DLK− cells or with cytokines alone for 2 weeks. (B) Numbers of cells expanded from one SLAM+ HSC after 2 weeks of coculture with DLK+ or DLK− cells or cytokines alone. (C) Comparison of donor-derived reconstitution of peripheral blood cells 1 and 4 months after transplantation with HSCs cocultured with DLK+ cells or DLK− cells for 2 weeks.
Discussion
Because hematopoietic stem cells are mostly quiescent in adults, uncovering the cells that are supportive of HSC expansion at the fetal stage will likely provide keys toward understanding how HSCs are generated and how their self-renewal and expansion can be achieved.
The AGM region is a major site for de novo generation of adult-type HSCs. Oostendorp et al. [30] generated a large collection of immortalized cell lines from the AGM region and from E11 embryonic liver. Cells from the AGM region generated colonies that were capable of maintaining mouse HSCs in long-term in vitro culture [30] as well as expanding human cord blood cobblestone area-forming cells [31]. Importantly, the E11 AGM region generated HSC supportive colonies at a higher frequency than E11 embryonic liver, suggesting that the AGM region provides the most supportive microenvironment for HSCs in the midgestation mouse embryo.
Starting from embryonic day 12, HSCs begin to migrate into the fetal liver and undergo significant expansion. Similar approaches were used to generate more than 200 immortalized cell lines from E14 feta liver [32]. A cell line named AFT024 is capable of supporting transplantable HSCs after 4–7 weeks of ex vivo coculture [33]. AFT024 cells express a-fetoprotein and cytokeratin 8, suggesting that it might derive from a subset of fetal hepatic–endodermal cells [34]. These immortalized cell lines are able to sustain HSCs in culture, but are incapable of expanding their numbers. It is not known whether these cells are part of the HSC expansion niche in vivo and whether their HSC expanding ability is lost during ex vivo culture and immortalization.
Previously, we discovered that a subpopulation of fetal liver AFP+ cells are enriched for the expression of HSC-supportive cytokines such as SCF, TPO, and CXCL12, suggesting that subpopulations of fetal hepatic cells are prime candidates for the cells that are supportive of HSC expansion. Hepatic cells, which are a type of epithelial cells derived from the endoderm, have not been linked previously to HSC-supportive functions. However, previous work suggests that there is an intertwining relationship between hepatic–endodermal cells and hematopoietic cells.
Fetal hepatic stem–progenitors derive from the definite endoderm, which emerges from the primitive steak around E7.0 [35]. Interestingly, primitive and visceral endodermal cells were shown to provide potent signals to promote the primitive wave of hematopoiesis in the yolk sac [36]. In contrast, no hematopoiesis was observed in the AGM region, although it is a major site for the emergence of HSCs [36]. These results suggest that cells of endodermal origin other than hepatic cells play pivotal roles in regulating HSC expansion and hematopoiesis in the early embryo.
There is also a distinct difference between the HSC niches in the fetal liver and adult bone marrow. Although most of the adult HSCs are quiescent, remain in the bone marrow throughout life, and are tightly regulated by their surrounding niches cells, HSCs remain only in the fetal liver for a short period. In the mouse, these HSCs migrate into the fetal liver, expand in numbers or differentiate into hematopoietic progenitors cells, and evacuate the fetal liver less than 1 week later. Although in vivo methods have been useful to identify HSC niches in the adult bone marrow, their application in studying fetal liver niches is far more limited. Therefore, we sought to use in vitro coculture methods to directly prove that fetal hepatic cells are capable of expanding HSCs.
As discussed earlier, establishing immortalized cell lines is the most frequently used method in study HSC supportive cells at the fetal stages, but this method has significant flaws for identification of cells that support HSC expansion. First, the immortalization process will cause significant changes to the cells and it is unknown whether the supportive cells will still possess HSC expanding capability. Second, although mesenchymal cells can be immortalized easily, the culture and immortalization of hepatic cells is far more difficult. Therefore, we decided to purify primary hepatic progenitors directly from the fetal liver and test their ability to expand HSCs.
The first issue we considered in developing this novel primary coculture method was the purity of the hepatic cells used for coculture. Fetal liver contains hepatic cells at multiple stages of development, and many of these subpopulations and their cell markers have not been characterized. Therefore, we focused on purifying the entire population of hepatic progenitors and examined whether HSC supportive cells exist within this group of cells. A potential drawback of using primary cells is that it is impossible to avoid contamination from other cell types. In our case, we could achieve approximately 70% purity using magnetic beads. Because most of the contaminating cells are hematopoietic, we incubated these cells on gelatin-coated plates for 2 days without adding supportive cytokines for hematopoietic cells. This method allowed the hepatic cells to attach and spread on the culture plate so that we could wash and remove all the nonadherent contaminating hematopoietic cells.
To ensure that the HSC expansion effect is from HSCs and not potential contaminating cells, we first used qPCR to show that only markers for hepatic cells are enriched in DLK+ cells versus DLK− cells (Fig. 1B). We then compared the ability of DLK+ and DLK− cells to expand HSCs. Although DLK+ cells supported significant HSC expansion, proportional numbers of DLK− cells failed completely to expand HSCs or hematopoietic progenitors in either serum-containing or serum-free media (Fig. 5, Supplementary Figure 4, online only, available at www. exphem.org). These results gave us confidence that the principle supportive cells for HSC expansion are indeed of hepatic origin.
The second issue we dealt with is whether hepatic progenitors can retain their ability to support HSC expansion in ex vivo culture. Because hepatic cells are difficult to culture, we carefully examined their survival in different conditions. We made the key observation that cultured hepatic cells could sustain hematopoietic cells without added cytokines (Fig. 1C). We also found that fetal hepatic cells maintain their expression of key HSC-supportive factors, including SCF, TPO, and CXCL12 (Supplementary Figure 2, online only, available at www.exphem.org), suggesting that they could at least maintain part of their HSC supportive ability in vitro. The expression of other factors such as DLK1, Angptl3, and IGF2 were greatly decreased in ex vivo culture, and it is possible that these factors are not essential for HSC expansion.
To develop this novel coculture system, we had to continuously adjust and improve our methods. For example, initially we purified DLK+ cells without collagenase treatment (Fig. 2). However, we found that collagenase treatment not only improved the purity of isolated DLK+ cells, but also increased their ability to attach to the culture plates. Therefore, far fewer DLK+ cells were needed for the later experiments. One key to achieving significant HSC expansion is to have as many DLK+ cells in the coculture as possible. When purified DLK+ cells were cultured in serum-containing medium, their mass increased significantly after 1 week, and it was a difficult task to consistently have sufficient numbers of DLK+ cells at the beginning of the coculture without overcrowding the culture at later stages. In contrast, when DLK+ cells were cultured in serum-free StemSpan medium, there was little change in their mass during the coculture; therefore, higher numbers of DLK+ cells could be plated without overcrowding the coculture. As a result, the coculture experiment was simplified and became more consistent.
In our study, three separate sets of coculture experiments in serum-free medium were performed, and HSCs were consistently expanded to similar levels. This method opens the possibility that this coculture system can be used to characterize signaling molecules that are important for HSC expansion. Finally, we optimized the cytokines that were added into the coculture and found that a low concentration of added SCF is sufficient for the expansion of HSCs (Fig. 4). An additional low concentration of TPO could slightly help with ex vivo HSC expansion; however, a high concentration of TPO should be avoided for the long-term expansion of HSCs because TPO is the major cytokine that drives HSCs into cycling and could be the main reason for the myeloid reconstituting defect that we observed using a high concentration of cytokines (Supplementary Figure 3, online only, available at www.exphem.org).
The establishment of a coculture system with fetal hepatic progenitors could also provide a major boost to the ex vivo culture and expansion of HSCs. Despite the fact that ex vivo–cultured DLK+ hepatic progenitors lose expression of many cytokines and that only a fraction of DLK+ hepatic cells are likely to be supportive of HSCs, we have consistently observed significant expansion of HSCs in both serum-containing and serum-free media using our coculture system. Moreover, we suspect that the true potential of hepatic stromal cells to expand HSCs in ex vivo coculture is much higher. The fact that many progenitors and short-term HSCs were generated in our long-term coculture indicates that many expanded HSCs underwent differentiation during coculture. Insufficient protection of expanded HSCs through direct contact with hepatic stromal cells is possibly the main reason for this finding. Better maintenance of long-term HSCs will also allow the duration of the coculture to be further extended, thus expanding more HSCs. Therefore, promoting direct contact between HSCs and their hepatic stromal cells, thus maintaining them in an undifferentiated state, is a key to successful long-term culture and expansion of HSCs.
One remaining key question is which cell surface proteins on the surface of DLK+ cells are important for the expansion of HSCs. A natural candidate is DLK1, a homolog to the notch ligands. However, DLK1 expression is rapidly diminished in ex vivo culture, suggesting that it is dispensable for the ex vivo expansion of HSCs. Other candidates include Notch ligands because Notch pathway was suggested to play an important role in the regulation of HSCs by endothelial cells. We examined the expression of all the Notch ligands and found none of them is enriched by DLK+ cells. Therefore, fetal hepatic cells can use multiple signaling pathways to control the growth and differentiation of HSCs.
Supplementary Material
Supplementary Figure 1. Ex vivo–cultured fetal DLK+ cells can survive for an extend period in both serum containing medium and serum-free stem span medium. (A) Magnetic bead purification of DLK+ cells from E15.5 liver. Total fetal liver cells were stained with FITC-conjugated DLK1 antibody followed by anti-FITC magnetic beads. The DLK− (left) and DLK+ (middle) cells were separated using an automatic cell separator. Collagenase treatment before purification further improved the purity of DLK1+ cells. (B) Comparison of the survival of DLK+ cells in serum-containing medium, serum-free StemSpan medium, both of which are also capable of supporting hematopoietic cells and hepatocyte-defined medium (HDM; BD Biosciences), a serum-free medium specifically formulated for adult hepatocytes. GFP+ hepatic remained alive after 7 days in both serum and StemSpan medium after 7 days, but died in HDM (top row). Culturing DLK+ cells on gelatin-coated plates improved their survival, and hepatic cells spread and formed monolayers (bottom row). On both regular culture plates and gelatin-coated plates, hepatic cells survived the best in serum medium. (C) Very little hematopoietic cell growth surrounding DLK+ hepatic cells in StemSpan medium after 14 days.
Supplementary Figure 2. The expression of many growth factors by DLK+ cells is downregulated during ex vivo coculture. Indicated is the relative abundance of mRNA for each gene before culture and after 3 and 7 days of culture in serum-containing medium. The relative expression of each gene at each time point was calculated by setting the level of each mRNA before culture as one. Although expression of several HSC supportive factors such as SCF, TPO, and CXCl12 was maintained during culture, expression levels of DLK1, Angptl3, and IGF2 dropped quickly.
Supplementary Figure 3. Long-term culture of HSCs in serum containing STF medium causes a temporary myeloid reconstituting defect. Representative FACS analysis of donor-derived (CD45.1+) myeloid (CD11b+ or Gr-1+) reconstitution of recipient mice at 1, 2, and 4 months after transplantation. Mice transplanted with HSCs expanded by 2 weeks (A) and 3 weeks (B) of coculture showed a myeloid reconstituting defect at month 2. This reconstituting defect is limited to myeloid lineage (and possible erythroid lineage) only; B and T lineages appear to be completely normal (C). A similar defect was also observed in mice transplanted of HSCs cultured in cytokines only (D, E). Coculture in serum-free, low-cytokine medium completely eliminated this problem (F).
Supplementary Figure 4. DLK− cells failed to expand hematopoietic cells in serum containing STF medium after 3 weeks of coculture. HSCs were cocultured with DLK+ cells, a proportional amount of DLK− cells, or cytokines only in serum medium for 2 weeks. The numbers of hematopoietic cells expanded were counted using a hemocytometer. Fibroblastic cells from DLK− cell population grew rapidly, and a small portion of hematopoietic progenitors remained and expanded in serum medium; therefore, significant amount of cells were present in control wells with only DLK− cells (B, bottom right). We therefore subtracted the number of cells from control wells without HSCs added from the total number of cells after coculture. As (A) shows, coculture with DLK− cells did not expand hematopoietic cells when compared with cytokines only. For week 3, coculture with DLK+ cells expanded sixfold more cells than coculture with DLK− cells. FACS analysis shows that 88.5% of total cells expanded by coculture with DLK+ cells are donor-derived CD45.1+ cells (C, left panel). In contrast, only 2.2% of cells expanded by coculture with DLK− cells are CD45.1+ (C, right panel). The rest of the cells were mainly CD45.2+ contaminating hematopoietic cells from the fetal liver. Therefore, coculture with DLK+ cells expanded more than 200-fold of hematopoietic cells than coculture with DLK− cells.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants P01 HL 32262 and DK067356 (to H.F.L.), an NIH National Research Service Award Fellowship (to S.C.), and a grant from the Diamond-Blackfan Anemia Foundation, a fellowship from the Swedish Research Council, and stipends from Maja och Hjalmar Leanders Stiftelse and The Sweden-America Foundation (to J.F.). We thank Wenquian Hu, Beiyan Zhou, and Christine Patterson for critically reading the manuscript.
Footnotes
Author contributions: S.C. designed research, performed all the experiments except Figure 2F, analyzed data, and wrote the paper; J.F. performed the experiments for Figure 2F; H.F.L. supervised the project and edited the paper.
Conflict of interest disclosure No financial interest/relationships with financial interest relating to the topic of this article have been declared.
References
- 1.Mikkola HK, Orkin SH. The journey of developing hematopoietic stem cells. Development. 2006;133:3733–3744. doi: 10.1242/dev.02568. [DOI] [PubMed] [Google Scholar]
- 2.Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132:631–644. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Medvinsky A, Dzierzak E. Definitive hematopoiesis is autonomously initiated by the AGM region. Cell. 1996;86:897–906. doi: 10.1016/s0092-8674(00)80165-8. [DOI] [PubMed] [Google Scholar]
- 4.Gekas C, Dieterlen-Lievre F, Orkin SH, Mikkola HK. The placenta is a niche for hematopoietic stem cells. Dev Cell. 2005;8:365–375. doi: 10.1016/j.devcel.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 5.Samokhvalov IM, Samokhvalova NI, Nishikawa S. Cell tracing shows the contribution of the yolk sac to adult haematopoiesis. Nature. 2007;446:1056–1061. doi: 10.1038/nature05725. [DOI] [PubMed] [Google Scholar]
- 6.Inman KE, Downs KM. The murine allantois: emerging paradigms in development of the mammalian umbilical cord and its relation to the fetus. Genesis. 2007;45:237–258. doi: 10.1002/dvg.20281. [DOI] [PubMed] [Google Scholar]
- 7.Ema H, Nakauchi H. Expansion of hematopoietic stem cells in the developing liver of a mouse embryo. Blood. 2000;95:2284–2288. [PubMed] [Google Scholar]
- 8.Arai F, Suda T. Maintenance of quiescent hematopoietic stem cells in the osteoblastic niche. Ann N Y Acad Sci. 2007;1106:41–53. doi: 10.1196/annals.1392.005. [DOI] [PubMed] [Google Scholar]
- 9.Wilson A, Laurenti E, Oser G, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135:1118–1129. doi: 10.1016/j.cell.2008.10.048. [DOI] [PubMed] [Google Scholar]
- 10.Spradling A, Drummond-Barbosa D, Kai T. Stem cells find their niche. Nature. 2001;414:98–104. doi: 10.1038/35102160. [DOI] [PubMed] [Google Scholar]
- 11.Moore KA, Lemischka IR. Stem cells and their niches. Science. 2006;311:1880–1885. doi: 10.1126/science.1110542. [DOI] [PubMed] [Google Scholar]
- 12.Zhang J, Niu C, Ye L, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425:836–841. doi: 10.1038/nature02041. [DOI] [PubMed] [Google Scholar]
- 13.Calvi LM, Adams GB, Weibrecht KW, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841–846. doi: 10.1038/nature02040. [DOI] [PubMed] [Google Scholar]
- 14.Kiel MJ, Yilmaz OH, Iwashita T, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 15.Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. 2012;481:457–462. doi: 10.1038/nature10783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25:977–988. doi: 10.1016/j.immuni.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 17.Mendez-Ferrer S, Michurina TV, Ferraro F, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466:829–834. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamazaki S, Ema H, Karlsson G, et al. Nonmyelinating Schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell. 2011;147:1146–1158. doi: 10.1016/j.cell.2011.09.053. [DOI] [PubMed] [Google Scholar]
- 19.Wilson A, Trumpp A. Bone-marrow haematopoietic-stem-cell niches. Nat Rev Immunol. 2006;6:93–106. doi: 10.1038/nri1779. [DOI] [PubMed] [Google Scholar]
- 20.Avraham H, Scadden DT, Chi S, Broudy VC, Zsebo KM, Groopman JE. Interaction of human bone marrow fibroblasts with megakaryocytes: role of the c-kit ligand. Blood. 1992;80:1679–1684. [PubMed] [Google Scholar]
- 21.Heissig B, Hattori K, Dias S, et al. Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of kit-ligand. Cell. 2002;109:625–637. doi: 10.1016/s0092-8674(02)00754-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chou S, Lodish HF. Fetal liver hepatic progenitors are supportive stromal cells for hematopoietic stem cells. Proc Nat Acad Sci U S A. 2010;107:7799–7804. doi: 10.1073/pnas.1003586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang CC, Lodish HF. Cytokines regulating hematopoietic stem cell function. Curr Opin Hematol. 2008;15:307–311. doi: 10.1097/MOH.0b013e3283007db5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wright DE, Bowman EP, Wagers AJ, Butcher EC, Weissman IL. Hematopoietic stem cells are uniquely selective in their migratory response to chemokines. J Exp Med. 2002;195:1145–1154. doi: 10.1084/jem.20011284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dabeva MD, Petkov PM, Sandhu J, et al. Proliferation and differentiation of fetal liver epithelial progenitor cells after transplantation into adult rat liver. Am J Pathol. 2000;156:2017–2031. doi: 10.1016/S0002-9440(10)65074-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oertel M, Menthena A, Chen YQ, Teisner B, Jensen CH, Shafritz DA. Purification of fetal liver stem/progenitor cells containing all the repopulation potential for normal adult rat liver. Gastroenterology. 2008;134:823–832. doi: 10.1053/j.gastro.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 27.Butler JM, Nolan DJ, Vertes EL, et al. Endothelial cells are essential for the self-renewal and repopulation of Notch-dependent hematopoietic stem cells. Cell Stem Cell. 2010;6:251–264. doi: 10.1016/j.stem.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang CC, Kaba M, Ge G, et al. Angiopoietin-like proteins stimulate ex vivo expansion of hematopoietic stem cells. Nat Med. 2006;12:240–245. doi: 10.1038/nm1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Himburg HA, Muramoto GG, Daher P, et al. Pleiotrophin regulates the expansion and regeneration of hematopoietic stem cells. Nat Med. 2010;16:475–482. doi: 10.1038/nm.2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oostendorp RA, Harvey KN, Kusadasi N, et al. Stromal cell lines from mouse aorta-gonads-mesonephros subregions are potent supporters of hematopoietic stem cell activity. Blood. 2002;99:1183–1189. doi: 10.1182/blood.v99.4.1183. [DOI] [PubMed] [Google Scholar]
- 31.Kusadasi N, Oostendorp RA, Koevoet WJ, Dzierzak EA, Ploemacher RE. Stromal cells from murine embryonic aorta-gonad-mesonephros region, liver and gut mesentery expand human umbilical cord blood-derived CAFC(week6) in extended long-term cultures. Leukemia. 2002;16:1782–1790. doi: 10.1038/sj.leu.2402615. [DOI] [PubMed] [Google Scholar]
- 32.Moore KA, Pytowski B, Witte L, Hicklin D, Lemischka IR. Hematopoietic activity of a stromal cell transmembrane protein containing epidermal growth factor-like repeat motifs. Proc Natl Acad Sci USA. 1997;94:4011–4016. doi: 10.1073/pnas.94.8.4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nolta JA, Thiemann FT, Arakawa-Hoyt J, et al. The AFT024 stromal cell line supports long-term ex vivo maintenance of engrafting multipotent human hematopoietic progenitors. Leukemia. 2002;16:352–361. doi: 10.1038/sj.leu.2402371. [DOI] [PubMed] [Google Scholar]
- 34.Chagraoui J, Lepage-Noll A, Anjo A, Uzan G, Charbord P. Fetal liver stroma consists of cells in epithelial-to-mesenchymal transition. Blood. 2003;101:2973–2982. doi: 10.1182/blood-2002-05-1341. [DOI] [PubMed] [Google Scholar]
- 35.Zhao R, Duncan SA. Embryonic development of the liver. Hepatology. 2005;41:956–967. doi: 10.1002/hep.20691. [DOI] [PubMed] [Google Scholar]
- 36.Baron MH. Embryonic origins of mammalian hematopoiesis. Exp Hematol. 2003;31:1160–1169. doi: 10.1016/j.exphem.2003.08.019. [DOI] [PubMed] [Google Scholar]
- 37.Flygare J, Rayon Estrada V, Shin C, Gupta S, Lodish HF. HIF1alpha synergizes with glucocorticoids to promote BFU-E progenitor self-renewal. Blood. 2011;117:3435–3444. doi: 10.1182/blood-2010-07-295550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Ex vivo–cultured fetal DLK+ cells can survive for an extend period in both serum containing medium and serum-free stem span medium. (A) Magnetic bead purification of DLK+ cells from E15.5 liver. Total fetal liver cells were stained with FITC-conjugated DLK1 antibody followed by anti-FITC magnetic beads. The DLK− (left) and DLK+ (middle) cells were separated using an automatic cell separator. Collagenase treatment before purification further improved the purity of DLK1+ cells. (B) Comparison of the survival of DLK+ cells in serum-containing medium, serum-free StemSpan medium, both of which are also capable of supporting hematopoietic cells and hepatocyte-defined medium (HDM; BD Biosciences), a serum-free medium specifically formulated for adult hepatocytes. GFP+ hepatic remained alive after 7 days in both serum and StemSpan medium after 7 days, but died in HDM (top row). Culturing DLK+ cells on gelatin-coated plates improved their survival, and hepatic cells spread and formed monolayers (bottom row). On both regular culture plates and gelatin-coated plates, hepatic cells survived the best in serum medium. (C) Very little hematopoietic cell growth surrounding DLK+ hepatic cells in StemSpan medium after 14 days.
Supplementary Figure 2. The expression of many growth factors by DLK+ cells is downregulated during ex vivo coculture. Indicated is the relative abundance of mRNA for each gene before culture and after 3 and 7 days of culture in serum-containing medium. The relative expression of each gene at each time point was calculated by setting the level of each mRNA before culture as one. Although expression of several HSC supportive factors such as SCF, TPO, and CXCl12 was maintained during culture, expression levels of DLK1, Angptl3, and IGF2 dropped quickly.
Supplementary Figure 3. Long-term culture of HSCs in serum containing STF medium causes a temporary myeloid reconstituting defect. Representative FACS analysis of donor-derived (CD45.1+) myeloid (CD11b+ or Gr-1+) reconstitution of recipient mice at 1, 2, and 4 months after transplantation. Mice transplanted with HSCs expanded by 2 weeks (A) and 3 weeks (B) of coculture showed a myeloid reconstituting defect at month 2. This reconstituting defect is limited to myeloid lineage (and possible erythroid lineage) only; B and T lineages appear to be completely normal (C). A similar defect was also observed in mice transplanted of HSCs cultured in cytokines only (D, E). Coculture in serum-free, low-cytokine medium completely eliminated this problem (F).
Supplementary Figure 4. DLK− cells failed to expand hematopoietic cells in serum containing STF medium after 3 weeks of coculture. HSCs were cocultured with DLK+ cells, a proportional amount of DLK− cells, or cytokines only in serum medium for 2 weeks. The numbers of hematopoietic cells expanded were counted using a hemocytometer. Fibroblastic cells from DLK− cell population grew rapidly, and a small portion of hematopoietic progenitors remained and expanded in serum medium; therefore, significant amount of cells were present in control wells with only DLK− cells (B, bottom right). We therefore subtracted the number of cells from control wells without HSCs added from the total number of cells after coculture. As (A) shows, coculture with DLK− cells did not expand hematopoietic cells when compared with cytokines only. For week 3, coculture with DLK+ cells expanded sixfold more cells than coculture with DLK− cells. FACS analysis shows that 88.5% of total cells expanded by coculture with DLK+ cells are donor-derived CD45.1+ cells (C, left panel). In contrast, only 2.2% of cells expanded by coculture with DLK− cells are CD45.1+ (C, right panel). The rest of the cells were mainly CD45.2+ contaminating hematopoietic cells from the fetal liver. Therefore, coculture with DLK+ cells expanded more than 200-fold of hematopoietic cells than coculture with DLK− cells.