

Abstract
Endothelial senescence may contribute to the pathogenesis of age-related vascular disorders. Furthermore, chronic exposure to risk factors for cardiovascular disease (CVD) accelerates the effects of chronological aging by generating stress-dependent damages, including oxidative stress, therefore promoting stress-induced premature senescence. Our objective was to determine whether a chronic treatment with an antioxidant (N-acetyl-cystein, NAC) could delay senescence of endothelial cells (EC) isolated and cultured from arterial segments of patients with severe coronary artery disease. If EC were considered as one population (n = 26), chronic NAC treatment slightly shortened telomere attrition rate associated with senescence but did not significantly delay the onset of endothelial senescence. However, in a subgroup of NAC-treated EC (n = 15) cellular senescence was significantly delayed, NAC decreased lipid peroxidation (HNE), activated the catalytic subunit of telomerase (hTERT) and inhibited telomere attrition. In contrast, in another subgroup of EC (n = 11) characterized by initial short telomeres, no effect of NAC on HNE and high levels of DNA damages, the antioxidant was not beneficial on senescence, suggesting an irreversible stress-dependent damage. In conclusion, chronic exposure to NAC can delay senescence of diseased EC via hTERT activation and transient telomere stabilization, unless oxidative stress-associated cell damage has become irreversible.
Keywords: Endothelium, Cardiovascular disease, Oxidative stress, Antioxidant, Cellular senescence
1. Introduction
The free radical theory of aging hypothesizes that the accumulation of oxidative damage is a central mediator of the aging process and age-related disorders (Harman, 1956; Humphries et al., 2006; Donato et al., 2007). At the cellular level, aging of healthy vascular endothelial cells (EC) leads to replicative senescence, a state of permanent growth arrest due to cumulative telomere attrition (Ben-Porath and Weinberg, 2005; Chen and Goligorsky, 2006). Oxidative stress associated with aging and risk factors for cardiovascular diseases (CVDs) (Csiszar et al., 2002) may induce DNA damage (Lorenz et al., 2001; von Zglinicki, 2002), accelerate reparative cell division, promote telomere instability and lead to premature stress-induced senescence (SIS) (Kurz et al., 2004; Voghel et al., 2007). Excessive telomere shortening has been reported in circulating white blood cells from patients with various CVD (Jeanclos et al., 1998, 2000; Samani et al., 2001; Brouilette et al., 2003). Evidences for the potential impact of vascular cellular senescence on aging and age-related disorders are emerging: endothelial dysfunction and senescence measured in vivo in the animal model of metabolic syndrome/type 2 diabetes could be prevented by ebselen, a peroxinitrite scavenger (Brodsky et al., 2004). Furthermore, in vitro studies in healthy human umbilical vein EC and fibroblasts in culture showed that cellular senescence could be prevented when oxidative stress is controlled (Furumoto et al., 1998; Haendeler et al., 2003a,b; Wang et al., 2003; Bode-Böger et al., 2005). It is not known however, if such manipulations can delay stress-induced senescence of EC isolated from patients exposed to risk factors for CVD (Voghel et al., 2007). EC from atherosclerotic patients are most likely damaged, and thus expected to respond differently from EC isolated from healthy donors.
The objective of the present study was to elucidate whether a chronic antioxidant treatment could reverse premature stress-induced senescence in EC isolated from patients with severe coronary artery disease (CAD). We demonstrate that chronic antioxidant treatment can only delay the appearance of EC senescence – via lessening of stress-induced senescence and telomerase-induced telomere stabilization – unless EC have undergone an irreversible ROS-associated damage. This contrasts with previous reports showing that antioxidant significantly delays senescence in healthy cells exposed to an exogenous stress.
2. Experimental procedures
2.1. Clinical profile of the donors
Segments of human distal (close to the bifurcation) internal mammary artery (IMA, n = 26, Table 1) harvested with low electrocautery energy and excised with cold scissors, discarded during primary CABG were used in this study. This study was approved by our institutional ethical committee.
Table 1.
Clinical profile of patients undergoing CABG (men (n = 21), women (n = 5))
| Age (years) | 60 ± 2 |
| Age range (years) | [39–80] |
| History of disease (years) | 8 ± 2 |
| History of disease range (years) | [0.1–29] |
| BMI (kg/m2) | 30 ± 1 |
| Dyslipidemia | 88% (22/25) |
| Hypertension | 68% (17/25) |
| Diabetes | 12% (3/26) |
| Nicotine use (active or recent ex-smokers) | 85% (22/26) |
| Left ventricular ejection fraction (%) | 50 ± 3% |
| Number of grafts | 3 ± 0 |
Patients were treated with similar medications including aspirin, angiotensin-converting enzyme inhibitors, β-blockers, calcium channel blockers, statins and nitrates.
2.2. Isolation and culture of EC
Endothelial cells were isolated and cultured by an explant technique from segments of human IMA, as previously described (Voghel et al., 2007). Cells were incubated in Dubelcco’s modified Eagle medium (DMEM) supplemented with 10% foetal bovine serum, 10% calf serum, 1% penicillin–streptomycin, 90 μg/ml sodium heparin salt (Sigma–Aldrich), 60 μg/ml EC growth supplement (Becton Dickinson), and 100 U/ml fungizone (Gibco), at 37 °C in a 95% air/5% CO2 incubator. Starting at passage 2, primary cells isolated from the same donor were divided into two lots and grown throughout the duration of the culture, in the presence or absence of the antioxidant N-acetyl-cystein (NAC) (10 μM) (Sigma). NAC was replaced every 2 days. At each passage, cells were collected for senescence-associated β galactosidase (SA-β-Gal), DNA (Southern blotting), RNA (real-time RT-PCR) and protein (Western blotting) extraction and some cells were plated on coverslips for immunostaining. Before replating, cells were counted using a hemocytometer and population doubling levels (PDL) were calculated.
2.3. Senescence-associated β-galactosidase staining
SA-β-Gal was used as a marker of senescence (Dimri et al., 1995; Voghel et al., 2007).
2.4. Telomere length measurement
Endothelial cells were grown in 75 cm2 flasks at early and subsequent passages until senescence was reached, in the presence or absence of NAC. DNA was extracted from EC with a phenol/chloroform/isoamyl alcohol technique, and restriction fragments lengths (RFL) were quantified using a Southern blot technique (Zhang et al., 2000; Voghel et al., 2007).
2.5. Immunofluorescence
Immunostaining was used to assess the expression and sub-cellular localization of 4-hydroxy-nonenal (HNE) (rabbit polyclonal anti-HNE, 1:200, Alpha Diagnostics), p53 (mouse monoclonal anti-p53, 1:200, Upstate), phospho H2AX (mouse monoclonal anti-H2AX, 1:200, Upstate), ATM (rabbit polyclonal anti-ATM, 1:100, Santa Cruz), promyelocytic leukemia (PML) nuclear bodies (rabbit polyclonal anti-PML, 1:200, generous gift of Dr. G. Ferbeyre, University of Montreal, Canada) and hTERT (mouse monoclonal anti-hTERT, 1:200, Abcam) at different levels of senescence, in the presence or absence of NAC. DNA counterstaining was performed by incubating EC with propidium iodide (20 μM, Molecular Probe) or TOPRO-3 (2 μM, Molecular Probes). Negative controls consisted in omitting the primary antibodies during the protocol. Cells were visualized using a confocal microscope (Zeiss LSM 510). Semi-quantitative analysis (LSM-510, Zeiss) was performed by measuring the HNE-fluorescence intensity in whole cells from four to five different pictures from the same coverslip. Quantification of the nuclear and cytosolic fluorescent signals obtained with H2AX, ATM, p53, PML was also performed. The ratio nucleus/cytosol was calculated and normalized by the number of cells. Values are expressed in arbitrary units of fluorescence (a.u.).
2.6. Telomerase activity
Real-time-TRAPassay was used to measure hTERT activity in hIMA EC (Ohuchida et al., 2005). Serial dilutions (0.1 ng to 1000 pg) of HEK293 (Invitrogen) were used as standard curve. Real-time-TRAPassay was performed using 5 pmol of M2-TS forward primer (5′-AATCCGTCGAGCAGAGTT-3′), 1.25 pmol of ACX reverse primer (5′-GCGCGG(CTTACC)3CTAACC-3′) and EC nuclear or cytosolic proteins (20 ng) or serially diluted HEK293 proteins. Nuclear and cytosolic proteins were extracted using a nuclear and cytoplasmic extraction kit (Pierce Biotechnology) in the presence of 10× protease inhibitor cocktail (Pierce Biotechnology). All samples were run in triplicate and either lysis buffer or heat-inactivated samples were used as a negative control. Enzymatic activity was reported using arbitrary units: one unit is equivalent to the activity of 1 ng of HEK293. Telomerase activity was quantified at low senescence in the presence or absence of NAC, and the average values are presented (Fig. 7). The detection limit of endogenous hTERT activity was estimated to be the background value of hTERT activity in samples with only lysis buffer.
Fig. 7.

Modulation of hTERT by NAC. Confocal images of hTERT-immunostained cells in control and NAC-treated EC. Translocation of the enzyme from the cytosol to the nucleus was observed if the antioxidant delayed the appearance of senescence (B) but not if NAC did not affect the time course of senescence (A). Cytosolic and nuclear hTERT activity (a.u.) detected by real-time-TRAP assay, in control and NAC-treated cells (C) not affected (n = 5) or positively affected (n = 5) by NAC. The bar scale represents 10 μm. *p < 0.05 compared to control cells. †p < 0.05 compared to nuclear hTERT activity in NAC-treated cells from the “unaffected senescence” group.
2.7. Real-time RT-PCR
Total RNA was isolated using a RNeasy kit (Qiagen) at early and subsequent passages until senescence was reached, in the presence and absence of NAC. Real-time PCR was carried out on diluted RT products using the DNA-binding dye SYBR Green I for the detection of PCR products (Mx3005P system, Stratagene) according to the manufacturer’s instruction. Serial dilutions (100 ng to 1 pg) of human aortic EC (hAoEC) (Cambrex) total RNA were used as standard. The following primers designed by primer express (Version 2.0) were used in order to quantify gene expression of Caveolin-1 and ATM:
| Primer | (5′–3′) Forward | (5′–3′) Reverse |
|---|---|---|
| GAPDH | TGAAGGTCGGAGTCAACGGA | CATTGATGACAAGCTTCCCG |
| Caveolin-1 | GCTGAGCGAGAAGCAAGTGT | TGGTGAAGCTGGCCTTCCAA |
| ATM | GGCAGCTGATATTCGGAGGA | CATCTTGGTCACGACGATAC |
mRNA levels in each sample were calculated relative to GAPDH which did not vary with senescence or NAC treatment.
2.8. Statistical analysis of the data
Data are presented as mean ± S.E.M. for continuous variables, with n indicating the number of patients. Unpaired t-tests and χ2 tests were used to study the clinical profile of EC donors (Tables 1 and 2).
Table 2.
Clinical profile of EC donors according to the cellular response to the chronic treatment with the antioxidant NAC
| Positive response to NAC (n = 15) | Negative response to NAC (n = 11) | |
|---|---|---|
| Sex | 10/5 (men/women) | 11/0 (men/women)* |
| Age (years) | 63 ± 3 | 57 ± 3 |
| History of disease (years) | 9 ± 2 | 5 ± 3 |
| BMI (kg/m2) | 29 ± 2 | 30 ± 1 |
| Dyslipidemia | 93% (13/14) | 82% (9/11) |
| Hypertension | 67% (10/15) | 70% (7/10) |
| Diabetes | 13% (2/15) | 27% (3/11) |
| Nicotine use | 73% (11/15) | 100% (11/11) |
| LVEF (%) | 49 ± 3 | 52 ± 5 |
| Number of grafts | 3 ± 0 | 3 ± 0 |
p < 0.05 vs. positive response to NAC, χ2 test.
Two-tailed paired t-tests were used to compare NAC-treated and matched–control untreated cells. Unpaired t-tests were used to compare data in EC responding positively or not to the antioxidant treatment. The significance level was fixed to p < 0.05. EC were considered as positive responders if the time to reach 50% of senescence was greater than in matched–control untreated cells. In order to predict whether risk factors for CVD could have an impact on the response to NAC (dependent dichotomic variable), we used logistic regression models. Only univariate analyses could be performed because of the small n (26 patients: 11 with negative response and 15 with positive).
3. Results
3.1. Effect of a chronic treatment with an antioxidant on cell senescence
While cells were serially passaged, in the presence or absence of NAC, the first parameter routinely available was the level of senescence. Thus, it rapidly appeared that EC from different donors responded differently to the chronic treatment with the antioxidant.
When all EC are considered, NAC did not significantly affect the onset of senescence: the number of days needed to reach 50% of positive β-Gal cells was similar in NAC-treated cells (124 ± 9 days) and in their matched–control cells (115 ± 6 days) (n = 26, p = 0.0789, paired t-test). NAC also did not improve the replication potential of the cells (cumulative PDL in NAC-treated cells 13.7 ± 1.3 versus 13.4 ± 1.2 in untreated cells, p = 0.6686). Similarly, final telomere length only tended to be higher in NAC-treated cells (9.4 ± 0.2 kbp) compared to their control cells (9.1 ± 0.2 kbp, p = 0.1090), while initial telomere length was not different between NAC-treated (9.4 ± 0.2 kbp) and untreated cells (9.7 ± 0.2 kbp, p = 0.0951). Globally, however, NAC-lowered telomere attrition observed in control untreated EC; NAC significantly reduced the telomere (RFL) shortening rate (ΔRFL by replication) associated with senescence, decreasing from −56.6 ± 14.0 bp/PDL in untreated EC to +3.3 ± 19.7 bp/PDL in NAC-treated cells ( p = 0.0277).
Because NAC clearly had opposite effects on cell senescence depending on the donor, i.e. either had no effect (n = 11) or in contrast delayed significantly (n = 15) EC senescence, cells were split in two categories and analyzed separately. The parameter used to categorize the cells was the number of days to reach 50% of senescence, as it characterizes the senescence profile of a given cell culture and allows comparisons between the different donors (Voghel et al., 2007). For a given donor, if the number of days to reach 50% of senescence was higher in EC treated with NAC than in paired untreated EC, cells were considered in the “delayed senescence” group (Fig. 1C). In contrast, if NAC-treated EC reached 50% of senescence slightly faster or at a similar rate than their control, cells were classified in the “unaffected senescence” group (Fig. 1A). In the category where NAC did not affect senescence, the time to reach 50% of senescent EC slightly decreased from 115 ± 9 to 107 ± 7 days (p = 0.0132, n = 11), cell replication potential was not improved (Fig. 1A) and senescence-associated changes in cell morphology were not restored (Fig. 1B). In contrast, in the group where NAC postponed the appearance of senescence, the time to reach 50% of senescence increased from 116 ± 8 to 148 ± 17 days ( p = 0.0063, n = 15) (Fig. 1C) while cells underwent more cell divisions (increase in PDL at 50% of senescent EC; p = 0.0334) and normal EC morphology was partially restored (Fig. 1D).
Fig. 1.

Dual effect of chronic treatment with N-Acetyl-Cystein on the time course of senescence. Two populations of EC were isolated according to their response to NAC (10 μM) on senescence: time course of senescence was either unaffected (n = 11) (A and B) or delayed (n = 15) (C and D). Unaffected senescence was evidenced by a slightly shorter time needed to reach 50% of senescence (SA-β-Gal positive cells) ( *p < 0.05 compared to control untreated cells) with no change in the replication potential (A) or cell morphology (B). Delayed senescence was demonstrated by a longer time needed to reach 50% of β-Gal positive cells ( p < 0.05) and an increased replication potential (C) ( *p < 0.05 compared to control untreated cells) as well as an improvement in cell morphology (D).
3.2. Effect of a chronic treatment with an antioxidant on telomere length
In untreated EC, RFL significantly decreased with senescence (Fig. 2). Most importantly, our results show that when NAC did not affect senescence, EC tended to have shorter ( p = 0.0552) initial telomere length (8.9 ± 0.3 kbp, n = 11) than EC in which NAC postponed senescence (9.8 ± 0.3 kbp, n = 15) (Fig. 2A). This strongly suggests that a critical telomere length may exist, beneath which NAC no longer provides benefit, possibly reflecting an irreversible advanced state of cell damage. In EC where NAC-delayed senescence, chronic antioxidant treatment prevented RFL-shortening associated with cellular senescence (from 9.8 ± 0.3 to 9.8 ± 0.5 kpb, p = 0.9437, n = 15) (Fig. 2B). In EC where NAC-delayed senescence, in 7 out of 15 donors, NAC even increased telomere length, but the final average RFL of control and NAC-treated cells were not significantly different ( p = 0.0881) (Fig. 2B). This can be explained by the large heterogeneity of telomere shortening/lengthening rate in the NAC-treated EC (−155 to +171 bp/PDL) and by the fact that we have taken into account only the initial and final RFL but not the intermediate telomere RFL. Our results therefore, cannot describe the complex telomeric dynamic in the presence of NAC but only provide a picture of the final consequence of NAC on telomere shortening.
Fig. 2.

Dual effect of chronic treatment with N-Acetyl-Cystein on telomere length. Telomere length (RFL, bp) was measured by Southern blot in control and NAC-treated cells, initially (initial passage) and when cells reached senescence (final passage). (A) In NAC-unaffected EC, RFL were not affected by the antioxidant. (B) In cells where NAC-delayed senescence, telomere shortening was prevented by NAC. Representative Southern blot and the distribution (%) of short (5–10 kbp), intermediate (10–15 kbp) and long (15–20 kb) telomeric fragments length are shown in control non-treated EC (C), NAC-treated unaffected EC (D) and NAC-delayed EC. (E) *p < 0.05 final passage vs. initial passage in control cells.
When the distribution of long and short fragments of RFL are analyzed, it appears that in EC where NAC-delayed senescence, the percentage of RFL with long fragments increased. The opposite was observed in NAC-unaffected EC or untreated EC (Fig. 2 C–E).
3.3. Effect of a chronic treatment with an antioxidant on oxidative stress and DNA damage markers
In cells in which NAC-delayed senescence, the antioxidant significantly decreased the levels of lipid peroxidation, i.e. HNE expression (Fig. 3A and B), an index of oxidative stress, compared to their matched–control cells and to NAC-treated EC in the unaffected category.
Fig. 3.
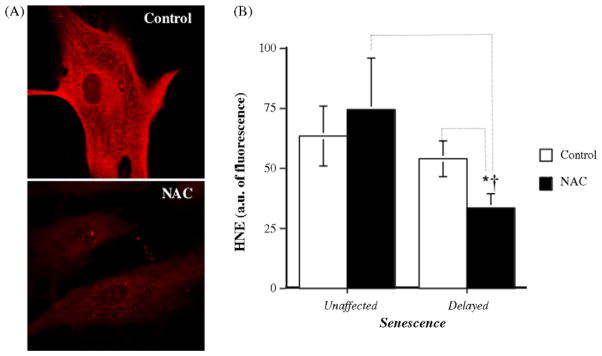
Modulation of oxidative stress by NAC. (A) Typical paired measurement (Control/NAC) of lipid peroxidation in fixed EC by HNE immunostaining in control and NAC-treated cells. (B) Average levels of HNE (intensity of fluorescence detected by immunofluorescent staining, a.u.) in control (n = 7–9) and NAC-treated (n = 11) EC. *p < 0.05 compared to untreated cells in the “delayed senescence” group. †p < 0.05 compared to NAC-treated cells in “unaffected senescence” group.
Oxidative damages engender double-strand breaks, activate ATM and activated ATM is one of the mediators of histone H2AX phosphorylation (Tanaka et al., 2006a,b). Semi-quantitative analysis shows that, at senescence, NAC-unaffected EC-displayed significant higher levels of both markers of DNA damage (Fig. 4). In addition, activated ATM and phosphorylated H2AX strongly positively correlate (Fig. 5). Although predominantly localized in the nucleus, ATM and phosphorylated H2AX were also observed in the cytoplasmic compartment as punctuate structures, likely reflecting an increase in the biosynthesis of these cell damage-induced proteins that can be mobilized to the nucleus.
Fig. 4.

Modulation of DNA damage makers by NAC. Immunostaining of H2AX and ATM in NAC-treated EC, at low passage (P4) (A) and at senescence (final P) (B), in cells in which senescence was either unaffected or delayed by the chronic antioxidant treatment. Bar graphs of H2AX (C) and ATM (D) staining of EC in the “delayed” group (n = 10) and in the “unaffected” group (n = 11). Data represent the ratio of nuclear/cytosolic fluorescent signal corrected by the number of cells. Bars = 20 μm. *p < 0.05 compared to EC at passage 4, in the “unaffected” or “delayed” category. †p < 0.05 compared to EC in the “unaffected” category.
Fig. 5.

Positive correlation between ATM and H2AX. Immunostaining of H2AX (red), ATM (green) and TOPRO-3 (nuclear staining in blue) in NAC-treated EC, at senescence (final P), in cells in which senescence was either unaffected or delayed by the chronic antioxidant treatment. H2AX and ATM positively correlate (n = 11). Data represent the ratio of nuclear/cytosolic fluorescent signal corrected by the number of cells. Bars = 20 μm.
Following cell stress, it has been reported that p53-dependent senescence is associated with a DNA damage/repair process in which PML bodies intervene (Moiseeva et al., 2006). Our results show that p53 strongly positively correlate with PML expression and that NAC significantly decreased nuclear expression of both PML and p53 (Fig. 6A–C). Interestingly, the PML bodies observed in senescent EC were large and exhibited similar nucleolar rim staining as reported in human primary senescent fibroblasts (Condamine et al., 2007), supporting the nucleolar targeting of PML during senescence.
Fig. 6.

Modulation cellular damage makers by NAC. Immunostaining of PML bodies (red), p53 (green) and TOPRO-3 (nuclear staining in blue) (A) in control senescent EC and in NAC-treated EC in which senescence was delayed by the chronic antioxidant treatment. (B) Bar graphs of PML and p53 staining of EC (n = 14). (C) p53 and PML positively correlate. Data represent the ratio of nuclear/cytosolic fluorescent signal corrected by the number of cells. Bars = 20 μm. *p < 0.05 compared to control EC.
Western blot and immunohistochemical staining of p53 and TRF2 revealed higher average levels of nuclear p53 and TRF2 protein expression in untreated cells in the category where NAC had no effect on senescence (data not shown). Although p53 and TRF2 are not direct markers of DNA damage, their accumulation reflects global cumulative cell damage and telomere instability.
3.4. Effect of a chronic treament with an antioxidant on molecular senescence markers
We previously reported that in EC isolated from human IMA from patients with severe CAD, caveolin-1 gene expression positively correlates with the lipid peroxidation index HNE and that caveolin-1 gene expression predicts the onset of stress-induced senescence (Voghel et al., 2007). In contrast, ATM gene expression is a marker of DNA damage and rather reflects replicative senescence (Voghel et al., 2007). In EC treated chronically with the antioxidant, NAC abrogated the expression of caveolin-1 (mRNA level under the detection limit, n = 12) compared with untreated control EC (1.411 ± 0.416, p = 0.0281, n = 26). In contrast, the average expression of ATM increased from 0.530 ± 0.045 (n = 26) in untreated EC to 3.341 ± 0.969 (n = 12) in EC treated chronically with NAC ( p = 0.0001). These changes in gene expression were observed whether or not EC responded positively or not to NAC. These data suggest that in EC from coronary patients, senescence is mainly triggered by oxidative stress and can be partly delayed by an antioxidant treatment. However, because cells are not immortalized by NAC, DNA and cell damages still occur, leading eventually to senescence.
3.5. Effect of NAC on telomerase
Since NAC prevented telomere attrition in EC in which senescence was delayed, the effect of the antioxidant on hTERT activity and its sub-cellular localization were assessed. NAC triggered some hTERT translocation from the cytosol to the nucleus when senescence was delayed but not when senescence was unaffected (Fig. 7A and B). Telomerase activity in normal EC is extremely low and was therefore detected with the very sensitive real-time-TRAP assay. Fig. 7C shows that, in EC partially rescued from senescence, NAC increased nuclear hTERT activity by ~95% (from 16.4 ± 2.7 a.u. in control cells to 31.9 ± 4.6 a.u. in NAC-treated cells, p = 0.0401, n = 5). Such increase in hTERT activity was not found in EC showing unaffected senescence (Fig. 7C, 16.0 ± 2.3 a.u. versus 17.9 ± 2.7 a.u., p = 0.6573, n = 5). Hence, the tendency for telomere elongation observed in EC in which NAC-delayed senescence could be at least partly due to telomerase translocation and activation.
4. Discussion
An abundant literature suggests that risk factors for CVD accelerate the normal aging process of the endothelium (Cooper et al., 1994; Cohen, 1995; Lakatta, 2002; Serrano and Andres, 2004; Chen and Goligorsky, 2006). We previously reported that in EC isolated and cultured from segments of IMA from atherosclerotic patients, the duration of exposure to risk factors for CVD is predictive of time to senescence in culture, independently of the age of the donor, and that in this context of high endogenous oxidative stress, senescence was stress-induced rather than replicative (Voghel et al., 2007). In the present study, we demonstrate that a chronic treatment with an antioxidant could only delay the onset of senescence in a subgroup of diseased EC.
In 58% (15/26) of EC chronically treated with NAC, the lipid peroxidation marker HNE decreased DNA and cellular damage markers decreased telomere length was maintained (and elongated in 7/15 patients), and thus senescence was significantly delayed. NAC is a source of sulfhydryl groups and is physiologically converted to metabolites that will stimulate glutathione synthesis, an important source of endogenous antioxidant. Thus, NAC is an indirect antioxidant agent by increasing intracellular glutathione, but it is also known as a direct scavenger of ROS (Zafarullah et al., 2003). Chronic treatment with NAC activated and led to some translocation of the catalytic subunit of the telomerase from the cytosol to the nucleus. This effect might be responsible for the lack of telomere shortening and thus, delayed senescence. The role of this NAC-induced hTERT activation in the delay of senescence is in accordance with data collected in healthy human umbilical vein EC exposed to exogenous H2O2 (Haendeler et al., 2003a,b). Although the response to NAC cannot be linked with the clinical history of the patient, due to the low number, this is the first demonstration that human EC exposed for years to risk factors for CVD, a condition with no equivalent experimental model, can be partially rescued from senescence by an antioxidant. The beneficial effect remains modest, and restricted to a particular subgroup of patients, in contrast to what was previously reported in healthy EC exposed to an exogenous oxidative stress (Furumoto et al., 1998; Haendeler et al., 2003a,b; Bode-Böger et al., 2005).
We found indeed that hIMA EC from another subset of patients (11/26) did not benefit from a chronic NAC treatment: oxidative stress (HNE) was not reduced, DNA and cell damage markers accumulated, hTERT was neither translocated to the nucleus nor activated, telomeres did not elongate and senescence was not affected. This may suggest that the beneficial effect of NAC on senescence was hTERT-dependent, but the level of NAC-induced hTERT activation remains low compared to HEK tumoral cells (data not shown), and NAC-treated cells eventually entered in senescence. Thus, it is unlikely that such modest increase in hTERT activity per se is sufficient to bypass EC damage and prolong cell survival. Indeed, EC that did not benefit from NAC treatment had shorter initial telomere and higher accumulation of DNA and cell damage markers such as H2AX, ATM, PML, p53 and TRF2. This suggests that the beneficial effect of NAC on senescence of EC that have been exposed to risk factors for CVD in vivo is primarily dependent on a reduction of oxidative stress that will subsequently favor some hTERT activity. These changes should limit further cellular damage, reduce telomere attrition and delay temporarily the onset of senescence. One hypothesis for the lack of effect of NAC on HNE levels in unaffected–treated cells is that in these particularly damaged EC, endogenous antioxidant capacities are extremely limited and that glutathione synthesis is not stimulated (Lapenna et al., 2004). Consequently, HNE levels are not decreased and senescence not delayed.
Telomerase enzymatic activity can be regulated at multiple levels, including hTERT transcription, alternative splicing, chaperon-mediated folding, phosphorylation and nuclear translocation. The major control mechanism of hTERT activity, however, seems to be the regulation of hTERT expression. Indeed, the hTERT promoter contains a binding site for transcription factors such as SP-1 and c-Myc, both activators of hTERT transcription and for Mad-1, an inhibitor of hTERT expression (Cong et al., 1999). Recent studies revealed that HNE can repress directly hTERT expression. Indeed, Pizzimenti et al. showed that HNE is able to inhibit c-Myc-binding complex and induce Mad-1 in human leukemic cell lines (Pizzimenti et al., 2006). Furthermore, HNE is also able to repress cyclin D2, a cell cycle regulatory protein (Pizzimenti et al., 1999). This could explain why EC unaffected by NAC and presenting higher levels of HNE exhibit no increase in hTERT activity. Another explanation for HNE-induced hTERT repression could be mediated by p53; HNE enhances p53 expression and this protein has the ability to inhibit the binding of SP-1 to the proximal promoter of hTERT and thus, repress hTERT activity (Stampfer et al., 2003; Shats et al., 2004). In addition, Shats et al. found an atypical E2F site responsible for the repression of hTERT as well as other cell cycle molecules such as cdc2 (Shats et al., 2004; Taylor et al., 2001). This transcription factor can be activated via the p53–p21–pRb pathway and repress the endogenous expression of hTERT. As EC unaffected by NAC presented higher levels of p53 and HNE levels were unchanged by NAC, this could explain, at least in part, why NAC-unaffected EC did not exhibit an increase in hTERT activity. In contrast, in the delayed group, NAC decreased both HNE and p53 proteins levels, in accordance with the increase in nuclear hTERT activity.
Since senescence can be triggered either by telomere instability (replicative senescence, ATM/p53–p21 pathway) or by multiple types of stresses including oxidative stress (SIS, caveolin/p53 pathway) (Voghel et al., 2007), we assessed which pathways were modified by the antioxidant by quantifying the genic expression of caveolin-1 and ATM. Caveolin-1, which is one of the principal protein of caveolae, invaginations enriched in cholesterol within the plasma membrane, is also known to increase in the presence of oxidative stress and to trigger senescence (Galbiati et al., 2001; Voghel et al., 2007). Our results show that caveolin-1 gene expression was abrogated in NAC-treated EC whether or not NAC delayed the onset of senescence. This strongly suggests that stress-induced senescence was lowered by the antioxidant. In contrast, we observed that the level of ATM gene expression, a protein acting as a controller of cellular responses to DNA damage (Pandita, 2002; Herbig et al., 2004), increased in EC treated with NAC in all cells compared to their matched untreated EC. This suggests that even in the presence of NAC, DNA and cellular damage accumulate and trigger some replicative senescence (a schematic representation of the different pathways of senescence is summarized in Fig. 8).
Fig. 8.
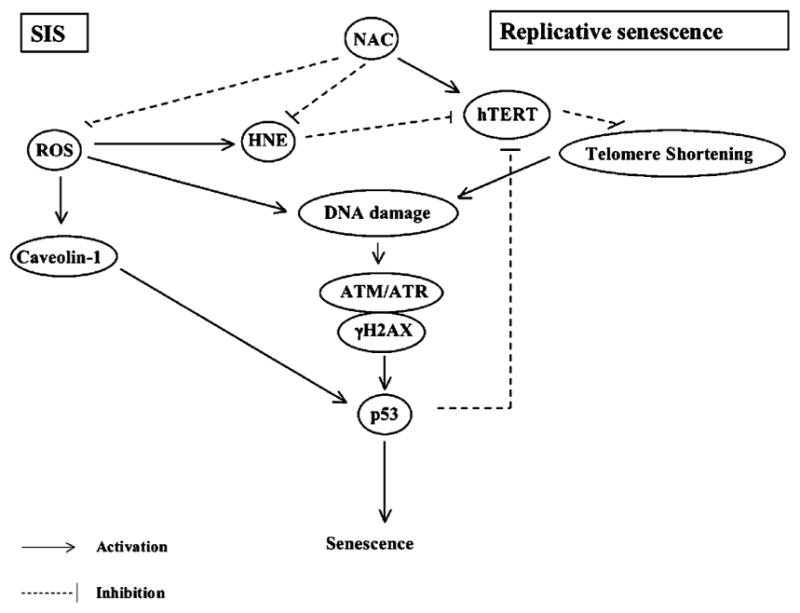
Schematic representation of molecular pathways of senescence. In control untreated EC, p53-dependent stress-induced senescence (SIS) can be triggered either by ROS-dependent caveolin-1 activation, or by ROS-dependent accumulation of DNA damage. Replicative senescence is mainly triggered by excessive telomere shortening which is recognized as DNA damage as reflected by markers such as ATM/H2AX. When treated chronically with NAC, caveolin-1 expression was abolished in both the unaffected and the delayed senescence group. We showed however, that when the levels of HNE and p53, both suppressors of hTERT activity, are too high, hTERT is not activated, telomere shortening is not prevented, DNA damage accumulates and senescence is unaffected. In contrast, when HNE and p53 levels are lowered, NAC activated hTERT and this was associated with a delay in the onset of senescence. These EC however, still accumulate DNA damages associated with a loss of endogenous DNA repair mechanism and thus, finally, reach senescence.
In order to predict statistically the positive (delayed senescence) or negative (unaffected senescence) response of EC treated with NAC, we used logistic regression model. The only risk factor for CVD that significantly predicted the response to NAC is sex (Table 2): negative response to NAC was never observed in EC from female donors ( p = 0.0331). However, it could only reflect the larger proportion of male donors (n = 21) compared to female (n = 5). Neither age (p = 0.1842), BMI ( p = 0.8448), duration of exposure to CVD ( p = 0.2807), dyslipidemia ( p = 0.4142), hypertension ( p = 0.8611), diabetes ( p = 0.3812), smoking ( p = 0.3938), nor LVEF ( p = 0.6812) could predict the response of EC to NAC treatment, clearly demonstrating the multifactorial origin of CAD. We previously showed, however, that in these EC isolated from coronary patients, long exposure to CVD and particularly to hypertension, lead to precocious senescence and low proliferation (Voghel et al., 2007).
In conclusion, our results show that chronic antioxidant treatment can delay stress-induced senescence of cultured EC isolated from patients with extensive CAD, partially by decreasing cellular and genic oxidative stress markers, by maintaining telomere length and by activating hTERT. There is no beneficial effect of NAC however, in EC that appear to have undergone an insurmountable damage possibly due to uncontrolled free radical production and lower endogenous antioxidant capacities.
Acknowledgments
This work has been supported in part by the Foundation of the Montreal Heart Institute, the Heart and Stroke Foundation of Quebec, and the Canadian Institute for Health Research (MOP 14496). Guillaume Voghel is a fellow of the Fonds de la Recherche en Santé du Québec. We thank Guy Charron (Montreal Heart Institute) for fruitful discussions and technical advises, and the biological tissue bank (RETEB) of the Fonds de la Recherche en Santé du Québec for technical support.
References
- Ben-Porath I, Weinberg RA. The signals and pathways activating cellular senescence. Int J Biochem Cell Biol. 2005;37:961–976. doi: 10.1016/j.biocel.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Bode-Böger SM, Martens-Lobenhoffer J, Täger M, Schröder H, Scalera F. Aspirin reduces endothelial senescence. Biochem Biophys Res Commun. 2005;334:1226–1232. doi: 10.1016/j.bbrc.2005.07.014. [DOI] [PubMed] [Google Scholar]
- Brodsky SV, Gealekman O, Chen J, Zhang F, Togashi N, Crabtree M, Gross SS, Nasjletti A, Goligorsky MS. Prevention and reversal of premature endothelial cell senescence and vasculopathy in obesity-induced diabetes by ebselen. Cir Res. 2004;94:377–384. doi: 10.1161/01.RES.0000111802.09964.EF. [DOI] [PubMed] [Google Scholar]
- Brouilette S, Singh RK, Thompson JR, Goodall AH, Samani NJ. White cell telomere length and risk of premature myocardial infarction. Arterioscler Thromb Vasc Biol. 2003;23:842–846. doi: 10.1161/01.ATV.0000067426.96344.32. [DOI] [PubMed] [Google Scholar]
- Chen J, Goligorsky MS. Premature senescence of endothelial cells: Methusaleh’s dilemma. Am J Physiol. 2006;290:H1729–H1739. doi: 10.1152/ajpheart.01103.2005. [DOI] [PubMed] [Google Scholar]
- Condamine W, Takahashi Y, Le Bras M, de Thé H. A nucleolar targeting signal in PML-I addresses PML to nucleolar caps in stressed or senescent cells. J Cell Sci. 2007;120:3219–3227. doi: 10.1242/jcs.007492. [DOI] [PubMed] [Google Scholar]
- Cohen RA. The role of nitric oxide and other endothelium-derived vasoactive substances in vascular disease. Prog Cardiovasc Dis. 1995;38:105–128. doi: 10.1016/s0033-0620(05)80002-7. [DOI] [PubMed] [Google Scholar]
- Cong YS, Wen J, Bacchetti S. The human telomerase catalytic subunit hTERT: organization of the gene and characterization of the promoter. Hum Mol Genet. 1999;8:137–142. doi: 10.1093/hmg/8.1.137. [DOI] [PubMed] [Google Scholar]
- Cooper LT, Cooke JP, Dzau VJ. The vasculopathy of ageing. J Gerontol. 1994;49:B191–B196. doi: 10.1093/geronj/49.5.b191. [DOI] [PubMed] [Google Scholar]
- Csiszar A, Ungvari Z, Edwards JG, Kaminski P, Wolin MS, Koller A, Kaley G. Ageing-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ Res. 2002;90:1159–1166. doi: 10.1161/01.res.0000020401.61826.ea. [DOI] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, Peacock M, Campisi J. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato AJ, Eskurza I, Silver AE, Levy AS, Pierce GL, Gates PE, Seals DR. Direct evidence of endothelial oxidative stress with aging in humans. Relation to impaired endothelium-dependent dilation and upregulation of nuclear factor-kB. Circ Res. 2007;100:1659–1666. doi: 10.1161/01.RES.0000269183.13937.e8. [DOI] [PubMed] [Google Scholar]
- Furumoto K, Inoue E, Nagao N, Hiyama E, Miwa N. Age-dependent telomere shortening is slowed down by enrichment of intracellular vitamin C via suppression of oxidative stress. Life Sci. 1998;63:935–948. doi: 10.1016/s0024-3205(98)00351-8. [DOI] [PubMed] [Google Scholar]
- Galbiati F, Volonte D, Liu J, Capozza F, Frank PG, Zhu L, Pestell RG, Lisanti MP. Caveolin-1 expression negatively regulates cell cycle progression by inducing G0/G1 arrest via p53/p21WAF/Cip1-dependent mechanism. Mol. Biol. Cell. 2001;12:2229–2244. doi: 10.1091/mbc.12.8.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haendeler J, Hoffmann J, Diehl JF, Vasa M, Spyridopoulos I, Zeiher AM, Dimmeler S. Antioxidant inhibit nuclear export of telomerase reverse transcriptase and delay replicative senescence of endothelial cells. Circ Res. 2003a;94:768–775. doi: 10.1161/01.RES.0000121104.05977.F3. [DOI] [PubMed] [Google Scholar]
- Haendeler J, Hoffmann J, Brandes RD, Zeiher AM, Dimmeler S. Hydrogen peroxide triggers nuclear export of telomerase reverse transcriptase via Src kinase family-dependent phosphorylation of tyrosine 707. Mol Cell Biol. 2003b;23:4598–4610. doi: 10.1128/MCB.23.13.4598-4610.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- Herbig U, Jobling WA, Chen BPC, Chen DJ, Sedivy JM. Telomere shortening triggers senescence of human cells trough a pathway involving ATM, p53 and p21CIP1, but not p16INK4a. Mol. Cell. 2004;14:501–513. doi: 10.1016/s1097-2765(04)00256-4. [DOI] [PubMed] [Google Scholar]
- Humphries KM, Sweda PA, Sweda LI. Ageing: a shift from redox regulation to oxidative damage. Free Radic Res. 2006;40:1239–1243. doi: 10.1080/10715760600913184. [DOI] [PubMed] [Google Scholar]
- Jeanclos E, Krolewski A, Skurnick J, Kimura M, Aviv H, Warram JH, Aviv A. Shortened telomere length in white blood cells of patients with IDDM. Diabetes. 1998;47:482–486. doi: 10.2337/diabetes.47.3.482. [DOI] [PubMed] [Google Scholar]
- Jeanclos E, Schork NJ, Kyvik KO, Kimura M, Skurnick JH, Aviv A. Telomere length inversely correlates with pulse pressure and is highly familial. Hypertension. 2000;36:195–200. doi: 10.1161/01.hyp.36.2.195. [DOI] [PubMed] [Google Scholar]
- Kurz DJ, Decary S, Hong Y, Trivier E, Akhmedov A, Erusalimsky JD. Chronic oxidative stress compromises telomere integrity and accelerates the onset of senescence in human endothelial cells. J Cell Sci. 2004;117:2417–2426. doi: 10.1242/jcs.01097. [DOI] [PubMed] [Google Scholar]
- Lakatta EG. Age-associated cardiovascular changes in health: impact on cardiovascular disease in older persons. Heart Fail Rev. 2002;7:29–49. doi: 10.1023/a:1013797722156. [DOI] [PubMed] [Google Scholar]
- Lapenna D, Pierdomenico SD, Ciofani G, Giamberardino MA, Cuccurullo F. Aortic glutathione metabolic status: time-dependent alterations in fat-fed rabbits. Atherosclerosis. 2004;173:19–25. doi: 10.1016/j.atherosclerosis.2003.11.014. [DOI] [PubMed] [Google Scholar]
- Lorenz M, Saretzki G, Sitte N, Metzkow S, von Zglinicki T. BJ fibroblasts display high antioxidant capacity and slow telomere shortening independent of hTERT transfection. Free Radic Biol Med. 2001;31:824–831. doi: 10.1016/s0891-5849(01)00664-5. [DOI] [PubMed] [Google Scholar]
- Moiseeva O, Mallette FA, Mukhopadhyay UK, Moores A, Ferbeyre G. DNA damage signaling and p53-dependent senescence after prolonged beta-interferon stimulation. Mol Biol Cell. 2006;17:1583–1592. doi: 10.1091/mbc.E05-09-0858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohuchida K, Mizumoto K, Ogura Y, Ishikawa N, Nagai E, Yamaguchi K, Tanaka M. Quantitative assessment of telomerase activity and human telomerase reverse transcriptase messenger RNA levels in pancreatic juice samples for the diagnosis of pancreatic cancer. Clin Cancer Res. 2005;11:2285–2292. doi: 10.1158/1078-0432.CCR-04-1581. [DOI] [PubMed] [Google Scholar]
- Pandita TK. ATM function and telomere stability. Oncogene. 2002;21:611–618. doi: 10.1038/sj.onc.1205060. [DOI] [PubMed] [Google Scholar]
- Pizzimenti S, Barrera G, Dianzani MU, Brusselbach S. Inhibition of D1, D2 and A-cyclin expression in HL-60 cells by the lipid peroxydation product 4-hydroxynonenal. Free Radic Biol Med. 1999;26:1578–1586. doi: 10.1016/s0891-5849(99)00022-2. [DOI] [PubMed] [Google Scholar]
- Pizzimenti S, Briatore F, Laurora S, Toaldo C, Maggio M, De Grandi M, Meaglia L, Menegatti E, Giglioni B, Dianzani MU, Barrera G. 4-Hydroxynonenal inhibits telomerase activity and hTERT expression in human leukemic cell lines. Free Radic Biol Med. 2006;409:1578–1591. doi: 10.1016/j.freeradbiomed.2005.12.024. [DOI] [PubMed] [Google Scholar]
- Samani NJ, Boultby R, Butler R, Thompson JR, Goodall AJ. Telomere shortening in atherosclerosis. Lancet. 2001;358:472–473. doi: 10.1016/S0140-6736(01)05633-1. [DOI] [PubMed] [Google Scholar]
- Serrano AL, Andres V. Telomeres and cardiovascular disease: does size matter? Circ Res. 2004;94:575–584. doi: 10.1161/01.RES.0000122141.18795.9C. [DOI] [PubMed] [Google Scholar]
- Shats I, Milyavsky M, Tang X, Stambolsky P, Erez N, Brosh R, Kogan I, Braunstein I, Tzukerman M, Ginsberg D, Rotter V. p53-dependent down-regulation of telomerase is mediated by p21waf1. J Biol Chem. 2004;279:50976–50985. doi: 10.1074/jbc.M402502200. [DOI] [PubMed] [Google Scholar]
- Stampfer MR, Garbe J, Nijjar T, Wigington K, Swisshelm K, Yaswen P. Loss of p53 function accelerates acquisition of telomerase activity in indefinite lifespan human mammary epithelial cell lines. Oncogene. 2003;22:5238–5251. doi: 10.1038/sj.onc.1206667. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Kurose A, Huang X, Dai W, Darzynkiewicz Z. ATM activation and histone H2AX phosphorylation as indicators of DNA damage by DNA topoisomerase I inhibitor topecan and during apoptosis. Cell Prolif. 2006a;39:49–60. doi: 10.1111/j.1365-2184.2006.00364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Halicka HD, Huang X, Traganos F, Darzynkiewicz Z. Constitutive histone H2AX phosphorylation and ATM activation, the reporters of DNA damage by endogenous oxidants. Cell Cycle. 2006b;5:1940–1945. doi: 10.4161/cc.5.17.3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor WR, Schonthal AH, Galante J, Stark GR. p130/E2F4 binds to and represses the cdc2 promoter in response to p53. J Biol Chem. 2001;276:1998–2006. doi: 10.1074/jbc.M005101200. [DOI] [PubMed] [Google Scholar]
- Voghel G, Thorin-Trescases N, Farhat N, Nguyen A, Villeneuve L, Mamarbachi AM, Fortier A, Perrault LP, Carrier M, Thorin E. Cellular senescence in endothelial cells from atherosclerotic patients is accelerated by stress associated with cardiovascular risk factors. Mech Ageing Dev. 2007;128:662–671. doi: 10.1016/j.mad.2007.09.006. [DOI] [PubMed] [Google Scholar]
- von Zglinicki T. Oxidative stress shortens telomeres. Trends Biochem Sci. 2002;27:339–344. doi: 10.1016/s0968-0004(02)02110-2. [DOI] [PubMed] [Google Scholar]
- Wang P, Zhang Z, Ma X, Huang Y, Liu X, Tu P, Tong T. HDTIC-1 and HDTIC-2, two compounds extracted from Astragali Radix, delay replicative senescence of human diploid fibroblasts. Mech Ageing Dev. 2003;124:1025–1034. doi: 10.1016/j.mad.2003.08.005. [DOI] [PubMed] [Google Scholar]
- Zafarullah M, Li WQ, Sylvester J, Ahmad M. Molecular mechanisms of N-acetylcystein actions. Cell Mol Life Sci. 2003;60:6–20. doi: 10.1007/s000180300001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Aviv H, Gardner JP, Okuda K, Patel S, Kimura M, Bardeguez A, Aviv A. Loss of chromosome 13 in cultured human vascular endothelial cells. Exp Cell Res. 2000;260:357–364. doi: 10.1006/excr.2000.4997. [DOI] [PubMed] [Google Scholar]