

Abstract
There is continuing interest in the discovery and development of new κ opioid receptor antagonists. We recently reported that N-substituted 3-methyl-4-(3-hydroxyphenyl)piperazines were a new class of opioid receptor antagonists. In this study we report the syntheses of two piperazine JDTic-like analogues. Evaluation of the two compounds in an in vitro [35S]GTPγS binding assay showed that neither compound showed the high potency and κ opioid receptor selectivity of JDTic. A library of compounds using the core scaffold 21 was synthesized and tested for their ability to inhibit [35S]GTPγS binding stimulated by the selective κ opioid agonist U69,593. These studies led to N-[(1S)-1-{[(3S)-4-(3-hydroxyphenyl)-3-methylpiperazin-1-yl]methyl}-2-methylpropyl]-4-phenoxybenzamide (11a), a compound that showed good κ opioid receptor antagonist properties. An SAR study based on 11a provided 28 novel analogues. Evaluation of these 28 compounds in the [35S]GTPγS binding assay showed that several of the analogues were potent and selective κ opioid receptor antagonists.
The opioid receptors, μ, δ, κ, and the opioid-like receptor ORL-1 belong to the superfamily of G-protein coupled receptors (GPCRs) that possess seven helical trans-membrane spanning domains in their architecture.1 These opioid receptor systems have been extensively studied, and thousands of compounds have been synthesized and evaluated by in vitro binding and functional assays as well as by animal models.2 An integral part of the effort to characterize the opioid receptor system has been the discovery of potent, pure antagonists. Naloxone and naltrexone (Chart 1), both competitive antagonists at μ, δ, and κ opioid receptors,3 have been extensively used as pharmacological tools to identify and characterize opioid systems. Additionally, naloxone is approved to treat heroin overdose and to reverse respiratory depression caused by morphine.3 Naltrexone is used to treat heroin and alcohol abuse.
Chart 1.

In 1978, Zimmerman and co-workers reported the discovery of a structurally unique series of opioid receptor pure antagonists based on N-substituted analogues of 3,4-dimethyl-4-(3-hydroxyphenyl)piperidine (2a, LY272922) (Chart 1).4 Unlike naloxone and naltrexone where the antagonist activity is dependent on the N-allyl or N-cyclopropylmethyl substituent, all N-substituted trans-3,4-dimethyl-4-(3-hydroxyphenyl)piperidines (2) including the N-methyl analogue 2b are opioid receptor pure antagonists (Chart 1).4–8 A few of the more interesting analogues include alvimopan (3), which is an FDA-approved drug for GI motility disorder,9 2d (Chart 1),7,10 which was developed to treat obesity, and the selective κ opioid receptor antagonist JDTic (Chart 1),11–14 which shows activity in rat models of depression,15 anxiety,16 and stress-induced cocaine relapse.15 Recently, 5,17,18 6,19 and 7 (Chart 2)20,21 have been reported as selective κ opioid receptor antagonists (see reference 22 for a review).
Chart 2.

Studies with nor-BNI (Chart 2) and JDTic as well as 5, 6, and 7 have shown that this system is intimately involved in brain processes that relate to stress, fear, and anxiety as well as reward-seeking behavior.22 Studies have shown that JDTic and nor-BNI dose-dependently reduce fear and stress-induced responses in multiple behavioral paradigms with rodents (immobility in the forced-swim assay,15,23 reduction of exploratory behavior in the elevated plus maze, and fear-potentiated startle).16 Furthermore, selective κ antagonists have been shown to reduce stress-induced reinstatement of cocaine self-administration in rats,15 to block the stress-induced potentiation of cocaine place preference conditioning,24–26 to decrease dependence-induced ethanol self-administration,27 to diminish deprivation-induced eating in rats,28 and to prevent pre-pulse inhibition mediated by the selective κ opioid receptor agonist U50,488.29 These observations regarding the behavioral consequences of receptor blockade in several animal tests suggest that κ antagonists will be useful for treating depression, anxiety, schizophrenia, addiction, and eating disorders.
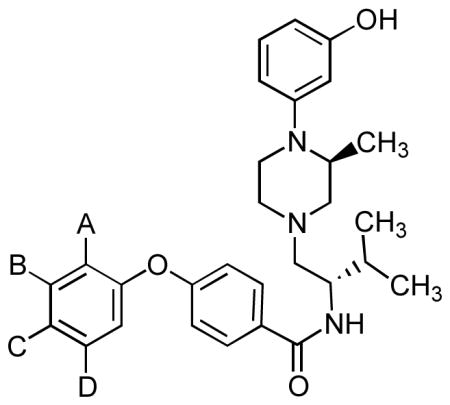
In view of the above, there is continuing interest in the discovery and development of new κ opioid receptor antagonists. In addition, there is need for additional κ opioid receptor antagonists to further characterize the recently reported structure of the human κ opioid receptor.30 We recently reported the discovery of 3-(4-substituted piperazin-1-yl)phenols (9) (Chart 2) as a new class of opioid receptor antagonists.31 These compounds were found to be relatively nonselective opioid receptor antagonists. Thus, their opioid receptor properties are more like those of naloxone (1a), naltrexone (1b), and the originally reported N-substituted 3,4-dimethyl-4-(3-hydroxyphenyl)piperidines (2b–d).7
In this study we report the syntheses and evaluation of the in vitro efficacy properties using the [35S]GTPγS assay of the two piperazine JDTic-like analogues 10a–b. In addition, a library of compounds was synthesized and tested for their ability to inhibit [35S]GTPγS binding stimulated by the selective κ opioid agonist U69,593 which led to N-[(1S)-1-{[(3S)-4-(3-hydroxyphenyl)-3-methylpiperazin-1-yl]methyl}-2-methylpropyl]-4-phenoxybenzamide (11a). An in vitro [35S]GTPγS binding assay efficacy study based on analogues of 11a provided the N-{4-[(3-hydroxyphenyl)-3-methylpiperazin-1-yl]methyl-2-methylpropyl}-4-phenoxybenzamide analogues (11b–q (Table 3), and 12a–l, 13, and 14 (Table 4)). The two piperazine JDTic-like analogues 10a–b did not retain the high potency and κ opioid receptor selectivity of JDTic. However, several of the 11a–q and/or 12a–l analogues were potent and selective κ opioid receptor antagonists.
Table 3.
Inhibition of Agonist Stimulated [35S]GTPγS Binding in Cloned Human μ, δ, and κ-Opioid Receptors for 11a–q
| ||||||
|---|---|---|---|---|---|---|
| compd | A, B, C, D | μ, DAMGO Ke (nM)a | δ, DPDPE Ke (nM)a | κ, U69,593 Ke (nM)a | μ/κ | δ/κ |
| 11a | H, H, H, H | 51 ± 15 | 570 ± 79 | 0.85 ± 0.35 | 60 | 671 |
| 11b | OCH3, H, H, H | 127 ± 78 | 869 ± 200 | 5.60 ± 1.41 | 23 | 155 |
| 11c | OH, H, H, H | 125 ± 10 | 960 ± 220 | 1.06 ± 0.26 | 146 | 906 |
| 11d | F, H, H, H | 21.1 ± 4 | 96 ± 18 | 0.76 ± 0.21 | 28 | 126 |
| 11e | H, CH3, H, H | 13 ± 4 | 131 ± 44 | 0.17 ± 0.04 | 77 | 771 |
| 11f | H, CF3, H, H | 21 ± 7 | 54 ± 19 | 1.2 ± 0.4 | 18 | 45 |
| 11g | H, OCH3, H, H | 33 ± 14 | 1502 ± 400 | 0.63 ± 0.13 | 52 | 2384 |
| 11h | H, OH, H, H | 69 ± 14 | 625 ± 120 | 1.85 ± 0.51 | 37 | 338 |
| 11i | H, F, H, H | 30 ± 9.0 | 173 ± 5 | 1.48 ± 0.51 | 20 | 117 |
| 11j | H, Cl, H, H | 18 ± 7 | 8.3 ± 0.9 | 0.42 ± 0.05 | 43 | 20 |
| 11k | H, Br, H, H | 17 ± 6 | 42 ± 19 | 0.48 ± 0.02 | 35 | 88 |
| 11l | H, H, CH3, H | 20 ± 6 | 188 ± 33 | 0.69 ± 0.25 | 29 | 272 |
| 11m | H, H, OCH3, H | 17 ± 5 | 1383 ± 1050 | 0.60 ± 0.18 | 28 | 2305 |
| 11n | H, H, OH, H | 71 ± 19 | 1696 ± 380 | 8.6 ± 3.7 | 8 | 197 |
| 11o | OH, CH3, H, H | 23.8 ± 8.9 | 93 ± 14 | 0.34 ± 0.16 | 70 | 272 |
| 11p | OH, H, H, CH3 | 65 ± 18 | 93 ± 27 | 0.61 ± 0.27 | 106 | 152 |
| 11q | H, CH3, H, CH3 | 25 ± 7 | 78 ± 20 | 0.98 ± 0.06 | 26 | 80 |
The data represent the means (SE) from at least three independent experiments.
Table 4.
Inhibition of Agonist Stimulated [35S]GTPγS Binding in Cloned Human μ, δ, and κ-Opioid Receptors for 12a–l, 13, and 14
| ||||||
|---|---|---|---|---|---|---|
| compd | X, Y, A, B | μ, DAMGO Ke (nM)a | δ, DPDPE Ke (nM)a | κ, U69,593 Ke (nM)a | μ/κ | δ/κ |
| 12a | CH3, H, H, H | 15 ± 2 | 435 ± 140 | 0.63 ± 0.19 | 24 | 691 |
| 12b | CH3, H, H, CH3 | 14.4 ± 3.9 | 21 ± 6 | 0.16 ± 0.03 | 89 | 131 |
| 12c | OCH3, H, H, CH3 | 35.1 ± 15 | 93 ± 4 | 0.25 ± 0.07 | 140 | 372 |
| 12d | OH, H, H, CH3 | 13.7 ± 6 | 81 ± 35 | 0.57 ± 0.14 | 24 | 142 |
| 12e | Cl, H, H, CH3 | 43 ± 10 | 1100 ± 260 | 0.29 ± 0.13 | 148 | 3793 |
| 12f | H, CH3, H, CH3 | 18 ± 3 | 14 ± 3 | 0.65 ± 0.2 | 28 | 22 |
| 12g | H, OCH3, H, CH3 | 49 ± 14 | 63 ± 20 | 1.3 ± 0.03 | 38 | 48 |
| 12h | H, OH, H, CH3 | 119 ± 28 | 880 ± 320 | 1.9 ± 0.4 | 63 | 463 |
| 12i | OCH3, H, OH, H | 74 ± 23 | 117 ± 60 | 16.3 ± 2.7 | 5 | 7 |
| 12j | Cl, H, OH, H | 36 ± 9 | 210 ± 82 | 5.0 ± 0.82 | 7 | 42 |
| 12k | OCH3, H, OH, CH3 | 10.2 ± 3 | 117 ± 80 | 6.2 ± 2.2 | 1.6 | 19 |
| 12l | Cl, H, OH, CH3 | 8.2 ± 1.7 | 15 ± 4 | 3.0 ± 1.1 | 3 | 5 |
| 13 | — | 101 ± 16 | 2023 ± 680 | 2.8 ± 1.2 | 36 | 723 |
| 14 | — | 60 ± 11 | 1790 ± 60 | 1.6 ± 0.6 | 38 | 1119 |
The data represent the means (SE) from at least three independent experiments.
CHEMISTRY
The JDTic-like piperazine analogue 10a was synthesized by the route shown in Scheme 1. Coupling (2S)-3-(2-methylpiperazine-1-yl)phenol (15)31 with tert-butoxycarbonyl-L-valine using (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP) in tetrahydrofuran containing triethylamine followed by reduction of the intermediate amide with borane-tetrahydrofuran (BH3•THF) in tetrahydrofuran and removal of the tert-butoxycarbonyl protecting group with concentrated hydrochloric acid gave 16. Coupling 16 with Boc-D-7-hydroxy-1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid using BOP in tetrahydrofuran containing triethylamine followed by removal of the tert-butoxycarbonyl protecting group with trifluoroacetic acid in methylene chloride afforded 10a.
Scheme 1.a.

Synthesis of 10a
aReagents: (a) N-Boc-L-valine, BOP, Et3N, THF; (b) BH3•THF, THF; (c) conc. HCl; (d) Boc-D-7-hydroxy-1,2,3,4- tetrahydroisoquinoline-3-carboxylic acid, BOP, THF, 0 °C; (e) CF3CO2H, CH2Cl2; (f) HBTU, RCO2H, CH3CN, Et3N.
The piperazine JDTic-like analogue 10b was synthesized by a procedure similar to that used for 10a starting with (2R)-1-tert-butoxycarbonyl-4-(3-methoxyphenyl)-3-methylpiperazine (17)31 as outlined in Scheme 2. Treatment of 1731 with 1 N hydrochloric acid in tetrahydrofuran gave 18. Coupling of 18 with tert-butoxycarbonyl-L-valine using BOP in tetrahydrofuran containing triethylamine followed by reduction of the intermediate amide with borane•dimethylsulfide [BH3•S(CH3)2] in tetrahydrofuran and removal of the tert-butoxycarbonyl protecting group with 6 N hydrochloric acid yielded 19. Treatment of 19 with 48% hydrobromic acid gave 20. Coupling of 20 with Boc-D-7-hydroxy-1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid using BOP in tetrahydrofuran containing triethylamine, followed by removal of the tert-butoxycarbonyl-protecting group with 6 N hydrochloric acid, afforded 10b.
Scheme 2.a.

Synthesis of 10b
aReagents: (a) 1 N HCl, THF; (b) N-Boc-L-valine, BOP, Et3N, THF; (c) BH3•S(CH3)2, THF; (d) 6 N HCl; (e) 48% HBr; (f) Boc- D-7-hydroxytetrahydroquinoline-3-carboxylic acid, BOP, Et3N, THF.
A library of compounds were synthesized by coupling 16 with commercially available carboxylic acids using N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU) as the coupling agent in acetonitrile containing triethylamine which provided very clean product 2l (Scheme 1; see Table S1 in supporting information for structures). In order to identify compounds that showed potent κ opioid receptor antagonism, the library compounds were evaluated at 10 nM concentration for percent inhibition of selective kappa agonist U69,593-stimulated κ receptors without further purification. Four of the compounds (11a and 22–24) showing greater than 60% inhibition of κ agonist-stimulated inhibition were resynthesized by conventional synthetic methodology as shown in Scheme 3. Coupling of 16 with the appropriate substituted benzoic acid using HBTU as the coupling agent in the presence of triethylamine yielded 11a and 22–24. Since 11a had the best efficacy for binding in the [35S]GTPγS assay and had the greatest selectivity relative to the μ receptor, compounds 11a–q, 12a–l, 13, and 14 were designed and synthesized for SAR studies. LogBB values were calculated for all 31 compounds.32,33 The values ranged from −0.52 to 0.20, which is well within the range −1 to 0.3 predicted for compounds to cross the blood-brain barrier (BBB). For comparison, JDTic has a logBB = −0.57. All compounds were characterized by MS, 1H NMR, and elemental analysis.
Scheme 3a.
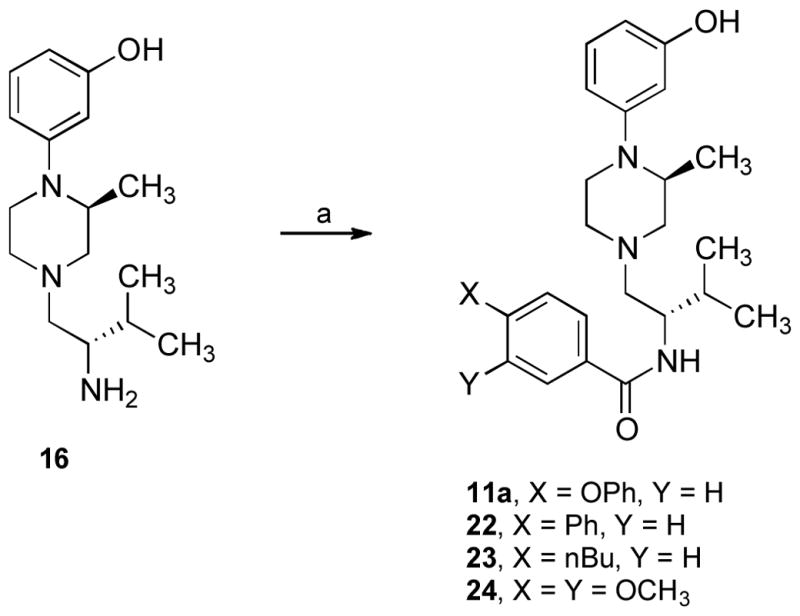
a Reagents and conditions: (a) ArCO2H, HBTU, Et3N, CH3CN.
The substituted 4-phenoxybenzoic acids (25b–q) required for the synthesis of 11b–q were prepared via nucleophilic aromatic substitution of 4-fluorobenzaldehyde or 4-fluorobenzonitrile with the appropriate phenol in dimethylformamide (Scheme 4). Potassium hydroxide was found to be a suitable base when the phenol was used in slight excess. The reactions proceeded very rapidly (15–20 min) when heated to 175 °C in a sealed tube. The benzaldehyde could then be oxidized with chromic acid or the benzonitrile could be hydrolyzed with potassium hydroxide to afford the desired benzoic acids (25b, 25d–g, 25i–m, 25o, and 25q). Hydroxy-substituted derivatives (25c, 25h, 25n, and 25p) were prepared from the corresponding methoxy compounds (e.g., 25b, 25g, or 25m) by refluxing in 48% hydrogen bromide in acetic acid. Condensation of the benzoic acid (25b–q) with amine 16 using BOP or N-ethylcarbodiimide•hydrochloride (EDC•HCl) with catalytic hydroxybenzotriazole (HOBt) afforded the desired products (11b–q).
Scheme 4a.
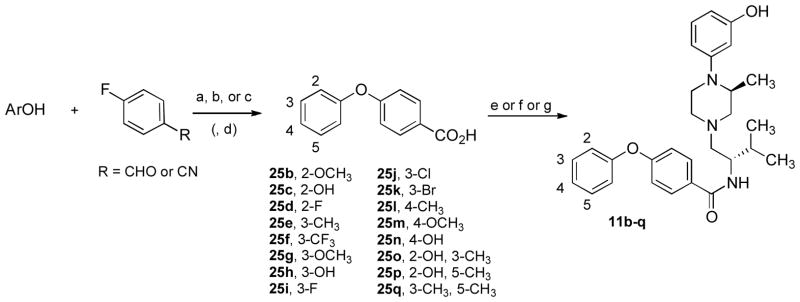
a Reagents and conditions: (a) KOH, DMF, 175 °C, 20 min; (b) CrO3, aq. H2SO4, acetone; (c) aq. KOH, reflux; (d) HBr, AcOH, reflux; (e) HBTU, NEt3, CH3CN; (f) 16, BOP, NEt3, CH2Cl2; (g) 16, EDC•HCl, cat. HOBt, NEt3, CH2Cl2.
Similarly, the substituted 4-phenoxybenzoic acids 26a–l were prepared from an appropriate phenol and a substituted fluorobenzaldehyde or fluorobenzonitrile (Scheme 5). Hydroxy-substituted benzoic acids 26d and 26h were prepared from the corresponding methoxy-derivatives (26c and 26g). Benzoic acid 26k was prepared from phenol 29 and thus required deprotection with dry hydrogen chloride in a tetrahydrofuran-isopropyl alcohol mixture. Phenol 29, in turn, was prepared by the oxidation of the pinacolborate 28 prepared from the aryl bromide 27 (Scheme 6).34
Scheme 5a.
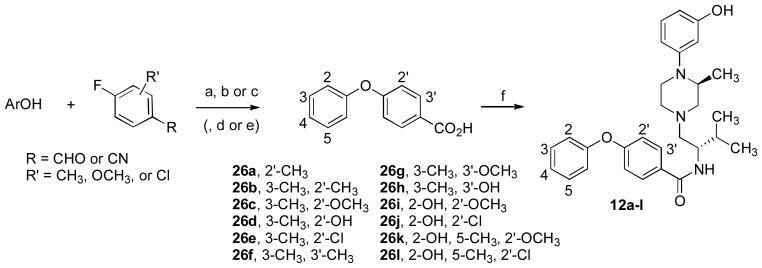
a Reagents and conditions: (a) KOH, DMF, 175 °C, 20 min; (b) CrO3, aq. H2SO4, acetone; (c) aq. KOH, reflux; (d) HBr, AcOH, reflux; (e) conc. HCl, THF/iPrOH; (f) 16, EDC•HCl, cat. HOBt, NEt3, CH2Cl2.
Scheme 6a.
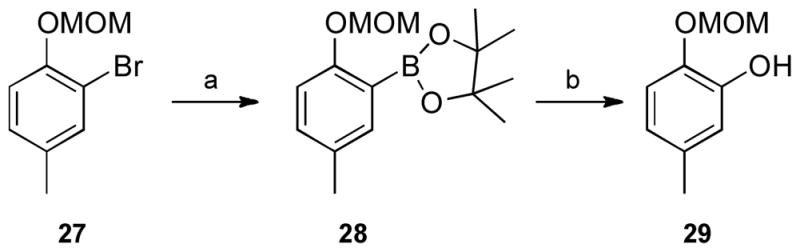
a Reagents and conditions: (a) bis(pinacolato)diborane, Pd(dppf)Cl2, KOAc; (b) Oxone, aq. acetone.
Compound 13 was synthesized as shown in Scheme 7. Pyridine 3035 was treated with m-chloroperoxybenzoic acid to yield 31. The resulting N-oxide rearranges in an acetic anhydride–acetic acid mixture to afford the acetate, which is readily hydrolyzed to the pyridyl methanol 32.36 Oxidation with potassium permanganate affords the picolinic acid 33, which was treated with amine 16 and HBTU to afford 13.
Scheme 7a.

a Reagents and conditions: (a) mCPBA, DCM; (b) Ac2O, AcOH, 150 °C, 5 min; (c) aq. K2CO3, MeOH; (d) KMnO4, acetone; (e) 16, HBTU, NEt3, CH3CN.
Scheme 8 outlines the synthetic route used to prepare 14. Conversion of nicotinic acid 34 to its methyl ester followed by nucleophilic aromatic substitution with phenol and methyl ester hydrolysis with lithium hydroxide afforded 35 which was then coupled with amine 16 using EDC•HCl as the coupling agent in the presence of catalytic HOBt to give 14.
Scheme 8a.
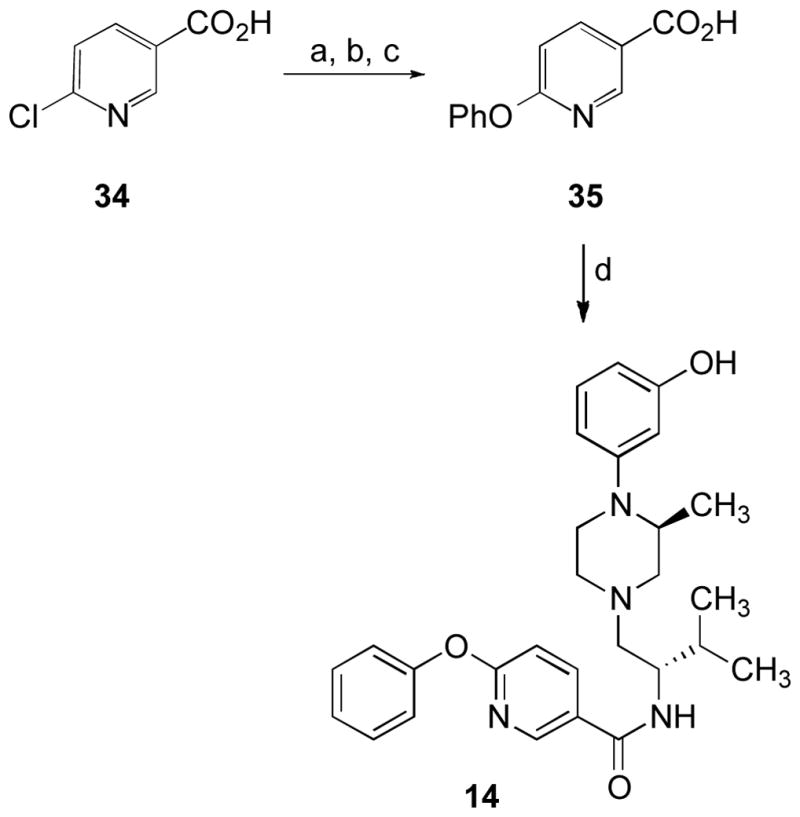
a Reagents and conditions: (a) TMSCHN2, tol., MeOH; (b) Cs2CO3, PhOH, CH3CN, reflux; (c) aq. LiOH, MeOH; (d) 16, EDC•HCl, cat. HOBt, NEt3, CH2Cl2.
Pharmacology
The test compounds were first evaluated at 10 μM for intrinsic activity at the human MOP, DOP, and KOP (over-expressed in CHO cells) using the [35S]GTPγS binding assay. Because none of these compounds displayed measurable intrinsic activity at this concentration, they were evaluated for antagonist efficacy and selectivity at these same receptors. These data were obtained by monitoring the ability of test compounds to inhibit [35S]GTPγS binding stimulated by the selective agonists (d-Ala2,MePhe4,Gly-ol5)enkephalin (DAMGO, μ receptor), cyclo[D-Pen2,D-Pen5]enkephalin (DPDPE, δ), and N-methyl-N-[(5R,7S,8S)-7-(l-pyrrolidinyl)-1-oxaspiro[4.5]dec-8-yl]benzeneacetarnide (U69,593). Agonist concentration response curves (eight different concentrations in duplicate) were run in the presence or absence of a single concentration of test compound. The Ke values were calculated using the formula Ke = [L]/DR-1, where [L] is the concentration of test compound and DR is the ratio of agonist EC50 value in the presence or absence of test compound, respectively. At least two different concentrations of test compound were used to calculate the Ke, and the concentrations were chosen such that the agonist EC50 exhibited at least a four-fold shift to the right, and there was a clear upper asymptote to the agonist + compound concentration response curve.
RESULTS AND DISCUSSION
Recently, we reported that 1-substituted 4-(3-hydroxyphenyl)piperazines (9), like N-substituted trans-3,4-dimethy-4-(3-hydroxyphenyl)piperidines (2), were pure opioid receptor antagonists.31 The piperazine analogues of (S)-1,3-dimethyl-4-(3-hydroxyphenylpiperazine (36a) (Chart 3), which is a piperazine analogue of 2b, was a pure non-selective opioid receptor antagonist. Zimmerman and co-workers reported that replacement of the N-methyl group in 2b with an N-phenylpropyl group resulted in the more potent non-selective pure opioid receptor antagonist 2c.6 We found that (S)-1-phenylpropyl-3-methy-4-(3-hydroxyphenyl)piperazine (36b), which has the N-methyl in 36a replaced with an N-phenylpropyl group, was also a more potent, pure non-selective opioid receptor antagonist.31 These results suggested that 10a, which is an N-substituted (S)-3-methyl-4-(3-hydroxyphenyl)piperazine analogue having the same N-substituent as the potent and selective κ opioid receptor antagonist JDTic, might be a potent and selective κ opioid receptor antagonist. An overlay of 10a on the structure of the human κ opioid receptor in complex with JDTic30 showed that the two compounds were essentially superimposable (see Figure 1). Unfortunately, even though 10a had a Ke = 3.37 nM at the κ receptor, this was 169-times less potent than the Ke = 0.02 nM for JDTic (Table 2). Moreover, its 22- and 388-fold selectivity relative to the μ and δ receptors, respectively, was much less than the 1255- and 3820-fold selectivity for the μ and δ receptors, respectively, for JDTic (Table 2).
Chart 3.
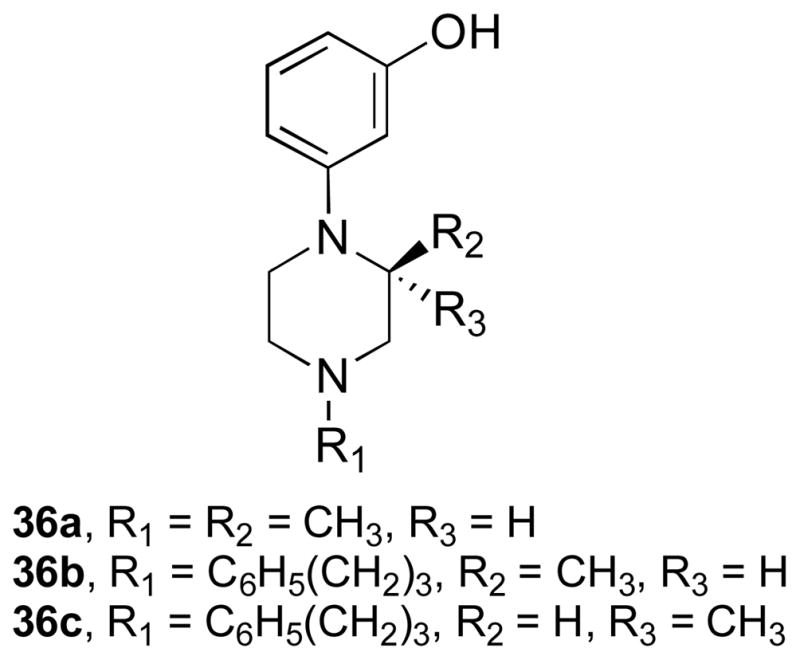
Figure 1.
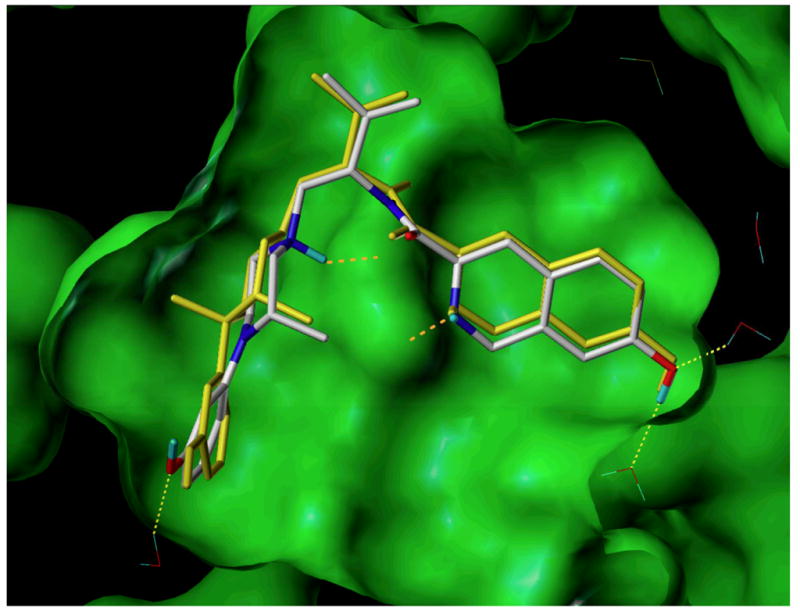
Compound 10a (CPK color scheme) docked into the kappa-opioid receptor ligand binding site (PDB: 4DJH). The observed configuration of JDTic (yellow) shown for comparison. Electrostatic interactions indicated by yellow (hydrogen-bonds involving water) or orange (interactions with Asp 238) dashed lines.
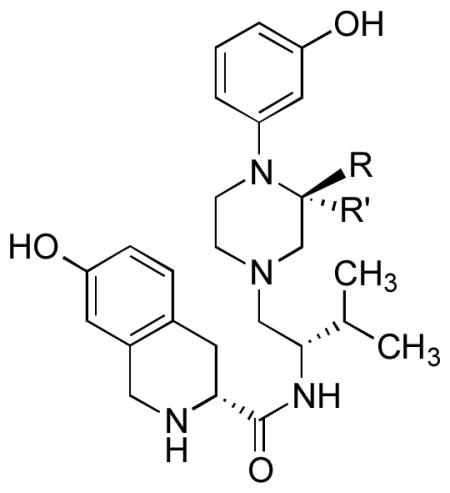
Table 2.
Comparison of Inhibition of Agonist Stimulated [35S]GTPγS Binding in Cloned Human μ, δ, and κ-Opioid Receptors for 10a and 10b to JDTic and norBNI
| |||||||
|---|---|---|---|---|---|---|---|
| compd | R | R′ | μ, DAMGO Ke (nM)a | δ, DPDPE Ke (nM)a | κ, U69,593 Ke (nM)a | μ/κ | δ/κ |
| norBNIb | 26.7 ± 7 | 29 ± 8 | 0.05 ± 0.01 | 534 | 580 | ||
| JDTicb | CH3 | H | 25.1 ± 3.5 | 76.4 ± 2.7 | 0.02 ± 0.01 | 1255 | 3820 |
| 10a | CH3 | H | 74.2 ± 7.6 | 1312 ± 317 | 3.37 ± 0.6 | 22 | 388 |
| 10b | H | CH3 | 49.4 ± 2.7 | 1546 ± 251 | 2.04 ± 0.6 | 25 | 594 |
The data represent the means (SE) from at least three independent experiments.
The Ke values for JDTic supplied by the NIDA Opioid Treatment Discovery Program (OTDP) were 3.41, 79.3, and 0.01 nM for the μ, δ, and κ receptors, respectively (ref. 12).
Based on our findings that (R)-1-phenylpropyl-3-methyl-4-(3-hydroxypheny)piperazine (36c) had Ke values at the μ, δ, and κ receptors similar to those of the (S)-isomer 36b, we synthesized 10b, which has an (R)-3-methyl substitutent, (see Table 2 for structure) with the hope that it might have opioid receptor efficacy properties similar to JDTic. However, with Ke values of 49.4, 1546, and 2.04 nM at the μ, δ, and κ receptors, respectively, 10b was also a much less potent κ opioid receptor antagonist with much less κ selectivity relative to the μ and δ receptors than JDTic, particularly at the μ receptor.
As another strategy for obtaining a potent and κ selective compound from the (S)-1-substituted 3-methyl-4-(3-hydroxyphenyl)piperazine class of compounds, a library of 79 analogues with general structure 21 was synthesized, and their % inhibitions of binding stimulated by the κ selective agonist U69,593 at 10 nM using the [35S]GTPγS assay were determined. The structures of the compounds synthesized and their % inhibitions are given in Table S1 in supporting information. The results revealed that the four compounds 11a, 22, 23, and 24 had greater than 60% inhibition. These four compounds were synthesized in pure form, and their Ke values at the μ, δ, and κ receptors were determined (Table 1). Three of the compounds, 11a, 22, and 23, had Ke values at the κ receptor of 0.85, 1.87, and 2.8 nM, respectively, with 24 having a much larger Ke value of 81.7 nM. Compound 11a was not only the most potent κ antagonist, it also had 60- and 671-fold κ selectivity relative to the μ and δ receptors. Compound 22 had 4.7- and 137-fold selectivity for the κ relative to the μ and δ receptors, and compound 23 had only 7.5- and 83-fold selectivity for the κ relative to the μ and δ receptors, respectively. Based on these results, an SAR study was directed toward analogues of 11a as a means of obtaining even more potent antagonists with greater selectivity for the κ receptor. The compounds synthesized and their Ke values at μ, δ, and κ receptors are listed in Tables 3 and 4. Compounds 11b–q, which have substituents added to only the end phenoxy ring of 11a, are given in Table 3. Compounds 11b–n have only one substituent, whereas 11o–q each have two substituents on the phenoxy ring. With Ke values ranging from 0.17 to 1.85 nM, with the exception of the 2-methoxyphenoxy analogue 11b (Ke = 5.6 nM) and 4-hydroxyphenoxy analogue 11n (Ke = 8.6 nM), all the compounds had good κ binding affinity in the functional [35S]GTPγS assay. Thirteen of the compounds had subnanomolar Ke values at the κ receptor. Twelve of the compounds had 126-fold or greater selectivity for the κ receptor relative to the δ receptor. Five of the 11a analogues had 50-fold selectivity for the κ receptor relative to the μ receptor, and two had greater than 100-fold selectivity. The 3-methylphenoxy analogue 11e with a Ke = 0.17 nM was the most potent κ antagonist of all the compounds tested. This compound also had 77- and 771-fold selectivity for κ receptor relative to the μ and δ receptors, respectively. The only compounds showing greater than 100-fold selectivity for κ relative to both the μ and δ receptor, respectively, were the 2-hydroxyphenoxy analogue 11c with a κ Ke = 1.06 nM and the 2-hydroxy-5-methylphenoxy analogue 11p, which had a κ Ke = 0.61 nM. However, note that the 2-hydroxy-3-methylphenoxy analogue 11o with a Ke = 0.34 nM for the κ receptor, which was the second most potent analogue in Table 3 had 70- and 272-fold selectivity for the κ relative to the μ and δ receptors, respectively.
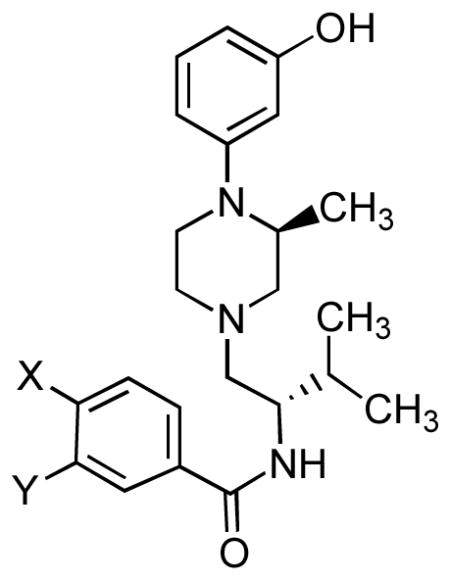
Table 1.
Inhibition of Agonist-Stimulated [35S]GTPγS Binding in Cloned Human μ, δ, and κ Opioid Receptors for 11a and 22–24
| |||||||
|---|---|---|---|---|---|---|---|
| Compd | X | Y | μ, DAMGO Ke (nM)a | δ, DPDPE Ke (nM)a | κ, U69,593 Ke (nM)a | μ/κ | δ/κ |
| 11a | C6H5O | H | 51 ± 14.9 | 570 ± 79 | 0.85 ± 0.35 | 60 | 671 |
| 22 | C6H5 | H | 8.69 ± 3 | 258 ± 88 | 1.87 ± 0.47 | 4.7 | 137 |
| 23 | C4H9 | H | 21.1 ± 2 | 230 ± 3 | 2.8 ± 1 | 7.5 | 83 |
| 24 | CH3O | CH3O | 334 ± 110 | >10,000 | 81.7 ± 30 | 4 | >122 |
The data represent the means (SE) from at least three independent experiments.
It is somewhat interesting that compound 11e, which does not have a second amino or a phenol group in a position similar to the 7′-hydroxytetrahydroquinoline group in JDTic, is the most potent κ opioid receptor antagonist. An overlay of 11e and JDTic in the human κ opioid receptor30 (see Figure 2) illustrates that these two compounds may have different binding modes or bind to different states of the receptor. Both compounds wrap around Asp138 in a similar fashion and occupy the same general region of the ligand-binding pocket. In addition, the 2′-isopropyl of 11e and the 2′-isopropyl group of JDTic lay in the same region and have similar hydrophobic interactions with Trp287. Interestingly, the 7′-hydroxytetrahydroisoquinoline group of JDTic and the 3-methylphenoxyphenyl group of 11e lay in completely different regions of the receptor. Thus, the predicted docking pose of 11e is oriented 180° away from the observed arrangement of JDTic.
Figure 2.
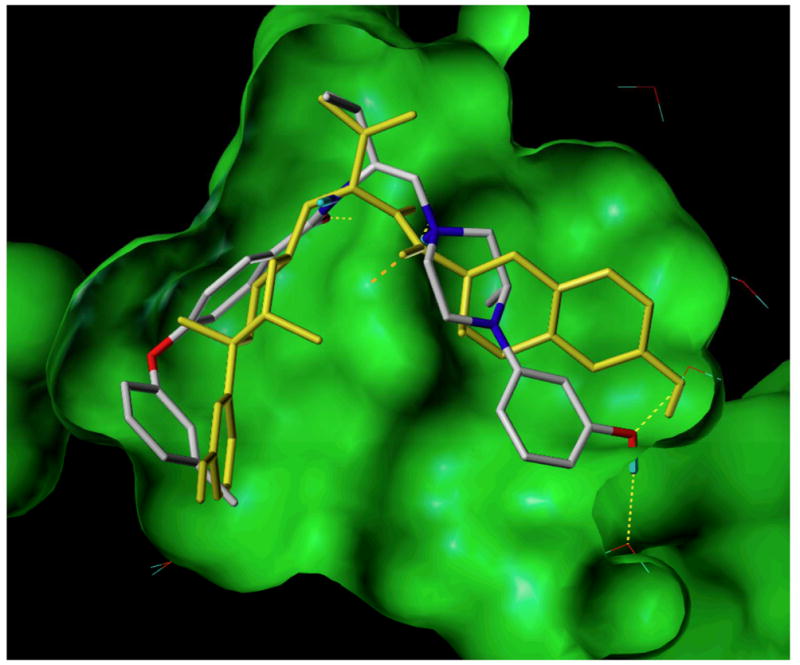
Compound 11e (CPK color scheme) docked into the kappa-opioid receptor ligand binding site (PDB: 4DJH). The observed configuration of JDTic (yellow) shown for comparison. Electrostatic interactions indicated by yellow (hydrogen-bonds involving water) or orange (interaction with Asp 238) dashed lines.
The Ke values for compounds 12a–l, which have substituents on the benzamide group and/or the phenoxy group of the phenoxybenzamide substituent, are listed in Table 4. Analogues 12a–f with Ke values of 0.16 to 0.65 nM at the κ receptor all had subnanomolar potency for the κ receptor. Five of the six compounds 12a–e had 131-fold or greater selectivity for the κ receptor relative to the δ receptor and three of the compounds 12b, 12c, and 12e had 89-fold or greater κ receptor selectivity relative to the μ receptor. Compound 12b, which had a methyl group on the 3-position of the benzamide phenyl ring and the 3-position of the phenoxy ring, with a Ke = 0.16 nM was the most potent κ antagonist listed in Table 4. This compound also had 89- and 131-fold selectivity for the κ receptor relative to the μ and δ receptors, respectively. The only two compounds in Table 4 that had κ selectivity of greater than 100-fold relative to the μ and δ receptors was 12c which has a 3-methoxy substituent on the benzamide phenyl ring and a 3-methyl substituent on the phenoxy ring, and 12e, which has a 3-chloro substituent on the benzamide phenyl ring, and a 3-methyl substituent on the phenoxy ring. Compound 12c had a Ke = 0.25 nM at the κ receptor and was 140- and 372-fold selective for the κ relative to the μ and δ receptors, respectively. Compound 12e had a Ke = 0.29 nM at the κ receptor with 148- and 3793-fold selectivity for the κ relative to the μ and δ receptors, respectively, was the most κ selective compound listed in Table 4. Even though 12h, which has a 2-hydroxy substituent on the benzamide phenyl ring and a 3-methyl substituent on the phenoxy ring only had a Ke = 1.9 nM at the κ receptor, it was 63- and 463-fold selective for the κ relative to the μ and δ receptors. Compounds 12i–l, all of which had a hydroxy substituent in the phenoxy ring combined with a methoxy or chloro substituent in the benzamide phenyl ring, had Ke values of 3 to 16 nM and very low selectivity for the κ receptor relative to both the μ and δ receptors. Compounds 13 and 14, which have the phenyl ring in the benzamide part of 11a replaced by a pyridine ring, have Ke values of 2.8 and 1.6 nM, respectively, at the κ receptor. With 723- and 1119-fold selectivity for the κ receptor relative to the δ receptor, both compounds were highly selective for the κ relative to the δ receptor. However, compounds 13 and 14 have only 36- and 38-fold selectivity for the κ receptor relative to the μ receptor.
In summary, the synthesis and evaluation of a library of N-{4-[(3-hydroxyphenyl)-3-methylpiperazin-1-yl]methylpropyl}-4-phenoxybenzamides in a [35S]GTPγS functional assay led to N-{[(1S)-1-{[(3S)-4-(3-hydroxyphenyl)-3-methylpiperazine-1-yl]methyl}-2-methylpropyl]-4-phenoxybenzamide (11a), a compound with good potency and selectivity as an opioid receptor κ antagonist. An SAR study built around the 11a structure led to several analogues that were more potent κ antagonists than 11a and six analogues, 11e, 11o, 11p, 12b, 12c, and 12e, that were both more potent and more κ selective relative to antagonist potency at the μ and/or δ receptors. N-(1S)-1-{[(3S)-4-(3-hydroxyphenyl)-3-methylpiperazine-1-yl]methyl}-2-methylpropyl]-4-(3-methylphenoxy)benzamide (11e) and N-{(2S)-1-[(3S)-4-(3-hydroxyphenyl)-3-methylpiperazin-1-yl]-3-methylbutan-2-yl}-3-methyl-4-(3-methylphenoxy)benzamide (12b) with Ke values of 0.17 and 0.16 nM, respectively, were the two most potent and selective κ antagonists.
An overlay of 11e and JDTic on the human X-ray structure of the κ opioid receptor shows that they likely have completely different binding modes to the κ receptor.
Compounds from this study provide additional information about structural requirements for interaction with the κ opioid receptor. In addition, one or more of the compounds will be useful as pharmacological tools and development of clinical candidates for treating depression, anxiety, schizophrenia, and addiction.
EXPERIMENTAL SECTION
1H NMR spectra were determined on a Bruker 300 spectrometer using tetramethylsilane as an internal standard. Mass spectral data were obtained using a Finnegan LCQ electrospray mass spectrometer in positive ion mode at atmospheric pressure. Medium-pressure flash column chromatography was done on a CombiFlash Companion system using Teledyne Isco prepacked silica gel columns or using EM Science silica gel 60 Å (230–400 mesh). All reactions were followed by thin-layer chromatography using Whatman silica gel 60 TLC plates and were visualized by UV. Optical rotations were measured on an Auto Pol III polarimeter. All solvents were reagent grade. HCl in dry diethyl ether was purchased from Aldrich Chemical Co. and used while fresh before discoloration. CMA80 is a mixture of 80% chloroform, 18% methanol, and 2% concentrated ammonium hydroxide. Purity of compounds (>95%) was established by elemental analysis. Elemental analyses were performed by Atlantic Microlab, Inc., Atlanta, GA. Care should be used when using BOP in coupling reactions as it yields the carcinogenic byproduct HMPA
(3R)-7-Hydroxy-N-[(1S)-1-{[(3S)-4-(3-hydroxyphenyl)-3-methylpiperazin-1-yl]methyl}-2-methylpropyl]-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (10a) Trihydrochloride
In a round-bottom flask, 120 mg (0.432 mmol) of 16 and 133 mg (0.454 mmol) of 7-hydroxy-Boc-D-Tic were dissolved in 15 mL of dry THF, and the solution was cooled to 0 °C. Into this solution 0.06 mL of Et3N was added followed by 201 mg (0.454 mmol) of BOP. The solution was warmed up to room temperature, stirred for 3 h, and then added to an ice-cold conc. NaHCO3 solution. The mixture was extracted three times with 5 mL of EtOAc. The pooled organic extracts were washed once with conc. NaHCO3 solution, once with brine, dried over MgSO4 and the solvents were removed under reduced pressure to yield a residue that was dissolved in 5 mL of CH2Cl2 and 3 mL of CF3COOH and stirred overnight. The solvents were removed under reduced pressure to yield a residue which was stirred with 10 mL of conc. NaHCO3 and 10 mL of EtOAc. The layers were separated, and the aqueous layer was extracted three times with 3 mL of EtOAc. The pooled organic extracts were washed once with brine, dried over MgSO4, filtered, and the solvents removed under reduced pressure to yield a residue that was purified by silica-gel flash-column chromatography eluting with a 2:1:1 mixture of CMA80–EtOAc–hexanes to yield a residue that was dissolved in 3 mL of a 2 M solution of HCl in EtOH. This solvent was removed under reduced pressure to leave a solid that was triturated under MeOH and collected by filtration to give 61 mg (31%) of 10a•3HCl: mp >220 °C (dec). [α] = +67.6 (c 0.21, CH3OH). 1H NMR (CD3OD) δ 8.75 (d,1H), 7.38 (b, 1H), 7.10 (b+d, 3H), 6.92 (b, 1H), 6.76 (dd, 1H), 6.67 (d, 1H), 4.44–4.33 (m, 6H), 3.91–3.67 (m, 3H), 3.67–3.50 (m, 2H), 3.50–3.35 (m, 2H), 3.31–3.21 (m, 1H), 2.81 (dd, 1H), 1.92 (m, 1H), 1.18 (b, 3H), 1.05 (t, 6H). ESIMS: m/z 453 (M+H+, 100). Anal. (C26H39Cl3N4O3•3H2O) C, H, N.
(3R)-7-Hydroxy-N-[(1S)-1-[[(3R)-4-(3-hydroxyphenyl)-3-methylpiperazin-1-yl]methyl}-2-methylpropyl]-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (10b) Trihydrochloride
Compound 20 (1.30 g, 0.00468 mol) was added to THF (100 mL) followed by Boc-D-7-hydroxy-1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid (1.37 g, 0.00468 mol), BOP reagent (2.07 g, 0.00468 mol), and finally Et3N (0.95 g, 0.00936 mol). The mixture was stirred at room temperature for 4.5 h, then was added in a mixture of ether (200 mL)–H2O (100 mL). The organic phase was separated and washed with water, 10% NaHCO3, and brine, then dried (Na2SO4) and concentrated in vacuo to give 2.56 g (99%) as a beige foam which was dissolved in a solution of THF (100 mL) and 6 N HCl (3 mL). The mixture was stirred at reflux for 4 h, cooled, and concentrated in vacuo. The residue was solubilized in 1 N HCl and extracted with EtOAc. The EtOAc was discarded, and the aqueous layer was basified with solid Na2CO3. The aqueous medium was then decanted, and the residue was extracted with CH2Cl2, which was dried (Na2SO4) and concentrated to give 1.71 g of a pink amorphous solid. The crude product was purified by silica gel chromatography, using CMA80–CH2Cl2 (1:1) as eluent, to give 1.57 g of the free base. The HCl salt was prepared by dissolving the free base in MeOH, adding excess 2 M ethereal HCl and concentrating the mixture in vacuo. The residue was dried on high vacuum to yield 1.40 g (51%) of 10b•3HCl as a beige solid: mp 230–235 °C; [α]25D = +79.9 (c 0.715, MeOH). 1H NMR (CD3OD) δ 7.03 (t, 1H, J = 9 Hz), 6.93 (d, 1H, J = 9 Hz), 6.60 (dd, 1H, J = 8, 3 Hz), 6.35–6.50 (m, 2H), 6.29 (dd, 1H, J = 8, 3 Hz), 3.90–4.07 (bm, 3H), 3.72 (bm, 1H), 3.57 (q, 1H, J = 5 Hz), 2.78–3.06 (m, 5H), 2.55 (bm, 2H), 2.40 (d, 2H, J = 6.0 Hz), 2.31 (m, 1H), 1.85 (m, 1H), 1.10 (d, 3H, J = 6.8 Hz), 0.94 (t, 6H, J = 6.8 Hz). Anal. (C26H39Cl2N4O3•1.5 H2O) C, H, N.
General Procedures for the Synthesis of 11b–q, 12a–l, 13, and 14
General Procedure A
To a solution of the appropriate acid (0.05 mmol) and BOP reagent (0.05 mmol) in CH2Cl2 (10 mL) was added piperazine 16 (0.05 mmol) in THF (2 mL) and Et3N (25 μL). After 12 h, the residue resulting from concentration was purified by flash column chromatography on silica gel using an EtOAc gradient in hexane. The residue from concentration of the combined desired fractions was dissolved in CH2Cl2 and treated with dry HCl in Et2O. Removal of the solvent, followed by trituration of the residue with Et2O yielded the desired product as the dihydrochloride salt.
General Procedure B
To a solution of the appropriate acid (0.12 mmol) and piperazine 16 (0.12 mmol) in CH2Cl2 (5 mL) was added HOBt (10 mol%), EDC•HCl (0.12 mmol) and Et3N (40 μL). After 12 h, the residue resulting from concentration was purified by flash column chromatography on silica gel using an EtOAc gradient in hexane. The residue from concentration of the combined desired fractions was dissolved in CH2Cl2 and treated with dry HCl in Et2O. Removal of the solvent, followed by trituration of the residue with Et2O yielded the desired product as the dihydrochloride salt.
General Procedure C
The appropriate phenol (5.10 mmol) and KOH (5.10 mmol) were dissolved in DMF (3 mL) before the appropriate 4-fluorobenzaldehyde (5.00 mmol) was added. The solution was heated in a sealed tube to 175 °C for 20 min, poured into H2O (25 mL) and extracted with Et2O (75 mL). The organic layer was washed with H2O (25 mL), brine (10 mL) and dried (Na2SO4). The crude residue resulting from concentration was dissolved in acetone (25 mL) and treated with Jones reagent (3 mL, 0.1 M CrO3 in aqueous H2SO4). Upon completion (monitored by TLC), isopropanol (3 mL) was added, and the reaction mixture was concentrated. The residue was dissolved in 5% aqueous NaOH, filtered, and the filtrate was acidified with 50% H2SO4 and extracted with EtOAc (3 × 25 mL). The combined EtOAc extracts were dried (Na2SO4) and concentrated to afford the desired substituted 4-phenoxybenzoic acid.
General Procedure D37
The appropriate phenol (1.4 mmol) and KOH (1.1 mmol) were dissolved in DMF (1.5 mL) before the appropriate 4-fluorobenzaldehyde (1 mmol) was added. The solution was heated in a sealed tube to 175 °C for 20 min, poured into H2O (25 mL) and extracted with Et2O (75 mL). The organic layer was washed with H2O (25 mL), brine (10 mL) and dried (Na2SO4). The crude residue was dissolved in 5:1 acetonitrile:water (6 mL) along with NaH2PO4 (36 mg) and H2O2 (150 μL, 30%). In an ice bath, a solution of NaClO2 (158 mg) in water (1.5 mL) is slowly added. After 12 h at rt, the reaction was quenched with Na2S2O3, diluted with brine and extracted with EtOAc. The desired benzoic acid was isolated by extracting into aqueous base, acidification, and extraction into EtOAc.
N-(1S)-1-{[(3S)-4-(3-Hydroxyphenyl)-3-methylpiperazin-1-yl]methyl}-2-methylpropyl-4-phenoxybenzamide (11a) Dihydrochloride
To a solution of 16 (55.5 mg, 0.20 mmol), 4-phenoxybenzoic acid (48.6 mg, 0.22 mmol) and Et3N (0.056 mL, 0.40 mmol) in CH3CN (10 mL) at room temperature was added HBTU (91.0 mg, 0.24 mmol). The reaction was stirred for 3 h. The mixture was diluted with Et2O (50 mL), washed with saturated NaHCO3 (2 × 10 mL), brine (2 × 10 mL), dried (Na2SO4) and concentrated. The crude product was purified by preparative TLC (33% 80CMA–CH2Cl2) to afford 68 mg (72%) of 11a free base as a glassy solid. 1H NMR (CDCl3) δ 7.76 (d, 2H, J = 9.0 Hz), 7.36 (t, 2H, J = 9.0 Hz), 7.14 (d, 1H, J = 9.0 Hz), 7.10–6.90 (m, 5H), 6.50–6.30 (m, 4H), 4.30–4.22 (m, 1H), 3.80–3.65 (m, 1H), 3.20–2.94 (m, 2H), 2.82–2.70 (m, 2H), 2.68–2.52 (m, 1H), 2.50–2.30 (m, 3H), 2.11–1.94 (m, 1H), 0.99 (d, 3H, J = 6.0 Hz), 0.97 (d, 3H, J = 6.0 Hz), 0.88 (d, 3H, J = 6.0 Hz); 13C NMR (CDCl3) δ 167.5, 160.4, 157.5, 155.9, 151.3, 130.0, 129.8, 129.1, 128.9, 124.2, 119.8, 117.8, 108.5, 106.8, 103.9, 58.5, 57.9, 54.4, 51.4, 50.9, 43.8, 30.9, 18.9, 18.1, 12.8; MS (ESI) m/z 474.7 (M + H)+. The free base was converted to 11a•HCl as an off-white solid: mp 135 °C (fusion); [α]25D +77.5° (c 0.50, CH3OH); Anal. (C29H37Cl2N3O3) C, H, N.
N-(1S)-1-{[(3S)-4-(3-Hydroxyphenyl)-3-methylpiperazin-1-yl]methyl}-2-methylpropyl-4-(2-methoxyphenoxy)benzamide (11b) Dihydrochloride
General Procedure A using acid 25b afforded 24 mg (83%) of 11b•2HCl as a white solid: mp 145 °C (fusion); [α]25D +57.8° (c 0.86, CH3OH). Anal. (C30H39Cl2N3O4•2.5 H2O) C, H, N. 11b free base: 1H NMR (CDCl3) δ 7.76 (d, 2H, J = 8.8 Hz), 7.21–7.12 (m, 1H), 7.06–6.88 (m, 4H), 6.84 (d, 2H, J = 8.8 Hz), 6.39 (s, 1H), 6.38 (d, 1H, J = 7.5 Hz), 6.30 (d, 1H, J = 7.8 Hz), 4.37–4.23 (m, 1H), 3.82–3.69 (m, 1H), 3.74 (s, 3H), 3.20–2.82 (m, 5H), 2.74–2.47 (m, 3H), 2.07–1.93 (m, 1H), 0.99 (d, 6H, J = 6.9 Hz), 0.88 (d, 3H, J = 6.4 Hz); 13C NMR (CDCl3) δ 167.6, 161.0, 157.4, 151.7, 151.1, 143.7, 131.5, 129.9, 128.8, 128.1, 125.8, 125.6, 122.1, 121.2, 121.3, 121.2, 116.1, 115.8, 113.0, 64.4, 58.4, 57.9, 55.9, 53.9, 50.9, 50.7, 31.2, 19.0, 18.1, 13.2; MS (ESI) m/z 504.6 (M + H)+.
N-(1S)-1-{[(3S)-4-(3-Hydroxyphenyl)-3-methylpiperazin-1-yl]methyl}-2-methylpropyl-4-(2-hydroxyphenoxy)benzamide (11c) Dihydrochloride
General Procedure B using acid 25c afforded 31.5 mg (44%) of 11c•2HCl as a white solid: mp 173 °C (fusion); [α]25D +60.0° (c 1.50, CH3OH). Anal. (C29H37Cl2N3O4•CH3OH) C, H, N. 11c free base: 1H NMR (CD3OD) δ 7.8 (d, 2H, J = 8.8 Hz), 7.13–6.81 (m, 7H), 6.45–6.26 (m, 3H), 4.26–4.15 (m, 1H), 3.85–3.71 (m, 1H), 3.18–2.94 (m, 3H), 2.91–2.79 (m, 3H), 2.77–2.63 (m, 1H), 2.58–2.39 (m, 3H), 1.96–1.82 (m, 1H), 1.02 (d, 3H, J = 7.0 Hz), 0.99 (d, 3H, J = 6.9 Hz), 0.92 (d, 3H, J = 6.4 Hz); 13C NMR (CD3OD) δ 170.0, 162.5, 159.3, 152.9, 150.7, 143.8, 140.0, 130.8, 130.2, 129.8, 127.1, 123.2, 121.3, 118.5, 117.0, 110.3, 108.3, 105.7, 101.4, 60.8, 59.2, 55.3, 53.0, 52.8, 46.0, 32.8, 20.1, 18.8, 13.5; MS (ESI) m/z 490.7 (M + H)+.
4-(2-Fluorophenoxy)-N-[(1S)-1-{[(3S)-4-(3-hydroxyphenyl)-3-methylpiperazin-1-yl]methyl}-2-methylpropyl]benzamide (11d) Dihydrochloride
General Procedure B with acid 25d afforded 37 mg (64%) of 11d•2HCl as a white powder: mp 156–159 °C (fusion), [α]25D +64.6° (c 0.395, CH3OH). Anal. (C29H36Cl2FN3O3•H2O) C, H, N. 11d free base: 1H NMR (CDCl3) δ 7.75 (d, 2H, J = 8.7 Hz), 7.24–7.07 (m, 3H), 7.03 (t, 1H, J = 8.0 Hz), 6.94 (d, 2H, J = 8.7 Hz), 6.43–6.22 (m, 4H), 4.27–4.15 (m, 1H), 3.83–3.72 (m, 1H), 3.20–3.09 (m, 1H), 3.08–2.96 (m, 1H), 2.95–2.71 9(m, 2H), 2.63–2.51 (m, 1H), 2.45–2.26 (m, 3H), 2.11–1.98 (m, 1H), 1.02–0.94 (m, 6H), 0.88 (d, 3H, J = 6.4 Hz); 13C NMR (CDCl3) δ 167.2, 160.2, 157.1, 151.4, 129.9, 129.5, 128.8, 125.8, 125.0, 122.8, 117.4, 117.2, 116.4, 108.5, 106.3, 103.4, 58.5, 57.8, 54.5, 51.3, 50.9, 43.6, 30.8, 18.9, 18.0, 12.8; MS (ESI) m/z 492.5 (M + H)+.
N-(1S)-1-{[(3S)-4-(3-Hydroxyphenyl)-3-methylpiperazin-1-yl]methyl}-2-methylpropyl-4-(3-methylphenoxy)benzamide (11e) Dihydrochloride
General Procedure A using acid 25e afforded 92 mg (64%) of 11e•2HCl as a white solid: mp 165 °C (fusion); [α]25D +63.8° (c 0.58, CH3OH); Anal. (C30H39Cl2N3O3•H2O) C, H, N. 11e free base: 1H NMR (CDCl3) δ 7.77 (d, 2H, J = 8.6 Hz), 7.22 (t, 1H, J = 7.8 Hz), 7.05–6.75 (m, 5H), 6.91 (d, 2H, J = 8.7 Hz), 6.41–6.35 (m, 2H), 6.32 (d, 1H, J = 8 Hz), 4.34–4.20 (m, 1H), 3.81–3.70 (m, 3H), 3.18–2.96 (m, 2H), 2.91–2.76 (m, 2H), 2.65–2.41 (m, 3H), 2.31 (s, 3H), 2.09–1.95 (m, 1H), 1.00 (d, 3H, J = 6.8 Hz), 0.99 (d, 3H, J = 6.8 Hz), 0.89 (d, 3H, J = 6.7 Hz); 13C NMR (CDCl3) δ 167.4, 160.5, 157.3, 155.9, 151.2, 140.2, 131.5, 129.9, 129.6, 128.8, 125.0, 124.7, 120.4, 117.7, 117.2, 116.7, 107.2, 104.5, 58.4, 57.9, 54.0, 50.9, 31.5, 31.1, 22.6, 21.3, 19.0, 18.1, 14.1, 13.2; MS (ESI) m/z 488.6 (M + H)+.
N-[(1S)-1-{[(3S)-4-(3-Hydroxyphenyl)-3-methylpiperazin-1-yl]methyl}-2-methylpropyl]-4-[3-(trifluoromethyl)phenoxy]benzamide (11f)
General Procedure B with acid 25f afforded 45 mg (71%) of 11f as a white powder: mp 110–115 °C (fusion), [α]25D +45.1° (c 0.27, CH3OH). Anal. (C30H36Cl2F3N3O3•1.25H2O) C, H, N. 11f free base: 1H NMR (CDCl3) δ 7.80 (d, 2H, J = 8.7 Hz), 7.51–7.36 (m, 1H), 7.27 (s, 1H), 7.17 (d, 2H, J = 7.6 Hz), 7.10–6.96 (m, 3H), 6.43–6.23 (m, 4H). 4.30–4.14 (m, 1H), 3.84–3.74 (m, 1H), 3.22–3.11 (m, 1H), 3.09–2.97 (m, 1H), 2.85–2.72 (m, 2H), 2.66–2.54 (m, 1H), 2.49–2.29 (m, 3H), 2.13–1.98 (m, 1H), 1.03–0.95 (m, 6H), 0.88 (d, 3H, J = 6.5 Hz); 13C NMR (CDCl3) δ 167.2, 159.2, 157.1, 156.7, 151.4, 130.6, 130.4, 129.9, 129.1, 122.5, 120.6, 118.5, 116.2, 108.6, 106.5, 103.5, 58.5, 58.5, 57.8, 54.5, 51.4, 50.9, 43.6, 30.8, 18.9, 18.0, 12.8; MS (ESI) m/z 542.6 (M + H)+.
N-(1S)-1-{[(3S)-4-(3-Hydroxyphenyl)-3-methylpiperazin-1-yl]methyl}-2-methylpropyl-4-(3-methoxyphenoxy)benzamide (11g) Dihydrochloride
General Procedure A using acid 25g afforded 11.3 mg (55%) of 11g•2HCl as a beige solid: mp 145 °C (fusion); [α]25D +57.6° (c 0.59, CH3OH); Anal. (C30H39Cl2N3O4•2 H2O) C, H, N. 11g free base: 1H NMR (CDCl3) δ 7.77 (d, 2H, J = 8.7 Hz), 7.25 (t, 1H, J = 7.9 Hz), 7.05 (t, 1H, J = 8.0 Hz), 7.01 (d, 2H, J = 8.8 Hz), 6.74–6.56 (m, 3H), 6.43–6.28 (m, 3H), 4.27–4.15 (m, 1H), 3.85–3.74 (m, 1H), 3.77 (s, 3H), 3.23–3.11 (m, 1H), 3.11–2.98 (m, 1H), 2.84–2.71 (m, 2H), 2.66–2.54 (m, 1H), 2.51–2.30 (m, 3H), 2.11–1.94 (m, 1H), 0.99 (d, 2H, J = 6.7 Hz), 0.98 (d, 2H, J = 6.7 Hz), 0.89 (d, 2H, J = 6.5 Hz); 13C NMR (CDCl3) δ 167.3, 161.1, 160.1, 157.3, 157.0, 151.4, 130.4, 129.9, 129.5, 128.8, 128.7, 118.1, 118.0, 111.7, 109.9, 108.6, 106.2, 106.2, 105.7, 103.3, 58.6, 57.8, 55.4, 54.5, 51.3, 50.9, 43.5, 30.8, 18.9, 18.0, 12.8; MS (ESI) m/z 504.5 (M + H)+.
N-(1S)-1-{[(3S)-4-(3-Hydroxyphenyl)-3-methylpiperazin-1-yl]methyl}-2-methylpropyl-4-(3-hydroxyphenoxy)benzamide (11h) Dihydrochloride
General Procedure B using acid 25h afforded 23.7 mg (33%) of 11h•2HCl as a white solid: mp 132 °C (fusion); [α]25D +59.6° (c 1.51, CH3OH); Anal. (C29H37Cl2N3O4•2 H2O) C, H, N. 11h free base: 1H NMR (CD3OD) δ 7.85 (d, 2H, J = 8.7 Hz), 7.18 (t, 1H, J = 8.0 Hz), 7.07–6.97 (m, 3H), 6.65–6.59 (m, 1H), 6.53–6.37 (m, 4H), 6.34–6.29 (m, 1H), 4.28–4.17 (m, 1H), 3.85–3.74 (m, 1H), 3.19–2.94 (m, 3H), 2.93–2.66 (m 4H), 2.63–2.42 (m, 3H), 1.98–1.84 (m, 1H), 1.02 (d, 3H, J = 6.8 Hz), 1.00 (d, 3H, J = 6.9 Hz), 0.93 (d, 3H, J = 6.4 Hz); 13C NMR (CD3OD) δ 169.9, 161.9, 160.3, 159.3, 158.5, 152.8,131.6, 130.9, 130.5, 130.4, 118.7, 112.5, 111.7, 110.4, 108.4, 108.0, 105.8, 104.6, 98.2, 60.8, 59.2, 55.3, 53.0, 52.7, 32.8, 20.1, 18.8, 13.6; MS (ESI) m/z 490.7 (M + H)+.
N-(1S)-1-{[(3S)-4-(3-Hydroxyphenyl)-3-methylpiperazin-1-yl]methyl}-2-methylpropyl-4-(3-fluorophenoxy)benzamide (11i) Dihydrochloride
General Procedure A using acid 25i afforded 14.4 mg (51%) of 11i•2HCl as a white solid: mp 85 °C (fusion); [α]25D +61.3° (c 0.46, CH3OH). Anal. (C29H36Cl2FN3O3•2 H2O) C, H, N. 11i free base: 1H NMR (CDCl3) δ 7.81 (d, 2H, J = 8.8 Hz), 7.32–7.21 (m, 1H), 7.00 (t, 1H, J = 8.1 Hz), 6.93 (d, 2H, J = 8.6 Hz), 6.87–6.65 (m, 3H), 6.40 (s, 1H), 6.39 (d, 1H, J = 7.2 Hz), 6.30 (d, 1H, J = 8.0 Hz), 4.39–4.26 (m, 1H), 3.81–3.70 (m, 1H), 3.19–2.93 (m, 3H), 2.93–2.80 (m, 2H), 2.74–2.46 (m, 3H), 2.09–1.94 (m, 1H), 1.00 (d, 6H, J = 6.8 Hz), 0.90 (d, 3H, J = 6.5 Hz); 13C NMR (CDCl3) δ 167.3, 165.1, 161.9, 159.4, 157.3, 151.1, 131.6, 130.7, 130.6, 129.9, 129.7, 129.1, 118.3, 117.9, 114.8, 114.8, 111.0, 110.7, 110.4, 109.9, 107.7, 107.2, 106.9, 104.9, 58.3, 57.9, 53.8, 50.9, 50.7, 31.2, 19.0, 18.1, 13.4; MS (ESI) m/z 492.4 (M + H)+.
4-(3-Chlorophenoxy)-N-[(1S)-1-{[(3S)-4-(3-hydroxyphenyl)-3-methylpiperazin-1-yl]methyl}-2-methylpropyl]benzamide (11j) Dihydrochloride
General Procedure B with acid 25j afforded 39 mg (64%) of 11j•2HCl as a white powder: mp 103–105 °C (fusion), [α]25D +79.3° (c 0.145, CH3OH). Anal. (C29H36Cl3N3O3•1.5H2O) C, H, N. 11j free base: 1H NMR (CDCl3) δ 7.78 (d, 2H, J = 7.8 Hz), 7.26 (t, 1H, J = 8.0 Hz), 7.15–6.84 (m, 5H), 6.50 (d, 1H, J = 7.8 Hz), 6.43–6.25 (m, 3H), 4.31–4.16 (m, 1H), 3.82–3.70 (m, 1H); 13C NMR (CDCl3) δ 167.3, 159.4, 157.3, 157.1, 151.3, 135.3, 130.7, 130.0, 129.9, 129.0, 128.9, 124.2, 119.8, 118.5, 118.4, 117.6, 108.8, 106.7, 103.8, 58.5, 57.8, 54.4, 51.2, 50.9, 43.8, 30.9, 18.9, 18.1, 12.9; MS (ESI) m/z 508.5 (M + H)+.
4-(3-Bromophenoxy)-N-[(1S)-1-{[(3S)-4-(3-hydroxyphenyl)-3-methylpiperazin-1-yl]methyl}-2-methylpropyl]benzamide (11k) Dihydrochloride
General Procedure B with acid 25k afforded 40 mg (61%) of 11k•2HCl as a white powder: mp 106–109 °C (fusion), [α]25D +60.4° (c 0.23, CH3OH). Anal. (C29H36BrCl2N3O3•1.5H2O) C, H, N. 11k free base: 1H NMR (CDCl3) δ 7.78 (d, 2H, J = 8.7 Hz), 7.31–7.13 (m, 2H), 7.09–6.91 (m, 4H), 6.44–6.23 (m, 4H), 4.29–4.14 (m, 1H); 13C NMR (CDCl3) δ 167.2, 159.4, 157.1, 151.4, 131.0, 123.0, 129.9, 127.1, 123.0, 122.6, 118.4, 118.0, 108.5, 106.4, 103.5, 58.5, 57.8, 54.5, 51.4, 50.9, 30.8, 18.9, 18.1, 12.8; MS (ESI) m/z 552.5 (M + H)+.
N-(1S)-1-{[(3S)-4-(3-Hydroxyphenyl)-3-methylpiperazin-1-yl]methyl}-2-methylpropyl-4-(4-methylphenoxy)benzamide (11l) Dihydrochoride
General Procedure A using acid 25l afforded 11.8 mg (48%) of 11l•2HCl as a white solid: mp 160 °C (fusion); [α]25D +60.6° (c 0.33, CH3OH); Anal. (C30H39Cl2N3O3•1.5 H2O) C, H, N. 11l free base: 1H NMR (CDCl3) δ 7.74 (d, 2H, J = 8.7 Hz), 6.42–6.27 (m, 3H), 7.16 (d, 2H, J = 8.3 Hz), 7.04 (t, 1H, J = 8.1 Hz), 6.94 (d, 2H, J = 8.7 Hz), 6.92 (d, 2H, J = 8.4 Hz), 4.28–4.17 (m, 1H), 3.85–3.71 (m, 1H), 3.21–2.97 (m, 2H), 2.85–2.73 (m, 2H), 2.65–2.53 (m, 1H), 2.46–2.30 (m, 3H), 2.34 (s, 3H), 2.14–2.02 (m, 1H), 0.99 (d, 3H, J = 6.7 Hz), 0.97 (d, 3H, J = 6.8 Hz), 0.89 (d, 3H, J = 6.7 Hz); 13C NMR (CDCl3) δ 167.3, 160.9, 157.1, 153.5, 151.4, 133.9, 130.4, 129.9, 129.0, 128.7, 119.8, 117.3, 108.5, 106.3, 103.4, 58.6, 57.9, 54.5, 51.3, 50.9, 43.6, 31.6, 30.8, 22.6, 20.7, 18.9, 18.0, 14.1, 12.7; MS (ESI) m/z 488.6 (M + H)+.
N-(1S)-1-{[(3S)-4-(3-Hydroxyphenyl)-3-methylpiperazin-1-yl]methyl}-2-methylpropyl-4-(4-methoxyphenoxy)benzamide (11m) Dihydrochloride
General Procedure B using acid 25m afforded 27.6 mg (39%) of 11m•2HCl as a white solid: mp 125 °C (fusion); [α]25D +64.5° (c 1.01, CH3OH). Anal. (C30H39Cl2N3O4•H2O) C, H, N. 11m free base: 1H NMR (CDCl3) δ 7.74 (d, 2H, J = 8.8 Hz), 7.08–6.84 (m, 7H), 6.48 (br s, 1H), 6.41–6.24 (m, 3H), 4.30–4.17 (m, 1H), 3.81–3.70 (m, 1H), 3.80 (s, 3H), 3.19–2.93 (m, 2H), 2.86–2.56 (m, 4H), 2.54–2.30 (m, 3H), 2.10–1.94 (m, 1H), 1.01–0.93 (m, 6H), 0.87 (d, 3H, J = 6.4 Hz); 13C NMR (CDCl3) δ 167.4, 161.4, 157.2, 156.5, 151.3, 149.0, 129.9, 128.8, 121.4, 116.7, 115.0, 108.9, 106.7, 103.8, 58.44, 57.9, 57.8, 55.7, 54.3, 51.1, 50.9, 43.8, 43.8, 30.9, 18.9, 18.0, 12.9; MS (ESI) m/z 504.7 (M + H)+.
N-(1S)-1-{[(3S)-4-(3-Hydroxyphenyl)-3-methylpiperazin-1-yl]methyl}-2-methylpropyl-4-(4-hydroxyphenoxy)benzamide (11n) Dihydrochloride
General Procedure B using acid 25n afforded 33.8 mg (48%) of 11n•2HCl as a white solid: mp 185 °C (fusion); [α]25D +62.4° (c 1.60, CH3OH). Anal. (C29H37Cl2N3O4•1.5 H2O) C, H, N. 11n free base: 1H NMR (CD3OD) δ 7.81 (d, 2H, J = 8.8 Hz), 7.03 (t, 1H, J = 8.1 Hz), 6.95–6.87 (m, 4H), 6.85–6.79 (m, 2H), 6.46–6.26 (m, 3H), 4.26–4.16 (m, 1H), 3.85–3.72 (m, 1H), 3.18–2.93 (m, 3H), 2.90–2.78 (m, 3H), 2.76–2.62 (m, 1H), 2.58–2.40 (m, 3H), 1.98–1.93 (m, 1H), 3.54 (d, 3H, J = 7.1 Hz), 1.00 (d, 3H, J = 7.0 Hz), 0.92 (d, 3H, J = 6.6 Hz); 13C NMR (CD3OD) δ 170.0, 163.3, 159.3, 155.7, 152.9, 149.3, 130.8, 130.3, 122.7, 117.4, 117.3, 110.3, 108.3, 105.7, 60.8, 59.2, 55.3, 53.0, 52.8, 46.1, 46.1, 33.0, 32.8, 23.7, 20.1, 18.8, 14.5, 13.5; MS (ESI) m/z 490.7 (M + H)+.
4-(2-Hydroxy-3-methylphenoxy)-N-[(2S)-1-[(3S)-4-(3-hydroxyphenyl)-3-methylpiperazin-1-yl]-3-methylbutan-2-yl]benzamide (11o) Dihydrochloride
General Procedure B with acid 25o afforded 31.4 mg (52%) of 11o•2HCl as a white powder: mp 173 °C (fusion), [α]25D 63.8° (c 0.24, CH3OH). Anal. (C30H39Cl2N3O4•1.5 H2O) C, H, N. 11o free base: 1H NMR (CDCl3) δ 7.81–7.65 (m, 2H), 7.09–6.70 (m, 5H), 6.41–6.24 (m, 4H), 4.28–4.13 (m, 1H), 3.75–3.58 (m, 1H), 3.16–2.88 (m, 2H), 2.80–2.65 (m, 2H), 2.64–2.51 (m, 1H), 2.45–2.24 (m, 4H), 2.06–1.92 (m, 4H), 1.00–0.92 (m, 6H), 0.86–0.76 (m, 3H); 13C NMR (CDCl3) δ 167.5, 157.1, 151.3, 149.0, 139.6, 132.0, 129.9, 129.0, 128.9, 128.6, 126.2, 122.7, 119.9, 117.2, 114.9, 114.5, 108.8, 103.7, 58.5, 54.4, 51.3, 50.9, 30.9, 18.9, 18.0, 16.1, 12.9; MS (ESI) m/z 504.6 (M + H)+.
4-(2-Hydroxy-5-methylphenoxy)-N-[(2S)-1-[(3S)-4-(3-hydroxyphenyl)-3-methylpiperazin-1-yl]-3-methylbutan-2-yl]benzamide (11p) Dihydrochloride
General Procedure B using acid 25p afforded 43 mg (72%) of 11p•2HCl as a white powder: mp 179–183 °C (fusion), [α]25D +56.5° (c 1.35, CH3OH). Anal. (C30H39Cl2N3O4•H2O) C, H, N. 11p free base: 1H NMR (CDCl3) δ 7.70 (d, 2H, J = 8.8 Hz), 7.01 (t, 1H, J = 8.1 Hz), 6.96–6.82 (m, 4H), 6.72 (s, 1H), 6.41–6.26 (m, 4H), 4.27–4.14 (m, 1H), 3.70–3.58 (m, 1H), 3.06 (d, 1H, J = 11.7 Hz), 2.95 (t, 1H, J = 10.5 Hz), 2.71 (d, 2H, J = 10.6 Hz), 2.56 (t, 1H, J = 11.3 Hz), 2.45–2.18 (m, 3H), 2.21 (s, 3H), 2.07–1.93 (m, 1H), 1.00–0.91 (m, 6H), 0.81 (d, 3H, J = 6.4 Hz); 13C NMR (CDCl3) δ 167.5, 160.2, 157.2, 151.4, 145.7, 142.1, 130.5, 129.9, 129.2, 128.8, 128.8, 126.2, 120.9, 116.9, 116.7, 108.8, 106.7, 103.8, 58.6, 57.9, 54.4, 51.4, 51.0, 43.9, 30.9, 20.6, 18.9, 18.0, 12.9; MS (ESI) m/z 504.6 (M + H)+.
4-(3,5-Dimethylphenoxy)-N-[(1S)-1-{[(3S)-4-(3-hydroxyphenyl)-3-methylpiperazin-1-yl]methyl}-2-methylpropyl]benzamide (11q) Dihydrochloride
General Procedure B using acid 25q afforded 37 mg (61%) of 11q•2HCl as a white powder: mp 117–120 °C (fusion), [α]25D +66.4° (c 0.66, CH3OH). Anal. (C31H41Cl2N3O3•2H2O) C, H, N. 11q free base: 1H NMR (CDCl3) δ 7.75 (d, 2H, J = 8.7 Hz), 7.03 (t, 1H, J = 8.0 Hz), 6.95 (d, 2H, J = 8.7 Hz), 6.79 (s, 1H), 6.63 (s, 2H), 6.43–6.24 (m, 4H), 4.29–4.16 (m, 1H), 3.83–3.72 (m, 1H), 3.20–3.11 (m 1H), 3.09–2.97 (m, 1H), 2.95–2.72 (m, 2H), 2.61 t, 1H, J = 11 Hz), 2.51–2.30 (m, 3H), 2.28 (s, 6H), 2.11–1.98 (m, 1H), 1.03–0.93 (m, 6H), 0.88 (d, 3H, J = 6.5 Hz); 13C NMR (CDCl3) δ 166.2, 159.4, 156.0, 154.8, 150.2, 138.7, 128.7, 127.9, 127.6, 124.8, 116.6, 116.2, 107.4, 105.3, 102.4, 57.3, 56.7, 53.3, 50.1, 49.7, 42.4, 29.7, 20.1, 17.7, 16.9, 11.7; MS (ESI) m/z 502.8 (M + H)+.
N-(1S)-1-{[(3S)-4-(3-Hydroxyphenyl)-3-methylpiperazin-1-yl]methyl}-2-methylpropyl-3-methyl-4-phenoxybenzamide (12a) Dihydrochloride
General Procedure B using acid 26a afforded 31.9 mg (45%) of 12a•2HCl as a pale yellow solid: mp 88 °C (fusion); [α]25D +62.1° (c 1.51, CH3OH). Anal. (C30H39Cl2N3O3•H2O) C, H, N. 12a free base: 1H NMR (CDCl3) δ 7.70 (s, 1H), 7.56 (d, 1H, J = 8.5 Hz), 7.32 (t, 2H, J = 8.3 Hz), 7.09 (t, 1H, J = 7.5 Hz), 7.03 (t, 1H, J = 7.9 Hz), 6.92 (d, 2H, J = 8.1 Hz), 6.81 (d, 1H, J = 8.4 Hz), 6.46 (br s, 1H), 6.41–6.28 (m, 3H), 4.32–4.17 (m, 1H), 3.83–3.69 (m, 1H), 3.18–2.95 (m, 2H), 2.86–2.58 (m, 3H), 2.52–2.32 (m, 3H), 2.27 (s, 3H), 2.10–1.96 (m, 1H), 1.02–0.93 (m, 6H), 0.89 (d, 3H, J = 6.4 Hz); 13C NMR (CDCl3) δ 167.6, 157.9, 157.3, 151.3, 130.3, 129.9, 129.7, 129.7, 129.5, 126.0, 123.4, 118.4, 118.1, 108.7, 106.8, 103.9, 103.2, 96.8, 58.5, 57.9, 54.3, 51.2, 50.9, 30.9, 18.9, 18.0, 16.2, 12.9; MS (ESI) m/z 488.6 (M + H)+.
N-[(2S)-1-[(3S)-4-(3-Hydroxyphenyl)-3-methylpiperazin-1-yl]-3-methylbutan-2-yl]-3-methyl-4-(3-methylphenoxy)benzamide (12b) Dihydrochloride
General Procedure B with acid 26b afforded 42.9 mg (70%) of 12b•2HCl as a white powder: mp 124–130 °C (fusion), [α]25D 46.6° (c 0.50, CH3OH). Anal. (C31H41Cl2N3O3•2 H2O) C, H, N. 12b free base: 1H NMR (CDCl3) δ 7.68 (s, 1H), 7.55 (d, 2H, J = 8.4 Hz), 7.19 (t, 1H, J = 7.7 Hz), 7.03 (t, 1H, J = 8.0 Hz), 6.91 (d, 1H, J = 7.5 Hz), 6.81 (d, 1H, J = 8.4 Hz), 6.75 (s, 1H), 6.42–6.28 (m, 5H), 4.29–4.15 (m, 1H), 3.84–3.72 (m, 1H), 3.15 (d, 1H, J = 11.4 Hz), 3.03 (t, 1H, J = 10.3 Hz), 2.84–2.72 (m 2H), 2.65–2.53 (m, 1H), 2.47–2.32 (m, 3H), 2.31 (s, 3H), 2.27 (s, 3H), 2.12–1.98 (m, 1H), 1.02–0.95 (m, 6H), 0.90 (d, 3H, J = 6.4 Hz); 13C NMR (CDCl3) δ 167.6, 157.9, 157.2, 156.8, 151.4, 140.1, 130.3, 129.9, 129.7, 129.6, 129.5, 125.9, 124.2, 119.1, 118.1, 115.4, 108.5, 106.5, 103.6, 58.6, 57.9, 54.5, 51.3, 50.9, 43.7, 30.8, 21.4, 18.9, 18.1, 16.3, 12.8; MS (ESI) m/z 502.8 (M + H)+.
N-[(2S)-1-[(3S)-4-(3-Hydroxyphenyl)-3-methylpiperazin-1-yl]-3-methylbutan-2-yl]-3-methoxy-4-(3-methylphenoxy)benzamide (12c) Dihydrochloride
General Procedure B with acid 26c afforded 46.7 mg (75%) of 12c•2HCl as a white powder: mp 128–131 °C, [α]25D 54.0° (c 0.73, CH3OH). Anal. (C31H41Cl2N3O4•2 H2O) C, H, N. 12c free base: 1H NMR (CDCl3) δ 7.53 (d, 1H, J = 1.9 Hz), 7.27–7.14 (m, 2H), 7.03 (t, 1H, J = 8.0 Hz), 6.90 (d, 1H, J = 7.6 Hz), 6.86 (d, 1H, J = 8.3 Hz), 6.81–6.71 (m, 2H), 6.42–6.28 (m, 4H), 4.28–4.16 (m, 1H), 3.84 (s, 3H), 3.83–3.72 (m, 1H), 3.14 (d, 1H, J = 11.8 Hz), 3.01 (t, 1H, J = 10.7 Hz), 2.83–2.71 (m, 2H), 2.64–2.52 (m, 1H), 2.47–2.27 (m, 3H), 2.30 (s, 3H), 2.11–1.98 (m, 1H), 1.03–0.94 (m, 6H), 0.90 (d, 3H, J = 6.4 Hz); 13C NMR (CDCl3) δ 167.5, 157.3, 156.8, 151.4, 151.1, 148.7, 139.9, 130.8, 129.9, 129.4, 124.2, 119.1, 118.9, 118.9, 115.2, 112.3, 108.5, 106.6, 103.7, 58.6, 57.9, 56.6, 54.5, 51.5, 50.9, 43.7, 30.8, 21.4, 18.9, 18.1, 12.8; MS (ESI) m/z 518.7 (M + H)+.
3-Hydroxy-N-[(2S)-1-[(3S)-4-(3-hydroxyphenyl)-3-methylpiperazin-1-yl]-3-methylbutan-2-yl]-4-(3-methylphenoxy)benzamide (12d) Dihydrochloride
General Procedure B with acid 26d afforded 38.6 mg (64%) of 12d•2HCl as a pale yellow powder: mp 195–200 °C, [α]25D 58.5° (c 1.07, CH3OH). Anal. (C30H39Cl2N3O4•1.5 H2O) C, H, N. 12d free base: 1H NMR (CDCl3) δ 7.42 (s, 1H), 7.23–7.10 (m, 2H), 7.01 (t, 1H, J = 7.8 Hz), 6.93 (d, 1H, J = 7.4 Hz), 6.77 (s, 1H), 6.71 (t, 2H, J = 8.7 Hz), 6.55 (bs, 1H), 6.45–6.32 (m, 3H), 4.33–4.19 (m, 1H), 3.52 (bs, 1H), 3.12–3.02 (m, 1H), 2.96–2.83 (m, 1H), 2.67–2.47 (m, 4H), 2.39–2.25 (m, 3H), 2.29 (s, 3H), 2.01–1.88 (m, 1H), 1.00–0.91 (m, 6H), 0.81 (d, 3H, J = 6.3 Hz); 13C NMR (CDCl3) δ 168.0, 157.3, 156.1, 151.5, 147.4, 146.9, 140.3, 131.1, 129.8, 129.7, 124.9, 119.2, 119.1, 119.1, 118.1, 115.7, 115.6, 107.7, 58.9, 53.9, 51.4, 51.2, 31.2, 21.4, 19.1, 19.1, 17.9, 13.8; MS (ESI) m/z 504.5 (M + H)+.
3-Chloro-N-[(2S)-1-[(3S)-4-(3-hydroxyphenyl)-3-methylpiperazin-1-yl]-3-methylbutan-2-yl]-4-(3-methylphenoxy)benzamide (12e) Dihydrochloride
General Procedure B using acid 26e afforded 25 mg (41%) of 12e•2HCl as a white powder: mp 154–158 °C (fusion), [α]25D +63.2° (c 0.95, CH3OH). Anal. (C30H38Cl3N3O3•H2O) C, H, N. 12e free base: 1H NMR (CDCl3) δ 7.87 (s, 1H), 7.60 (d, 1H, J = 8.5 Hz), 7.23 (t, 1H, J = 7.7 Hz), 7.04 (t, 1H, J = 8.0 Hz), 6.97 (d, 1H, J = 7.4 Hz), 6.88 (d, 1H, J = 8.5 Hz), 6.84–6.76 (m, 2H), 6.40 (d, 1H, J = 8.2 Hz), 6.35–6.25 (m, 3H), 4.26–4.12 (m, 1H), 3.85–3.74 (m, 1H), 3.17 (d, 1H, J = 11.7 Hz), 3.04 (t, 1H, J = 10.0 Hz), 2.77 (t, 2H, J = 8.8 Hz), 2.64–2.52 (m, 1H), 2.48–2.29 (m, 3H), 2.33 (s, 3H), 2.11–1.97 (m, 1H), 1.05–0.93 (m, 6H), 0.89 (d, 3H, J = 6.4 Hz); 13C NMR (CDCl3) δ 166.2, 157.0, 155.7, 151.4, 140.3, 130.7, 129.9, 129.7, 129.5, 126.7, 125.2, 125.1, 119.7, 119.0, 116.0, 108.6, 106.3, 103.4, 58.5, 57.9, 54.5, 51.5, 50.9, 43.6, 30.8, 21.4, 18.9, 18.1, 12.8; MS (ESI) m/z 522.4 (M + H)+.
N-[(1S)-1-{[(3S)-4-(3-Hydroxyphenyl)-3-methylpiperazin-1-yl]methyl}-2-methylpropyl]-2-methyl-4-(3-methylphenoxy)benzamide (12f) Dihydrochloride
General Procedure B with acid 26f afforded 13 mg (22%) of 12f•2HCl as a white powder: mp 164–167 °C (fusion), [α]25D +46.6° (c 0.35, CH3OH). Anal. (C31H41Cl2N3O3•H2O) C, H, N. 12f free base: 1H NMR (CDCl3) δ 7.39 (d, 1H, J = 8.2 Hz), 7.22 (t, 1H, J = 8.0 Hz), 7.06 (t, 1H, J = 8.0 Hz), 6.95 (d, 1H, J = 7.4 Hz), 6.85–6.74 (m, 3H), 6.43 (dd, 1H, J = 8.3, 1.6 Hz), 6.36 (s, 1H), 6.29 (dd, 1H, J = 7.9, 1.8 Hz), 5.81 (bd, 1H, J = 6.8 Hz), 4.30–4.16 (m, 1H), 3.89–3.77 (m, 1H); 13C NMR (CDCl3) δ 169.8, 158.7, 156.9, 156.5, 151.5, 140.1, 138.7, 129.9, 129.6, 128.5, 124.6, 120.8, 120.0, 116.4, 115.4, 108.8, 106.3, 103.6, 59.4, 58.4, 54.6, 51.2, 51.0, 43.9, 30.6, 21.4, 20.2, 19.1, 17.8, 13.1; MS (ESI) m/z 502.8 (M + H)+.
N-[(1S)-1-{[(3S)-4-(3-Hydroxyphenyl)-3-methylpiperazin-1-yl]methyl}-2-methylpropyl]-2-methoxy-4-(3-methylphenoxy)benzamide (12g) Dihydrochloride
General Procedure B with acid 26g afforded 35 mg (58%) of 12g•2HCl as a white powder: mp 99–102 °C (fusion), [α]25D +82.3° (c 0.265, CH3OH). Anal. (C31H41Cl2N3O4•0.5H2O) C, H, N. 12g free base: 1H NMR (CDCl3) δ 8.11 (d, 1H, J = 8.7 Hz), 7.83 (d, 1H, J = 8.4 Hz), 7.29–7.19 (m, 1H), 7.07–6.95 (m, 2H, 6.89–6.80 (m, 2H), 6.64–6.53 (m, 2H), 6.42–6.26 (m, 3H), 4.37–4.25 (m, 1H), 3.89 (s, 3H), 3.82–3.70 (m, 1H); 13C NMR (CDCl3) δ 165.1, 161. 6, 158.9, 157.4, 155.7, 151.5, 140.2, 133.8, 129.8, 129.7, 125.2, 120.5, 116.9, 116.5, 110.2, 108.4, 106.4, 103. 7, 101.7, 59.5, 58.7, 56.1, 54.0, 51.5, 51.1, 43.8, 43.8, 30.0, 21.4, 19.4, 17.3, 12.7; MS (ESI) m/z 518.7 (M + H)+.
2-Hydroxy-N-[(1S)-1-{[(3S)-4-(3-hydroxyphenyl)-3-methylpiperazin-1-yl]methyl}-2-methylpropyl]-4-(3-methylphenoxy)benzamide (12h) Dihydrochloride
General Procedure B with acid 26h afforded 65 mg (43%) of 12h•2HCl as a white powder: mp 119–123 °C (fusion), [α]25D +81.8° (c 0.08, CH3OH). Anal. (C30H39Cl2N3O4•1.25 H2O) C, H, N. 12h free base: 1H NMR (CDCl3) δ 7.37 (d, 1H, J = 8.6 Hz), 7.24 (t, 1H, J = 7.7 Hz), 7.07 (t, 1H, J = 8.1 Hz), 6.99 (d, 1H, J = 7.6 Hz), 6.91–6.82 (m, 2H), 6.50–6.40 (m, 3H), 6.32 (t, 1H, J = 2.2 Hz), 6.27 (dd, 1H, J = 7.9, 2.0 Hz), 4.23–4.10 (m, 1H), 3.90–3.80 (m, 1H), 3.26–3.16 (m, 1H), 3.08 (td, 1H, J = 11.3, 2.5 Hz), 2.89–2.72 (m, 2H), 2.68–2.56 (m, 1H), 2.5–2.33 (m, 3H), 2.34 (s, 3H), 2.16–2.02 (m, 1H), 1.01–0.95 (m, 6H), 0.92 (d, 3H, J = 6.5 Hz); 13C NMR (CDCl3) 170.1, 163.6, 163.1, 156.8, 155.3, 151.6, 140.4, 130.3, 129.9, 127.1, 125.7, 121.3, 117.6, 109.4, 108.9, 108.9, 106.2, 106.2, 103.3, 58.2, 57.9, 54.8, 51.0, 50.9, 43.5, 30.8, 21.6, 18.9, 18.4, 12.9; MS (ESI) m/z 504.6 (M + H)+.
4-(2-Hydroxyphenoxy)-N-[(1S)-1-{[(3S)-4-(3-hydroxyphenyl)-3-methylpiperazin-1-yl]methyl}-2-methylpropyl]-3-methoxybenzamide (12i) Dihydrochloride
General Procedure B using acid 26i afforded 38.5 mg (42%) of 12i•2HCl as an off-white powder: mp 95–97 °C (fusion), [α]25D +55.2° (c 0.29, CH3OH). Anal. (C30H39Cl2N3O5•H2O) C, H, N. 12i free base: 1H NMR (CDCl3) δ 7.50 (s, 1H), 7.20 (d, 1H, J = 8.3 Hz), 7.06–6.98 (3H, m), 6.88–6.78 (m, 3H), 6.45–6.23 (m, 4H), 5.71 (bs, 2H), 4.29–4.14 (m, 1H), 3.79 (s, 3H), 3.75–3.62 (m, 1H), 3.15–2.24 (m, 8H), 2.07–1.93 (m, 1H), 1.01–0.93 (m, 6H), 0.85 (d, 3H, J = 6.3 Hz); 13C NMR (CDCl3) δ 167.4, 157.2, 151.4, 150.5, 148.6, 147.6, 143.7, 131.0, 129.9, 125.2, 120.5, 119.1, 119.0, 118.4, 116.7, 112.2, 108.8, 106.7, 103.8, 58.6, 57.9, 56.1, 54.4, 51.5, 50.9, 43.9, 30.9, 18.9, 18.1, 12.9; MS (ESI) m/z 520.6 (M + H)+.
3-Chloro-4-(2-hydroxyphenoxy)-N-[(1S)-1-{[(3S)-4-(3-hydroxyphenyl)-3-methylpiperazin-1-yl]methyl}-2-methylpropyl]benzamide (12j) Dihydrochloride
General Procedure B using acid 26j afforded 23.4 mg (26%) of 12j•2HCl as a white powder: mp 153–157 °C (fusion), [α]25D +68° (c 0.053, CH3OH). Anal. (C29H36Cl3N3O4•0.5 H2O) C, H, N. 12j free base: 1H NMR (CDCl3) δ 7.83 (s, 1H), 7.55 (d, 1H, J = 8.6 Hz), 7.12–6.99 (m, 3H), 6.88–6.78 (m, 3H), 6.44–6.21 (m, 4H), 4.26–4.11 (m, 1H), 3.74–3.62 (m, 1H), 3.16–2.91 (m, 2H), 2.78–2.65 (m, 2H), 2.64–2.50 (m, 1H), 2.49–2.26 (m, 3H), 2.09–1.94 (m, 1H), 1.00–0.93 (m, 6H), 0.82 (d, 3H, J = 6.5 Hz); 13C NMR (CDCl3) δ 166.1, 156.9, 155.2, 151.4, 147.6, 142.5, 130.0, 129.6, 126.8, 125.9, 120.8, 119.5, 117.9, 117.1, 109.0, 106.6, 58.4, 57.9, 54.4, 51.5, 50.9, 43.9, 30.9, 18.9, 18.1, 14.2, 13.0; MS (ESI) m/z 524.7 (M + H)+.
4-(2-Hydroxy-5-methylphenoxy)-N-[(1S)-1-{[(3S)-4-(3-hydroxyphenyl)-3-methylpiperazin-1-yl]methyl}-2-methylpropyl]-3-methoxybenzamide (12k) Dihydrochloride
General Procedure B with acid 26k afforded 49.8 mg (53%) of 12k•2HCl as a white powder: mp 100–103 °C (fusion), [α]25D +60.6° (c 0.18, CH3OH). Anal. (C31H41Cl2N3O5•H2O) C, H, N. 12k free base: 1H NMR (CDCl3) δ 7.49 (d, 1H, J = 1.7 Hz), 7.21 (dd, 1H, J = 8.3, 1.7 Hz), 7.01 (t, 1H, J = 8.3 Hz), 6.92–6.78 (m, 3H), 6.69–6.66 (m, 1H), 6.46–6.28 (m, 4H), 4.29–4.14 (m, 1H), 3.79 (s, 3H), 3.75–3.63 (m, 1H), 3.15–2.81 (m, 2H), 2.72 (d, 2H, J = 10.2 Hz), 2.57 (t, 1H, J = 11.2 Hz), 2.50–2.25 (m, 3H), 2.18 (s, 3H), 2.08–1.93 (m, 1H), 1.00–0.94 (m, 6H), 0.85 (d, 3H, J = 6.4 Hz); 13C NMR (CDCl3) δ 167.5, 157.2, 151.4, 150.5, 148.7, 145.2, 143.3, 130.9, 130.1, 129.9, 125.6, 119.7, 119.0, 118.3, 116.4, 112.2, 108.7, 106.7, 103.8, 58.6, 57.9, 56.1, 54.4, 51.5, 50.9, 43.9, 30.9, 20.6, 19.0, 18.1, 12.9; MS (ESI) m/z 534.3 (M + H)+.
3-Chloro-4-(2-hydroxy-5-methylphenoxy)-N-[(1S)-1-{[(3S)-4-(3-hydroxyphenyl)-3-methylpiperazin-1-yl]methyl}-2-methylpropyl]benzamide (12l) Dihydrochloride
General Procedure B using acid 26l afforded 29.0 mg (31%) of 12l•2HCl as a white powder: mp 145–148 °C (fusion), [α]25D +76° (c 0.073, CH3OH). Anal. (C30H38Cl3N3O4•H2O) C, H, N. 12l free base: 1H NMR (CDCl3) δ 7.89 (s, 1H), 7.62 (d, 1H, J = 8.2 Hz), 7.02 (t, 1H, J = 8.0 Hz), 6.94 (d, 1H, J = 7.9 Hz), 6.87 (d, 1H, J = 8.2 Hz), 6.81 (d, 1H, J = 8.6 Hz), 6.67 (s, 1H), 6.41–6.21 (m, 3H), 4.33–4.18 (m, 1H), 3.73–3.55 (m, 1H), 3.15–2.45 (m, 7H), 2.21 (s, 3H), 2.06–1.90 (m, 1H), 1.00–0.90 (m, 6H), 0.80 (d, 3H, J = 6.0 Hz); 13C NMR (CDCl3) δ 166.2, 157.0, 155.4, 151.4, 145.2, 142.1, 130.7, 130.6, 129.9, 129.5, 126.8, 126.4, 124.4, 120.1, 117.8, 116.8, 108.9, 106.6, 103.7, 58.5, 57.9, 54.4, 51.5, 50.9, 43.9, 30.9, 20.6, 18.9, 18.1, 13.0; MS (ESI) m/z 538.3 (M + H)+.
N-[(2S)-1-[(3S)-4-(3-Hydroxyphenyl)-3-methylpiperazin-1-yl]-3-methylbutan-2-yl]-6-phenoxypyridine-3-carboxamide (13) Trihydrochloride
General Procedure B using acid 33 afforded 33 mg (51%) of 13•3HCl as a white powder: mp 168–170 °C (fusion), [α]25D +59.7° (c 1.55, CH3OH). Anal. (C29H39Cl3N4O3•2.5H2O) C, H, N. 13 free base: 1H NMR (CDCl3) δ 8.57 (d, 1H, J = 2.3 Hz), 8.12 (dd, 1H, J = 8.6, 2.5 Hz), 7.41 (t, 2H, J = 7.9 Hz), 7.23 (t, 1H, J = 7.4 Hz), 7.16–7.10 (m, 2H), 7.04 (t, 1H, J = 8.0 Hz), 6.91 (d, 1H, J = 8.6 Hz), 6.38 (d, 1H, J = 8.0 Hz), 6.33–6.23 (m, 3H), 4.27–4.12 (m, 1H), 3.83–3.71 (m, 1H), 3.19–3.09 (m, 1H), 3.06–2.95 (m, 1H), 2.75 (d, 2H, J = 10.8 Hz), 2.62–2.49 (m, 1H), 2.46–2.25 (m, 3H), 2.11–1.96 (m, 1H), 1.01–0.93 (m, 6H), 0.86 (d, 3H, J = 6.4 Hz); 13C NMR (CDCl3) δ 165.7, 165.6, 157.1, 153.5, 151.4, 146.4, 139.0, 129.9, 129.8, 125.8, 125.3, 121.4, 111.1, 108.6, 106.4, 103.5, 58.5, 57.8, 54.5, 51.4, 50.9, 43.6, 30.7, 18.8, 18.0, 12.8; MS (ESI) m/z 475.7 (M + H)+.
N-(1S)-1-{[(3S)-4-(3-Hydroxyphenyl)-3-methylpiperazin-1-yl]methyl}-2-methylpropyl]-5-phenoxypyridine-2-carboxamide (14) Trihydrochloride
To a solution of 35 (182 mg, 0.85 mmol) in CH3CN (10 mL) at room temperature were added HBTU (355 mg, 0.94 mmol), Et3N (0.24 mL, 1.7 mmol), and 16 (230 mg, 0.85 mmol). THF (2 mL) was added for solubility. The reaction mixture was stirred for 12 h and concentrated. Flash column chromatography of the crude product on silica gel using an EtOAc gradient in hexane afforded 14 free base: 1H NMR (CDCl3) δ 8.29 (d, 1H, J = 2.6 Hz), 8.13 (d, 1H, J = 8.7 Hz), 7.94 (d, 1H, J = 9.0 Hz), 7.45–7.36 (m, 2H), 7.32 (dd, 1H, J = 8.6, 2.9 Hz), 7.21 (t, 1H, J = 7.3 Hz), 7.11–7.03 (m, 3H), 6.43 (dd, 1H, J = 8.2, 1.6 Hz), 6.32 (s, 1H), 6.25 (d, 1H, J = 7.2 Hz), 4.67 (br s, 1H), 4.26–4.12 (m, 1H), 3.92–3.78 (m, 1H), 3.27–3.14 (m, 1H), 3.06 (td, 1H, J = 11.4, 3.3 Hz), 2.89 (d, 1H, J = 11.4 Hz), 2.78 (d, 1H, J = 11.0 Hz), 2.62–2.52 (m, 1H), 2.48–2.28 (m, 3H), 2.15–1.95 (m, 1H), 1.01 (d, 3H, J = 4.7 Hz), 0.98 (d, 3H, J = 4.7 Hz), 0.93 (d, 3H, J = 6.3 Hz); 13C NMR (CDCl3) δ 164.1, 157.7, 156.5, 155.2, 151.4, 144.3, 138.8, 130.2, 129.7, 124.9, 124.9, 123.5, 119.7, 108.0, 106.3, 103.5, 59.3, 58.2, 54.3, 51.4, 50.9, 43.5, 30.7, 19.3, 17.7, 12.6. MS (ESI) m/z 476.0 (M + H)+. The free base was converted to 121 mg (31%) of 14•3HCl as a tan solid: mp 95 °C (fusion); [α]25D +73.1° (c 0.67, CH3OH). Anal. (C28H37Cl3N4O3•0.5 EtOAc) C, H, N.
3-{(2S)-4-[(2S)-2-Amino-3-methylbutyl]-2-methylpiperazin-1-yl}phenol (16)
In a round-bottom flask, 570 mg (2.49 mmol) of (S)-3-(2-methylpiperazin-1-yl)phenol (15)31 was dissolved in 30 mL of dry THF along with 542 mg (2.49 mmol) of N-Boc-L-valine. The solution was cooled to 0 °C in an ice-bath, and 1.38 mL (9.97 mmol) of Et3N was added followed by 1.10 g (2.49 mmol) of BOP. The flask was removed from the ice bath, and the reaction was stirred for 2 h. The solution was then added to a concentrated aqueous NaHCO3 solution, and the mixture extracted three times with 15 mL of EtOAc. The pooled organic extracts were washed with brine, dried over MgSO4, filtered, and the solution evaporated to leave a residue that was purified by silica-gel flash column chromatography to yield 415 mg (42%) of the intermediate amide. This amide was dissolved in 20 mL of THF, and 3.18 mL (3.18 mmol) of a 1 M solution of BH3•THF was added. The solution was stirred at reflux overnight, cooled to rt, and quenched with 5 mL of H2O. Into this solution was added 10 mL of conc. HCl, the mixture was stirred for 1 h, and 20 mL of water was added. Solid NaHCO3 was added to adjust the solution to a pH of 8, and the mixture was extracted three times with 5 mL of CH2Cl2, washed with brine, and dried over MgSO4. Removal of the solvent afforded a residue that was purified by silica-gel flash-column chromatography eluting with a 6:2:1 mixture of CMA80–hexanes–EtOAc to yield 241 mg (82%) of 16 as a white solid, which after recrystallization from EtOAc–hexanes had mp 210–212 °C. [α]D25 +48.8° (c 0.1, MeOH). 1H NMR (CD3OD) δ 7.46–7.40 (t, 1H), 7.16 (m, 2H), 6.98 (d, 1H), 4.14 (m, 1H), 3.96 (m, 1H), 3.65 (m, 1H), 3.30 (m, 3H), 2.95 (m, 3H), 2.00 (m, 1H), 1.23–1.03 (m, 9H). ESIMS: m/z 278 (M+H+, 100).
(2R)-4-(3-Methoxyphenyl)-3-methylpiperazine (18)
To compound 17 (7.5 g, 0.0244 mol) in THF (40 mL) was added 1 N HCl (40 mL). The reaction mixture was stirred at reflux for 2 h and cooled to room temperature. THF was removed in vacuo, and saturated Na2CO3 solution (20 mL) was added followed by extraction with CH2Cl2. The organic layer was separated, dried (Na2SO4), and concentrated to yield a yellow oil which was converted to the HCl salt with 1 N ethereal HCl to afford 4.72 g (69%) of 18 as a tan solid which was used in the next step without further purification.
3-{(2R)-4-[(2S)-2-Amino-3-methylbutyl-4-(methoxyphenyl)-3-methylpiperazine (19)
Compound 18 (2.00 g, 0.00716 mol) was added to THF (100 mL) followed by N-Boc-L-valine (1.56 g, 0.00716 mol), BOP reagent (3.36 g, 0.00716 mol), and finally Et3N (2.20 g, 0.0216 mol). The mixture was stirred at room temperature for 4.5 h, then was added to a mixture of ether (200 mL)–H2O (100 mL). The organic phase was separated and washed with water, 10% NaHCO3 solution, and brine then dried (Na2SO4) and concentrated in vacuo to give 2.73 g of a dark red oil.
Without further purification the oil (2.70 g, 0.0066 mol) was dissolved in THF (30 mL), borane dimethylsulfide complex (5M, 6.6 mL, 0.033 mol) was added dropwise, and the reaction mixture was stirred at reflux for 2 h. The mixture was cooled to room temperature, treated with 6 N HCl (5 mL) and refluxed for 2 h. Water (100 mL) and EtOAc (100 mL) were added, and the reaction was stirred for an additional 15 min. The aqueous layer was separated, basified with Na2CO3, and extracted with CH2Cl2. The organic layer was separated, dried (Na2SO4), and evaporated to yield 1.64 g (80%) of 19 as a thick orange oil. 1H NMR (CDCl3) δ 7.16 (t, 1H, J = 8.3 Hz), 6.52 (d, 1H, J = 7.2 Hz), 6.43 (s, 1H), 6.38 (dd, 1H, J = 6.0, 2.1 Hz), 3.20 (m, 1H), 3.79 (s, 1H), 3.10 (t, 1H, J = 7.2 Hz), 2.90 (d, 1H, J = 7.2 Hz), 2.75 (bm, 1H), 2.51–2.61 (m, 1H), 2.10–2.13 (m, 3H), 1.57 (m, 3H), 1.11 (d, 3H, J = 6.3 Hz), 0.93 (d, 6H, J = 6.3 Hz).
3-{(2R)-4-[(2S)-2-Amino-3-methylbutyl]-2-methylpiperazine-1-yl}phenol (20)
Compound 19 (1.60 g, 0.00549 mol) was added to 48% HBr (15 mL) and stirred at reflux for 4 h. The reaction was concentrated to dryness, added water (100 mL), and basified with solid Na2CO3 to a pH = 10. The mixture was extracted with CH2Cl2, dried (Na2SO4), and concentrated to afford 1.33g (87%) of 20 as a beige foam. It was used as is for the next step.
N-[(1S)-1-{[(3S)-4-(3-Hydroxyphenyl)-3-methylpiperazin-1-yl]methyl}-2-methylpropyl]biphenyl-4-carboxamide (22) Dihydrochloride
To a solution of 16 (42 mg, 0.15 mmol), 4-phenylbenzoic acid (33 mg, 0.17 mmol) and Et3N (0.073 mL, 53 mmol) in CH2Cl2 (10 mL) at room temperature was added EDC•HCl (32 mg, 0.17 mmol). The reaction was stirred for 12 h. The mixture concentrated and the residue purified by chromatography on silica gel using a gradient up to 40% CMA80 in CH2Cl2 to afford 22 free base: 1H NMR (CDCl3) δ 7.86 (d, 2H, J = 8.2 Hz), 7.66–7.55 (m, 4H), 7.48–7.33 (m, 3H), 7.03 (t, 1H, J = 8.4 Hz), 6.48 (d, 1H, J = 8.2 Hz), 6.41–6.30 (m, 3H), 4.33–4.18 (m, 1H), 3.83–3.69 (m, 1H), 3.21–2.98 (m, 3H), 2.94–2.82 (m, 2H), 2.78–2.62 (m, 1H, 2.60–2.39 (m, 3H), 2.14–1.96 (m, 1H), 1.03–0.96 (m, 6H), 0.89 (d, 3H, J = 6.4 Hz); 13C NMR (CDCl3) δ 168.1, 157.3, 151.1, 144.4, 140.0, 133.1, 130.0, 128.9, 128.0, 127.5, 127.3, 127.2, 108.8, 106.9, 104.0, 58.8, 57.9, 54.4, 51.4, 50.9, 30.9, 19.0, 18.1, 12.8; MS (ESI) m/z 458.6 (M + H)+. The free base was converted to 34.8 mg (76%) of 22•2HCl as a white powder: mp 97–100 °C (fusion); [α]25D +72.9° (c 1.00, CH3OH). Anal. (C29H37Cl2N3O2•2.5H2O) C, H, N.
4-Butyl-N-[(1S)-1-{[(3S)-4-(3-hydroxyphenyl)-3-methylpiperazin-1-yl]methyl}-2-methylpropyl]benzamide (23) Dihydrochloride
To a solution of 16 (28 mg, 0.10 mmol), 4-butylbenzoic acid (48.6 mg, 0.12 mmol) and Et3N (0.050 mL, 36 mmol) in CH3CN (10 mL) at room temperature was added HBTU (45 mg, 0.12 mmol). The reaction was stirred for 12 h. The mixture was diluted with Et2O (50 mL), washed with saturated NaHCO3 (2 × 10 mL), brine (2 × 10 mL), dried (Na2SO4) and concentrated. The product was purified by chromatography on silica gel using a gradient up to 40% CMA80 in CH2Cl2 to afford 23 free base: 1H NMR (CDCl3) δ 7.74 (d, 2H, J = 8.2 Hz), 7.24 (d, 2H, J = 8.2 Hz), 7.04 (t, 1H, J = 8.1 Hz), 6.59 (d, 1H, J = 8.1 Hz), 6.42–6.30 (m, 3H), 4.33–4.17 (m, 1H), 3.86–3.71 (m, 1H), 3.24–2.92 (m, 7H), 2.91–2.71 (m, 3H), 2.63 (t, 2H, J = 7.7 Hz), 2.11–1.92 (m, 1H), 1.64–1.52 (m, 2H), 1.40–1.27 (m, 5H), 1.05–0.95 (m, 6H), 0.95–0.85 (m, 6H); 13C NMR (CDCl3) δ 169.2, 156.9, 150.3, 147.4, 130.5, 129.9, 128.6, 126.9, 109.4, 107.7, 104.6, 59.2, 57.6, 53.9, 51.2, 50.6, 47.4, 35.3, 33.0, 30.7, 22.1, 18.7, 17.8, 13.6, 12.3, 8.5; MS (ESI) m/z 438.6 (M + H)+. The free base was converted to 4.7 mg (9%) of 23•2HCl as a white powder: mp 116–120 °C (fusion); [α]25D +59° (c 0.10, CH3OH). Anal. (C27H41Cl2N3O2•0.5H2O) C, H, N.
N-[(1S)-1-{[(3S)-4-(3-Hydroxyphenyl)-3-methylpiperazin-1-yl]methyl}-2-methylpropyl]-3,4-dimethoxybenzamide (24) Dihydrochloride
To a solution of 16 (56 mg, 0.20 mmol), 3,4-dimethoxybenzoic acid (48.6 mg, 0.3 mmol) and Et3N (0.10 mL, 72 mmol) in CH2Cl2 (10 mL) at room temperature was added HBTU (42 mg, 0.22 mmol). The reaction was stirred for 12 h. The mixture was diluted with Et2O (50 mL), washed with saturated NaHCO3 (2 × 10 mL), brine (2 × 10 mL), dried (Na2SO4) and concentrated. The product was purified by chromatography on silica gel using a gradient up to 40% CMA80 in CH2Cl2 to afford 24 free base: 1H NMR (CDCl3) δ 7.43 (s, 1H), 7.30 (dd, 1H, J = 8.4, 1.8 Hz), 7.04 (t, 1H, J = 8.1 Hz), 6.84 (d, 1H, J = 8.3 Hz), 6.44–6.24 (m, 4H), 4.27–4.14 (m, 1H), 3.93–3.85 (m, 6H), 3.85–3.72 (m, 1H), 3.23–2.96 (m, 2H), 2.87–2.72 (m, 2H), 2.66–2.54 (m, 1H), 2.47–2.30 (m, 3H), 2.13–1.99 (m, 1H), 1.02–0.95 (m, 6H), 0.89 (d, 3H, J = 6.1 Hz); 13C NMR (CDCl3) δ 168.1, 157.2, 152.0, 151.1, 149.1, 130.0, 119.6, 110.7, 110.5, 108.9, 107.0, 104.0, 58.8, 57.9, 56.0, 54.3, 51.4, 50.8, 47.6, 30.9, 18.9, 18.1, 12.7, 8.7; MS (ESI) m/z 442.6 (M + H)+. The free base was converted to 31 mg (32%) of 24•2HCl as a white powder: mp 105–108 °C (fusion); [α]25D +73.° (c 0.20, CH3OH). Anal. (C25H37Cl2N3O4•1.5H2O) C, H, N.
4-(2-Methoxyphenoxy)benzoic Acid (25b) was prepared according to General Procedure C. Yield 31%. 1H NMR (CDCl3) δ 7.99 (d, 2H, J = 8.5 Hz), 7.22 (t, 1H, J = 7.8 Hz), 7.10–6.95 (m, 3H), 6.91 (d, 2H, J = 8.4 Hz), 3.80 (s, 3H).
4-(2-Hydroxyphenoxy)benzoic Acid (25c) was prepared by refluxing 4-(2-methoxyphenoxy)benzoic acid (25b) (100 mg) in 48% HBr (4 mL) and AcOH (4 mL) for 12 h. Extraction with CH2Cl2, followed by concentration from toluene afforded 25c (46%). 1H NMR (CDCl3) δ 8.03 (d, 2H, J = 8.9 Hz), 7.17–7.04 (m, 2H), 7.01 (d, 2H, J = 8.9 Hz), 6.97 (d, 1H, J = 1.5 Hz), 6.94–6.86 (m, 1H).
4-(2-Fluorophenoxy)benzoic Acid (25d) was prepared by heating a mixture of KOH (219 mg, 3.3 mmol), 2-fluorophenol (310 μL, 3.5 mmol), and 4-fluorobenzonitrile (377 mg, 3.1 mmol) in DMF (1.5 mL) to 175 °C for 20 min in a sealed tube. Ether extraction gave the intermediate diaryl ether. Refluxing in 30% KOH aq. resulted in incomplete hydrolysis of the nitrile, so the material was refluxed in 50% aq. H2SO4 (10 mL) and AcOH (5 ml). Silica chromatography (gradient 5–100% EtOAc in hexanes) gave the desired acid (25d) (405 mg, 56%). 1H NMR (CDCl3) δ 8.07 (d, 2H, J = 8.9 Hz), 7.29–7.13 (m, 4H), 6.99 (d, 2H, J = 8.8 Hz); MS (ESI) m/z 231.6 (M - H)−.
4-(3-Methylphenoxy)benzoic Acid (25e) was prepared according to General Procedure C. Yield 46%. 1H NMR (CDCl3) δ 8.06 (d, 2H, J = 8.0 Hz), 7.28 (t, 1H, J = 7.7 Hz), 7.02 (d, 1H, J = 7.7 Hz), 7.00 (d, 2H, J = 8.1 Hz), 6.90 (s, 1H), 6.88 (d, 1H, J = 8.0 Hz), 2.37 (s, 3H).
4-(3-(Trifluoromethyl)phenoxy)benzoic Acid (25f) was prepared by heating a mixture of KOH (203 mg, 3.0 mmol), 3-(trifluoromethyl)phenol (389 μL, 3.2 mmol), and 4-fluorobenzonitrile (348 mg, 2.9 mmol) in DMF (1.5 mL) to 175 °C for 20 min in a sealed tube. Ether extraction gave the intermediate diaryl ether. Refluxing in 30% KOH aq. resulted in incomplete hydrolysis of the nitrile, so the material was refluxed in 50% aq. H2SO4 (10 mL) and AcOH (5 ml). Silica chromatography (gradient 5–100% EtOAc in hexanes) gave the desired acid (25f) (190 mg, 23%). 1H NMR (CDCl3) δ 8.11 (d, 2H, J = 8.9 Hz), 7.56–7.43 (m, 2H), 7.34 (s, 1H), 7.15 (t, 1H, J = 8.6 Hz), 7.05 (d, 2H, J = 8.8 Hz); MS (ESI) m/z 281.4 (M - H)−.
4-(3-Methoxyphenoxy)benzoic Acid (25g) was prepared according to General Procedure C. Yield 12%. 1H NMR (CDCl3) δ 8.08 (d, 2H, J = 7.7 Hz), 7.29 (t, 1H, J = 8.1 Hz), 7.02 (d, 2H, J = 7.7 Hz), 6.75 (d, 1H, J = 7.4 Hz), 6.66 (d, 1H, J = 7.4 Hz), 6.64 (s, 1H).
4-(3-Hydroxyphenoxy)benzoic Acid (25h) was prepared by refluxing 4-(3-methoxyphenoxy)benzoic acid 25g (120 mg) in 48% HBr (5 mL) and AcOH (5 mL) for 12 h. Extraction with CH2Cl2, concentrated, followed by flash column chromatography on silica gel using an EtOAc gradient in hexane afforded 96 mg (79%) of 25h. 1H NMR (CD3OD) δ 8.00 (d, 2H, J = 9.0 Hz), 7.20 (t, 1H, J = 8.1 Hz), 6.99 (d, 2H, J = 8.8 Hz), 6.67–6.60 (m, 1H), 6.55–6.47 (m, 2H).
4-(3-Fluorophenoxy)benzoic Acid (25i) was prepared from 4-(3-fluorophenoxy) benzaldehyde according to General Procedure C. Yield 80%. 1H NMR (CDCl3) δ 8.08 (d, 2H, J = 8.8 Hz), 7.35 (q, 1H, J = 7.7 Hz), 7.05 (d, 2H, J = 8.7 Hz), 6.96–6.76 (m, 3H).
4-(3-Chlorophenoxy)benzoic Acid (25j) was prepared by heating a mixture of KOH (199 mg, 3.0 mmol), 3-chlorophenol (337 μL, 3.2 mmol), and 4-fluorobenzonitrile (348 mg, 2.9 mmol) in DMF (1.5 mL) to 175 °C for 20 min in a sealed tube. Ether extraction gave the intermediate crude diaryl ether, which was refluxed 12 h in 30% KOH aq. The resulting solution was extracted with EtOAc, acidified, then extracted to yield 766 mg (99%) of the desired acid (25j). 1H NMR (CDCl3) δ 8.10 (d, 2H, J = 8.7 Hz), 7.32 (t, 1H, J = 8.1 Hz), 7.21–7.16 (m, 1H), 7.08 (t, 1H, J = 2.0 Hz), 7.04 (d, 2H, J = 8.8 Hz), 7.00–6.95 (m, 1H); MS (ESI) m/z 247.3 (M - H)−.
4-(3-Bromophenoxy)benzoic Acid (25k) was prepared by heating a mixture of KOH (220 mg, 3.3 mmol), 3-bromophenol (605 mg, 3.5 mmol), and 4-fluorobenzonitrile (377 mg, 3.1 mmol) in DMF (1.5 mL) to 175 °C for 20 min in a sealed tube. Ether extraction gave the intermediate crude diaryl ether. Refluxing in 30% KOH aq. resulted in incomplete hydrolysis of the nitrile, so the material was refluxed in 50% aq. H2SO4 (10 mL) and AcOH (5 ml). Silica chromatography (gradient 5–100% EtOAc in hexanes) gave 371 mg (61%) of the desired acid 25k. 1H NMR (CDCl3) δ 8.10 (d, 2H, J = 8.7 Hz), 7.37–7.21 (m 3H), 7.07–6.99 (m, 3H); MS (ESI) m/z 291.1 (M - H)−.
4-(4-Methylphenoxy)benzoic Acid (25l) was prepared according to the general procedure of Evans et al. from 4-tolylboronic acid and 4-hydroxybenzoic acid.38 Yield 13%. 1H NMR (CDCl3) δ 8.05 (d, 2H, J = 8.8 Hz), 7.20 (d, 2H, J = 8.4 Hz), 6.98 (d, 4H, J = 8.7 Hz), 2.37 (s, 3H).
4-(4-Methoxyphenoxy)benzoic Acid (25m) was prepared according to General Procedure C. Yield 23%. 1H NMR (CDCl3) δ 8.05 (d, 2H, J = 8.3 Hz), 7.11–6.85 (m, 6H), 3.83 (s, 2H).
4-(4-Hydroxyphenoxy)benzoic Acid (25n) was prepared by refluxing 4-(4-methoxyphenoxy)benzoic acid 25m (100 mg) in 48% HBr (4 mL) and AcOH (4 mL) for 12 h. Extraction with CH2Cl2, followed by concentration from toluene afforded 25n (29%). 1H NMR (CDCl3) δ 7.99 (d, 2H, J = 9.0 Hz), 7.00–6.80 (m, 6H).
4-(2-hydroxy-3-methylphenoxy)benzoic Acid (25o) was prepared by heating a solution of 4-fluorobenzonitrile (1 mmol), 3-methylcatechol (1.1 mmol), and Cs2CO3 (1.1 mmol) in CH3CN (2 mL) to 100 °C in a sealed tube for 5 min, then again to 125 °C to an additional 5 min. The resulting mixture was concentrated, dissolved in 30% KOH aq. and refluxed. When TLC analysis indicated hydrolysis was complete, the solution was acidified with 50% H2SO4 and extracted with EtOAc. Silica gel chromatography (gradient up to 100% EtOAc in hexanes) gave 25o (50 mg, 20%). 1H NMR (CDCl3) δ 7.98 d, 2H, J = 8.8 Hz), 7.10–6.95 (m, 2H), 6.89 (d, 2H, J = 8.8 Hz), 6.82–6.75 (m, 1H), 2.08 (s, 3H); MS (ESI) m/z 243.3 (M – H)−.
4-(2-Hydroxy-5-methylphenoxy)benzoic Acid (25p) was prepared by refluxing crude 4-(5-methyl-2-methoxyphenoxy)benzoic acid (prepared from General Procedure C in 45% yield) (288 mg) in 48% HBr (10 mL for 8 h. Extraction with EtOAc, concentrated, followed by flash column chromatography on silica gel using an CH3OH gradient(1–5%) in CH2Cl2 afforded 25p (220 mg, 81%). 1H NMR (CD3OD) δ 7.97 (d, 2H, J = 8.0 Hz), 6.96–6.80 (m, 5H), 2.25 (s, 3H); MS (ESI) m/z 243.3 (M - H)−.
4-(3,5-Dimethylphenoxy)benzoic Acid (25q) was prepared by heating a mixture of KOH (189 mg, 2.9 mmol), 3,5-dimethylphenol (380 mg, 3.1 mmol), and 4-fluorobenzonitrile (333 mg, 2.7 mmol) in DMF (2mL) to 175 °C in a sealed tube for 20 min. The crude material from ether extraction was refluxed in 50% H2SO4 (10 mL) and AcOH (5 mL) for 12 h. The product was extracted with EtOAc (3 × 25 mL), washed with water then brine, dried with Na2SO4 and concentrated to yield 641 mg (96%) of 25q. 1H NMR (CDCl3) δ 8.06 (d, 2H, J = 9.0 Hz), 6.99 (d, 2H, J = 9.0 Hz), 6.84 (s, 1H), 6.70 (s, 2H), 2.32 (s, 3H).
3-Methyl-4-phenoxybenzoic Acid (26a) was prepared according to General Procedure C. Yield 30%. 1H NMR (CDCl3) δ 7.93 (d, 2H, J = 9.0 Hz), 7.44–6.74 (m, 6H), 2.35 (s, 3H).
3-Methyl-4-(3-methylphenoxy)benzoic Acid (26b) was prepared according to General Procedure C. Yield 28%. 1H NMR (CDCl3) δ 8.01 (s, 1H), 7.88 (d, 1H, J = 8.4 Hz), 7.24 (t, 1H, J = 8.1 Hz), 6.97 (d, 1H, J = 7.4 Hz), 6.86–6.77 (m, 3H), 2.35 (s, 3H), 2.34 (s, 3H).
3-Methoxy-4-(3-methylphenoxy)benzoic Acid (26c). Methyl 4-hydroxy-3-methyoxybenzoate was prepared in quantitative yield by Fischer esterification of the corresponding benzoic acid (3.04 g, 18.1 mmol) refluxed 12 h in methanol (50 mL) with catalytic dry HCl (2 mL, 2 M in ether). The phenol (546.6 mg, 3 mmol) was combined with 3-iodotoluene (654 mg, 1.0 eq) and Cu2O (515 mg, 1.2 eq) in collidine (1 mL). The mixture was heated to 200 °C for 1 h in a sealed tube. The resulting solution was extracted with ether and washed with 2 N HCl. Chromatography on silica gel (using up to 50% EtOAc in hexanes) gave 435 mg (53%) of 26c. The methyl ester was saponified with LiOH (3 eq) in dioxane–water (1:1, 9.6 mL) at rt The resulting solution was acidified with 2 N H2SO4, concentrated, filtered and washed with water to give 26c (355 mg, 46%). 1H NMR (CDCl3) δ, 3.96 (s, 3H).
3-Hydroxy-4-(3-methylphenoxy)benzoic Acid (26d) was prepared from 26c by refluxing in 48% HBr (4 mL) and AcOH (4 mL) for 4 h. Extraction with CH2Cl2, followed by concentration from toluene afforded 22d (99+%). 1H NMR (CDCl3) δ 7.77 (d, 1H, J = 2.0 Hz), 7.59 (dd 1H, J = 8.5, 2.1 Hz), 7.26 (t, 1H, J = 7.9 Hz), 6.99 (d, 1H, J = 7.3 Hz), 6.88 (s, 1H), 6.87–6.84 (m, 1H), 6.82 (d, 1H, J = 8.5 Hz), 2.35 (s, 3H).
3-Chloro-4-(3-methylphenoxy)benzoic Acid (26e) was prepared according to General Procedure C. Yield 27%. 1H NMR (CDCl3) δ 8.20 (d, 1H, J = 2.0 Hz), 7.89 (dd, 1H, J = 8.7, 2.1 Hz), 7.29 (t, 1H, J = 7.9 Hz), 7.03 (d, 1H, J = 7.5 Hz), 6.91–6.84 (m, 3H), 2.37 (s, 3H).
2-Chloro-4-(3-methylphenoxy)benzoic Acid (26f) was prepared from m-cresol (1.5 mmol), KOH (1.1 mmol) and 4-fluoro-2-methylbenzonitrile (1 mmol) in DMF (1.5 mL) heated to 175 °C for 20 min. The phenoxybenzonitrile from ether extraction was converted to the benzamide with NaOH in 50% ethylene glycol with careful addition of H2O2 (1.5 mL, 50%). The residue from EtOAc extraction was then dissolved in CH3CN (7.5 mL) to which chilled 70% sulfuric acid was added (37 mL). The flask was wrapped with foil and NaNO2 (0.6 g) was added in portions over 1 h. After 4 h, the solution was poured onto ice and filtered to yield 134 mg (55% over 3 steps) of 26f. 1H NMR (CD3OD) δ 7.93 (d, 1H, J = 8.6 Hz), 7.27 (t, 1H, J = 7.8 Hz), 7.02 (d, 1H, J = 7.7 Hz), 6.89–6.73 (m, 4H), 2.54 (s, 3H), 2.34 (s, 3H).
2-Methoxy-4-(3-methylphenoxy)benzoic Acid (26g) was prepared by heating a mixture of m-cresol (0.64 mL, 6.1 mmol), KOH (386 mg, 5.8 mmol), and 4-fluoro-2-methoxybenzonitrile (830 mg, 5.5 mmol) in DMF (2 mL) to 175 °C for 20 min. The resulting solution was extracted with ether, washed with water and dried (Na2SO4) to yield the crude intermediate phenoxybenzonitrile, which was refluxed in 30% KOH for 12 h. Following acidification and extraction with EtOAc, the residue was purified by silica gel (EtOAc gradient in hexanes) to yield 415 mg (29% over 2 steps) of 26g. 1H NMR (CDCl3) δ 8.10 (d, 1H, J = 8.8 Hz), 7.30 (t, 1H, J = 8.2 Hz), 7.05 (d, 1H, J = 7.7 Hz), 6.93–6.84 (m, 2H), 6.66 (d, 1H, J = 2.1 Hz), 6.60 (dd, 1H, J =8.7, 2.2 Hz), 4.00 (s, 3H), 2.37 (s, 3H).
2-Hydroxy-4-(3-methylphenoxy)benzoic Acid (26h) was prepared from acid 26g (177 mg, 0.7 mmol) in CH2Cl2 (10 mL) treated with BBr3 (3.5 mL, 1 M in CH2Cl2) at −20 °C, warming to room temperature overnight. The reaction was quenched with and concentrated from methanol to yield 175 mg (99%) of 26h, used in the next reaction without further purification. 1H NMR (CDCl3) δ 10.51 (bs, 1H), 7.86 (d, 1H, J = 8.9 Hz), 7.28 (t, 1H, J = 7.7 Hz), 7.04 (d, 1H, J = 7.5 Hz), 6.93–6.86 (m, 2H), 6.55 (dd, 1H, J = 8.9, 2.2 Hz), 6.44 (d, 1H, J = 2.3 Hz), 2.37 (s, 3H).
4-(2-Hydroxyphenoxy)-3-methoxybenzoic Acid (26i) was prepared according to General Procedure D in 24% yield following silica gel chromatography (methanol–CH2Cl2 gradient). 1H NMR (CDCl3) δ 7.69 (s, 1H), 7.62 (d, 1H, J = 8.4 Hz), 7.12–6.70 (m, 6H), 3.97 (s, 3H).
3-Chloro-4-(2-hydroxyphenoxy)benzoic Acid (26j) was prepared according to General Procedure D in 20% yield following a silica preparatory plate (5% isopropanol in CH2Cl2). 1H NMR (CDCl3) δ 8.22 (d, 1H, J = 2.1 Hz), 7.92 (dd, 1H, J = 8.5, 2.0 Hz), 7.21–7.05 (m, 2H), 6.98–6.77 (m, 4H).
4-(2-Hydroxy-5-methylphenoxy)-3-methoxybenzoic acid (26k) was prepared from phenol 29 and the appropriate 4-fluorobenzalde using General Procedure D. Silica chromatography (gradient up to 40% EtOAc–hexanes) gave the intermediate aldehyde (481 mg, 47%). The oxidation was followed by MOM cleavage with conc. HCl (0.25 mL) in 50% THF–iPrOH (10 mL) to yield 391 mg (89% over two steps) of 26k. 1H NMR (CDCl3) δ 7.73–7.66 (m, 2H), 6.99–6.85 (m, 3H), 6.76 (s, 1H), 3.97 (s, 3H), 2.24 (s, 3H).
3-Chloro-4-(2-hydroxy-5-methylphenoxy)benzoic Acid (26l) was prepared according to General Procedure D from 2-methoxy-5-methylphenol and 3-chloro-4-fluorobenzaldehyde. The intermediate 3-chloro-4-(2-methoxy-5-methylphenoxy)benzoic acid product was treated with excess BBr3 (1 M in CH2Cl2) at room temperature overnight. The resulting solution was quenched with and concentrated from methanol. The resulting oil was subjected to a silica preparatory plate (5% isopropanol in CH2Cl2) to yield 162 mg (58% over 3 steps) of 26l. 1H NMR (CDCl3) δ 8.21 (s, 1H), 7.92 (d, 1H, J = 8.0 Hz), 7.01–6.89 (m, 3H), 6.76 (s, 1H), 2.26 (s, 3H).
2-[2-(Methoxymethoxy)-5-methylphenyl]-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (28)
The bromination of p-cresol, carried out via the method of Narender, et al.39 was followed by MOM protection40 (96% yield over two steps). A solution of bis(pinacolato)diborane (2.5 g, 9.8 mmol), KOAc (2.5 g, 25 mmol), and the aryl bromide (1.5 g, 6.5 mmol) in dioxane (40 mL) was purged with nitrogen before Pd(dppf)Cl2 (0.47 g, 0.56 mmol) was added. The mixture was refluxed overnight. Silica chromatography (gradient to 20% EtOAc–hexanes) gave the boronate ester (1.93 g). 1H NMR (CDCl3) δ 7.50 (d, 1H, J = 2.1 Hz), 7.17 (dd, 1H, J = 8.4, 2.1 Hz), 6.92 (d, 1H, J = 8.4 Hz), 5.15, (s, 2H), 3.51 (s, 3H), 2.28 (s, 3H), 1.35 (s, 12H).
2-(Methoxymethoxy)-5-methylphenol (29)
A solution of 28 (1.93 g) was dissolved in acetone (20 mL) and treated with a solution of oxone (4 g, 26 mmol) in H2O (20 mL). After 10 min, NaHSO3 was added and the resulting solution was extracted with EtOAc. The concentrated residue was subjected to chromatography on silica gel using a gradient up to 15% EtOAc in hexanes to afford 578 mg (53% over two steps) of 29. 1H NMR (CDCl3) δ 6.96 (d, 1H, J = 8.2 Hz), 6.77 (d, 1H, J = 2.0 Hz), 6.62 (dd, 1H, J = 8.2, 2.0 Hz), 5.92 (s, 1H), 5.16 (s, 2H), 3.52 (3, 3H), 2.26 (s, 3H).
2-Methyl-5-phenoxypyridine N-Oxide (31)
KOH (605 mg, 9.20 mmol) and 5-hydroxy-2-methylpyridine (1.00 g, 9.20 mmol) were dissolved in EtOH then concentrated to dryness. DMF (20 mL), copper dust (640 mg, 10.0 mmol), and iodobenzene (0.95 mL, 8.50 mmol) were added. The heterogeneous reaction was refluxed for 48 h then filtered through Celite and concentrated. Flash column chromatography of the crude product on silica gel using an EtOAc gradient in hexane afforded 0.94 g (55%) of 5-phenoxy-2-picoline (30) as an oil. To a solution of 5-phenoxy-2-picoline in CH2Cl2 (70 mL) at room temperature was slowly added mCPBA (1.15 g) in portions over 1 h. Sodium metabisulfite was added to quench excess oxidant. The resulting suspension was filtered, treated with K2CO3, filtered again, then concentrated to afford 390 mg (31%) of crude 2-methyl-5-phenoxypyridine N-oxide (31), which was used in the next step without purification. 1H NMR (CDCl3) δ 8.05 (d, 1H, J = 2.3 Hz), 7.43–7.35 (m, 2H), 7.24–7.19 (m, 1H), 7.17 (d, 1H, J = 8.2 Hz), 7.07–7.02 (m, 2H), 6.89 (dd, 1H, J = 8.7, 2.2 Hz), 2.48 (s, 3H).
(5-Phenoxypyridin-2-yl)methanol (32)
A solution of 31 (390 mg, 1.90 mmol) in acetic anhydride (2.5 mL) and AcOH (1 mL) was heated to 150 °C for 5 min in a sealed tube. The resulting solution was concentrated then diluted with H2O (5 mL) and CH3OH (5 mL). K2CO3 (4.35 g) was added to adjust the solution to pH 8.5 and the mixture was extracted with EtOAc (3 × 25 mL). The combined EtOAc extracts were dried (Na2SO4) and concentrated to afford 296 mg (75% over two steps) of 32, which was used in the next step without purification. 1H NMR (CDCl3) δ 8.35 (d, 1H, J = 1.9 Hz), 7.41–7.30 (m, 3H), 7.24 (d, 1H, J = 8.5 Hz), 7.16 (t, 1H, J = 7.4 Hz), 7.02 (d, 2H, J = 7.7 Hz), 4.75 (s, 2H).
5-Phenoxypyridine-2-carboxylic Acid (33)
Potassium permanganate (715 mg, 4.50 mmol) was added portion-wise over 2 h to a solution of 32 (296 mg, 1.50 mmol) in acetone (10 mL), keeping the temperature at 40–50 °C. The resulting black suspension was filtered and retentate washed with 0.1 N aqueous NaOH. The resulting aqueous solution was corrected to pH 4 with 2 N aqueous HCl and extracted with CHCl3 (3 × 25 mL). The combined CHCl3 extracts were dried (Na2SO4) and concentrated to afford 182 mg (57%) of 33, which was used in the next step without purification. 1H NMR (CDCl3) δ 8.38 (d, 1H, J = 2.5 Hz), 8.17 (d, 1H, J = 8.6 Hz), 7.50–7.24 (m, 4H), 7.11 (d, 2H, J = 7.6 Hz).
6-Phenoxynicotinic Acid (35)
The methyl ester of 6-chloronicotinic acid was prepared from 34 (0.78 g, 5 mmol) in toluene (50 mL) and methanol (10 mL) with dropwise addition of a solution of trimethylsilyldiazomethane (2.75 mL, 2.0 M in ether). After 30 min, acetic acid (0.5 mL) was added, and the solution was concentrated to dryness to yield crude 6-chloronicotinic acid methyl ester. Acetonitrile (50 mL), phenol (1.1 g, 11 mmol), Cs2CO3 (0.98 g), and K2CO3 (0.74 g) were added, and the mixture was refluxed overnight. The reaction showed incomplete conversion, so the solvent was replaced with DMF (10 mL), K2CO3 (0.91 g) and phenol (0.64 g) were added, and the mixture was refluxed 1 h. The mixture was poured into ice and extracted with ether (3 × 25 mL). The ether layer was washed with bicarbonate and brine, dried (Na2SO4), and concentrated. The crude 6-phenoxynicotinic acid methyl ester was combined with LiOH (0.49 g), methanol (15 mL), and water (5 mL) and stirred overnight. Following adjustment to pH 5 with 1 M NaHSO4, extraction with EtOAc and concentration gave 312 mg (29% over three steps) of 35 as a white powder. 1H NMR (CD3OD) δ 8.72 (d, 1H, J = 2.2 Hz), 8.34 (dd, 1H, J = 8.7, 2.4 Hz), 7.48–7.41 (m, 2H), 7.27 (t, 1H, J = 7.5 Hz), 7.15 (d, 2H, J = 8.4 Hz), 7.00 (d, 1H, J = 8.7 Hz).
Synthesis of Library Compounds
Amine 16 (0.02 mmol × the number of derivatives being prepared) was dissolved in anhydrous THF (0.5 mL × the number of derivatives being prepared). HBTU (0.024 mmol × the number of derivatives being prepared) was dissolved in anhydrous CH3CN (1 mL × the number of derivatives being prepared). To the prelabeled 20-mL scintillation vial containing a stir bar was added one of the chosen carboxylic acid (0.022 mmol) solutions in CH3CN (0.5 mL). To this was added the appropriate fraction of amine solution (0.5 mL) followed by Et3N (0.006 mL, 0.04 mmol) and the appropriate fraction of HBTU solution (1 mL). The vial was then capped with a Teflon-lined lid and stirred at room temperature. After TLC analysis indicated no amine left, Et2O (4 mL) and H2O (2 mL) were added to the vial. After the vial was shaken and the layers were allowed to settle, the aqueous layer was withdrawn with a pipet. Next, saturated NaHCO3 (2 × 2 mL) was added and the procedure repeated. This was followed by similar wash with brine (2 × 2 mL). Na2SO4 was added to the vial, and after drying, the mixture was pipeted into a preweighed, prelabeled 20-mL scintillation vial via a 6 in Pasteur pipet containing a small cotton plug. Following this, Et2O (2 mL) was added to the drying agent, and the vial was shaken, after which the Et2O rinse was filtered as above. The collecting vials were placed under a nitrogen outlet and allowed to evaporate. Once the solvent was removed, the vials were placed in a high vacuum desiccator and allowed to remain overnight. The vials were reweighed, and the crude yield was determined by difference. The products were used without further purification.
Docking Studies and Calculations
A. Ligand Preparation
Models of each ligand were constructed using the Tripos SYBYL41 fragment library. Nitrogen atoms predicted to be protonated at pH 7.4 (using the ChemAxon Marvin pKa calculator)42 were modified to reflect the corresponding atom type and valency. The structural models were then energy-minimized using the MMFF94 force field, Gasteiger-Hückel charges, and SYBYL default minimization conditions.
B. Receptor Preparation
Chain A of the X-ray crystallographic structure of the kappa-opioid receptor co-crystallized with JDTic30 (PDB: 4DJH) was extracted for the docking calculations. The receptor model was prepared by adding hydrogen atoms and assigning Gasteiger-Hückel charges. The JDTic molecule and nearby waters within the receptor binding pocket were left in place but all other non-peptide molecules were removed from the model.
C. Docking Calculations
Docking calculations were performed using the SYBYL Surflex43 flexible molecular docking module. A Surflex “protomol” was generated using the ligand-based method based on the observed binding pose of JDTic, a “threshold” value of 0.5, and “bloat” value of 1. Docking poses for 10a and 11e were then calculated using the JDTic-based protomol, the Surflex-Dock molecular fragmentation method combined with full side-chain flexibility, the Surflex-GEOM parameter, and 100 starting conformations per molecule. The CScore consensus scoring method was used to select a maximum of 20 optimal docking poses. The docking pose with the highest CScore value was selected for comparison to the observed binding pose of JDTic.
Supplementary Material
Acknowledgments
This research was supported by the National Institute on Drug Abuse (Grant DA09045). The authors thank Dr. Ann Decker, Ms. Tiffany Langston, and Mr. Keith Warner for conducting the [35S]GTPγS studies.
ABBREVIATIONS
- GPCRs
G-protein-coupled receptors
- cDNAs
complementary deoxyribonucleic acid
- SAR
structure-activity relationship
- [35S]GTPγS
sulfur-35 guanosine-5′-O-(3-thio)triphosphate
- DAMGO
[D-Ala2,MePhe4,Gly-ol5]enkephalin
- DPDPE
[D-Pen2,D-Pen5]enkephalin
- U69
593, (5α,7α,8β)-(−)-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro[4,5]dec-8-yl]benzeneacetamide
- CHO
Chinese hamster ovary
- GDP
guanosine diphosphate
- BOP
(benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate
- HBTU
O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- HMPA
hexamethylphosphoramide
- LDA
lithium diisopropylamide
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Any additional relevant notes should be placed here.
The authors declare no competing financial interest.
Supporting Information. Elemental analysis data. Table S1 listing the library of compounds synthesized and their inhibition of [35S]GTPγS binding at the κ opioid receptor. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Dhawan BN, Cesselin F, Raghubir R, Reisine T, Bradley PB, Portoghese PS, Hamon M. International Union of Pharmacology. XII. Classification of opioid receptors. Pharmacol Rev. 1996;48:567–592. [PubMed] [Google Scholar]
- 2.Aldrich JV, Vigil-Cruz SC. Narcotic Analgesics. In: Abraham DJ, editor. Burger’s Medicinal Chemistry and Drug Discovery. 6. Vol. 6. John Wiley & Sons; New York, NY: 2003. pp. 329–481. Chapter 7. [Google Scholar]
- 3.Kreek MJ, LaForge KS, Butelman E. Pharmacotherapy of addictions. Nat Rev Drug Discov. 2002;1:710–726. doi: 10.1038/nrd897. [DOI] [PubMed] [Google Scholar]
- 4.Zimmerman DM, Nickander R, Horng JS, Wong DT. New structural concepts for narcotic antagonists defined in a 4-phenylpiperidine series. Nature. 1978;275:332–334. doi: 10.1038/275332a0. [DOI] [PubMed] [Google Scholar]
- 5.Thomas JB, Mascarella SW, Rothman RB, Partilla JS, Xu H, McCullough KB, Dersch CM, Cantrell BE, Zimmerman DM, Carroll FI. Investigation of the N-substituent conformation governing potency and mu receptor subtype-selectivity in (+)-(3R, 4R)-dimethyl-4-(3-hydroxyphenyl)piperidine opioid antagonists. J Med Chem. 1998;41:1980–1990. doi: 10.1021/jm980063g. [DOI] [PubMed] [Google Scholar]
- 6.Zimmerman DM, Leander JD, Cantrell BE, Reel JK, Snoddy J, Mendelsohn LG, Johnson BG, Mitch CH. Structure-activity relationships of the trans-3,4-dimethyl-4-(3-hydroxyphenyl)piperidine antagonists for μ and κ opioid receptors. J Med Chem. 1993;36:2833–2841. doi: 10.1021/jm00072a001. [DOI] [PubMed] [Google Scholar]
- 7.Mitch CH, Leander JD, Mendelsohn LG, Shaw WN, Wong DT, Cantrell BE, Johnson BG, Reel JK, Snoddy JD, Takemori AE, Zimmerman DM. 3,4-Dimethyl-4-(3-hydroxyphenyl)piperidines: Opioid antagonists with potent anorectant activity. J Med Chem. 1993;36:2842–2850. doi: 10.1021/jm00072a002. [DOI] [PubMed] [Google Scholar]
- 8.Zimmerman DM, Gidda JS, Cantrell BE, Schoepp DD, Johnson BG, Leander JD. Discovery of a potent, peripherally selective trans-3,4-dimethyl-4-(3-hydroxyphenyl)piperidine opioid antagonist for the treatment of gastrointestinal motility disorders. J Med Chem. 1994;37:2262–2265. doi: 10.1021/jm00041a003. [DOI] [PubMed] [Google Scholar]
- 9.Delaney CP, Yasothan U, Kirkpatrick P. Alvimopan. Nat Rev Drug Discov. 2008;7:727–728. doi: 10.1038/nrd2668. [DOI] [PubMed] [Google Scholar]
- 10.Statnick MA, Suter TM, Gackenheimer SL, Emmerson PJ, Quimby SJ, Gehlert DR, Wheeler WJ, Mitch CH. Na+-dependent high affinity binding of [3H]LY515300, a 3,4-dimethyl-4-(3-hydroxyphenyl)piperidine opioid receptor inverse agonist. Eur J Pharmacol. 2003;482:139–150. doi: 10.1016/j.ejphar.2003.09.043. [DOI] [PubMed] [Google Scholar]
- 11.Carroll FI, Thomas JB, Dykstra LA, Granger AL, Allen RM, Howard JL, Pollard GT, Aceto MD, Harris LS. Pharmacological properties of JDTic: A novel κ-opioid receptor antagonist. Eur J Pharmacol. 2004;501:111–119. doi: 10.1016/j.ejphar.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 12.Thomas JB, Atkinson RN, Vinson NA, Catanzaro JL, Perretta CL, Fix SE, Mascarella SW, Rothman RB, Xu H, Dersch CM, Cantrell BE, Zimmerman DM, Carroll FI. Identification of (3R)-7-hydroxy-N-((1S)-1-[[(3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethyl-1-piperidinyl]methyl]-2-methylpropyl)-1,2,3,4-tetrahydro-3-isoquinolinecarboxamide as a novel potent and selective opioid kappa receptor antagonist. J Med Chem. 2003;46:3127–3137. doi: 10.1021/jm030094y. [DOI] [PubMed] [Google Scholar]
- 13.Thomas JB, Atkinson RN, Rothman RB, Fix SE, Mascarella SW, Vinson NA, Xu H, Dersch CM, Lu Y, Cantrell BE, Zimmerman DM, Carroll FI. Identification of the first trans-(3R,4R)-dimethyl-4-(3-hydroxyphenyl)piperidine derivative to possess highly potent and selective opioid kappa receptor antagonist activity. J Med Chem. 2001;44:2687–2690. doi: 10.1021/jm015521r. [DOI] [PubMed] [Google Scholar]
- 14.Thomas JB, Fall MJ, Cooper JB, Rothman RB, Mascarella SW, Xu H, Partilla JS, Dersch CM, McCullough KB, Cantrell BE, Zimmerman DM, Carroll FI. Identification of an opioid κ receptor subtype-selective N-substituent for (+)-(3R,4R)-dimethyl-4-(3-hydroxyphenyl)piperidine. J Med Chem. 1998;41:5188–5197. doi: 10.1021/jm980511k. [DOI] [PubMed] [Google Scholar]
- 15.Beardsley PM, Howard JL, Shelton KL, Carroll FI. Differential effects of the novel kappa opioid receptor antagonist, JDTic, on reinstatement of cocaine-seeking induced by footshock stressors vs cocaine primes and its antidepressant-like effects in rats. Psychopharmacology (Berl) 2005;183:118–126. doi: 10.1007/s00213-005-0167-4. [DOI] [PubMed] [Google Scholar]
- 16.Knoll AT, Meloni EG, Thomas JB, Carroll FI, Carlezon WA., Jr Anxiolytic-Like Effects of κ-Opioid Receptor Antagonists in Models of Unlearned and Learned Fear in Rats. J Pharmacol Exp Ther. 2007;323:838–845. doi: 10.1124/jpet.107.127415. [DOI] [PubMed] [Google Scholar]
- 17.Brugel TA, Smith RW, Balestra M, Becker C, Daniels T, Hoerter TN, Koether GM, Throner SR, Panko LM, Folmer JJ, Cacciola J, Hunter AM, Liu R, Edwards PD, Brown DG, Gordon J, Ledonne NC, Pietras M, Schroeder P, Sygowski LA, Hirata LT, Zacco A, Peters MF. Discovery of 8-azabicyclo[3.2.1]octan-3-yloxy-benzamides as selective antagonists of the kappa opioid receptor. Part 1. Bioorg Med Chem Lett. 2010;20:5847–5852. doi: 10.1016/j.bmcl.2010.07.113. [DOI] [PubMed] [Google Scholar]
- 18.Peters MF, Zacco A, Gordon J, Maciag CM, Litwin LC, Thompson C, Schroeder P, Sygowski LA, Piser TM, Brugel TA. Identification of short-acting kappa-opioid receptor antagonists with anxiolytic-like activity. Eur J Pharmacol. 2011;661:27–34. doi: 10.1016/j.ejphar.2011.04.017. [DOI] [PubMed] [Google Scholar]
- 19.Verhoest PR, Sawant Basak A, Parikh V, Hayward M, Kauffman GW, Paradis V, McHardy SF, McLean S, Grimwood S, Schmidt AW, Vanase-Frawley M, Freeman J, Van Deusen J, Cox L, Wong D, Liras S. Design and Discovery of a Selective Small Molecule kappa Opioid Antagonist (2-Methyl-N-((2′-(pyrrolidin-1-ylsulfonyl)biphenyl-4-yl)methyl)propan-1-amine, PF-4455242) J Med Chem. 2011;54:5868–5877. doi: 10.1021/jm2006035. [DOI] [PubMed] [Google Scholar]
- 20.Buezo ND, Pedregal-Tercero C, McKinzie DL, Mitch CH Eli Lilly and Company. 7,709,522. Kappa selective opioid receptor antagonist. 2010:B2.
- 21.Mitch CH, Quimby SJ, Diaz N, Pedregal C, de la Torre MG, Jimenez A, Shi Q, Canada EJ, Kahl SD, Statnick MA, McKinzie DL, Benesh DR, Rash KS, Barth VN. Discovery of aminobenzyloxyarylamides as kappa opioid receptor selective antagonists: application to preclinical development of a kappa opioid receptor antagonist receptor occupancy tracer. J Med Chem. 2011;54:8000–8012. doi: 10.1021/jm200789r. [DOI] [PubMed] [Google Scholar]
- 22.Carroll FI, Carlezon J, William A. Development of Kappa Opioid Receptor Antagonists. J Med Chem. 2013;56:2178–2195. doi: 10.1021/jm301783x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mague SD, Pliakas AM, Todtenkopf MS, Tomasiewicz HC, Zhang Y, Stevens WC, Jr, Jones RM, Portoghese PS, Carlezon WA., Jr Antidepressant-like effects of kappa-opioid receptor antagonists in the forced swim test in rats. J Pharmacol Exp Ther. 2003;305:323–330. doi: 10.1124/jpet.102.046433. [DOI] [PubMed] [Google Scholar]
- 24.McLaughlin JP, Marton-Popovici M, Chavkin C. Kappa opioid receptor antagonism and prodynorphin gene disruption block stress-induced behavioral responses. J Neurosci. 2003;23:5674–5683. doi: 10.1523/JNEUROSCI.23-13-05674.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Redila VA, Chavkin C. Stress-induced reinstatement of cocaine seeking is mediated by the kappa opioid system. Psychopharmacology (Berl) 2008;200:59–70. doi: 10.1007/s00213-008-1122-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carey AN, Borozny K, Aldrich JV, McLaughlin JP. Reinstatement of cocaine place-conditioning prevented by the peptide kappa-opioid receptor antagonist arodyn. Eur J Pharmacol. 2007;569:84–89. doi: 10.1016/j.ejphar.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walker BM, Koob GF. Pharmacological evidence for a motivational role of κ-opioid systems in ethanol dependence. Neuropsychopharmacology. 2007;33:643–652. doi: 10.1038/sj.npp.1301438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bodnar RJ, Glass MJ, Ragnauth A, Cooper ML. General, mu and kappa opioid antagonists in the nucleus accumbens alter food intake under deprivation, glucoprivic and palatable conditions. Brain Res. 1995;700:205–212. doi: 10.1016/0006-8993(95)00957-r. [DOI] [PubMed] [Google Scholar]
- 29.Bortolato M, Aru GN, Frau R, Orru M, Fa M, Manunta M, Puddu M, Mereu G, Gessa GL. Kappa opioid receptor activation disrupts prepulse inhibition of the acoustic startle in rats. Biol Psychiatry. 2005;57:1550–1558. doi: 10.1016/j.biopsych.2005.02.030. [DOI] [PubMed] [Google Scholar]
- 30.Wu H, Wacker D, Mileni M, Katritch V, Han GW, Vardy E, Liu W, Thompson AA, Huang XP, Carroll FI, Mascarella SW, Westkaemper RB, Mosier PD, Roth BL, Cherezov V, Stevens RC. Structure of the human kappa-opioid receptor in complex with JDTic. Nature. 2012;485:327–332. doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carroll FI, Cueva JP, Thomas JB, Mascarella SW, Runyon SP, Navarro HA. 1-Substituted 4-(3-hydroxyphenyl)piperazines are pure opioid receptor antagonists. ACS Med Chem Lett. 2010;1:365–369. doi: 10.1021/ml100126b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clark DE. Rapid calculation of polar molecular surface area and its application to the prediction of transport phenomena. 2. Prediction of blood-brain barrier penetration. J Pharm Sci. 1999;88:815–821. doi: 10.1021/js980402t. [DOI] [PubMed] [Google Scholar]
- 33.Clark DE, Pickett SD. Computational methods for the prediction of ‘drug-likeness’. Drug Discov Today. 2000;5:49–58. doi: 10.1016/s1359-6446(99)01451-8. [DOI] [PubMed] [Google Scholar]
- 34.Nakamura E, Aoki S, Sekiya K, Oshino H, Kuwajima I. Carbon-carbon bond-forming reactions of zinc homoenolate of esters. A novel three-carbon nucleophile with general synthetic utility. J Am Chem Soc. 1987;109:8056–8066. [Google Scholar]
- 35.Butler DE, Poschel BP, Marriott JG. Cognition-activating properties of 3-(Aryloxy)pyridines. J Med Chem. 1981;24:346–350. doi: 10.1021/jm00135a020. [DOI] [PubMed] [Google Scholar]
- 36.Faivre V, Roizard D, Brembilla A, Lochon P. Synthesis of long-chain picolinic derivatives. Bull Soc Chim Fr. 1991;128:278–285. [Google Scholar]
- 37.Dalcanale E, Montanari F. Selective oxidation of aldehydes to carboxylic acids with sodium chlorite-hydrogen peroxide. J Org Chem. 1986;51:567–569. [Google Scholar]
- 38.Evans DA, Katz JL, West TR. Synthesis of Diaryl Ethers through the Copper-Promoted Arylation of Phenols with Arylboronic Acids. An Expedient Synthesis of Thyroxine. Tetrahedron Lett. 1998;39:2937–2940. [Google Scholar]
- 39.Narender N, Srinivasu P, Prasad M, Kulkarni S, Raghavan K. An efficient and regioselective oxybromination of aromatic compounds using potassium bromide and oxone. Synth Commun. 2002;32:2313–2318. [Google Scholar]
- 40.Maekawa T, Yamamoto S, Igata Y, Ikeda S, Watanabe T, Shiraishi M. Synthesis and biological activity of novel 2-(alpha-alkoxyimino)benzylpyridine derivatives as K+ channel openers. Chem Pharm Bull. 1997;45:1994–2004. doi: 10.1248/cpb.45.1994. [DOI] [PubMed] [Google Scholar]
- 41.SYBYL-X 2.0. Tripos International; 1699 South Hanley Rd., St. Louis, Missouri, 63144, USA: 2012. [Google Scholar]
- 42.Marvin 5.11.5, ChemAxon. 2013 ( http://www.chemaxon.com)
- 43.Jain AN. Surflex-Dock 2.1: robust performance from ligand energetic modeling, ring flexibility, and knowledge-based search. J Comput Aided Mater Des. 2007;21:281–306. doi: 10.1007/s10822-007-9114-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.