

Abstract
Adequate therapies are lacking for Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, amyotrophic lateral sclerosis, and other neurodegenerative diseases. The ability to use antisense oligonucleotides (ASOs) to target disease-associated genes by means of RNA may offer a potent approach for the treatment of these, and other, neurodegenerative disorders. In modifying the basic backbone chemistry, chemical groups, and target sequence, ASOs can act through numerous mechanisms to decrease or increase total protein levels, preferentially shift splicing patterns, and inhibit microRNAs, all at the level of the RNA molecule. Here, we discuss many of the more commonly used ASO chemistries, as well as the different mechanisms of action that can result from these specific chemical modifications. When applied to multiple neurodegenerative mouse models, ASOs that specifically target the detrimental transgenes have been shown to rescue disease associated phenotypes in vivo. These supporting mouse model data have moved the ASOs from the bench to the clinic, with two neuro-focused human clinical trials now underway and several more being proposed. Although still early in development, translating ASOs into human patients for neurodegeneration appears promising.
Electronic supplementary material
The online version of this article (doi:10.1007/s13311-013-0194-5) contains supplementary material, which is available to authorized users.
Keywords: Antisense oligonucleotides, Neurodegeneration, RNA, Knockdown, Alternative splicing, MicroRNA
Antisense Oligonucleotides
Introduction
“Antisense oligonucleotide” or ASO, is a broad term, encompassing any relatively short string of nucleic acids. For this specific review, we focus only on those ASOs that are single-stranded sequences, 8–50 base pairs in length, and bind to the target RNA by means of standard Watson–Crick base pairing. Upon binding, the ASOs can alter the original function of the target RNA through an array of different mechanisms, many of which we discuss here.
ASO Chemical Modifications
As they are found in nature, short oligonucleotides possess weak intrinsic binding affinities and are readily degraded by nucleases. However, there are several ways to apply chemical modifications to ASOs in order to overcome these weaknesses. Both the backbone chemistry and sugar moieties are premier targets for ASO design enhancement.
The phosphorothioate (PS) backbone modification is one of the earliest and still, to date, one of the most widely used modifications for ASO drugs (Fig. 1). The nonbridging phosphate oxygen atoms in natural nucleic acids are replaced with sulfur atoms, equipping ASOs with several important properties that support their use as a systemic drug [1]. The PS backbone increases the stability of the ASO against nuclease degradation [2], ensuring that PS–ASOs can reach their target RNA in cells and tissues. Of equal importance, PS-ASOs can recruit the enzyme RNaseH to promote cleavage of the target RNA, a crucial mechanism of action for many ASOs. In fact, most ASO drugs in development use this exact mechanism to reduce total levels of the target gene [3]. In addition to increasing stability and recruiting RNaseH, the PS modifications enhance the attachment of ASOs to plasma proteins, facilitating binding to cells and uptake into specific tissues [4]. A similar backbone modification, thiophsophoramidate (Fig. 1), also promotes high nuclease resistance. However, this substitution abrogates any RNaseH activity [5], making the backbone more suited for nondegrading RNA manipulations, including alternative splicing changes, translation inhibition, and microRNA hindrance.
Fig. 1.

Antisense oligonucleotide (ASO) chemical structures. Schematic of unmodified DNA/RNA base pair (left). Different backbone modifications that can be applied (top row) and different 2’-sugar modifications that can be used (bottom row) to increase nuclease resistance and RNA binding affinity of the ASO. Deviations from the original unmodified DNA/RNA are highlighted by circles
ASO backbone chemistries have also been modified through replacing the sugar phosphate backbone with an isostere, such as with a Morpholino ASO (Fig. 1). Morpholino ASOs are neutral and, like the phosphoramidates, do not activate RNaseH. Thus, Morpholinos are often used in translation inhibition or other steric blocking mechanisms, including mostly alteration of gene splicing [6, 7]. Morpholinos have been reviewed in detail by others [8] and show promise for altering splicing and inhibiting translation in vivo [7–9].
While the backbone of the ASOs is an excellent target for manipulation, modifications at the 2’-position of the sugar moiety have also proven to be equally valuable for enhancing drug-like properties of ASOs. Modifications at the 2’ position enhance ASO potency by facilitating target binding. Of the 2’-modifications currently used, the 2’-O-methyl and 2’-O-methoxyethyl (MOE) sugar modifications are the most popular and are in multiple ongoing human clinical trials. MOE modifications not only increase resistance to nucleases but also reduce nonspecific protein binding, which can, in turn, reduce the ASO toxicity profile [10]. Another sugar modification, termed locked nucleic acid (LNA), is an analog of the 2’-O-methyl RNA [11, 12] (Fig. 1). The main advantage of LNA ASOs is the robust binding improvement and nuclease resistance compared with other 2’-modifications. LNA ASOs demonstrate much better potency, though at the price of increased toxicity liabilities [13]. In more recent years, several analogs of LNA have been synthesized. Some show improved activity and/or toxicity profiles in animals [14, 15], demonstrating potential for LNA ASOs in future years. Additional 2’-sugar modifications exist, including the 2’-fluoro additive [16], all with varying degrees of nuclease resistance and target binding affinities.
Although 2’-sugar modifications enhance binding to the mRNA target, almost all significantly reduce or even completely obstruct RNaseH from cleaving the target RNA. One of the most popular strategies used to circumvent this limitation has been to adopt the “gapmer” design, whereby regions of 2’-modified residues flank a longer central unmodified region (Fig. 2). These 2’-modified “wings” further increase binding affinity and nuclease resistance while still allowing the center gap region to recruit RNaseH.
Fig. 2.
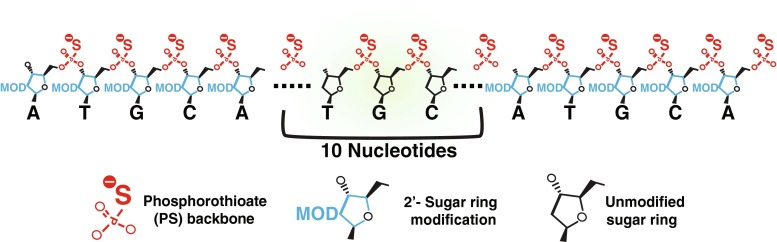
Antisense oligonucleotide (ASO) ‘gapmer’ design. Example of a 20-base pair ASO that would be designed to support RNaseH activity. The phosphorothioate backbone is used along the entire length of the ASO to provide nuclease resistance, while the 2’-sugar modification is used exclusively on the first and last 5 nucleotides, leaving the middle 10 nucleotides unmodified at the 2’-sugar position. This provides increased target RNA binding affinity on the outer portions of the ASO, while still allowing RNaseH cleavage at the central region of the ASO
While we have briefly highlighted several of the modifications used to develop ASOs, the first ASOs to be studied did not have the same advanced chemical alterations. First-generation ASOs only have the PS backbone, limiting ASO use to RNaseH degradation. Additionally, these first-generation ASOs produce nonspecific effects in vivo by interacting significantly with cell surface and intracellular proteins [17]. To improve on this design, second-generation ASOs incorporated 2’-sugar modifications, including the previously discussed 2’-O-methyl and MOE additions. As medicinal chemistry continued to evolve, more advanced modifications, including the LNA and Morpholino chemistries, were incorporated, resulting in the third generation ASO class.
The development of these modifications has helped with the potency and toxicology profiles of ASOs. As with most drugs, ASOs demonstrate dose-dependent transient and mild-to-moderate toxicities in rodents, primates, and humans. Some of the most common toxicities associated with ASOs include inhibition of the clotting cascade and enhanced liver enzymes, typically seen when the concentration of PS-ASOs is high in the plasma [18–20]. In addition, a common subchronic toxicity associated with ASO administration is the activation of the immune system. Specifically, ASOs interact with Toll-like receptor 9 [21], which can result in splenomegaly, though it appears that rodents are much more sensitive to this particular immune response than primates [22]. It should be noted that more is known in regard to the toxicities of ASOs in rodents and when delivered to humans peripherally. Thus far, only one study has been published that has tested the safety of ASOs when delivered directly to the central nervous system (CNS) of humans. It found no major side effects of the second-generation ASO in the treated human patients at any of the administered doses [23]. This bodes well for future second- and third-generation ASO studies in the CNS, though new modifications are continuously being investigated in an effort to achieve even better tolerated ASOs. While the second- and third-generation chemical modifications have made it possible for ASOs to enter into human clinical studies, the single best chemistry for the human application of ASOs to the CNS may not yet be known. It is an ongoing process to more fully understand the tolerability and potency of both old and new ASO chemistries in an effort to find the most suitable ASO for human delivery.
ASO Mechanisms of Action
ASOs work through a variety of different mechanisms to achieve target manipulation (Fig. 3), though these can be broken into two broad categories: those that recruit RNaseH for degradation and those that do not. RNA-degrading ASOs cleave the target mRNA by activating the nuclear enzyme RNAseH [24]. As described earlier, these ASOs must have at least a portion of the ASO unmodified at the 2’ position (Fig. 2) [25]. RNaseH is a mammalian enzyme that recognizes RNA–DNA heteroduplexes, cleaves the RNA strand, and releases the intact DNA [3]. More specifically, RNaseH1 is responsible for mediating RNA cleavage as directed by ASO hybridization [26]. Because RNaseH1 releases the intact ASO upon cleavage of the bound RNA, it conveniently allows for a single ASO to catalytically cleave many RNA molecules, further increasing ASO potency.
Fig. 3.

Antisense oligonucleotide (ASO) mechanisms of action. ASOs have been proposed to enter into cells through high- and low-binding plasma protein receptors on the cell surface, resulting in ASO compartmentalization into lysosomes and endosomes [58]. Through a largely unknown mechanism, ASOs are released from the vesicles into the cytoplasm where they can freely move in and out of the nucleus [24, 60]. Upon entry into the nucleus, ASOs can bind directly to mRNA structures and prevent the formation of the 5’-mRNA cap (1), modulate alternative splicing (2), dictate the location of the polyadenylation site (3), and recruit RNaseH1 to induce cleavage (4). ASOs in the cytoplasm can bind directly to the target mRNA and sterically block the ribosomal subunits from attaching and/or running along the mRNA transcript during translation (5). ASOs can also be designed to directly bind to microRNA (miRNA) sequences (6) and natural antisense transcripts (NATs) (7), thereby prohibiting miRNAs and NATs from inhibiting their own specific mRNA targets. Ultimately, this leads to gene upregulation of the miRNA and NAT targets
Activation of RNAseH is extremely sequence specific, as a single mismatch results in a 3- to 5-fold decrease in RNaseH cleavage [27–29], and 3 or more mismatches results in a complete loss of RNaseH degrading activity [30]. When designing ASOs, an ideal ASO would have a perfect match to the target and more than 3 base pair mismatches to all other genes (Fig. 4). Designing ASOs with at least 2 mismatches to other genes is often achievable.
Fig. 4.

Designing antisense oligonucleotides (ASOs). Designing ASOs for a new genetic target from scratch can be an overwhelming task. However, there are certain guidelines that can be taken into account that can increase the chances of generating a successful ASO. Certain ASO characteristics need to be first identified (Step 1) and then specific ASO sequences can be generated using the listed guidelines as a starting point (Step 2). ASOs should first be screened in vitro (Step 3) and then the most successful ASOs can be moved forward for in vivo screening, both in the brain parenchyma (Step 4) and through intraventricular delivery (Step 5). These steps do not guarantee success as there are many exceptions to the guidelines when designing ASOs. Instead, this figure provides a starting guide for designing basic ASO sequences that can be translated to the central nervous system of in vivo rodent models
Nondegrading ASOs, however, are typically fully modified at the 2’ position and do not activate RNaseH. Instead, they exert their effect by binding tightly to the target mRNA, making them extremely useful for inhibiting translation, modulating splicing, and inhibiting microRNAs (miRNAs). Translation inhibitors can theoretically stick directly to the mRNA and prevent the movement of ribosomes down the transcript, and/or inhibit the physical assembly of the 40S and 60S ribosomal subunits onto the mRNA sequence. That being said, translation inhibition has not been a primary focus of current ASO therapies.
ASOs that bind, but do not destroy mRNA, may also be used to preferentially modulate processing of mRNA transcripts. After transcription from the genomic template, newly formed RNA undergoes several complex processing events in the nucleus, including alternative splicing, polyadenylation at the 3’ end, and the addition of a 5’-cap [31]. From some estimates, as much as 90 % of mRNA transcripts undergo alternative splicing in human cells [32], affording numerous opportunities for genetic diseases to manifest from aberrant splicing events [33]. There have now been several studies showing that ASOs have the ability to bind pre-mRNA structure and directly modulate splicing, both in vitro and in vivo, by masking splicing enhancers and repressor sequences, skipping exons, and/or forcing inclusion of otherwise alternatively spliced exons [6, 34–39]. In addition to forcing exon inclusion/exclusion, ASOs can modulate polyadenylation selection in those transcripts with more than one polyA site in the 3’-UTR [40], as well as inhibit or, surprisingly, promote expression of a target by binding the target RNA promoter sequence [41–43].
More recently, ASOs have been developed against miRNA sequences in an effort to inhibit miRNAs from binding their own target mRNA molecules. The ASO is designed such that it binds the full length of the miRNA and blocks its ability to interact with mRNAs. The ASO can robustly interfere with the miRNA function while not promoting degradation of the miRNA itself [44]. Inhibiting miRNA function with ASOs has now been successfully shown in vitro and in vivo [45–48], and holds great therapeutic promise as the role of miRNAs in neurodegenerative diseases continues to develop [49].
Pharmacokinetic Properties of ASOs
Intraperitoneal, subcutaneous, or intravenous delivery of ASOs results in widespread delivery of the ASOs to many peripheral tissues [50, 51] through binding both low- and high-affinity plasma proteins [15, 52, 53]. As might be expected, the highly charged ASOs do not cross the blood–brain barrier (BBB) [54], thus complicating delivery for neurodegenerative diseases. To circumvent the BBB, ASOs can be delivered directly into the cerebral spinal fluid (CSF) that, in turn, bathes the brain and spinal cord, allowing for surprisingly efficient distribution of the ASOs in the CNS, both in rodents and Rhesus monkeys [54, 55]. Staining for ASOs in the CNS reveals a relatively uniform distribution, suggesting an active ASO uptake mechanism rather than simple diffusion, which would be expected to show the highest concentration near the ventricles. When tested in vitro, accumulation of ASOs inside cultured cells far exceeded the concentration of ASOs in the media by more than 100-fold after just 1 h of ASO exposure [56]. Further, when administered to both rodents and human patients, only 10–30 % of the administered ASO was excreted through the urine [57]. Taken together, these data strongly suggest that ASOs are efficiently and readily taken up by cells and tissues, supporting an active uptake mechanism, though the exact mechanism remains unknown.
As a first pass to understand the mechanism of entry, it was found that vesicular pathways can either deliver ASOs to lysosomes or release them directly into the cytoplasm in cultured hepatocytes (Fig. 3), though the route whereby ASOs escape from vesicles in not yet well understood [58, 59]. Once in the cytoplasm, ASOs can readily move between cytoplasmal and nuclear compartments [60], and PS–ASOs appear to be sequestered in both the nuclei and mitochondria fractions following subcellular fractionation [56]. These studies were done in cultured cells and need to be done in vivo to confirm the mechanism.
Upon successful entry into cells, ASOs demonstrate a relatively long duration of action. Following termination of a short ASO infusion period using RNaseH ASOs, target mRNA levels can be suppressed for up to 12–16 weeks [55] and even longer with the fully MOE-modified ASOs [61], suggesting that the effect of ASOs is extremely long-lived in tissues, likely owing to a lengthy ASO half-life. When administering ASOs into the CSF, a constant infusion or single bolus injection can be used, directed to either the lateral ventricles or intrathecal (IT) space. Both the ventricular bolus and constant infusion methods have been used in mouse models [62]. Bolus injections typically result in better uniform distribution of ASOs [63], though the duration of target modulation may not last as long. In rodents, ASO delivery is often achieved through the lateral ventricles owing to the relative ease of surgery compared with an IT infusion. In larger animals, such as nonhuman primates, IT delivery is more practical and successfully results in therapeutic doses of ASO throughout the CNS [54, 55], though deeper brain structures may not receive the same concentration of ASO as more superficial layers through this route [55]. This is an important consideration for diseases such as Huntington’s disease (HD) where the primary affected brain regions are deeper structures.
How these methods of delivery affect pharmacokinetics and distribution of ASOs are important variables to consider in the delivery of ASOs to human patients. The first two human clinical trials for CNS disorders have both used IT dosing, and thus a safety precedent will be set for IT delivery. Though IT is more invasive than a peripheral injection, IT delivery is a common, generally well-tolerated procedure that is currently used in the clinic for a number of other uses, including steroid, analgesia, and anesthesia delivery [64–66]. The first CNS human ASO clinical trial targeting superoxide dismutase 1 (SOD1) in familial ALS patients used a slow IT infusion, designed to mimic longer, chronic infusion [23]. In an ongoing human trial for spinal muscular atrophy (SMA), the survival of motor neuron (SMN) ASO is delivered through a single rapid IT injection, which may be more comparable to bolus injections. Repeat CSF injections, if deemed necessary, may be more practical with IT delivery, though an intraventricular Ommaya reservoir could also be placed.
While current practices for ASO delivery to the CNS of human patients involves an IT injection, future methods may be designed to be significantly less invasive as a lumbar puncture is more uncomfortable and cumbersome than a peripheral injection. There is growing interest in using ASOs tethered to nanoparticles in an effort to traffic ASOs across the BBB [67, 68] so that a simple systemic injection can be used. In turn, this could make ASO therapies, especially for nonlife-threatening diseases, a more attractive option.
ASOs in Human Neurodegeneration
Using ASOs to Decrease Total Protein
There are numerous neurodegenerative disorders that could potentially benefit from a knockdown therapeutic strategy—primarily those disorders that are a direct result of a single aberrant protein. The first in vivo treatment for a CNS-neurodegenerative disorder using ASOs with a PS backbone and 2’-O-methyl modification targeted the human SOD1 transgene in the transgenic SOD1G93A rat model of amyotrophic lateral sclerosis (ALS) [54], a fatal motor neuron disease. The SOD1 ASOs reduced total human SOD1 mRNA levels by 50 % throughout the brain and spinal cord of the rats, resulting in a 37 % extension of survival after the disease onset in vivo. These studies, along with promising toxicology studies in nonhuman primates, prompted the advancement of human SOD1 ASOs into phase I human clinical trials for those familial ALS patients with SOD1 mutations [23]. Results from this study showed an excellent safety profile after a single 11.5-hour IT infusion of ASOs [23] and strongly suggest that CSF delivery of ASOs may be a viable strategy for other neurologic disorders.
With the success of the SOD1 in vivo work, additional neurodegenerative disorders were explored, including Huntington's disease (HD). HD is an autosomal dominant neurodegenerative disorder that results from a CAG expansion in exon 1 of the huntingtin gene. Those with HD display progressive hyperkinetic involuntary movements, chorea, dystonia, and cognitive deficits. Owing to the direct link between mutated huntingtin and development of HD, a likely therapy may be to decrease the causative mutant huntingtin. When delivered to human huntingtin transgenic mice, human huntingtin ASOs were able to significantly reduce total huntingtin mRNA and protein levels, and were able to rescue the detrimental motor and anxiety phenotypes classically seen in the transgenic line [55]. Interestingly, even after total human huntingtin levels had returned to baseline following ASO treatment, the rescue in behavior persisted, suggesting that a single bolus of huntingtin ASO may have long-lasting beneficial effects as a therapy. With such a striking rescue in both the transgenic mouse behavior and poly-glutamine aggregates, plans for a phase I trial are underway for the treatment of HD with human huntingtin ASOs. Additionally, ASOs have been developed to preferentially bind to the CAG repeat in exon 1 of huntingtin mRNA, allowing for selective reduction of mutated huntingtin while leaving wild-type huntingtin untouched [69]. This would eliminate the need to knockdown all huntingtin in the event that too much huntingtin knockdown is detrimental [70, 71].
The most prevalent neurodegenerative disease that demands therapeutic intervention is Alzheimer’s disease (AD), with 5.4 million Americans currently afflicted with the disease and, if a treatment is not found, 13.2 million by 2050 [72]. AD is pathologically characterized by two primary hallmarks—diffuse amyloid-beta plaques and intraneuronal neurofibrillary tangles, both of which are potential ASO targets. ASOs against the amyloid precursor protein (APP), the precursor to the toxic amyloid-beta peptides that are generated in the development and progression of AD, have shown in vivo promise. When injected in a mouse model that over expresses hAPP, the APP ASOs resulted in a significant reduction in hAPP protein levels in the brain, and a rescue in learning and memory in both young and aged mice [73–75].
In addition to targeting amyloid-beta, ASOs against tau, the protein that composes intraneuronal neurofibrillary tangles, may also provide therapeutic benefits. When crossed to amyloid-beta depositing mouse lines, tau knockout significantly rescued the amyloid-beta-induced cognitive deficits [76–80], strongly suggesting that a tau reduction therapy may be beneficial for the treatment of AD. Importantly, tau knockout in vivo appears to be surprisingly normal [81–83], though some have recently reported that a complete lack of tau for long periods of time may result in a Parkinsonism phenotype [83, 84]. These data suggest that tau knockdown for short periods of time may be safe, though a total decrease of tau for extended periods of time is less advised.
Using ASOs to Increase Total Protein
When altering total levels of a target, ASOs are almost exclusively used to decrease overall target levels through RNaseH recruitment. However, there are emerging data that clearly demonstrate the ability of ASOs to upregulate genes by binding to and inhibiting natural antisense transcripts. Modarresi et al. [85] found that expression of the brain-derived neurotrophic factor (BDNF) is typically suppressed by a noncoding antisense RNA transcript termed BDNF-AS. Using a complementary ASO to directly bind and inhibit BDNF-AS activity, BDNF-AS was no longer able to repress BDNF expression and total BDNF mRNA levels increased 2–7-fold. More impressively, after suppressing BDNF-AS, BDNF proteins levels also increased and induced neuron outgrowth and differentiation, both in vitro and in vivo. While these results could provide a novel way of increasing total protein levels in vivo, it is too early to tell whether targeting an natural antisense transcript associated with a gene will be a routine way to increase a gene product or whether this will only be feasible for a relatively small subset of mRNAs.
Using ASOs to Modulate Splicing
ASOs as drugs are most often used to change total levels of a gene target, though using ASOs to preferentially modulate alternative splicing is emerging as an increasingly powerful tool in neurodegeneration. One premier example is SMA, a genetic neuromuscular disorder that presents with lower motor neuron loss in the spinal cord and, consequently, paralysis and muscular atrophy. The severest form of the disease can lead to early infant death. SMA is a direct result of either a homozygous loss of or a mutation in the SMN1 gene, which encodes the SMN protein. Complete loss of SMN would be lethal except for the presence of the SMN2 gene. The majority of SMN2 transcripts incorrectly splice out exon 7—a crucial exon for SMN function—thus producing very little functional protein [86]. It had long been recognized that forcing inclusion of exon 7 would create a functional SMN2 mRNA that would in turn express increased amounts of full-length SMN protein. Using mice that express the human SMN2 transcript, it has been demonstrated that ASOs can dramatically shift SMN2 splicing patterns, resulting in nearly complete inclusion of exon 7 and thus greatly increasing functional SMN protein. SMN-splicing ASOs delivered to the CNS of transgenic SMA newborn mice resulted in significant improvements in the righting response of mice, and delayed the onset of tail and ear necrosis [61, 87, 88]. Interestingly, a recent study showed that systemic delivery of SMN splicing ASOs also resulted in a significant phenotypic rescue in a severe SMA mouse model [50]. The previous studies were carried out with fully MOE-modified PS–ASOs to modulate SMN2 splicing, though recent reports have also shown a significant rescue in disease phenotype with Morpholino ASOs that force inclusion of exon 7 into SMN2, demonstrating that more than one ASO chemistry may be viable for therapy [89, 90]. SMN splicing ASOs are now in phase I human clinical trials for children with SMA (Clinicaltrials.gov NCT01703988, NCT01780246).
In addition to SMA there are several other neurodegenerative disorders that may benefit from a splicing therapy, including frontotemporal dementia with Parkinsonism linked to chromosome 17. Several mutations in the gene tau have been identified that lead to a stark change in tau protein isoform ratios. In adult human brains, there are six isoforms of tau that can be expressed [91], which differ by the presence or absence of 2 N-terminal exons and by the presence of either 3 (3R) or 4 (4R) C-terminal repeat domains, depending on the exclusion or inclusion of exon 10, respectively [92, 93]. In normal human brain, there is a 1:1 ratio of 4R:3R tau isoforms [94, 95], though mutations in tau can lead to a high 4R:3R ratio [96, 97], implicating 4R tau in the disease pathogenesis. Other tauopathies, including progressive supranuclear palsy, corticobasal degeneration, and, more controversially, AD, also experience a high 4R:3R tau ratio [98–102]. These disorders may benefit from a tau splicing strategy that leads to exclusion of exon 10, thus decreasing the high 4R:3R tau ratio without changing total tau levels. Tau-splicing ASOs have been developed that have the capability to alter tau splicing patterns in vitro [39], though in vivo work with a tau-splicing ASO treatment paradigm is still needed.
In loss of function diseases where a partial protein has been shown to demonstrate activity, it may be possible to create a splicing strategy that excludes a particular exon, and thus a mutation or early stop site. If this exon exclusion leaves the mRNA in frame, it may effectively cut out the aberrant portion of the final protein. Based on prior work demonstrating that expression of a mini-gene construct of dystrophin can function similarly to full-length dystrophin in vivo [103], this type of creative strategy was engineered for Duchenne’s muscular dystrophy (DMD), a severe genetic disorder involving a defect in the dystrophin gene that results in extreme and rapid muscle wasting such that immobility occurs between the age of 1–12 years and death in the third or fourth decade of life. Because the disease results from a loss-of-function of dystrophin, a splicing strategy was used to eliminate the defective portion of the gene giving rise to functional dystrophin. Dystrophin-splicing ASOs were developed and tested in DMD transgenic mice, resulting in significantly improved muscle function and restoration of dystrophin protein levels [7, 104]. Even when administered to dogs with DMD, the number of dystrophin-positive muscle fibers was restored to 80–97 % of normal levels, with significant improvements in motor performance [105]. Based on these positive results in vivo, splicing- ASOs have entered human DMD clinical trials [106–109] and have shown a successful increase in the percent of dystrophin-positive muscle fibers in phase II human studies; multinational phase III studies are currently underway. As proteins are restored in loss-of-function diseases, activation of the immune system may complicate some of these therapies [110], though this splicing strategy still may be a promising approach for some selected diseases.
Using ASOs to Inhibit miRNAs
miRNAs are small, noncoding RNAs that control the regulation of 200–300 genes, typically through binding the 3’-untranslated region and repressing the expression of mRNA targets [111, 112]. More than 33 % of human genes are suspected targets of miRNA inhibition [113]. miRNAs have increasingly been recognized as part of disease processes, including those related to neurodegeneration [49]. ASOs may be used to directly bind and inhibit miRNAs [44, 114]. Although miRNAs can target numerous mRNAs, inhibiting one miRNA may still be a well-tolerated and effective strategy. The first example of a miRNA ASO to show therapeutic promise in vivo targeted miR-122 in the setting of hepatitis C infection [115]. Inhibition of miR-122 was well tolerated in phase II human studies [116] and substantially decreased the hepatitis C viral load in monkeys [117, 118], as well as human patients measured at the completion of the phase II human trial [119].
In addition to miR-122 ASOs, ASOs against miR-206 have been tested in an amyloid-beta-depositing AD mouse model. By inhibiting miR-206, BDNF protein levels significantly increased and cognitive performance was improved [120]. Additionally, by targeting miR-34c, a negative regulator of memory consolidation, learning performance in AD mouse models was also rescued [121]. These preliminary results suggest that miRNA ASOs may also prove to be a viable therapeutic strategy for AD. As miRNAs become a larger field of research, more potential targets for a miRNA-based ASO neurodegenerative therapy are likely to emerge.
Conclusions
Numerous methods exist for ultimately achieving desired modifications at the protein level, including small molecules, immunization, and RNA targeted approaches. ASOs, the focus of the review herein, possess the capability to selectively manipulate RNA processing in order to increase or decrease total target levels, preferentially alter splicing, and inhibit intrinsic miRNAs. Advances in medicinal chemistry have equipped ASOs with the necessary chemical modifications to significantly increase resistance to nucleases and strengthen RNA binding affinities, both crucial in the design of ASOs as therapeutic drugs. Further, ASOs possess the capability to freely distribute throughout the brain and spinal cord following an IT injection, strongly supporting their use as a viable therapy for a variety of CNS disorders where an aberrant target can be identified. Two CNS–ASO human clinical trials are currently underway for ALS and SMA, and thus far appear to be well tolerated. With positive preclinical data and new RNA targets constantly being generated, there are seemingly endless possible ASO therapeutic approaches for neurodegenerative disease.
Electronic Supplementary Material
(PDF 510 kb)
Acknowledgments
TMM is supported by NIH/NINDS K08NS074194 and NIH/NINDS R01NS078398. Isis Pharmaceuticals has supplied antisense oligonucleotides for studies conducted by the authors. Other than those mentioned, we declare no real or perceived conflicts of interest.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article
Contributor Information
Sarah L. DeVos, Email: sldevos@wustl.edu
Timothy M. Miller, Email: millert@neuro.wustl.edu
References
- 1.Eckstein F. Phosphorothioate oligodeoxynucleotides: what is their origin and what is unique about them? Antisense Nucleic Acid Drug Dev. 2000;10:117–121. doi: 10.1089/oli.1.2000.10.117. [DOI] [PubMed] [Google Scholar]
- 2.Stein CA, Subasinghe C, Shinozuka K, Cohen JS. Physicochemical properties of phosphorothioate oligodeoxynucleotides. Nucleic Acids Res. 1988;16:3209–3221. doi: 10.1093/nar/16.8.3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cerritelli SM, Crouch RJ. Ribonuclease H: the enzymes in eukaryotes. FEBS J. 2009;276:1494–1505. doi: 10.1111/j.1742-4658.2009.06908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown D, Kang S, Gryaznov S, DeDionisio L, Heidenreich O, Sullivan S, et al. Effect of phosphorothioate modification of oligodeoxynucleotides on specific protein binding. J Biol Chem. 1994;269:26801–26805. [PubMed] [Google Scholar]
- 5.Gryaznov S, Skorski T, Cucco C, Nieborowska-Skorska M, Chiu CY, Lloyd D, et al. Oligonucleotide N3’-- > P5’ phosphoramidates as antisense agents. Nucleic Acids Res. 1996;24:1508–1514. doi: 10.1093/nar/24.8.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sazani P, Gemignani F, Kang S-H, Maier MA, Manoharan M, Persmark M, et al. Systemically delivered antisense oligomers upregulate gene expression in mouse tissues. Nat Biotechnol. 2002;20:1228–1233. doi: 10.1038/nbt759. [DOI] [PubMed] [Google Scholar]
- 7.Alter J, Lou F, Rabinowitz A, Yin H, Rosenfeld J, Wilton SD, et al. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat Med. 2006;12:175–177. doi: 10.1038/nm1345. [DOI] [PubMed] [Google Scholar]
- 8.Summerton J. Morpholino antisense oligomers: the case for an RNase H-independent structural type. Biochim Biophys Acta. 1999;1489:141–158. doi: 10.1016/s0167-4781(99)00150-5. [DOI] [PubMed] [Google Scholar]
- 9.Wu B, Moulton HM, Iversen PL, Jiang J, Li J, Li J, et al. Effective rescue of dystrophin improves cardiac function in dystrophin-deficient mice by a modified morpholino oligomer. Proc Natl Acad Sci U S A. 2008;105:14814–14819. doi: 10.1073/pnas.0805676105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Teplova M, Minasov G, Tereshko V, Inamati GB, Cook PD, Manoharan M, et al. Crystal structure and improved antisense properties of 2’-O-(2-methoxyethyl)-RNA. Nat Struct Biol. 1999;6:535–539. doi: 10.1038/9304. [DOI] [PubMed] [Google Scholar]
- 11.Koshkin AA, Singh SK, Nielsen P, Rajwanshi VK, Kumar R, Meldgaard M, et al. LNA (locked nucleic acids): synthesis of the adenine, cytosine, guanine, 5-methylcytosine, thymine and uracil bicyclonucleoside monomers, oligomerisation, and unprecedented nucleic acid recognition. Tetrahedron. 1998;54:3607–3630. [Google Scholar]
- 12.Wengel J. Synthesis of 3’-C- And 4’-C- branched oligodeoxynucleotides and the development of locked nucleic acid (LNA) Acc Chem Res. 1999;32:301–310. [Google Scholar]
- 13.Swayze EE, Siwkowski AM, Wancewicz EV, Migawa MT, Wyrzykiewicz TK, Hung G, et al. Antisense oligonucleotides containing locked nucleic acid improve potency but cause significant hepatotoxicity in animals. Nucleic Acids Res. 2007;35:687–700. doi: 10.1093/nar/gkl1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seth PP, Siwkowski A, Allerson CR, Vasquez G, Lee S, Prakash TP, et al. Short antisense oligonucleotides with novel 2’-4’ conformationaly restricted nucleoside analogues show improved potency without increased toxicity in animals. J Med Chem. 2009;52:10–13. doi: 10.1021/jm801294h. [DOI] [PubMed] [Google Scholar]
- 15.Koizumi M. ENA oligonucleotides as therapeutics. Curr Opin Mol Ther. 2006;8:144–149. [PubMed] [Google Scholar]
- 16.Kawasaki AM, Casper MD, Freier SM, Lesnik EA, Zounes MC, Cummins LL, et al. Uniformly modified 2’-deoxy-2’-fluoro phosphorothioate oligonucleotides as nuclease-resistant antisense compounds with high affinity and specificity for RNA targets. J Med Chem. 1993;36:831–841. doi: 10.1021/jm00059a007. [DOI] [PubMed] [Google Scholar]
- 17.Kurreck J. Antisense technologies. Improvement through novel chemical modifications. Eur J Biochem. 2003;270:1628–1644. doi: 10.1046/j.1432-1033.2003.03555.x. [DOI] [PubMed] [Google Scholar]
- 18.Smith RA, Miller TM. Targeting neurological disorders with antisense oligonucleotides. In: Crooke ST, editor. Antisense drug technology: principles, strategies, and applications. 2. Boca Raton, FL, USA: CRC Press; 2008. pp. 721–745. [Google Scholar]
- 19.Chan JHP, Lim S, Wong WSF. Antisense oligonucleotides: from design to therapeutic application. Clin Exp Pharmacol Physiol. 2006;33:533–540. doi: 10.1111/j.1440-1681.2006.04403.x. [DOI] [PubMed] [Google Scholar]
- 20.Jason TLH, Koropatnick J, Berg RW. Toxicology of antisense therapeutics. Toxicol Appl Pharmacol. 2004;201:66–83. doi: 10.1016/j.taap.2004.04.017. [DOI] [PubMed] [Google Scholar]
- 21.Senn JJ, Burel S, Henry SP. Non-CpG-containing antisense 2’-methoxyethyl oligonucleotides activate a proinflammatory response independent of Toll-like receptor 9 or myeloid differentiation factor 88. J Pharmacol Exp Ther. 2005;314:972–979. doi: 10.1124/jpet.105.084004. [DOI] [PubMed] [Google Scholar]
- 22.Miller TM, Smith RA, Kordasiewicz H, Kaspar BK. Gene-targeted therapies for the central nervous system. Arch Neurol. 2008;65:447–451. doi: 10.1001/archneur.65.4.nnr70007. [DOI] [PubMed] [Google Scholar]
- 23.Miller TM, Pestronk A, David W, Rothstein J, Simpson E, Appel SH, et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol. 2013;12:435–442. doi: 10.1016/S1474-4422(13)70061-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lorenz P, Baker BF, Bennett CF, Spector DL. Phosphorothioate antisense oligonucleotides induce the formation of nuclear bodies. Mol Biol Cell. 1998;9:1007–1023. doi: 10.1091/mbc.9.5.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suzuki Y, Holmes JB, Cerritelli SM, Sakhuja K, Minczuk M, Holt IJ, et al. An upstream open reading frame and the context of the two AUG codons affect the abundance of mitochondrial and nuclear RNase H1. Mol Cell Biol. 2010;30:5123–5134. doi: 10.1128/MCB.00619-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu H, Lima WF, Zhang H, Fan A, Sun H, Crooke ST. Determination of the role of the human RNase H1 in the pharmacology of DNA-like antisense drugs. J Biol Chem. 2004;279:17181–17189. doi: 10.1074/jbc.M311683200. [DOI] [PubMed] [Google Scholar]
- 27.Carroll JB, Warby SC, Southwell AL, Doty CN, Greenlee S, Skotte N, et al. Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the Huntington disease gene/allele-specific silencing of mutant huntingtin. Mol Ther. 2011;19:2178–2185. doi: 10.1038/mt.2011.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Basilion JP, Schievella AR, Burns E, Rioux P, Olson JC, Monia BP, et al. Selective killing of cancer cells based on loss of heterozygosity and normal variation in the human genome: a new paradigm for anticancer drug therapy. Mol Pharmacol. 1999;56:359–369. doi: 10.1124/mol.56.2.359. [DOI] [PubMed] [Google Scholar]
- 29.Lima WF, Rose JB, Nichols JG, Wu H, Migawa MT, Wyrzykiewicz TK, et al. Human RNase H1 discriminates between subtle variations in the structure of the heteroduplex substrate. Mol Pharmacol. 2007;71:83–91. doi: 10.1124/mol.106.025015. [DOI] [PubMed] [Google Scholar]
- 30.Monia BP, Johnston JF, Ecker DJ, Zounes MA, Lima WF, Freier SM. Selective inhibition of mutant Ha-ras mRNA expression by antisense oligonucleotides. J Biol Chem. 1992;267:19954–19962. [PubMed] [Google Scholar]
- 31.Sharp PA. The centrality of RNA. Cell. 2009;136:577–580. doi: 10.1016/j.cell.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 32.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cooper TA, Wan L, Dreyfuss G. RNA and disease. Cell. 2009;136:777–793. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dominski Z, Kole R. Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotides. Proc Natl Acad Sci U S A. 1993;90:8673–8677. doi: 10.1073/pnas.90.18.8673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roberts J, Palma E, Sazani P, Ørum H, Cho M, Kole R. Efficient and persistent splice switching by systemically delivered LNA oligonucleotides in mice. Mol Ther. 2006;14:471–475. doi: 10.1016/j.ymthe.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 36.Vickers TA, Zhang H, Graham MJ, Lemonidis KM, Zhao C, Dean NM. Modification of MyD88 mRNA splicing and inhibition of IL-1beta signaling in cell culture and in mice with a 2’-O-methoxyethyl-modified oligonucleotide. J Immunol. 2006;176:3652–3661. doi: 10.4049/jimmunol.176.6.3652. [DOI] [PubMed] [Google Scholar]
- 37.Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet. 2008;82:834–848. doi: 10.1016/j.ajhg.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jearawiriyapaisarn N, Moulton HM, Buckley B, Roberts J, Sazani P, Fucharoen S, et al. Sustained dystrophin expression induced by peptide-conjugated morpholino oligomers in the muscles of mdx mice. Mol Ther. 2008;16:1624–1629. doi: 10.1038/mt.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peacey E, Rodriguez L, Liu Y, Wolfe MS. Targeting a pre-mRNA structure with bipartite antisense molecules modulates tau alternative splicing. Nucleic Acids Res. 2012;40:9836–9849. doi: 10.1093/nar/gks710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vickers TA, Wyatt JR, Burckin T, Bennett CF, Freier SM. Fully modified 2’ MOE oligonucleotides redirect polyadenylation. Nucleic Acids Res. 2001;29:1293–1299. doi: 10.1093/nar/29.6.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morris KV, Chan SW-L, Jacobsen SE, Looney DJ. Small interfering RNA-induced transcriptional gene silencing in human cells. Science. 2004;305:1289–1292. doi: 10.1126/science.1101372. [DOI] [PubMed] [Google Scholar]
- 42.Janowski BA, Kaihatsu K, Huffman KE, Schwartz JC, Ram R, Hardy D, et al. Inhibiting transcription of chromosomal DNA with antigene peptide nucleic acids. Nat Chem Biol. 2005;1:210–215. doi: 10.1038/nchembio724. [DOI] [PubMed] [Google Scholar]
- 43.Janowski BA, Younger ST, Hardy DB, Ram R, Huffman KE, Corey DR. Activating gene expression in mammalian cells with promoter-targeted duplex RNAs. Nat Chem Biol. 2007;3:166–173. doi: 10.1038/nchembio860. [DOI] [PubMed] [Google Scholar]
- 44.Davis S, Propp S, Freier SM, Jones LE, Serra MJ, Kinberger G, et al. Potent inhibition of microRNA in vivo without degradation. Nucleic Acids Res. 2009;37:70–77. doi: 10.1093/nar/gkn904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Esau C, Kang X, Peralta E, Hanson E, Marcusson EG, Ravichandran LV, et al. MicroRNA-143 regulates adipocyte differentiation. J Biol Chem. 2004;279:52361–52365. doi: 10.1074/jbc.C400438200. [DOI] [PubMed] [Google Scholar]
- 46.Krützfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, et al. Silencing of microRNAs in vivo with “antagomirs”. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 47.Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006;3:87–98. doi: 10.1016/j.cmet.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 48.Elmén J, Lindow M, Schütz S, Lawrence M, Petri A, Obad S, et al. LNA-mediated microRNA silencing in non-human primates. Nature. 2008;452:896–899. doi: 10.1038/nature06783. [DOI] [PubMed] [Google Scholar]
- 49.Junn E, Mouradian MM. MicroRNAs in neurodegenerative diseases and their therapeutic potential. Pharmacol Ther. 2012;133:142–150. doi: 10.1016/j.pharmthera.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hua Y, Sahashi K, Rigo F, Hung G, Horev G, Bennett CF, et al. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature. 2011;478:123–126. doi: 10.1038/nature10485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yin H, Moulton HM, Seow Y, Boyd C, Boutilier J, Iverson P, et al. Cell-penetrating peptide-conjugated antisense oligonucleotides restore systemic muscle and cardiac dystrophin expression and function. Hum Mol Genet. 2008;17:3909–3918. doi: 10.1093/hmg/ddn293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Srinivasan SK, Tewary HK, Iversen PL. Characterization of binding sites, extent of binding, and drug interactions of oligonucleotides with albumin. Antisense Res Dev. 1995;5:131–139. doi: 10.1089/ard.1995.5.131. [DOI] [PubMed] [Google Scholar]
- 53.Watanabe TA, Geary RS, Levin AA. Plasma protein binding of an antisense oligonucleotide targeting human ICAM-1 (ISIS 2302) Oligonucleotides. 2006;16:169–180. doi: 10.1089/oli.2006.16.169. [DOI] [PubMed] [Google Scholar]
- 54.Smith RA, Miller TM, Yamanaka K, Monia BP, Condon TP, Hung G, et al. Antisense oligonucleotide therapy for neurodegenerative disease. J Clin Invest. 2006;116:2290–2296. doi: 10.1172/JCI25424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kordasiewicz HB, Stanek LM, Wancewicz EV, Mazur C, McAlonis MM, Pytel KA, et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron. 2012;74:1031–1044. doi: 10.1016/j.neuron.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iversen PL, Zhu S, Meyer A, Zon G. Cellular uptake and subcellular distribution of phosphorothioate oligonucleotides into cultured cells. Antisense Res Dev. 1992;2:211–222. doi: 10.1089/ard.1992.2.211. [DOI] [PubMed] [Google Scholar]
- 57.Arora V, Devi GR, Iversen PL. Neutrally charged phosphorodiamidate morpholino antisense oligomers: uptake, efficacy and pharmacokinetics. Curr Pharm Biotechnol. 2004;5:431–439. doi: 10.2174/1389201043376706. [DOI] [PubMed] [Google Scholar]
- 58.Koller E, Vincent TM, Chappell A, De S, Manoharan M, Bennett CF. Mechanisms of single-stranded phosphorothioate modified antisense oligonucleotide accumulation in hepatocytes. Nucleic Acids Res. 2011;39:4795–4807. doi: 10.1093/nar/gkr089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Geary RS, Wancewicz E, Matson J, Pearce M, Siwkowski A, Swayze E, et al. Effect of dose and plasma concentration on liver uptake and pharmacologic activity of a 2’-methoxyethyl modified chimeric antisense oligonucleotide targeting PTEN. Biochem Pharmacol. 2009;78:284–291. doi: 10.1016/j.bcp.2009.04.013. [DOI] [PubMed] [Google Scholar]
- 60.Lorenz P, Misteli T, Baker BF, Bennett CF, Spector DL. Nucleocytoplasmic shuttling: a novel in vivo property of antisense phosphorothioate oligodeoxynucleotides. Nucleic Acids Res. 2000;28:582–592. doi: 10.1093/nar/28.2.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hua Y, Sahashi K, Hung G, Rigo F, Passini MA, Bennett CF, et al. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010;24:1634–1644. doi: 10.1101/gad.1941310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.DeVos SL, Miller TM. Direct intraventricular delivery of drugs to the rodent central nervous system. JoVE 2013;e50326. [DOI] [PMC free article] [PubMed]
- 63.Southwell AL, Skotte NH, Bennett CF, Hayden MR. Antisense oligonucleotide therapeutics for inherited neurodegenerative diseases. Trends Mol Med. 2012;18:634–643. doi: 10.1016/j.molmed.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 64.Benoist M, Boulu P, Hayem G. Epidural steroid injections in the management of low-back pain with radiculopathy: an update of their efficacy and safety. Eur Spine J. 2012;21:204–213. doi: 10.1007/s00586-011-2007-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Freise H, Van Aken HK. Risks and benefits of thoracic epidural anaesthesia. Br J Anaesth. 2011;107:859–868. doi: 10.1093/bja/aer339. [DOI] [PubMed] [Google Scholar]
- 66.Hayek SM, Deer TR, Pope JE, Panchal SJ, Patel VB. Intrathecal therapy for cancer and non-cancer pain. Pain Physician. 2011;14:219–248. [PubMed] [Google Scholar]
- 67.Schneider T, Becker A, Ringe K, Reinhold A, Firsching R, Sabel BA. Brain tumor therapy by combined vaccination and antisense oligonucleotide delivery with nanoparticles. J Neuroimmunol. 2008;195:21–27. doi: 10.1016/j.jneuroim.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 68.Wohlfart S, Gelperina S, Kreuter J. Transport of drugs across the blood–brain barrier by nanoparticles. J Control Release. 2012;161:264–273. doi: 10.1016/j.jconrel.2011.08.017. [DOI] [PubMed] [Google Scholar]
- 69.Hu J, Matsui M, Gagnon KT, Schwartz JC, Gabillet S, Arar K, et al. Allele-specific silencing of mutant huntingtin and ataxin-3 genes by targeting expanded CAG repeats in mRNAs. Nat Biotechnol. 2009;27:478–484. doi: 10.1038/nbt.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nasir J, Floresco SB, O’Kusky JR, Diewert VM, Richman JM, Zeisler J, et al. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81:811–823. doi: 10.1016/0092-8674(95)90542-1. [DOI] [PubMed] [Google Scholar]
- 71.Dragatsis I, Levine MS, Zeitlin S. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat Genet. 2000;26:300–306. doi: 10.1038/81593. [DOI] [PubMed] [Google Scholar]
- 72.Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol. 2003;60:1119–1122. doi: 10.1001/archneur.60.8.1119. [DOI] [PubMed] [Google Scholar]
- 73.Kumar VB, Farr SA, Flood JF, Kamlesh V, Franko M, Banks WA, et al. Site-directed antisense oligonucleotide decreases the expression of amyloid precursor protein and reverses deficits in learning and memory in aged SAMP8 mice. Peptides. 2000;21:1769–1775. doi: 10.1016/s0196-9781(00)00339-9. [DOI] [PubMed] [Google Scholar]
- 74.Banks WA, Farr SA, Butt W, Kumar VB, Franko MW, Morley JE. Delivery across the blood–brain barrier of antisense directed against amyloid beta: reversal of learning and memory deficits in mice overexpressing amyloid precursor protein. J Pharmacol Exp Ther. 2001;297:1113–1121. [PubMed] [Google Scholar]
- 75.Erickson MA, Niehoff ML, Farr SA, Morley JE, Dillman LA, Lynch KM, et al. Peripheral administration of antisense oligonucleotides targeting the amyloid-β protein precursor reverses AβPP and LRP-1 overexpression in the aged SAMP8 mouse brain. J Alzheimer Dis. 2012;28:951–960. doi: 10.3233/JAD-2011-111517. [DOI] [PubMed] [Google Scholar]
- 76.Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316:750–753. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 77.Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, Van Eersel J, et al. Dendritic function of tau mediates amyloid-β toxicity in Alzheimer’s disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 78.Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, et al. Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J Neurosci. 2011;31:700–711. doi: 10.1523/JNEUROSCI.4152-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Leroy K, Ando K, Laporte V, Dedecker R, Suain V, Authelet M, et al. Lack of Tau Proteins rescues neuronal cell death and decreases amyloidogenic processing of APP in APP/PS1 mice. Am J Pathol. 2012;181:1940–1928. doi: 10.1016/j.ajpath.2012.08.012. [DOI] [PubMed] [Google Scholar]
- 80.Andrews-Zwilling Y, Bien-Ly N, Xu Q, Li G, Bernardo A, Yoon SY, et al. Apolipoprotein E4 causes age- and Tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J Neurosci. 2010;30:13707–13717. doi: 10.1523/JNEUROSCI.4040-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tucker KL, Meyer M, Barde Y. Neurotrophins are required for nerve growth during development. Nat Neurosci. 2001;4:29–37. doi: 10.1038/82868. [DOI] [PubMed] [Google Scholar]
- 82.Dawson HN, Ferreira A, Eyster MV, Ghoshal N, Binder LI, Vitek MP. Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J Cell Sci. 2001;114:1179–1187. doi: 10.1242/jcs.114.6.1179. [DOI] [PubMed] [Google Scholar]
- 83.Morris M, Hamto P, Adame A, Devidze N, Masliah E, Mucke L. Age-appropriate cognition and subtle dopamine-independent motor deficits in aged Tau knockout mice. Neurobiol Aging. 2013;34:1523–1529. doi: 10.1016/j.neurobiolaging.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lei P, Ayton S, Finkelstein DI, Spoerri L, Ciccotosto GD, Wright DK, et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat Med. 2012;18:291–295. doi: 10.1038/nm.2613. [DOI] [PubMed] [Google Scholar]
- 85.Modarresi F, Faghihi MA, Lopez-Toledano MA, Fatemi RP, Magistri M, Brothers SP, et al. Inhibition of natural antisense transcripts in vivo results in gene-specific transcriptional upregulation. Nat Biotechnol. 2012;30:453–459. doi: 10.1038/nbt.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A. 1999;96:6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Passini MA, Bu J, Richards AM, Kinnecom C, Sardi SP, Stanek LM, et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med. 2011;3:72ra18. doi: 10.1126/scitranslmed.3001777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Williams JH, Schray RC, Patterson CA, Ayitey SO, Tallent MK, Lutz GJ. Oligonucleotide-mediated survival of motor neuron protein expression in CNS improves phenotype in a mouse model of spinal muscular atrophy. J Neurosci. 2009;29:7633–7638. doi: 10.1523/JNEUROSCI.0950-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhou H, Janghra N, Mitrpant C, Dickinson RL, Anthony K, Price L, et al. A novel morpholino oligomer targeting ISS-N1 improves rescue of severe spinal muscular atrophy transgenic mice. Hum Gene Ther. 2013;24:331–342. doi: 10.1089/hum.2012.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Porensky PN, Mitrpant C, McGovern VL, Bevan AK, Foust KD, Kaspar BK, et al. A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum Mol Genet. 2012;21:1625–1638. doi: 10.1093/hmg/ddr600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1989;3:519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- 92.Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J. 1989;8:393–399. doi: 10.1002/j.1460-2075.1989.tb03390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Andreadis A, Brown WM, Kosik KS. Structure and novel exons of the human tau gene. Biochemistry. 1992;31:10626–10633. doi: 10.1021/bi00158a027. [DOI] [PubMed] [Google Scholar]
- 94.Goedert M, Jakes R. Expression of separate isoforms of human tau protein: correlation with the tau protein pattern in brain and effects on tubulin polymerization. EMBO J. 1990;9:4225–4230. doi: 10.1002/j.1460-2075.1990.tb07870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kosik KS, Orecchio LD, Bakalis S, Neve RL. Developmentally regulated expression of specific tau sequences. Neuron. 1989;2:1389–1397. doi: 10.1016/0896-6273(89)90077-9. [DOI] [PubMed] [Google Scholar]
- 96.Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, et al. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science. 1998;282:1914–1917. doi: 10.1126/science.282.5395.1914. [DOI] [PubMed] [Google Scholar]
- 97.D’Souza I, Poorkaj P, Hong M, Nochlin D, Lee VM, Bird TD, et al. Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc Natl Acad Sci U S A. 1999;96:5598–5603. doi: 10.1073/pnas.96.10.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Buee L, Delacourte A. Comparative biochemistry of tau in progressive supranuclear palsy, corticobasal degeneration, FTDP-17 and Pick’s disease. Brain Pathol. 1999;693:681–693. doi: 10.1111/j.1750-3639.1999.tb00550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yagishita S, Beach T, Rogers J, Schwab C, Mcgeer PL. Distinct isoforms of tau aggregated in neurons and glial cells in brains of patients with Pick’s disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol. 2001;101:167–173. doi: 10.1007/s004010000283. [DOI] [PubMed] [Google Scholar]
- 100.Glatz DC, Rujescu D, Tang Y, Berendt FJ, Hartmann AM, Faltraco F, et al. The alternative splicing of tau exon 10 and its regulatory proteins CLK2 and TRA2-BETA1 changes in sporadic Alzheimer’s disease. J Neurochem. 2006;96:635–644. doi: 10.1111/j.1471-4159.2005.03552.x. [DOI] [PubMed] [Google Scholar]
- 101.Conrad C, Zhu J, Conrad C, Schoenfeld D, Fang Z, Ingelsson M, et al. Single molecule profiling of tau gene expression in Alzheimer’s disease. J Neurochem. 2007;103:1228–1236. doi: 10.1111/j.1471-4159.2007.04857.x. [DOI] [PubMed] [Google Scholar]
- 102.Ingelsson M, Ramasamy K, Cantuti-Castelvetri I, Skoglund L, Matsui T, Orne J, et al. No alteration in tau exon 10 alternative splicing in tangle-bearing neurons of the Alzheimer’s disease brain. Acta Neuropathol. 2006;112:439–449. doi: 10.1007/s00401-006-0095-3. [DOI] [PubMed] [Google Scholar]
- 103.Phelps SF, Hauser MA, Cole NM, Rafael JA, Hinkle RT, Faulkner JA, et al. Expression of full-length and truncated dystrophin mini-genes in transgenic mdx mice. Hum Mol Genet. 1995;4:1251–1258. doi: 10.1093/hmg/4.8.1251. [DOI] [PubMed] [Google Scholar]
- 104.Lu QL, Rabinowitz A, Chen YC, Yokota T, Yin H, Alter J, et al. Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in body-wide skeletal muscles. Proc Natl Acad Sci U S A. 2005;102:198–203. doi: 10.1073/pnas.0406700102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yokota T, Lu Q-L, Partridge T, Kobayashi M, Nakamura A, Takeda S, et al. Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann Neurol. 2009;65:667–676. doi: 10.1002/ana.21627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Van Deutekom JC, Janson AA, Ginjaar IB, Frankhuizen WS, Aartsma-Rus A, Bremmer-Bout M, et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N Engl J Med. 2007;357:2677–2686. doi: 10.1056/NEJMoa073108. [DOI] [PubMed] [Google Scholar]
- 107.Goemans NM, Tulinius M, Van den Akker JT, Burm BE, Ekhart PF, Heuvelmans N, et al. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N Engl J Med. 2011;364:1513–1522. doi: 10.1056/NEJMoa1011367. [DOI] [PubMed] [Google Scholar]
- 108.Kinali M, Arechavala-Gomeza V, Feng L, Cirak S, Hunt D, Adkin C, et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009;8:918–928. doi: 10.1016/S1474-4422(09)70211-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K, et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011;378:595–605. doi: 10.1016/S0140-6736(11)60756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mendell JR, Campbell K, Rodino-Klapac L, Sahenk Z, Shilling C, Lewis S, et al. Dystrophin immunity in Duchenne’s muscular dystrophy. N Engl J Med. 2010;363:1429–1437. doi: 10.1056/NEJMoa1000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11:597–610. doi: 10.1038/nrg2843. [DOI] [PubMed] [Google Scholar]
- 112.Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet. 2011;12:99–110. doi: 10.1038/nrg2936. [DOI] [PubMed] [Google Scholar]
- 113.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 114.Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metabol. 2006;3:87–98. doi: 10.1016/j.cmet.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 115.Fabani MM, Gait MJ. miR-122 targeting with LNA/2’-O-methyl oligonucleotide mixmers, peptide nucleic acids (PNA), and PNA-peptide conjugates. RNA. 2008;14:336–346. doi: 10.1261/rna.844108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Janssen HL, Reesink HW, Zeuzem S, Lawitz E, Rodriguez-Torres M, Chen A, et al. A randomized, double-blind, placebo (plb) controlled safety and anti-viral proof of concept study of miravirsen (MIR), an oligonucleotide targeting miR-122, in treatment naïve patients with genotype 1 (gt1) chronic HCV infection. Hepatology. 2011;54:LB-6. [Google Scholar]
- 117.Lanford RE, Hildebrandt-Eriksen ES, Petri A, Persson R, Lindow M, Munk ME, et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010;327:198–201. doi: 10.1126/science.1178178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hildebrandt-Eriksen ES, Aarup V, Persson R, Hansen HF, Munk ME, Ørum H. A locked nucleic acid oligonucleotide targeting microRNA 122 is well-tolerated in cynomolgus monkeys. Nucleic Acid Ther. 2012;22:152–161. doi: 10.1089/nat.2011.0332. [DOI] [PubMed] [Google Scholar]
- 119.Janssen HLA, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, et al. Treatment of HCV Infection by Targeting MicroRNA. N Engl J Med. 2013;368:1685–1694. doi: 10.1056/NEJMoa1209026. [DOI] [PubMed] [Google Scholar]
- 120.Lee S-T, Chu K, Jung K-H, Kim JH, Huh J-Y, Yoon H, et al. miR-206 regulates brain-derived neurotrophic factor in Alzheimer disease model. Ann Neurol. 2012;72:269–277. doi: 10.1002/ana.23588. [DOI] [PubMed] [Google Scholar]
- 121.Zovoilis A, Agbemenyah HY, Agis-Balboa RC, Stilling RM, Edbauer D, Rao P, et al. microRNA-34c is a novel target to treat dementias. EMBO J. 2011;30:4299–4308. doi: 10.1038/emboj.2011.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Freier SM, Watt AT. Basic principles of antisense drug discovery. In: Crooke ST, editor. Antisense drug technology: principles, strategies, and applications. Boca Raton, FL, USA: CRC Press; 2008. pp. 117–141. [Google Scholar]
- 123.Ho SP, Bao Y, Leshner T, Malhotra R, Ma LY, Fluharty SJ, Sakai RR. Mapping of RNA accessible sites for antisense experiments with oligonucleotide libraries. Nat Biotechnol. 1999;16:59–83. doi: 10.1038/nbt0198-59. [DOI] [PubMed] [Google Scholar]
- 124.Ding Y, Lawrence CE. Statistical prediction of single-stranded regions in RNA secondary structures and application to predicting effective target sites and beyond. Nucleic Acids Res. 2001;29:1034–1046. doi: 10.1093/nar/29.5.1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Far RK, Nedbal W, Sczakiel G. Concepts to automate the theoretical design of effective antisense oligonucleotides. Bioinformatics. 2001;17:1058–1061. doi: 10.1093/bioinformatics/17.11.1058. [DOI] [PubMed] [Google Scholar]
- 126.Yang SP, Song ST, Tang ZM, Song HF. Optimization of antisense drug design against conservative local motif in simulant secondary structures of HER-2 mRNA and QSAR analysis. Acta Pharmacol. 2003;24:897–902. [PubMed] [Google Scholar]
- 127.Vickers TA, Koo S, Bennett CF, Crooke ST, Dean NM, Baker BF. Efficient reduction of target RNAs by small interfering RNA and RNase H-dependent antisense agents. A comparative analysis. J Biol Chem. 2003;278:7108–7118. doi: 10.1074/jbc.M210326200. [DOI] [PubMed] [Google Scholar]
- 128.Bennett CF, Condon TP, Grimm S, Chan H, Chiang MY. Inhibition of endothelial cell adhesion molecule expression with antisense oligonucleotides. J Immunol. 1994;152:3530–3540. [PubMed] [Google Scholar]
- 129.Matveeva OV, Tsodikov AD, Giddings M, Freier SM, Wyatt JR, Spiridonov AN. Identification of sequence motifs in oligonucleotides whose presence is correlated with antisense activity. Nucleic Acids Res. 2000;28:2862–2865. doi: 10.1093/nar/28.15.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ho SP, Britton DH, Stone BA, Behrens DL, Leffet LM, Hobbs FW. Potent antisense oligonucleotides to the human multidrug resistance-1 mRNA are rationally selected by mapping RNA-accessible sites with oligonucleotide libraries. Nucleic Acids Res. 1996;24:1901–1907. doi: 10.1093/nar/24.10.1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Johnson E, Srivastava R. Volatility in mRNA secondary structure as a design principle for antisense. Nucleic Acids Res. 2013;41:e43. doi: 10.1093/nar/gks902. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 510 kb)